

Abstract
At early disease stages, Lewy body disorders are characterized by limbic vs. brainstem α‐synucleinopathy, but most preclinical studies have focused solely on the nigrostriatal pathway. Furthermore, male gender and advanced age are two major risk factors for this family of conditions, but their influence on the topographical extents of α‐synucleinopathy and the degree of cell loss are uncertain. To fill these gaps, we infused α‐synuclein fibrils in the olfactory bulb/anterior olfactory nucleus complex—one of the earliest and most frequently affected brain regions in Lewy body disorders—in 3‐month‐old female and male mice and in 11‐month‐old male mice. After 6 months, we observed that α‐synucleinopathy did not expand significantly beyond the limbic connectome in the 9‐month‐old male and female mice or in the 17‐month‐old male mice. However, the 9‐month‐old male mice had developed greater α‐synucleinopathy, smell impairment and cell loss than age‐matched females. By 10.5 months post‐infusion, fibril treatment hastened mortality in the 21.5‐month‐old males, but the inclusions remained centered in the limbic system in the survivors. Although fibril infusions reduced the number of cells expressing tyrosine hydroxylase in the substantia nigra of young males at 6 months post‐infusion, this was not attributable to true cell death. Furthermore, mesencephalic α‐synucleinopathy, if present, was centered in mesolimbic circuits (ventral tegmental area/accumbens) rather than within strict boundaries of the nigral pars compacta, which were defined here by tyrosine hydroxylase immunolabel. Nonprimate models cannot be expected to faithfully recapitulate human Lewy body disorders, but our murine model seems reasonably suited to (i) capture some aspects of Stage IIb of Lewy body disorders, which displays a heavier limbic than brainstem component compared to incipient Parkinson’s disease; and (ii) leverage sex differences and the acceleration of mortality following induction of olfactory α‐synucleinopathy.
Keywords: Lewy body, Lewy body disorder, limbic, Parkinson’s disease, olfactory, synuclein
Introduction
Lewy body disorders are a family of neurodegenerative conditions linked by the deposition of insoluble α‐synuclein and other proteins in hallmark inclusions 12, 56. Parkinson’s disease (PD) is the most extensively studied Lewy body disorder and is characterized by motor deficits due in large measure to retrograde degeneration of the nigrostriatal pathway 33, 42, 132. However, cells in many other brain regions also exhibit degeneration, including those of the anterior olfactory nucleus (AON), amygdala, nucleus basalis, locus coeruleus, pedunculopontine nucleus, dorsal raphe nucleus and ventral tegmental area, culminating in a series of nonmotor deficits 35, 65, 71, 72, 73, 76, 82, 87, 120, 134, 173. For example, PD patients may develop Parkinson’s disease dementia (PDD) years after the appearance of motor deficits, perhaps because of telencephalic pathology 1, 21, 27, 29, 88. However, if cognitive deficits appear before or within 1 year of motor impairments, patients are diagnosed with dementia with Lewy bodies (DLB), a condition distinct from PDD 110, 111, 118. DLB is the second most common dementia in the elderly and has been associated with Lewy pathology in the cranial nerve nuclei IX and X of the medulla, the pontine locus coeruleus, the ventral mesencephalon, neocortical regions of the telencephalon, and, in particular, the limbic system (eg, amygdala, transentorhinal cortex, cingulate gyrus) 49, 50, 75, 108, 118. Alzheimer’s disease with Lewy bodies (ADLB) is another subtype of this family of conditions, in which Lewy pathology is also particularly dense in the limbic system 16, 100, 164.
Most of the animal and cellular models of Lewy body disorders are focused on dopaminergic neurodegeneration, and limbic pathology is not typically addressed, although Hubbard and colleagues highlighted the limbic system in their study of olfactory Lewy pathology in dementia victims over a decade ago 84. Beach’s unified staging system for Lewy body disorders identified two distinct limbic stages in a cohort of 417 subjects: Stage IIb (limbic predominant) and Stage III (brainstem and limbic) 16. According to Beach et al., Stage IIb does not encompass PD patients, but is most closely associated with ADLB, DLB and incidental Lewy body disease (iLBD) Type 2. Toledo and colleagues employed a data‐driven and unsupervised clustering approach to classify Lewy pathology in the brains of patients from two independent autopsy cohorts and stated that demented subjects with AD and coincident α‐synucleinopathy display pathology that seems to “start in the olfactory bulb (OB), progressing to the amygdala and limbic system, with overall sparing of the brainstem” (p. 403, see 164). In this latter category, classified by the authors as “Dem‐ADLB,” limbic‐predominant α‐synucleinopathy was accompanied by preservation of the nigrostriatal pathway and sparing of the brainstem and extracranial sites (eg, enteric nervous system and gastrointestinal‐associated salivary glands). These collective findings support the separate categorization of subjects with limbic‐predominant Lewy pathology and highlight the importance of developing animal models that capture at least some of the key histological distinctions in this heterogeneous family of conditions.
Some of the earliest α‐synuclein+ inclusions in Lewy body disorders arise in the olfactory bulb (OB) 10, 16, 18, 26, 28, 77, 78. Indeed, among subjects with loss of pigmentation of the substantia nigra, all reportedly display Lewy bodies in the OB 156. Beach and colleagues stated that α‐synucleinopathy in the OB is a diagnostic predictor of PD or DLB with >90% specificity and sensitivity, and that Lewy inclusions are present in the OB of 95% of patients who died with PD, 97% of cases diagnosed with DLB, 67% of iLBD cases and only 7% of healthy elderly controls 16, 18. It is important to note that the AON displays even denser Lewy pathology than the OB but is often classified as part of the OB because it bulges rostrally into the latter structure 134, 165. Thus, the OB/AON complex is a major sentinel of impending Lewy body disorders and was often highlighted in the original Braak staging system for PD 25, 26, 28, 45, 46. For these reasons, we chose to infuse the murine OB/AON with preformed α‐synuclein fibrils in our previous work 109. In that study, dense α‐synucleinopathy emerged in the limbic connectome and overlapped with the pattern of retrograde label following infusions of the tract‐tracer FluoroGold at the same stereotaxic coordinates. Although we reported little to no Lewy‐like pathology in the brainstem, the survival period in that study was limited to 3 months to emphasize early disease processes and potential “first‐order” passage of pathology 109. Therefore, a major goal of the present study was to increase the survival period in the murine OB/AON fibril infusion model and determine if longer incubation or survival periods shift the center of gravity of Lewy‐related pathology from limbic‐predominant to other phenotypes that represent more advanced disease stages. Parkkinen’s postmortem examination of 1720 brains revealed that 55% of subjects with widespread α‐synucleinopathy at autopsy did not display overt signs of dementia or extrapyramidal symptoms before death 130. Furthermore, Jellinger argued that the “detection and staging of Lewy pathology without assessment of neuronal loss in specific areas may not have clinical impact and its predictive validity is questionable” 91. Therefore, aside from performing counts of α‐synucleinopathic inclusions, we measured both NeuN+ neuron numbers and Hoechst+ cell numbers (neurons plus glia) in 16 key brain regions following infusions of preformed α‐synuclein inclusions in the OB/AON.
In her dissertation, Parkkinen concluded that the prevalence of α‐synuclein+ inclusions and the “α‐synuclein index” are not influenced by gender (see Table 5, page 43) 129. However, men are roughly twice as likely to develop Lewy body disorders as women 14, 153. For example, men comprised ~61%–69% of all subjects with PD, DLB and iLBD in the Beach et al. study 16. Of the subjects beginning to show cognitive impairments, the best predictor of cognitive decline in PD is the male sex 39. After accounting for age, smoking history and education, Nelson and colleagues reported that male subjects are threefold more likely to develop the neocortical or diffuse subtype of DLB than female subjects and twice as likely to develop the intermediate/limbic subtype of DLB, whereas both genders are equally likely to develop the brainstem‐predominant subtype 121. Previous preclinical reports demonstrate that female rodents are more resistant than males to motor deficits and loss of nigrostriatal cells in response to dopaminergic neurotoxicants 52, 117, 119. In addition, estrogen, raloxifene and the estrogen receptor‐α agonist propyl‐pyrazole‐triol have been shown to protect dopaminergic neurons against MPTP toxicity in vivo 102. However, dopaminergic neurotoxicant studies do not recapitulate the early stages of Lewy body disorders and are not typically characterized by α‐synucleinopathic lesions 67. Therefore, these studies cannot inform us about potential gender dimorphisms in the transmission of α‐synucleinopathy as an explanation for the discrepancies in disease risk between the sexes. α‐synuclein transgenic female mice are known to outperform males on sensorimotor tests 7, 51, 66 and display changes in the expression of greater numbers of genes than male mice 171. However, sex‐dependent differences in α‐synuclein histopathology in the brain remain poorly understood, partly because females express approximately half as much wild‐type human α‐synuclein mRNA in the substantia nigra as male mice when α‐synuclein expression is driven by the Thy1 promoter 66. Thus, a second goal of the present study was to test the impact of gender on the regional distribution and density of α‐synuclein+ inclusions and on cell loss in 16 key brain regions, 6 months following infusions of the same dose of preformed α‐synuclein fibrils into the OB/AON of 3‐month‐old male and female nontransgenic mice. This survival period and age at infusion was chosen because Rey and colleagues reported that Lewy‐like pathology is widespread as early as 6 months following fibril infusions in the rostral OB of the 3‐month‐old mice, perhaps mimicking prodromal PD 143.
Aside from gender, a prominent risk factor for Lewy body disorders is age 20, 80, 166. Nigrostriatal α‐synuclein expression is known to be regulated by aging in mice, monkeys and humans 40, 106. In our 2016 study, aged male mice did not respond to fibril infusions in the OB/AON with systematically higher levels of α‐synucleinopathy than the young males at 3 months post‐surgery 109. However, the aged males did display somewhat greater pathology in the nucleus accumbens than the young cohort (see Table S2 from that study). In order to determine whether the expanse of the pathology widens beyond limbic sites at longer survival periods in aged animals, we infused 11‐month‐old male mice with vehicle or fibrils and intended to sacrifice them at both 6 and 12 months post‐infusion. The passage of additional time between 6 and 12 months post‐infusion is ineluctably linked with additional natural aging as well as a longer incubation period for the “seeding” of pathology. However, the majority of fibril‐infused animals died by 10.5 months post‐infusion and the experiment had to be terminated with ~30% of mice (n = 3) in the fibril group surviving till 21.5 months of age. Nevertheless, the center of gravity of α‐synucleinopathy remained firmly rooted in the limbic connectome even in these aged survivors.
Methods
Animals and surgeries
Experimental procedures were conducted after approval by the Duquesne University IACUC (Protocols 1403‐03 and 1604‐05) and in accordance with the NIH Guide for the Care and Use of Laboratory Animals. Forty‐eight male and 16 female CD‐1 mice (Charles River, Wilmington, MA, USA) were housed under a 12:12 photoperiod at room temperature with ad libitum access to food and water.
At the time of surgery, all the females and 16 of the 48 males were 3 months old, while the remaining male mice were 11 months of age. All mice were deeply anesthetized with 2% vaporized isoflurane and infused with 1 μL phosphate‐buffered saline (PBS) or 5 μg preformed α‐synuclein fibrils in 1 μL PBS after sonication in a water bath for 1 h (Bransonic series model M1800, Branson Ultrasonics Corporation, Danbury, CT, USA) immediately before use, according to our previously published methods 109. The OB/AON was infused at the following stereotaxic coordinates: AP +4.0 mm, ML ‒1.0 mm and DV ‒2.5 mm from Bregma and the top of the skull, based on the successful placement of blue food dye injections. The AP coordinate was increased to +4.1 mm in the 11‐month‐old mice to accommodate a slight increase in rostrocaudal cranial size with age. Infusions were performed with a motorized injection pump (Stoelting, Wood Dale, IL, USA) at a rate of 0.25 μL/minute and were followed by a 4‐minute rest period before the Hamilton syringe was withdrawn (Cat. no. 80330, Hamilton, Reno, NV, USA). All mice were subcutaneously injected with buprenorphine immediately after surgery and lidocaine was applied to the sutures for the subsequent 3 days. One week later, the animals were reanesthetized and their sutures were removed.
Anesthetized mice were sacrificed by cardiac perfusion through the left ventricle with 50 mL of saline followed by 100 mL of 4% formaldehyde (Thermo Fisher Scientific, Pittsburgh, PA, USA) in 0.1 M phosphate buffer. At 6 months post‐infusion, we sacrificed 16 males (8 PBS and 8 fibril‐infused males) and 14 females from the younger cohort of mice (7 PBS and 7 fibril‐infused females; 1 PBS‐infused female and 1 fibril‐infused female had died prematurely)—all were 9 months old at the time of sacrifice. We also perfused 6 PBS‐infused and 5 fibril‐infused mice from the older cohort (11 months old at surgery and 17 months of age at sacrifice) at 6 months post‐infusion. At 10.5 months post‐infusion (ie, 21.5 months of age), all remaining older animals had to be perfused, as 8 of the 11 fibril‐infused mice had died prematurely (discussed in the Results section). In contrast, only two of the PBS‐infused older animals had died by this time. The brain of one of the older PBS‐infused animals was lost during dissection. Therefore, the final count of animals used for the histological analyses was 51. However, the medial sections of one of the fibril‐infused 17‐month‐old animals were lost while sectioning the brains in the sagittal plane on a sliding/freezing microtome, leaving us with 50 animals for medial brain regions and 51 animals for lateral brain regions.
Behavior testing
Animals were subjected to memory, motor and olfactory tests subsequently rated by blinded observers. Tests were conducted at survival periods that are indicated on the behavior graphs (see Figure 13). Behavior tests were videotaped within a diffusely lit arena (45 cm × 60 cm × 60 cm) with minimal shadows. To examine learning and memory, the novel object and novel position tests were employed. Animals were subjected to a habituation phase consisting of 10 minutes in the arena 34. On each of the following 2 days, the animals were familiarized with two identical objects placed in opposite corners of the arena, each 10 cm away from the wall. The familiarization phase lasted 5 minutes and was followed by recognition testing on the fourth day, during which animals were exposed to only one of the two familiar objects from the previous days and a second novel object. The novel object exploration ratio was defined as the time spent exploring the novel object as a fraction of total exploration time (ie, novel + familiar). Exploration was defined as the animal facing the object within 4 cm at a 45° angle. On the day after the novel object test, the animal was subjected to the novel place recognition test, in which one object from the familiarization phase was moved to a new location in the arena. The novel object/place exploration ratio was defined as the percentage of time spent interacting with the novel object compared to time spent interacting with both (ie, novel + familiar) objects. Mice spending less than 10 s interacting with both objects were classified as “non‐responders” (n = 8/51) and excluded from the analyses 34.
Figure 13.
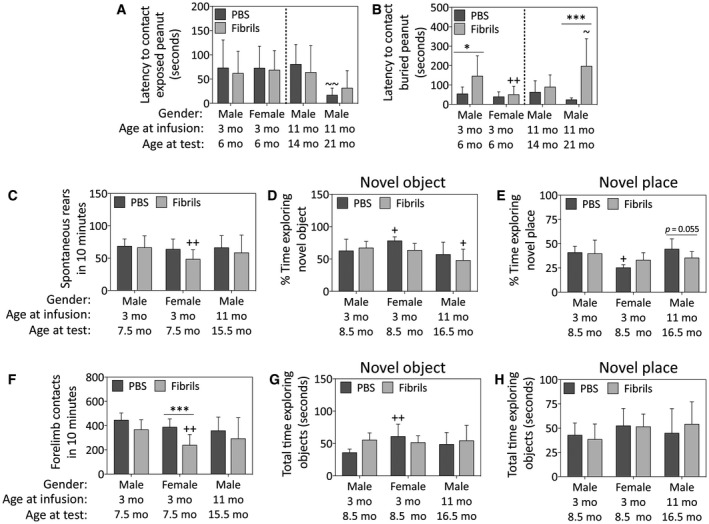
α‐Synuclein fibril injections in the OB/AON elicit sex and age‐dependent behavioral deficits. Mice were infused in the right olfactory bulb/anterior olfactory nucleus with either preformed α‐synuclein fibrils (5 μg/1 μL) or an equivalent volume of PBS (1 μL). The buried pellet test for olfactory capacity was conducted at 3 months post‐infusion in all groups, and again at 10 months post‐infusion in only the oldest group of mice (the only survivors at that time). A blinded observer calculated the latency to approach an exposed peanut (A) and the latency to approach a buried peanut (B). The cylinder test was performed at 4.5 months post‐infusion (C‐D). Shown are the number of rears (C) and forelimb contacts (D) with the walls of the cylinder in 10 minutes. The novel object/place recognition tests were performed at 5.5 months post‐infusion (E‐H). Note that the oldest group of mice were not subjected to any behavior assay after 6 months post‐infusion, other than the buried pellet test. Two‐way ANOVAs were followed by the Bonferroni post hoc correction. Shown are the mean and SD from an n of 3–16 mice per group (see Methods and Figure 1 for animal numbers). *P ≤ 0.05, ***P ≤ 0.001 PBS vs. fibrils; +P ≤ 0.05, ++P ≤ 0.01 vs. 3‐month male group. ~P ≤ 0.05 vs. 11‐month male group. Abbreviations are defined in Table S2. To view the original, higher resolution Adobe Illustrator or EPS files, please link to https://dsc.duq.edu/pharmacology/ or https://www.dropbox.com/sh/a6r5ylg1tco6trm/AAC9Mb2gWuP29ABbmdGuNoaJa?dl=0
To measure spontaneous motor activity, animals were placed in a transparent Plexiglass cylinder (8.9 cm diameter, 15.2 cm height) for 10 minutes 22. A blinded observer recorded spontaneous rears (defined as both forelimbs leaving the ground while bodyweight is placed on the hindlimbs) and spontaneous left or right forelimb contacts with the walls of the cylinder. No animals were excluded from the motor function tests.
The buried pellet test was performed to assess olfactory function 61. The animal was first habituated to a fresh cage in the testing arena for 5 minutes. On this first day of testing, a peanut was placed on top of the corncob bedding and the latency to contact the exposed pellet immediately following entry of the animal into the cage was measured. On the next day, the latency to contact a peanut buried ~1 cm deep in clean corncob bedding was measured. Animals expressing no interest in the exposed treat were excluded from the analysis to eliminate any confounding lack of motivation; thus, 15 of 64 animals were excluded at 3 months post‐infusion and no animals were excluded at 10.5 months post‐infusion.
Immunohistochemistry
After cardiac perfusion, brains were immersed for 24–48 h in 30% sucrose in 10 mM PBS, sectioned on a sliding/freezing microtome in the sagittal plane as a 1‐in‐5 free‐floating series, and stored at ‒20°C in cryoprotectant 168. Immediately prior to the histological staining, all tissue sections were washed 3 times in 10 mM PBS and then heated to 80°C in a 10 mM sodium citrate tribasic dihydrate solution (pH 8.5, Sigma‐Aldrich, St. Louis, MO, USA) for 30 minutes to expose antigen‐binding sites 92. The sections were then allowed to cool to room temperature, rinsed 3 times in PBS and blocked in 50% Odyssey Block (LI‐COR, Lincoln, NE, USA) in PBS for 1 h at room temperature on a shaker. This was followed by overnight incubation at 4°C with primary antibodies (see Table S1) diluted in the same blocking solution, with the addition of 0.3% Triton X‐100. The next day, sections were washed 3 times in 10 mM PBS and exposed to fluorescent secondary antibodies (see Table S1) for 1 h at room temperature. The sections were also exposed to Hoechst 33258 (0.005 μM) during the secondary antibody incubation period to stain all nuclei.
To visualize aggregated proteins in the aggresome 98, 157, the Proteostat aggresome detection kit was employed after the secondary antibody step, according to manufacturer’s instructions (Enzo Life Sciences, Farmingdale, NY, USA). At the end of all staining procedures, sections were washed 3 times in 10 mM PBS and mounted onto glass slides (SuperFrost Plus, ThermoFisher Scientific). The slides were coverslipped with Krystallon (EMD Chemicals, Gibbstown, NJ, USA). For all immunostaining procedures, negative control sections from the same group of animals were exposed to secondary but not primary antibodies (in parallel with the rest of the tissue) to determine background fluorescence.
Data from all groups were analyzed in parallel by blinded observers. Sections from control and experimental groups were processed in the same solutions and photographed at the same exposure and fixed intensity scaling, except for the qualitative aggresome analyses displayed in Figure 7, as the Proteostat signal bleached unexpectedly fast under high‐power illumination with a 100× oil objective.
Figure 7.
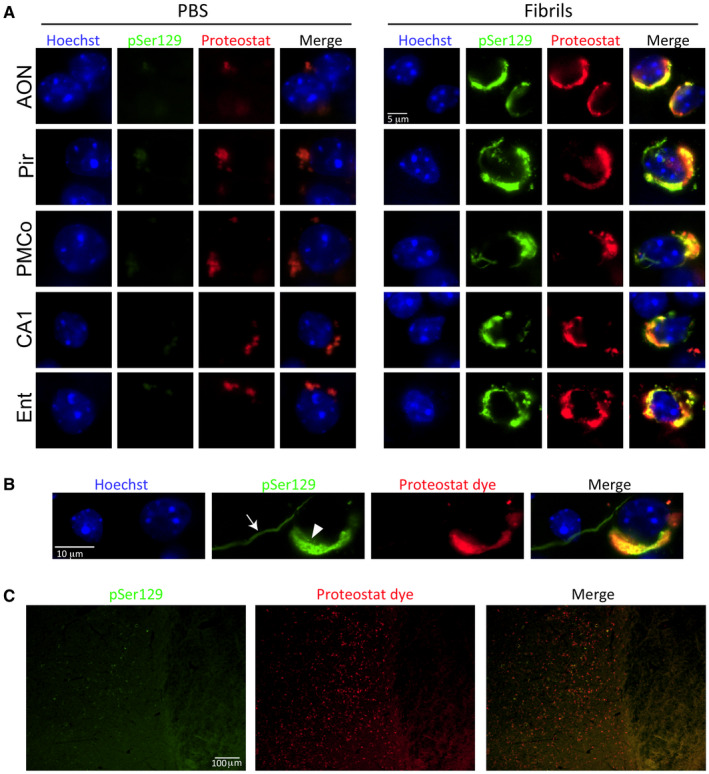
Preformed α‐synuclein fibril injections in the mouse OB/AON induce the formation of aggresome‐like structures. Mice were infused in the right olfactory bulb/anterior olfactory nucleus with either preformed α‐synuclein fibrils (5 μg/1 μL) or an equivalent volume of PBS (1 μL). Sagittal brain sections from the young male mice were immunostained for pathologically phosphorylated α‐synuclein (monoclonal 81A pSer129 antibody; see Table S1). The Proteostat detection reagent was applied to the tissue and the Hoechst reagent was used to stain cellular nuclei. Images were captured with a 100× objective under oil immersion. (A) Perinuclear pSer129+ inclusions were stained with the Proteostat dye in all affected brain regions, including the anterior olfactory nucleus (AON), piriform cortex (Pir), posteromedial cortical nucleus of the amygdala (PMCo), hippocampal CA1 and entorhinal cortex (Ent), in young male mice 6 months after fibril infusions. See text for details on background staining in PBS group. (B) The arrow points to a pSer129+ neuritic inclusion that does not colocalize with the Proteostat dye, in contrast with the dual‐labeled perinuclear inclusion in the soma (arrowhead). (C) Mild pSer129 but dense Proteostat label was observed in the tenia tecta. Abbreviations are defined in Table S2. To view the original, higher resolution Adobe Illustrator or EPS files, please link to https://dsc.duq.edu/pharmacology/ or https://www.dropbox.com/sh/a6r5ylg1tco6trm/AAC9Mb2gWuP29ABbmdGuNoaJa?dl=0
Image analyses
Immunostaining was visualized on an Olympus epifluorescent microscope (Olympus IX73, B&B Microscopes, Pittsburgh, PA, USA) and pseudocolored images were captured with a 12‐bit grayscale camera (CS‐S‐CCD High‐End Flash 4.0 Camera). Stitched images of the entire tissue section were generated with the 4× or 20× objective. Higher resolution epifluorescence images were captured with a 40× or 100× oil (numerical aperture (NA) = 1.40) objective. Confocal images were captured on a Nikon A1R LU‐NV Confocal Imaging System (Nikon Instruments Inc., Melville, NY, USA) with a 20× and a 60× oil (NA 1.40) objective on a Nikon Eclipse Ti2 microscope. For the confocal analyses, Z‐stacks were captured at 0.31 μm per pixel and 1.391 μm per step in the Z plane. Franklin and Paxinos’ “The Mouse Brain in Stereotaxic Coordinates” was continuously consulted during all photographic procedures (Franklin and Paxinos, 2013).
Monoclonal mouse and rabbit antibodies raised against pSer129 were employed to visualize Lewy‐related pathology, as Ser129 phosphorylation of α‐synuclein is the most frequent posttranslational modification of this protein within Lewy aggregates 8, 63, 169. Manual counts of the number of pSer129 inclusions were performed on tissue sections stained with the mouse monoclonal antibody, as this antibody was employed before we found that the rabbit monoclonal was even better, as confirmed in a recent study 47. A blinded observer manually counted the total number of pSer129+ inclusions under 200× magnification (field of view = 447.1 μm × 337.0 μm) in 2–3 sagittal sections per animal. It is important to note that the mouse brain is narrower in the sagittal plane than the coronal plane—the former plane therefore yields fewer sections than the latter. Furthermore, depending upon the starting location of the microtome sectioning, each brain subregion might span across varying numbers of sections per animal; for our specific subregions, this was 2–3 sections per region. An exception is the mediolaterally expansive piriform cortex, which we divided into two (rostromedial and caudolateral) main subregions. Hoechst+ nuclei (all cells) and NeuN+ nuclei (neurons only) were counted with the Olympus cellSens software (Version 1.17) in the same fields of view as the pSer129+ inclusions. To automatically count Hoechst+ and NeuN+ cells, images were converted to binary images with the thresholding tool and any cell clusters were split into individual cells using the “auto split” function. In both the NeuN and Hoechst images, noncellular background staining with surface areas below 10 μm2 were excluded from the analyses. A total of 7296 total images were analyzed in the above manners.
Aside from the above measurements, a blinded observer also calculated the widths of the pyramidal cell layer of the rostromedial and caudolateral piriform cortices and the hippocampal fields CA1, CA2, and CA3, as well as the widths of the strata granulosum of the dentate gyrus with the cellSens software “arbitrary line” measurement tool in two sagittal sections per animal.
For the qualitative heatmap, pathology was scored based on the relative abundance of pSer129+ inclusions only after first examining all of the tissue, including all the PBS‐infused mice. Each section was screened at 200× magnification and a score assigned to each brain corresponding to the maximal inclusion density in that brain region (0 = no aggregates, 1 = sparse, 2 = mild, 3 = medium, 4 = dense, 5 = very dense), as described in Supplemental Files of our previous work 109.
In addition to the aforementioned counts of inclusions, NeuN+ neurons, and Hoechst+ cells in 16 brain regions, measurements of hippocampal and piriform cortical layer width, and the qualitative heatmap, a blinded observer also counted dopaminergic neurons within the ventral mesencephalon. For the latter measurements, images of tyrosine hydroxylase (TH) and Hoechst labeling were captured at 200× magnification and combined into giant high‐resolution stitches of the entire ventral midbrain encompassing the A8 (retrorubral field), A9 (substantia nigra) and A10 dopaminergic cell groups (defined here as the ventral tegmental area according to Oades and Halliday 125). TH+ cells were manually counted by a blinded observer only if cytosolic TH label overlapped (ie, encircled) a clearly visible Hoechst+ nucleus in the DAPI channel. The anatomical boundaries of the TH+ areas in the ventral midbrain were defined as regions of interest (ROI) in ImageJ and overlaid on the Hoechst+ images of the same field of view, in order to selectively count Hoechst+ nuclei only within the defined boundaries of the dopaminergic cell groups. For the counts of Hoechst+ nuclei in the stitched images, the DAPI channel was saved as a TIFF file and then exported to ImageJ software for automatic offline counting. The midbrain stitches were first converted into binary images and the “watershed” tool was performed to automatically separate any cell clusters. Particles with areas <50 pixels (ie, background staining only) or >1000 pixels (indivisible clusters containing closely apposed cells) were excluded a priori. Using these methods, every visible “TH plus Hoechst‐positive” or “Hoechst‐positive” cell was counted and expressed as TH+ or Hoechst+ cell numbers in (i) the ventral tegmental area; (ii) the entire substantia nigra, pars compacta; (iii) the anterior substantia nigra, pars compacta; (iv) the posterior substantia nigra, pars compacta; (v) the lateral substantia nigra, pars compacta; or (vi) the retrorubral field. A total of 624 total images were analyzed in the above manner.
The expression levels of the dopamine transporter (DAT) and TH in the dorsal striatum, substantia nigra, nucleus accumbens, and ventral tegmental area were determined by scanning the infrared‐immunolabeled tissue on a 16‐bit imager (Odyssey Classic, LI‐COR Biosciences), following application of the appropriate primary and infrared secondary antibodies (see Table S1). The anatomical boundaries of these four brain regions were traced by a blinded observer in Image Studio software (Version 5.2, LI‐COR). Due to occasionally high interindividual variations in infrared immunostaining, the background signal for each individual animal was assessed by calculating the fluorescence signal per unit area of occipital cortex, which exhibits the lowest endogenous levels of dopaminergic markers in the brain 30, 86. Background signal for the region of interest was calculated for each animal according to the formula (IB/AB) × AX, where IB is the total fluorescence intensity of the background region, AB is the area of the background region and AX is the area of the target region of interest. Background signal was then subtracted from the raw TH and DAT signal in the regions of interest—the caudoputamen, substantia nigra, nucleus accumbens and ventral tegmental area 124.
Statistics
Two‐tailed logrank survival analyses were performed on all groups. As described in each figure legend, data are displayed as raw, unnormalized numbers or as ratios of raw numbers, with the exception of the qualitative heatmap data in Figure 2. Two‐way ANOVAs were followed by the Bonferroni post hoc correction (SPSS Version 22, IBM, Armonk, NY, USA). We were unable to perform three‐way ANOVAs, as we could not afford to include older females in the study. Thus, the oldest males were not contrasted with the younger, 9‐month‐old animals although they are plotted on the same graphs for ease of presentation (see dotted lines between young and older animals). Differences between groups were only deemed significant when p ≤ 0.05. Data are presented as the mean + SD in all but Figure 1.
Figure 2.
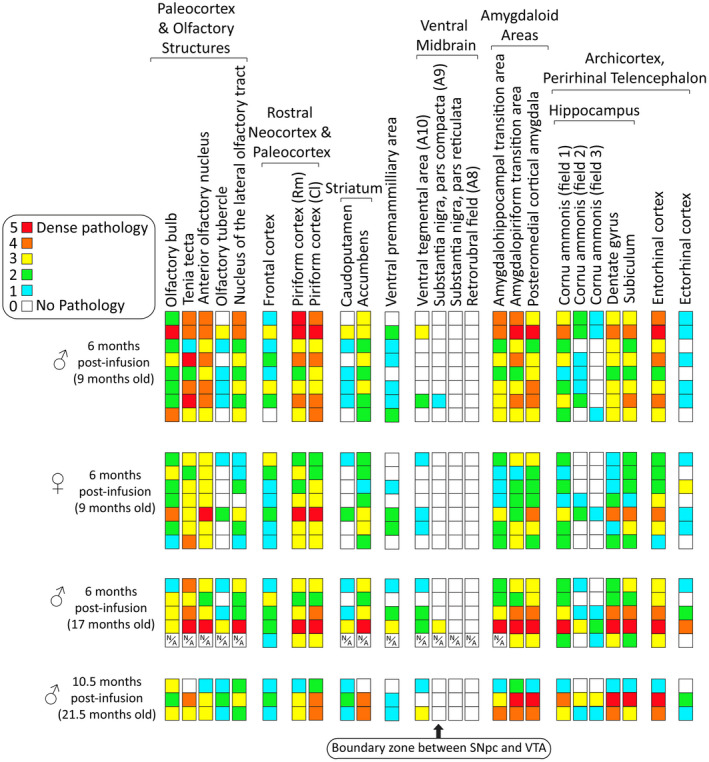
Heat map of α‐synucleinopathy following preformed α‐synuclein fibril injections in the mouse OB/AON. Qualitative assessments of the density of pSer129+ inclusions in the indicated brain regions of all fibril‐infused animals that survived until the planned assay time (see Methods). Each row represents one animal. A blinded investigator systematically examined the entire 1‐in‐5 series of sagittal brain sections of the ipsilateral, right hemisphere following immunostaining of pathologically phosphorylated α‐synuclein with the rabbit monoclonal EP1536Y pSer129 antibody (see Table S1). Boxes with a N/A label denote regions that were unavailable (see Methods). Brain regions with pSer129+ immunolabeling are arranged rostrocaudally and/or by anatomical/functional groupings. Most of the midbrain pathology was confined to the ventral tegmental area; only two male animals exhibited pSer129+ inclusions at the boundary zone between the substantia nigra and the ventral tegmental area at 6 months post‐infusion. Abbreviations are defined in Table S2. https://dsc.duq.edu/pharmacology/ or https://www.dropbox.com/sh/a6r5ylg1tco6trm/AAC9Mb2gWuP29ABbmdGuNoaJa?dl=0
Figure 1.
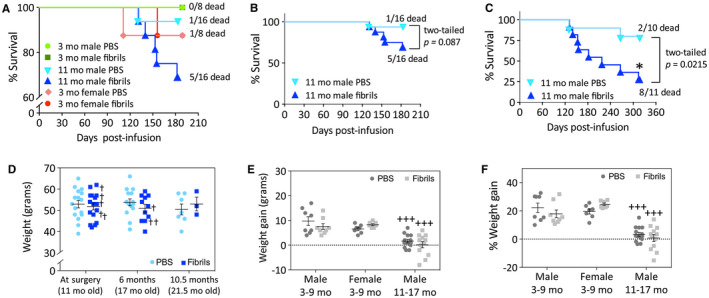
Preformed α‐synuclein fibril injections in the mouse OB/AON accelerate mortality in older mice without significant changes in body weight. Sixty‐four mice were infused in the right olfactory bulb/anterior olfactory nucleus with either preformed α‐synuclein fibrils (5 μg/1 μL) or an equivalent volume of PBS (1 μL) at the indicated ages. Logrank survival analyses were performed for all animals at 6 months post‐infusion (A) and for the older males at 6 months (B) or 10.5 months (C) post‐infusion. The two‐tailed P‐values of the logrank survival analyses are shown. (D) The body weights of the older males at surgery (11 months old), 6 months post‐surgery (17 months old), and 10.5 months post‐surgery (21.5 months old) are displayed as scatter plots with mean and SEM bars. In panel D, crosses are placed next to mice that died before the planned time of assay. (E‐F) Weight gained in both raw numbers (E) and percentage weight gain (F) 6 months after surgery in all experimental groups. Note the broken Y axis in panels A and D. For panels D‐F, two‐way ANOVAs were followed by the Bonferroni post hoc correction for multiple comparisons. In panels E‐F, +++ P ≤ 0.001 vs. males that were infused at 3 months of age and perfused at 9 months of age (3–9 month males).
Results
α‐synuclein fibril infusions in the murine OB/AON dramatically increase mortality in aged mice without loss of body weight
At the commencement of this study, 64 CD‐1 mice were infused with either PBS (n = 32) or preformed α‐synuclein fibrils (n = 32). Using the lowest number of mice and resources possible, the experiment was designed to (i) assess the effects of gender as an independent variable in male and female mice infused with PBS or fibrils (n = 8 per group, 32 total) at 3 months of age and sacrificed at 6 months post‐infusion; and (ii) assess the passage of time as an independent variable in male mice infused with PBS or fibrils at 11 months of age (n = 16 per group, 32 total) and sacrificed 6 and 12 months later, noting that incubation time or survival period is inextricably linked with additional natural aging. Originally, we planned to sacrifice 8 of the 16 older male mice in each group at 6 months post‐infusion, and to follow the remaining 8 for 12 months post‐infusion. At the 6‐month survival time point, no significant differences in mortality were observed between the groups (Figure 1A—note the broken Y axis). However, there was a slight trend toward increased mortality after fibril infusions when only the older mice were analyzed—5 of 16 older males from the fibril‐infused group had died prematurely, compared to only 1 out of 16 in the age‐matched PBS group (Figure 1B). Therefore, we only sacrificed 6 of the older PBS‐infused male mice and 5 of the fibril‐infused male mice at the 6‐month survival time point, leaving us with 10 PBS‐infused older male mice and 11 fibril‐infused older male mice to follow for another 6 months. However, by 10.5 months post‐infusion, only 3 of the 11 older fibril‐infused male mice were still alive, compared to 8 age‐matched males in the PBS group (Figure 1C). Given the significant lethality of our model in aged male mice, we perfused all remaining animals immediately, at 21.5 months of age.
Because of the significant increase in mortality in the fibril‐infused, older group of mice, we determined whether the fibril treatment was associated with loss of body weight (Figure 1D). In Figure 1D, crosses are placed next to the body weights of the eight fibril‐infused older males that subsequently died before the planned sacrifice date. In addition, graphs of the body weights of all animals are displayed in Figures 1E‐F. Older animals gained significantly less weight over the course of 6 months post‐infusion than the 3‐month‐old group, but fibril infusions per se did not exert significant effects in any group (Figure 1E‐F). These results fail to support the view that loss of body weight (and perhaps lack of food intake or dysphagia) contributed to mortality.
α‐synucleinopathy is centered within the limbic connectome following preformed fibril infusions in the murine OB/AON
After all the brains were harvested, they were immunostained together and a blinded observer scored the density of the ipsilateral α‐synucleinopathy in the fibril‐infused mice from 0 to 5 (Figure 2). PBS‐infused brains were also carefully examined to ascertain background staining. Abbreviations for brain structures are defined in Table S2. As in our previous work, dense inclusions harboring pathologically phosphorylated α‐synuclein (pSer129) were observed in rostral olfactory structures of fibril‐infused mice, including in the AON, tenia tecta and piriform cortex, as well as caudal limbic brain regions known to harbor connections with the OB/AON, such as the hippocampus, amygdala and entorhinal cortex 109. The site of injection in the OB displayed less overall pathology compared to other olfactory structures, especially the AON, consistent with the greater density of Lewy bodies in the human AON than the OB 16, 18, 26, 134, 165. Previously, we reported that the olfactory tubercle failed to develop any α‐synucleinopathy at 3 months post‐infusion 109. In the present study, we observed the emergence of pathology in the olfactory tubercle, but in all cases the brunt of pSer129 pathology remained centered within limbic structures of the temporal lobe, even in males that were 11 months old at the time of infusion and sacrificed at 6 months or 10.5 months post‐infusion. Some animals displayed mild extralimbic pathology in the caudoputamen (Figure 2), and one older male mouse displayed limbic pathology outside of the circuitry assessed in the heatmap (eg, bed nucleus of stria terminalis and premammillary area). The latter animal also displayed somal pSer129+ inclusions at the boundary between the nigra and ventral tegmental area (discussed below). Another younger male also displayed sparse, neuritic inclusions in this boundary zone.
Fibril‐induced murine α‐synucleinopathy and cell loss in several limbic brain regions is dependent upon gender
Aside from the impact of fibril infusions on 16 brain regions, we have focused our discussion of gender and the passage of time mostly on the fibril‐treated groups rather than the PBS groups, for the sake of brevity. In both the OB and AON, female mice exhibited fewer pSer129+ inclusions than males of the same age (Figure 3A‐B). α‐synucleinopathy can be associated with cell death 127, 167, but not inescapably so (see Introduction); therefore, we also counted NeuN+ neurons and Hoechst+ cells in the same brain regions. Fibril infusions induced a mild increase in Hoechst+ cells in the OB in females at 6 months post‐infusion (Figure 3A). However, fibrils induced a loss of NeuN+ neurons in the AON and the nucleus of the lateral olfactory tract in the oldest males and the youngest males, respectively (Figure 3B,C). Fibril‐infused females displayed slightly lower counts of NeuN+ neurons in the AON compared to the fibril‐infused males at 6 months post‐infusion (Figure 3B). In the nucleus of the lateral olfactory tract, but not in the AON, fibril‐induced changes in NeuN+ cells were paralleled by loss of Hoechst+ cells in the youngest males (Figure 3B,C). In the oldest males, the additional passage of time post‐infusion elicited a loss of Hoechst+ nuclei in the OB, but the fibril infusions did not modify this response (Figure 3A). We were unable to achieve convincing, high‐quality immunostaining in aged mice that died unexpectedly and were not perfused with formalin through the heart; therefore, statistical power in the 21.5 fibril‐infused mice is low, making conclusions about survival period/aging somewhat more tentative than conclusions about gender.
Figure 3.
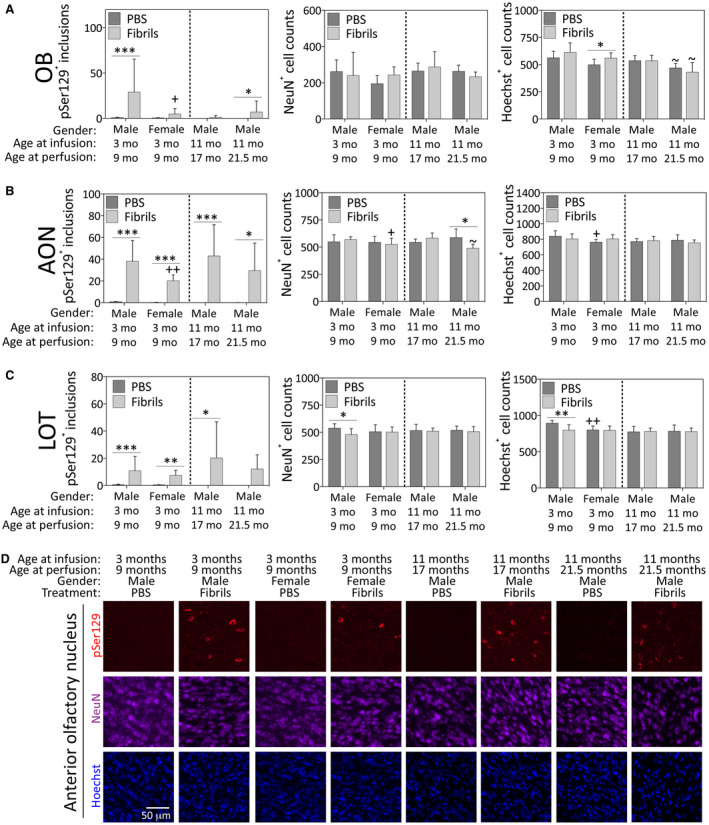
Impact of α‐synuclein fibril infusions in the mouse OB/AON on pSer129+ inclusion counts, NeuN+ neuron counts and Hoechst+ cell numbers in the rostral rhinencephalon. Mice were infused in the right olfactory bulb/anterior olfactory nucleus with either preformed α‐synuclein fibrils (5 μg/1 μL) or an equivalent volume of PBS (1 μL). A blinded observer manually counted the number of pSer129+ structures (monoclonal 81A pSer129 Ab; see Table S1) per field of view (200× magnification) and used cellSens software to count the number of NeuN+ and Hoechst+ nuclei in the olfactory bulb (OB; A), the anterior olfactory nucleus (AON; B, D) and the nucleus of the lateral olfactory tract (LOT; C) in the same field of view. Shown are the mean and SD of raw, unnormalized data. N = 3–8 mice per group (see Methods and Figure 1 for animal numbers). Two‐way ANOVAs were followed by the Bonferroni post hoc correction. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001 PBS vs. fibrils; +P ≤ 0.05, ++P ≤ 0.01 vs. 3–9 month males; ~P ≤ 0.05 vs. 11–17 month males. Abbreviations are defined in Table S2. To view the original, higher resolution Adobe Illustrator or EPS files, please link to https://dsc.duq.edu/pharmacology/ or https://www.dropbox.com/sh/a6r5ylg1tco6trm/AAC9Mb2gWuP29ABbmdGuNoaJa?dl=0
Female mice exhibited fewer pSer129+ inclusions than males in the caudolateral piriform cortex (Figure 4B), the amygdalopiriform transition area (Figure 4C) and the posteromedial cortical amygdala (Figure 4D). In the youngest males, fibril infusions elicited mild loss of Hoechst+ cells in the piriform cortices (Figure 4A,B), the posteromedial cortical amygdala (Figure 4D) and the nucleus accumbens (Figure 4F). Parallel loss of NeuN+ structures was only observed in the amygdala (Figure 4D). Notably, the caudoputamen only displayed significant fibril‐induced inclusions in the 17‐month‐old males sacrificed at 6 months post‐infusion (Figure 4E), and the number of inclusions in this group was significantly higher relative to the oldest males (21.5 months old). However, the dorsal striatum was relatively sparsely labeled compared to limbic sites, such as the nucleus accumbens (compare values on Y‐axes in Figure 4E vs. 4F).
Figure 4.
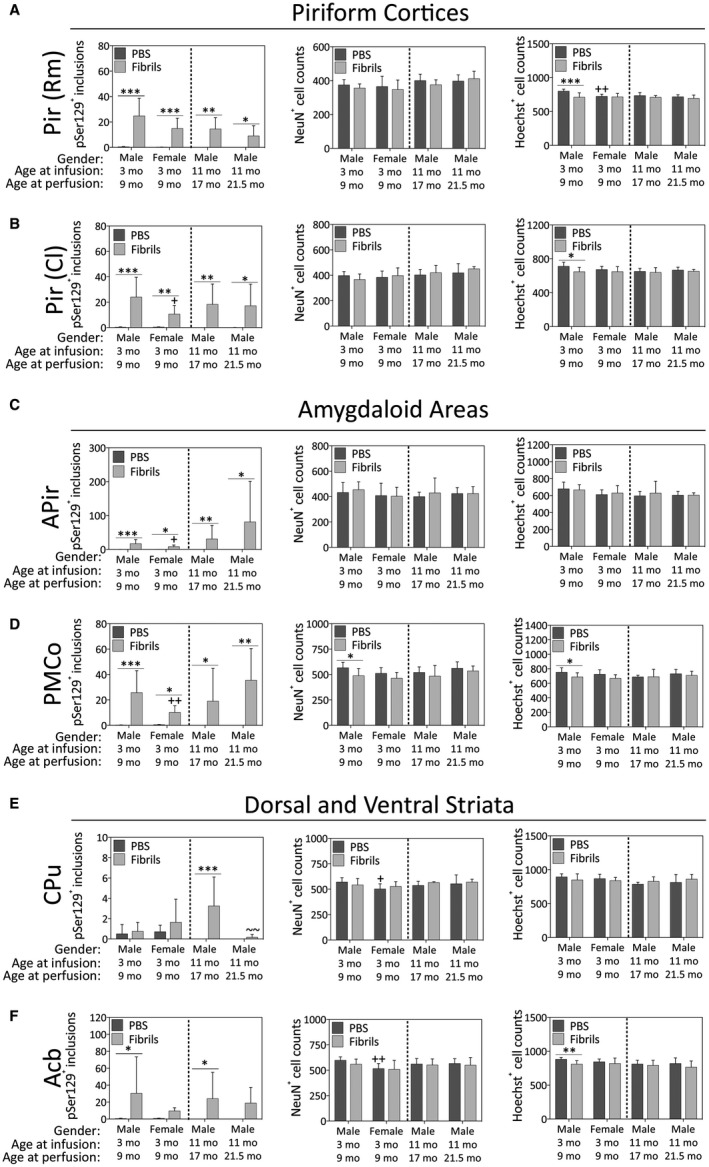
Impact of α‐synuclein fibril infusions in the mouse OB/AON on pSer129+ inclusion counts, NeuN+ neuron counts and Hoechst+ cell numbers in the paleocortices, amygdaloid complex, and the dorsal and ventral striata. Mice were infused in the right olfactory bulb/anterior olfactory nucleus with either preformed α‐synuclein fibrils (5 μg/1 μL) or an equivalent volume of PBS (1 μL). A blinded observer manually counted the number of pSer129+ structures (monoclonal 81A pSer129 Ab; see Table S1) per field of view (200× magnification) and used cellSens software to count the number of NeuN+ and Hoechst+ nuclei in the rostromedial piriform cortex (Pir Rm; A), caudolateral piriform cortex (Pir Cl; B), amygdalopiriform transition area (APir; C), posteromedial cortical amygdala (PMCo; D), caudoputamen (CPu; E) and nucleus accumbens (Acb; F). Shown are the mean and SD of raw, unnormalized data. N = 3–8 mice per group (see Methods and Figure 1 for animal numbers). Two‐way ANOVAs were followed by the Bonferroni post hoc correction. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001 PBS vs. fibrils; +P ≤ 0.05, ++P ≤ 0.01 vs. 3–9 month males; ~~P ≤ 0.01 vs. 11–17 month males. Abbreviations are defined in Table S2.
In the hippocampal formation, females developed similar numbers of inclusions as males in the CA1, CA2 and CA3 fields, but the overall number of inclusions in all groups was quite low in CA2 and CA3 (Figure 5B,C). In the dentate gyrus, females failed to develop a significant number of inclusions in response to fibril infusions, unlike all the male groups (Figure 5D). In the subiculum, the number of inclusions in fibril‐infused females was significantly lower than males (Figure 5E). In the ectorhinal cortex, the oldest males displayed no increase in the number of inclusions in response to fibrils (Figure 5G). Fibril infusions increased the numbers of NeuN+ cells in CA2 of the young males (Figure 5B) but significantly lowered the numbers of NeuN+ cells in both CA2 and the subiculum of the oldest males (Figure 5B and E). Fibril‐infused females displayed fewer NeuN+ neurons compared to fibril‐infused males in the CA2 region of the hippocampal formation (Figure 5B). Hoechst+ cell numbers in the ectorhinal cortex were lowered 6 months after fibril infusions in the 11‐month‐old males (Figure 5G).
Figure 5.
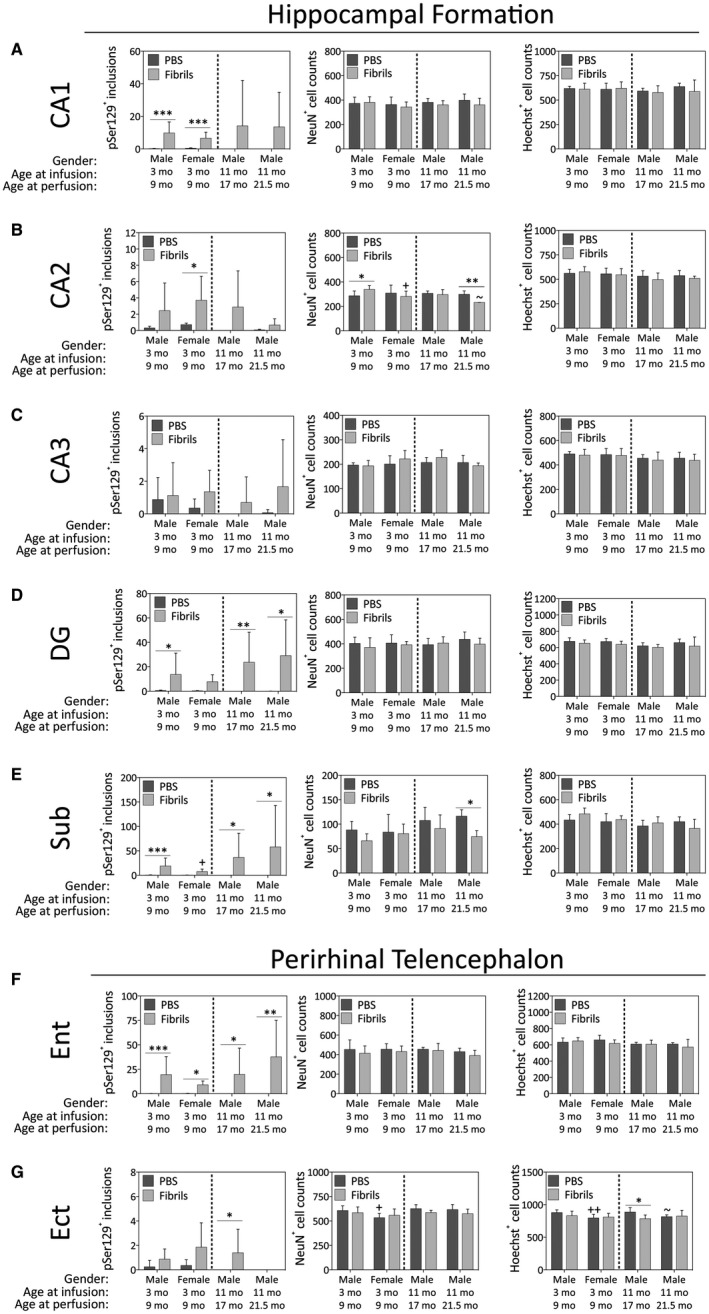
Impact of α‐synuclein fibril infusions in the mouse OB/AON on pSer129+ inclusion counts, NeuN+ neuron counts and Hoechst+ cell numbers in the hippocampal formation and perirhinal telencephalon. Mice were infused in the right olfactory bulb/anterior olfactory nucleus with either preformed α‐synuclein fibrils (5 μg/1 μL) or an equivalent volume of PBS (1 μL). A blinded observer manually counted the number of pSer129+ structures (monoclonal 81A pSer129 Ab; see Table S1) per field of view (200× magnification) and used cellSens software to count the number of NeuN+ and Hoechst+ nuclei in the pyramidal cell layers of hippocampal fields CA1 (A), CA2 (B) and CA3 (C), the stratum granulosum of the dentate gyrus (DG; D), the subiculum (Sub; E), entorhinal cortex (Ent; F), and the ectorhinal cortex (Ect; G). Shown are the mean and SD of raw, unnormalized data. N = 3–8 mice per group (see Methods and Figure 1 for animal numbers). Two‐way ANOVAs were followed by the Bonferroni post hoc correction. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001 PBS vs. fibrils, +P ≤ 0.05 vs. 3–9 month males, ~P ≤ 0.05 vs. 11–17 month males. Abbreviations are defined in Table S2.
The inclusion counts described above were performed on tissue immunolabeled with the mouse anti‐pSer129 antibody employed in previous work 109, 124. Subsequently, we employed a monoclonal rabbit pSer129 antibody and observed similar patterns as described above in all brain regions, including the AON (Figure 6A), tenia tecta (Figure 6B), dentate gyrus (Figure 6C), hippocampal CA1 (Figure 6D), amygdala (Figure 6E) and nucleus accumbens of the ventral striatum (Figure 6F). Sacino and colleagues cautioned that pSer129 antibodies (81A) can cross‐react with neurofilament L in white matter tracts 150. Delic et al. similarly observed nonspecific bands by Western blotting with all α‐synuclein phosphoantibodies, and reported that the rabbit monoclonal anti‐pSer129 antibody used here displays the highest sensitivity and specificity of commercially available anti‐pSer129 antibodies; there are simply no better markers of α‐synucleinopathy to date 47. It is also worth observing that, in those regions where the inclusions were quite dense per field of view (ie, more than one or two), statistically higher numbers of pSer129+ structures were always observed in the fibril‐infused mice, confirming that our blinded interpretations of genuine α‐synucleinopathic inclusions were indeed correct. Nevertheless, large stitched images of entire brain sections are provided in Figure S1, to allow the reader to independently verify the anatomical location and density of pSer129+ inclusions in male and female fibril‐infused mice, as well as the degree of background labeling. For example, these images reveal background label in the brainstem, which was present in both PBS and fibril groups. None of the background label displayed the tendril‐like appearance of genuine pSer129+ inclusions, as described in previous studies using the preformed fibril approach 127. It is important to note that the genuine pSer129+ inclusions only fluoresce in the appropriate channel whereas the minor, nonspecific brainstem label fluoresces in all channels, even when no primary or secondary antibodies are applied. We also employed the diaminobenzidine chromogen to assess pSer129 staining, but observed similar background labeling in the brainstem of the PBS group as well as in controls without application of primary antibody (not shown). Thus, a considerable advantage of the immunofluorescent approach was the ability to check whether the labeling appeared in “inappropriate” fluorescent channels in which no antibodies were applied. Given these collective observations, we hesitate to conclude that there is convincing pSer129 labeling in the brainstem of fibril‐infused mice.
Figure 6.
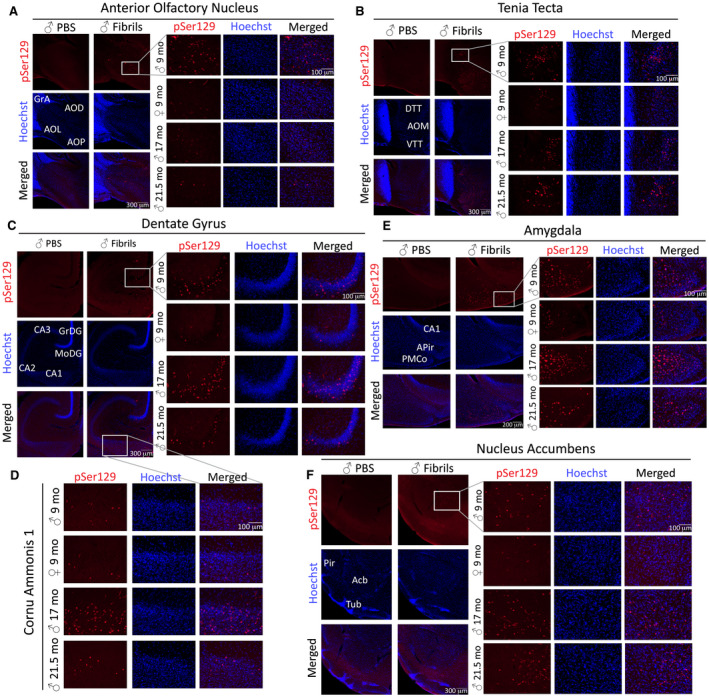
Preformed α‐synuclein fibril injections in the mouse OB/AON elicit α‐synucleinopathy in olfactory/limbic regions of the temporal lobe. Mice were infused in the right olfactory bulb/anterior olfactory nucleus with either preformed α‐synuclein fibrils (5 μg/1 μL) or an equivalent volume of PBS (1 μL). Sagittal brain sections were immunostained for pathologically phosphorylated α‐synuclein (EP1536Y rabbit monoclonal pSer129 antibody; see Table S1). The Hoechst reagent was used to stain nuclei and identify anatomical boundaries. Representative images were captured at 40× and 200× (insets) magnification in the anterior olfactory nucleus (A), tenia tecta (B), dentate gyrus (C), hippocampal CA1 (D), the posteromedial cortical amygdala (E) and the nucleus accumbens (F). Abbreviations are defined in Table S2. To view the original, higher resolution Adobe Illustrator or EPS files, please link to https://dsc.duq.edu/pharmacology/ or https://www.dropbox.com/sh/a6r5ylg1tco6trm/AAC9Mb2gWuP29ABbmdGuNoaJa?dl=0
Murine pSer129+ inclusions stain with an aggresome dye in the somata but not in neuritic processes
Our previous work demonstrates that some of the pSer129+ aggregates in the preformed fibril model colocalize with ubiquitin, an established marker of Lewy pathology 109. In that work, we conceded that ubiquitin antibodies do not recognize all the pSer129+ inclusions 109, 124, but this has also been observed in human post‐mortem material 159. We also noted that the Thioflavin S labeling in the hippocampus differed in conspicuous ways from the pSer129 immunostaining, and that these two markers were therefore not likely to exhibit much overlap 109, 124. As our attempts to colabel Thioflavin S and pathologically phosphorylated α‐synuclein on the very same sections failed, we relied on the well‐established Proteostat dye for aggresomes 157 in pSer129‐labeled brain tissue of the young males. Aggresomes originate from the centrosome, which serves as a scaffold to recruit proteasomal machinery, stress‐induced chaperones and other protein quality control systems 62, 154. Lewy bodies are thought to be related to aggresomes, as the centrosome expands into an aggresome to accommodate the influx of misfolded proteins under conditions of high proteotoxic stress 126. Many, but not all the somal pSer129+ structures bound the Proteostat dye, including in the tenia tecta, AON, piriform cortex, amygdala, hippocampus and entorhinal cortex (Figure 7A–C). Importantly, pSer129+ neuritic inclusions were not labeled by the Proteostat dye, consistent with the perinuclear localization of aggresomes, which supports the specificity of the staining methods (Figure 7B). However, in the PBS‐infused mice, the Proteostat dye sometimes weakly labeled a small structure—perhaps the centrosome—near the nucleus, which may reflect low levels of misfolded proteins under physiological conditions. In Figure 7A, we focused the photography on this potentially important “background” label rather than on the absence of staining, as was the case in the majority of brain cells in the PBS animals. Notably, more structures were stained with the Proteostat dye in fibril‐infused animals than were positive for pSer129 immunoreactivity, especially in the tenia tecta adjacent to the OB and AON (Figure 7c).
Fibril infusions in the murine OB/AON decrease the width of pyramidal or granule cell layers in the allocortex
The degree of cell loss noted above in key limbic structures was notably mild. In response to mild stress, cells may be atrophied or hypertrophied, and sufficient cellular atrophy may lead to overall shrinkage of the brain region without any measurable change in cell numbers. Hippocampal atrophy in DLB is associated with the severity of cognitive impairments 58. Thus, a blinded investigator measured the width of the pyramidal layers of the rostral medial piriform cortex, caudal lateral piriform cortex, CA1, CA2, CA3 and the granule cell layer of the ventral dentate gyrus (Figure 8A,B). Fibril infusions reduced the width of these layers in the piriform cortices, CA1 and the dentate gyri of the youngest males. CA1, CA2 and CA3 were significantly reduced in width 6 months after fibril infusions in the young females and in the 17‐month‐old males. Fibrils also reduced the pyramidal layer widths of the caudolateral piriform cortex in the young females. Compared to young males, young fibril‐infused females displayed significantly thicker pyramidal or granule cell layers in the rostromedial piriform cortex, CA1 and dentate gyrus.
Figure 8.
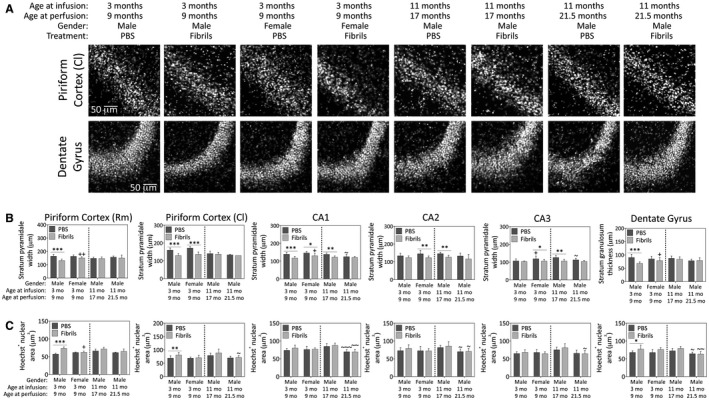
Preformed α‐synuclein fibril injections in the OB/AON induce atrophy of pyramidal and granule cell layers of the limbic allocortex in young male mice. Mice were infused in the right olfactory bulb/anterior olfactory nucleus with either preformed α‐synuclein fibrils (5 μg/1 μL) or an equivalent volume of PBS (1 μL). A blinded observer employed the “arbitrary line” tool in cellSens software to measure the width of the compact Hoechst+ pyramidal cell layer in the rostromedial piriform cortex, caudolateral piriform cortex, CA1, CA2 and CA3 of the hippocampus, or the stratum granulosum of the dentate gyrus. Representative grayscale micrographs for the caudolateral piriform cortex and dentate gyrus are displayed in panel (A) and the quantifications of pyramidal and granule cell layers width (B) and Hoechst+ nuclear area (C) are graphed below. Shown are the mean and SD of raw, unnormalized data. N = 3–8 mice per group (see Methods section and Figure 1 for animal numbers). Two‐way ANOVAs were followed by the Bonferroni post hoc correction. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001 PBS vs. fibrils; +P ≤ 0.05, ++P ≤ 0.01, +++P ≤ 0.001 vs. 3–9 month males; ~P ≤ 0.05, ~~P ≤ 0.01, ~~~P ≤ 0.001 vs. 11–17 month males. Abbreviations are defined in Table S2. To view the original, higher resolution Adobe Illustrator or EPS files, please link to https://dsc.duq.edu/pharmacology/ or https://www.dropbox.com/sh/a6r5ylg1tco6trm/AAC9Mb2gWuP29ABbmdGuNoaJa?dl=0
In order to determine whether the above changes reflect shrinkage of nuclear sizes, we also measured the area of Hoechst+ nuclei (Figure 8C). Fibril infusions significantly increased Hoechst+ nuclear sizes in the piriform cortices and the dentate gyri of the young males; these were the same brain regions that displayed shrinkage of strata width in this group. The hypertrophy of nuclei in young males was not evident in any of the other fibril‐infused groups, or in the sizes of the NeuN+ nuclei (data not shown), suggesting it may be attributable to glial hypertrophy in response to mild stress.
Anatomical range of α‐synucleinopathy after preformed fibril infusions in the murine OB/AON
Even in animals with extremely dense α‐synucleinopathy in the amygdaloid complex, piriform cortex, hippocampal formation, ectorhinal cortex and deeper layers of the entorhinal cortex, the pSer129‐labeled pathology did not extend widely out of the limbic connectome (Figure 9). The male depicted on the right of this figure was 11 months old at the time of fibril infusion and 17 months old at the time of perfusion, as was the PBS‐infused control in the left panels. Massive numbers of perinuclear and neuritic inclusions were observed in the AON and nucleus accumbens of this animal (Figure 10A–C). Some inclusions were also visible in the premammillary area, which is affected in human PD 45, the orbitofrontal and cingulate cortices, and the bed nucleus of stria terminalis—all part of the limbic system 99. A few lone inclusions were observed in the association motor cortex M2 (Figure 10A). Somal pSer129+ inclusions with perinuclear tendrils were found within the ventral midbrain (Figure 10D–H), but the cells harboring these inclusions did not exhibit convincing labeling for TH, the rate‐limiting enzyme for dopamine biosynthesis (video in Figure S2). In the vicinity of pSer129 inclusions, the TH label displayed a low signal to noise ratio and there was some pSer129‐/TH+ debris (Figure 10D–H; Figure S2). No pSer129+ inclusions were observed further lateral in the substantia nigra, pars compacta, including in the ventrolateral posterior subregion commonly associated with PD 42, 45, 60. In the ventral tegmental area of a number of other mice, the pSer129 label also did not convincingly colocalize with TH, with the potential exception of one neurite (see arrow in Figure S3).
Figure 9.
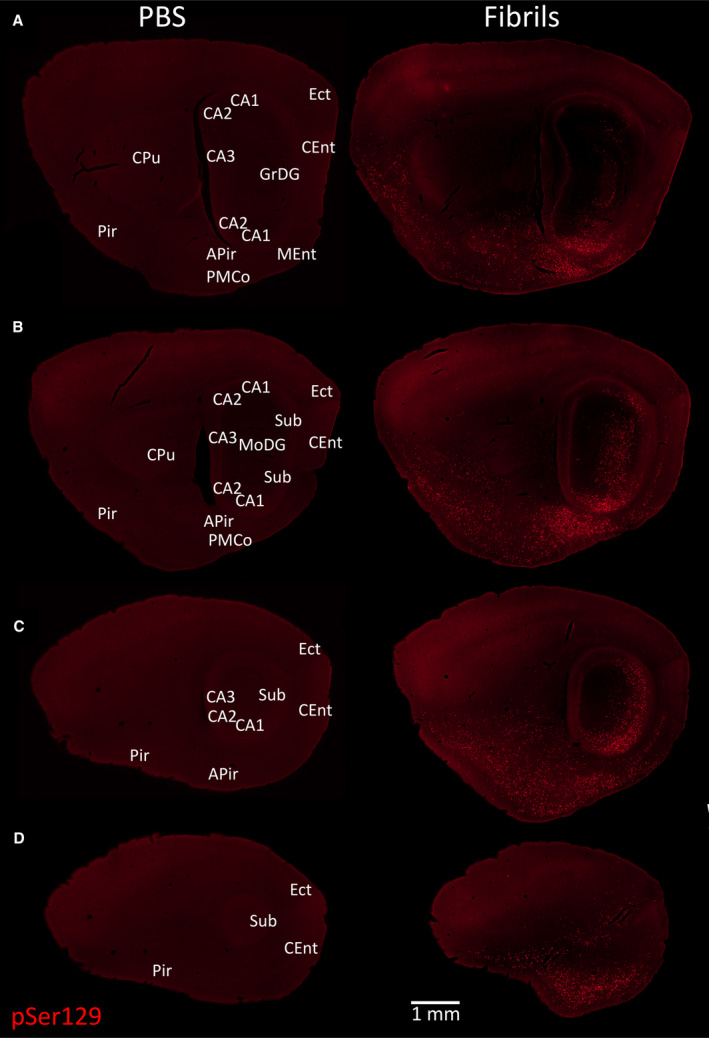
α‐synucleinopathy remains centered in the limbic allocortex 6 months following infusions of preformed α‐synuclein fibrils in the OB/AON. A series of stitched images of sagittal brain sections from two 17‐month‐old animals sacrificed 6 months after infusion of 1 µL PBS (left) or 5 µg/1 µL fibrils (right) into the OB/AON. All sections were stained in parallel with the monoclonal rabbit EP1536Y pSer129 antibody for pathologically phosphorylated α‐synuclein (see Table S1). Anatomical labels are based on cytoarchitectonic details provided by the Hoechst nuclear marker (not shown). All sections were processed in parallel and photographed at the same exposure and intensity scaling. Abbreviations are defined in Table S2. To view the original, higher resolution Adobe Illustrator or EPS files, please link to https://dsc.duq.edu/pharmacology/ or https://www.dropbox.com/sh/a6r5ylg1tco6trm/AAC9Mb2gWuP29ABbmdGuNoaJa?dl=0
Figure 10.
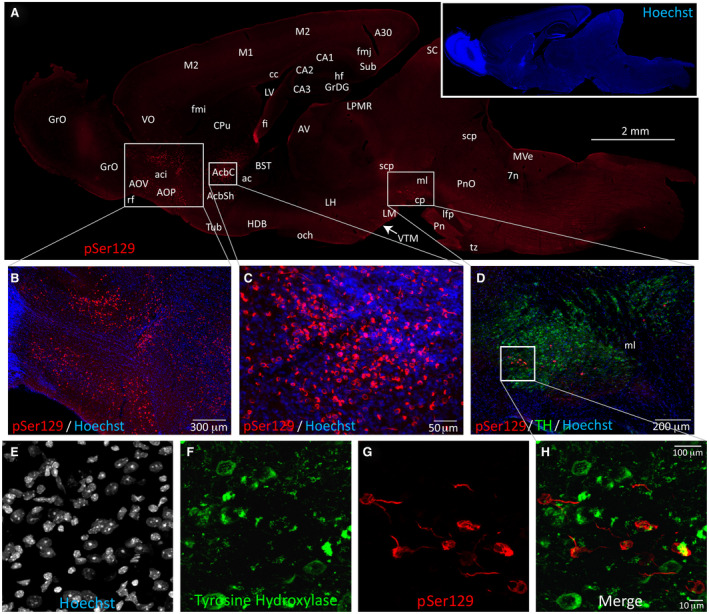
Mesencephalic α‐synucleinopathy is centered medially in the limbic‐related ventral tegmental area and does not colocalize with dopaminergic markers. A fibril‐injected 17‐month‐old mouse with the most widespread Lewy‐related pathology of all cases studied is displayed in this figure. (A) Stitched image of an entire brain section, with genuine α‐synucleinopathy at the boundary zone between the ventral tegmental area and substantia nigra, pars compacta (true boundaries were identified with antibodies against the dopaminergic marker tyrosine hydroxylase on the same section—see panel (D). Dense pSer129+ inclusions were observed in the anterior olfactory nucleus (AON) and nucleus accumbens (Acb), and sparse inclusions were observed in the olfactory bulb (OB), bed nucleus of stria terminalis (BST), orbitofrontal (VO) and cingulate cortices (A30), and ventral midbrain. A few sparse inclusions were observed in association neocortices (M2) of this one animal. Merged, higher magnification images of Hoechst+ cells (pseudocolored blue) and pSer129 immunostaining (pseudocolored red; EP1536Y rabbit monoclonal pSer129 antibody; see Table S1) in the AON (B) and accumbens (C). (D) Tyrosine hydroxylase (TH; pseudocolored green) and pSer129 (pseudocolored red) immunostaining in the ventral midbrain of the same animal. (E‐H) Lack of convincing colocalization of pSer129 and tyrosine hydroxylase in confocal images of the same animal. A movie of the rotating confocal Z‐stack for this animal can be viewed in Figure S2. The lack of convincing colocalization of pSer129 and tyrosine hydroxylase in additional animals is presented in Figure S3. Abbreviations are defined in Table S2. To view the original, higher resolution Adobe Illustrator or EPS files, please link to https://dsc.duq.edu/pharmacology/ or https://www.dropbox.com/sh/a6r5ylg1tco6trm/AAC9Mb2gWuP29ABbmdGuNoaJa?dl=0
Preformed fibril infusions in the OB/AON modify the expression of two dopaminergic markers in both the nigrostriatal and mesolimbic pathways of the old mice
To determine if fibril infusions affect the expression of dopaminergic markers, we immunostained the nigrostriatal and mesolimbic pathways with antibodies against TH and DAT (Figure 11). Fibril infusions increased the area of the caudoputamen in the 17‐month‐old males and decreased the area of the substantia nigra (includes the pars compacta and reticulata) in the 21.5 month‐old males (Figure 11A). The 21.5‐month‐old males also no longer displayed any significant increase in caudoputamenal area.
Figure 11.
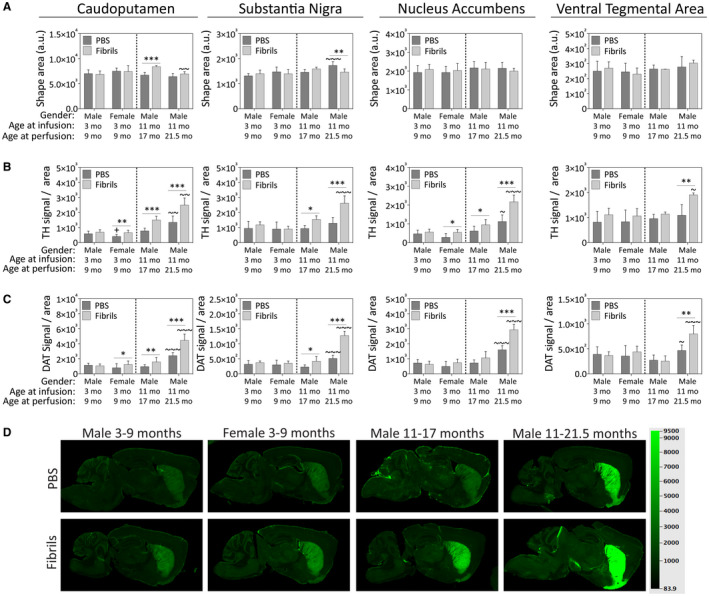
Preformed α‐synuclein fibril injections in the OB/AON increase the expression of dopaminergic markers in the nigrostriatal and mesolimbic pathways of older male mice. Mice were infused in the right olfactory bulb/anterior olfactory nucleus with either preformed α‐synuclein fibrils (5 μg/1 μL) or an equivalent volume of PBS (1 μL). Sagittal brain sections were immunostained with antibodies against the dopaminergic markers tyrosine hydroxylase (TH) and the dopamine transporter (DAT) and scanned on an ultrasensitive infrared imager (LiCor Odyssey). A blinded observer traced the anatomical boundaries of the dorsal striatum (caudoputamen), ventral striatum (nucleus accumbens), the substantia nigra and the ventral tegmental area in LiCor ImageStudio software. A. The shape areas drawn around the regions of interest are shown as raw data (unnormalized, arbitrary units) from ImageStudio software. B. TH signal is displayed after normalization to area, reflecting TH signal density. C. DAT signal per unit area is also shown, reflecting DAT signal density. D. Representative pseudocolored images of DAT immunostaining in all groups. Shown are the mean and SD. N = 3–8 mice per group (see Methods and Figure 1 for animal numbers). Two‐way ANOVAs were followed by the Bonferroni post hoc correction. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001 PBS vs. fibrils; +P ≤ 0.05 vs. 3–9 month males; ~P ≤ 0.05, ~~P ≤ 0.01, ~~~P ≤ 0.001 vs. 11–17 month males. Abbreviations are defined in Table S2. To view the original, higher resolution Adobe Illustrator or EPS files, please link to https://dsc.duq.edu/pharmacology/ or https://www.dropbox.com/sh/a6r5ylg1tco6trm/AAC9Mb2gWuP29ABbmdGuNoaJa?dl=0
We observed a dramatic fibril‐induced increase in TH signal in the oldest male mice in all four brain regions (Figure 11B). The 17‐month‐old mice also exhibited a fibril‐induced increase in TH signal in all but the ventral tegmental area. The increases in TH signal were age‐dependent, as they were absent from the young males. In all four regions, the passage of additional time after fibril infusions elicited a robust increase in TH signal in the 21.5‐month‐old males relative to the 17‐month‐old males. Fibril infusions also increased TH signal in the young female mice in the caudoputamen and nucleus accumbens. Measurements of DAT levels confirmed similar trends as the TH data (Figure 11C,D), confirming a robust increase in dopaminergic marker expression in response to fibril infusions in the oldest male mice.
Preformed fibril infusions in the murine OB/AON modify cell numbers and dopaminergic phenotype in the ventral mesencephalon
In total, 624 images of all midbrain sections were stitched and all TH+ cells that overlapped with a Hoechst+ nucleus were counted by a blinded observer. In these sagittal sections, the boundaries of dorsal and ventral tiers of the nigra, as originally defined by Fallon and Moore 59, were not clearly identifiable. Therefore, we only counted TH+ and Hoechst+ cells within the ventral tegmental area (A10), substantia nigra pars compacta (A9) and the retrorubral field (A8) (Figure 12). In the sagittal sections of the medial nigra, the TH+ area rostral to the medial lemniscus was readily subdivisible into anterior and posterior segments containing substantia nigra, pars compacta neurons and into clusters of TH+ cells in the retrorubral field caudal to the medial lemniscus (Figure 12E). The most lateral segments of the substantia nigra, pars compacta were not divisible along the rostrocaudal axis in the sagittal plane and were therefore counted as a single unit comprising part of the A9 group.
Figure 12.
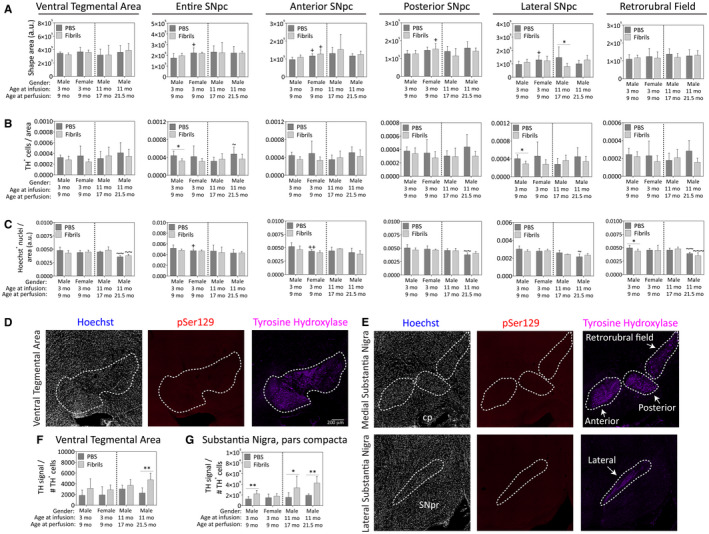
Impact of preformed α‐synuclein fibril injections in the OB/AON on dopaminergic cell numbers in the ventral mesencephalon. Mice were infused in the right olfactory bulb/anterior olfactory nucleus with either preformed α‐synuclein fibrils (5 μg/1 μL) or an equivalent volume of PBS (1 μL). Sagittal brain sections were stained with antibodies against the dopaminergic marker tyrosine hydroxylase (TH) and the nuclear marker Hoechst and 200× images were stitched together to form large, high‐resolution photomontages of the ventral mesencephalon from every section of a 1‐in‐5 sagittal series. A blinded observer traced the anatomical boundaries of the ventral tegmental area and the anterior, posterior and lateral subregions of the substantia nigra, pars compacta in cellSens software. A. The areas of the shapes drawn around the regions of interest are shown as raw data (unnormalized, arbitrary units per stitched section). B. Every single visible TH+ cell encompassing a Hoechst+ nucleus in all the ventral midbrain‐containing sections was manually counted by a blinded observer. In the medial substantia nigra, pars compacta, counts were made separately in the anterior and posterior nigra based on boundaries observed in the TH staining (see top row of panel E). The lateral substantia nigra was counted separately, also based on boundaries observed in the TH staining (see bottom row of panel E). Data are presented as area of the region of interest in arbitrary units (A), TH+ cell counts per unit area (B), and Hoechst+ cell counts per unit area (C), within the boundaries of the dotted lines, as shown in panels D‐E. Note that two of the panels in D‐E are from fibril‐infused mice exhibiting no pSer129 pathology in the ventral midbrain to illustrate the lack of background staining in this region. Raw TH+ cell counts per animal (see Figure S4 for raw data) were divided by the total TH signal generated on the LiCor imager for the ventral tegmental area (F) and substantia nigra, pars compacta (G) as a measure of “TH expression per cell.” Two‐way ANOVAs were followed by the Bonferroni post hoc correction. Shown are the mean and SD from an n of 3–8 mice per group (see Methods and Figure 1 for animal numbers). *P ≤ 0.05 for PBS vs. fibrils; +P ≤ 0.05 vs. 3–9 month male group; ~P ≤ 0.05, ~~P ≤ 0.01, ~~~P ≤ 0.001 vs. 11–17 month male group. Abbreviations are defined in Table S2. To view the original, higher resolution Adobe Illustrator or EPS files, please link https://dsc.duq.edu/pharmacology/ or https://www.dropbox.com/sh/a6r5ylg1tco6trm/AAC9Mb2gWuP29ABbmdGuNoaJa?dl=0
Fibril infusions elicited no major changes in TH+ regional areas, other than a reduction in the size of the lateral substantia nigra, pars compacta in the 17‐month‐old males (Figure 12A). Note that these regional areas differ from those in Figure 11 in that the latter traces also included the pars reticulata because of the lower resolution of the images captured on the Odyssey imager and our desire to include expression of dopaminergic markers in the perpendicular dendritic extensions of pars compacta neurons into the pars reticulata 93. The raw TH and Hoechst cell counts are presented to the reader in Figure S4. The cells per unit area presented in Figure 12B,C were calculated by dividing those raw numbers by the areas in which they were counted (see Figure 12A). Fibril infusions reduced TH+ cell density in the substantia nigra, pars compacta, when defined as a single structure, in the young males (Figure 12B). This effect may be explained by the focused fibril‐induced drop in TH+ cell density of the lateral substantia nigra, pars compacta, in the young males, an effect that was absent from the other groups. Fibril infusions elicited mild reductions in Hoechst+ cell density in the retrorubral field in young males (Figure 12C). There were no other fibril‐induced effects on Hoechst+ cell densities, suggesting a lack of true fibril‐induced cell death in any other part of the dopaminergic ventral mesencephalon.
In order to examine the impact of fibril infusions on TH expression level per TH+ cell number in the ventral midbrain, the raw TH signal generated on the Odyssey imager (Figure 11B) was divided by the raw TH+ cell counts calculated above for each animal. Fibril infusions raised TH protein expression per TH+ cell in the substantia nigra in all male groups and in the ventral tegmental area in the oldest male group (Figure 12F,G), perhaps as a compensatory response to mild proteotoxic stress.
Female mice infused with preformed fibrils in the OB/AON have fewer olfactory deficits and greater motor deficits than males
The primary goal of our study was to examine the histopathological effects of fibril infusions using the fewest animals possible. Because of limited resources, only a small number of behavioral tests were performed, with foci on olfactory, motor and cognitive abilities. Furthermore, individual behavioral tests were not repeated numerous times on each animal, to avoid undue influences of learning on performance 74, with the exception of the buried vs. exposed peanut test, which was the only test performed in the oldest male mice at any time point longer than 6 months post‐surgery.
Fibril infusions did not significantly influence the latency to contact an exposed peanut at 3 months post‐infusion, after excluding the mice that failed to show any interest in peanuts, suggesting equivalent motivation to find food in the remaining cohort (Figure 13A). However, by 10 months post‐surgery, the oldest PBS‐infused males contacted the exposed peanut faster than at 3 months post‐surgery, which may be the result of reexposure to and familiarity with the test.
The fibril‐treated young males were slower at locating buried food than PBS‐treated age‐matched controls, whereas female mice of the same age and the 14‐month‐old males were not similarly impaired after fibril treatment (Figure 13B). However, the oldest fibril‐infused males contacted buried food significantly slower at 10 months post‐surgery (at 21 months of age) than at 3 months post‐surgery (at 14 months of age; Figure 13B), consistent perhaps with the loss of NeuN+ cells in the AON of this group of animals (see Figure 3B).
In tests of motor function, female fibril‐infused animals exhibited less spontaneous rearing behavior and made fewer contacts with both forelimbs than the young fibril‐infused males (Figure 13C,D). Furthermore, fibril infusions significantly reduced the number of forelimb contacts in females, consistent with the reduction in spontaneous rearing behavior in the same group.
As Lewy pathology‐induced dysfunction in hippocampal neurons may contribute to symptoms of dementia 71, we performed the novel object and novel place recognition tests to assess spatial memory and learning. In the novel object test, the older fibril‐infused males spent significantly less time contacting the novel object than the young fibril‐infused males (Figure 13E). Compared to male mice, female PBS‐infused animals spent more time exploring the novel object (Figure 13E), both objects (Figure 13F), and spent less time exploring the novel place (Figure 13G). The older males tended to spend less time exploring the familiar object in the novel place in response to fibril infusions—these latter results were statistically significant by a two‐tailed t test, and almost reached statistical significance after the conservative Bonferroni correction for multiple comparisons was applied (p = 0.055 after Bonferroni correction; Figure 13G).
Discussion
The main finding of the present study is that the induction of α‐synucleinopathy in the OB/AON complex is associated with Lewy‐related pathology firmly positioned in the limbic system, spanning the piriform cortex, amygdala, entorhinal cortex and hippocampus of the temporal lobe, and extending in some cases to the nucleus accumbens and ventral tegmental area. Limbic‐centered pathology is evident for at least 6 months post‐infusion, regardless of sex in 9‐month old male and female mice, and irrespective of age in 17‐month old males. In the 21.5‐month old males, our results may be skewed toward “superager” survivors because of unexpected attrition of animals. In all animals, however, we failed to observe convincing pathology in the brainstem (pons and medulla oblongata) or within the anatomical boundaries of the substantia nigra, pars compacta, strictly defined by TH immunolabeling, despite the advanced age of the oldest survivors and their almost 11‐month‐long post‐infusion survival period.
The second main finding is the dramatic increase in mortality in the aged males. Our finding that fibril infusions accelerate mortality in male mice by 21.5 months of age is noteworthy, given the associations between human smell loss, olfactory bulb histopathology, neurodegenerative disease and mortality 10, 11, 53, 54, 55. Pinto and colleagues reported that 40% of older adults with complete smell loss were dead within 5 years (compared to only 10% of older adults with intact smell perception), and that smell loss was linked to higher mortality rates than heart failure, stroke, cancer and diabetes 138. Rey et al. did not report an increase in mortality from olfactory‐seeded α‐synucleinopathy, but their mice were 3 months old at the time of fibril injections 142, 143. In the present study, animals injected at 3 months of age also did not exhibit an increase in mortality, whereas infusions in the 11‐month‐old mice only exerted significant lethal effects by 10.5 months post‐infusion. In the latter 21.5‐month‐old males, fibril infusions elicited mild loss of NeuN+ neurons in the AON. In contrast, Rey and colleagues achieved severe cell loss in this structure 6 months after fibril infusions in the OB of young C57 mice 142, perhaps because of differences in fibril sonication protocols or mouse strain; they employed C57 mice, whereas we examined CD1 mice.
The third main finding is that female mice develop fewer inclusions than males in many subregions of the limbic system, supporting the view that the higher risk of Lewy body disorders in men may be partly related to gender‐dependent propensities to develop α‐synuclein protein aggregations. Fibril infusions failed to elicit any loss of NeuN+ or Hoechst+ cells in any brain region in females, whereas age‐matched male mice exhibited fibril‐induced losses of NeuN+ and Hoechst+ cells in the posteromedial cortical amygdala and the nucleus of the lateral olfactory tract, as well as loss of Hoechst+ cells in the piriform cortices and nucleus accumbens. In addition, the female mice were less impaired in finding buried food than males after fibril infusions, although additional olfactory tests must be completed to confirm this sex difference. The latter observation is not unexpected given our histological findings and clinical reports that olfactory dysfunction is more common in cases with limbic and neocortical‐predominant Lewy pathology than cases with brainstem‐predominant Lewy pathology 16, 18, 170.
The sex differences reported here seem consistent with clinical observations that women with PD perform better than male PD patients in the University of Pennsylvania Smell Identification Test 160. Other notable sex differences include greater rigidity and cognitive dysfunction in male PD patients, whereas women with PD are more likely than men to display severe tremors, postural imbalances and dyskinesias 64. Perhaps for the latter reasons, fibril‐infused females in the present study engaged in lower levels of spontaneous rearing behavior and made fewer forelimb contacts with the walls of a cylinder. However, the results of our motor tests are not readily explained by sex differences in TH or DAT expression in the nigrostriatal pathway and may therefore be mediated by extranigral neural circuitry, such as the sensorimotor cortex or cerebellum, or by sex‐dependent differences in electrophysiological activity that are not captured by simple histological assays. Kim and colleagues reported a lack of gender differences following infusions of preformed fibrils in the striatum of wild‐type mice, supporting the view that the induction site and the route of spread are critical 95. However, Cantuti‐Castelvetri and colleagues observed that nigral dopamine neurons in male subjects display higher baseline expression of α‐synuclein protein 36, suggesting that males may have more endogenous α‐synuclein molecules available as a substrate for seeding and aggregation.
Estrogen use beginning early in menopause reduces the risk of developing dementia in female subjects with PD 107 whereas bilateral oophorectomy increases the risk of parkinsonism 19, 148. Furthermore, women who are diagnosed with PD typically experience earlier menopause and curtailed reproductive lifespans 122. These findings are consistent with experimental observations that estrogens destabilize and/or de‐aggregate α‐synuclein fibrils 81. If sex steroids influence the emergence of inclusions and the risk of developing Lewy body disorders 116, 151, 152, then the neuroprotective effects of estrogens may extend beyond dopaminergic circuitry and suppress the density of telencephalic α‐synucleinopathy and its sphere of transmission. In our model, females failed to develop significantly more inclusions than males in any brain region examined, and fewer females than males displayed any inclusions in the olfactory tubercle and dorsal striatum (see heatmap in Figure 2). Given these collective observations, it seems plausible that females develop aggregations less readily and display more restricted spread. However, further studies are needed to test if the neuroanatomy of males and females differ at the macroscopic or microcircuitry level, which might lead to slight variations at the injection site or in spreading patterns, respectively.
A number of unexpected results in our study deserve mention. First, female mice displayed a slight increase in cells in the OB. It is worth establishing in future work whether this reflects a small increase in neurogenesis, which serves to repopulate OB cells via the rostral migratory stream 136 and is thought to be higher in females 48. Second, we observed a fibril‐induced loss of cells in the young males—but not the older mice—in a number of brain regions, such as the nucleus of the lateral olfactory tract and the piriform cortices (eg, Figures 3 and 4). Thus, it seems possible that the most vulnerable cellular populations in some brain regions are lost with aging, and that cells able to survive this attrition have sufficient fortifications to survive further proteotoxic stress; such resilience could underlie the slow progression of proteinopathic brain disorders. An additional explanation is that the group of older mice were skewed toward a superager phenotype, as alluded to above. Indeed, a superager phenotype would also explain why older mice do not always harbor more inclusions than the younger cohorts. In the caudoputamen, for example, the 21.5‐month group displays lower inclusion counts than the 17‐month‐old group.
A third unusual finding was that the 14 month‐old males did not display a higher latency to contact the buried peanut after fibril infusions. Odor naming capacity follows an inverted U‐shaped curve over the lifespan, being highest in the young adults and poorest in the very elderly 43, but stress levels are a confounding factor in virtually all rodent behavior studies, and can even be influenced by the gender of the human investigator 158. In human subjects, the SF‐36 mental health composite and the reverse‐coded Perceived Stress Scale scores display inverted U‐shapes across the lifespan, with the greatest resilience in middle age 163. Although all our animals are born and raised in the same facility, the middle‐aged males have experienced more human handling than the young males over the course of their longer lives. Perhaps for these reasons, the middle‐aged mice appeared less skittish than the other male groups. By 10 months post‐infusion, the oldest males were impaired in finding the peanut after fibril infusions, perhaps because significant loss of NeuN+ neurons had emerged in the AON by that time (Figure 3B).
Finally, we observed that PBS‐injected female mice explore novel objects more than male mice, but explore novel places less than males. In this context, it may be relevant that women appear to have superior memory measures 140, but worse spatial skills compared to men 23. There are too many other significant differences in the present report to discuss every single effect, but we caution that the effect sizes are often small and may not be biologically meaningful even if statistically significant. An added complication of murine behavior studies is the ~65–90 million years of evolutionary divergence between mice and humans 57, 104, 114, and the preferential reliance of rodents on olfactory rather than visual cues.
Olfactory‐induced α‐synucleinopathy does not reproduce PD‐related pathology within the substantia nigra, hippocampal formation or amygdaloid complex
At first glance, our findings seem consistent with the suppositions of Cersosimo, who recently posited an olfactory origin of the limbic α‐synucleinopathy in DLB 38. However, we hesitate to conclude that our OB/AON infusion model fully simulates DLB, PD or even iLBD, as these conditions are associated with loss of dopaminergic markers of the nigrostriatal pathway and true nigral cell death, and PD and DLB are associated with a more widespread extent of Lewy‐related pathology than in our murine model 16, 17, 85, 115, 137, 155. For example, a ~40% loss of pigmented nigral neurons and a ~50% reduction in TH is already evident in the striatum of iLBD patients 16, 17, 85. Furthermore, iLBD patients have fewer TH+ cells and reduced nigral neuron densities as early as Braak stages I and II, prior to the deposition of misfolded α‐synuclein in this brain region 115, further supporting the uncoupling of cell loss from Lewy pathology 41. In PD, the frequency and density of Lewy pathology in the medulla, pons and substantia nigra are similar to that in the limbic system (see Figure 2C,D in Beach’s work 16). Even in iLBD, pathology is evident in both the medulla and OB, and limbic pathology outside of the OB is observed less frequently than caudal brainstem pathology (see Figure 2A,B in Beach’s work 16).
As the brunt of α‐synucleinopathy in our material is centered in the limbic connectome, rather than taking firm root in both the brainstem and limbic system, we posit that our model captures some but not all aspects of the early limbic‐predominant Stage IIb of the unified staging system 16, 100, 164. Had we achieved immunostaining of sufficiently high quality in the 21.5‐month‐old animals that died prematurely, it is, of course, possible they would have exhibited some degree of brainstem pathology. On the other hand, according to other investigations in the preformed fibril model, Lewy‐related pathology is widespread even as early as 6 months post‐infusion 105, 133, 142, 144, at which time we failed to observe robust extralimbic α‐synucleinopathy. It is critical to note that Rey and colleagues noted a broader sphere of α‐synucleinopathy transmission following OB fibril infusions even through their mice were of the same age and survival period as in our study 141, 144. These facts contradict the argument that shorter survival periods per se and different ages at induction underlie the topographically restricted pathology in our hands. Rather, it seems more probable that fibril sonication protocols explain the differences across studies, as stated above 139. Our findings also differ significantly from a recent report that olfactory bulb α‐synucleinopathy causes similar pathology as in prodromal PD 123, but their model was generated by α‐synuclein overexpression.
In our previous report, retrogradely labeled FluoroGold+ cells and pSer129+ inclusions were reported in or near the parabrachial pigmented nucleus (part of the ventral tegmental area) following OB/AON infusions 109. At longer survival periods, approximately half of all the animals in the present study displayed inclusions in the ventral tegmental area or the boundary zone between the ventral tegmental area and the medial substantia nigra. The ventral tegmental area and medial substantia nigra are associated with the limbic system, whereas lateral and central subregions of the substantia nigra are involved with associative and striatal motor systems 68, 79. The dopaminergic subregions most vulnerable both to cell loss and the development of Lewy inclusions in human PD are concentrated caudally, ventrally and laterally within the tiers or nigrosomes of the substantia nigra 42, 45, 60. Although there are reports of mild cell loss in the A10 group in patients 4, 113, the foci of Lewy‐like pathology as well as cell loss in the ventral tegmental area (rather than the nigra proper) in our material seems at odds with the greater vulnerability of the nigral pars compacta in PD patients 26, 42, 97. Had the caudoputamen been more densely labeled in our material than the accumbens, we would have expected greater nigrostriatal than mesolimbic involvement. We did observe mild cell loss in the A8 group of the retrorubral field in young fibril‐infused males, but this area is resistant to degeneration in PD patients 113. Furthermore, our investigations failed to support convincing colocalization of the TH+ stain with pSer129 immunoreactivity in the midbrain. One explanation is that TH immunoreactivity may be suppressed in neurons with dense α‐synucleinopathy 5. Alternatively, non‐dopaminergic efferent projections from the substantia nigra, pars compacta and ventral tegmental area to the amygdaloid complex have been reported 103, as well as non‐dopaminergic projections from the ventral tegmental area to the nucleus accumbens 149. A recent tract‐tracing study also reported a direct nigro‐olfactory projection in rats, and the tracer appeared in the medial half of the substantia nigra, pars compacta (see Figure 2D in 83), which, as stated above, is more closely associated with the limbic system.
Of all cornu Ammonis hippocampal fields, the small CA2 subregion displays the earliest and densest Lewy pathology in both PD and DLB 2, 44, 45. According to our previous work and the present study, the earliest and densest hippocampal pathology in fibril‐infused mice was not in CA2, but centered in ventral CA1 and the subiculum, consistent with the dense retrograde labeling of these two structures following FluoroGold infusions in the OB/AON complex 109 and with a large body of work on the afferents of anterior olfactory brain regions (eg, see Table 2 in 31) 69, 70. If the spreading hypothesis for α‐synuclein transmission is correct, and the findings of our model eventually translate to humans, the initial transmission of Lewy pathology into CA2 of the hippocampal formation in humans may not originate at olfactory sites. However, such interpretations must be approached with caution, as macrosmatic mice may differ from microsmatic humans in the strengths and patterns of connections between olfactory and hippocampal structures.
The amygdala often harbors some of the densest Lewy pathology in human autopsy material 16. Atrophy of limbic structures such as the amygdala is strongly associated with olfactory dysfunction 13, and PD patients with severe hyposmia also display a significant decrease in the functional connectivity of the amygdala, as determined by magnetic resonance imaging 172. These clinical observations are consistent with first‐order inputs to the cortical amygdala from the OB 128, and with reports that the cortical amygdala displays both Lewy pathology and neuron loss in patients with PD 24, 76. The latter features were reproduced in our model, and are consistent with the view that the cortical amygdaloid nuclei form part of the greater olfactory paleocortex. On the other hand, Braak reported that the Lewy pathology in the amygdala initially arises in the central nucleus 24, 45, which is densely connected with vagus‐related nuclei of the brainstem 145, 146, 147. The early involvement of the cortical rather than central amygdala, of CA1 rather than CA2, and the mesolimbic rather than nigrostriatal pathway in our model therefore do not recapitulate the patterns of Lewy pathology in early PD with any degree of precision.
α‐synucleinopathic inclusions in the preformed fibril model are composed of aggregated proteins
The staining of pSer129+ inclusions with the Proteostat aggresomal dye is consistent with previous studies on similarities between Lewy bodies and aggresomes 112, 161. The formation of the aggresome is microtubule‐dependent 96 and α‐synuclein appears to interact with tubulin, perhaps serving as a microtubule dynamase 6, 37, 135. Aggresomes are hypothesized to serve as a protective mechanism facilitating cellular survival under proteotoxic conditions 162, as they are observed in 53%–60% of non‐apoptotic cells but only 10%–13% of apoptotic cells in vitro 161. In our material, not every single Proteostat+ aggregations overlapped with pSer129+ inclusions or vice versa. In this context, it is important to recognize that only a subset of somal inclusions in human tissue conform to the criteria for classic Lewy bodies 16. Thus, the multiple types of Lewy‐related aggregations with heterogeneous protein compositions may be an advantage of our model.
Additional caveats and conclusions
The present study includes three distinct measurements on 16 individual brain regions or subregions (spanning 7296 total images) and would not have been feasible had we only employed stereological counting tools. Obviously, there are caveats to our higher throughput approach, including that stereological methods account for changes in the overall size of the tissue and yield approximations of “absolute” numbers of labeled structures, whereas our measurements report density of labeling. However, many impactful postmortem studies on Lewy body disorders have relied on similar methods (ie, reporting density of α‐synucleinopathy and of nigral cells) and relied heavily on qualitative measures, such as heatmaps, because of the wide scope of the studies 16, 26, 164.
Evidence collected here strongly suggests that pathology induced at a single anatomical site (the OB/AON complex) differs from the fundamentally more complex human PD, DLB and iLBD phenotypes. We cannot expect a preclinical model in short‐lived and small creatures to capture all aspects of any human condition, but, if the hodological approach often applied in preformed fibril models translates to clinical studies, we believe our data agree with Braak’s original speculation that “ … the pathology in the anterior olfactory nucleus makes fewer incursions into related areas than that developing in the brainstem” 26. An alternative view is that there is no obligatory trigger or induction site or Lewy body disorders 9, 89, 90, 131, or even directional movement of α‐synucleinopathy; instead, pathology might arise simultaneously in many parts of the central and peripheral nervous systems caused by spontaneous mutations, which are quite common 94. Beach reasons that the emergence of α‐synuclein aggregates may be a stochastic process not related to cell‐to‐cell spread but to “probabilistic rather than deterministic factors” 16. In a study of >1000 brain autopsies, there were no cases with peripheral but not central Lewy pathology, which does not support the gut‐to‐brain transmission concept 3. Although there are important exceptions 16, clinical symptoms have not always been linked to the density and extent of Lewy pathology 32, 129. Indeed, Parkkinen et al. concluded that “The identification of cases with a reasonably high burden of alpha‐synuclein pathology in both brainstem and cortical areas without clinical symptoms suggests that there must be certain unrecognized pathologies of decisive importance that mediate the biological abnormality” 131. With current attention focused on α‐synuclein, it is also not widely appreciated that Lewy bodies may contain up to ~300 other proteins 101. In short, there are unexplained heterogeneities in Lewy body disorders that have impeded our understanding 15, 32, as well as heterogeneity in the physical structures of the Lewy‐related aggregations and in the proteins trapped in their clutches.
Despite some caveats, the murine preformed fibril model with injections in various brain locations seems reasonably suited as the first rough screen for sex and age differences, mortality risk, heterogeneities in induction sites, varying compositions and structures of protein inclusions, differences in patterns of spread, and divergent behavioral sequelae of unique regional patterns of Lewy‐related pathology. Our OB/AON infusion model is specifically noteworthy in the atypical density of some of the pSer129+ pathology, perhaps because our sonication protocol was previously optimized in vivo based on this very readout 109, or because the Lewy‐like aggregations in our hands are less toxic and more closely related to protective aggresomes and do not kill the host cells in massive numbers. To us, it seems important to test therapies in models with mild cell loss and limited topographic extents of pathology—as observed here—because our only hope at curing these conditions may be those interventions initiated before substantial, irreversible cell loss unfolds and the disease has spewed shoots into widespread neuroanatomical circuits.
Supporting information
Figure S1. Preformed α synuclein fibril injections in the olfactory peduncle induce a similar topographic extent of α synucleinopathy in males and females. Mice were infused in the right olfactory bulb anterior olfactory nucleus with either preformed α synuclein fibrils (5 μg 1 μL) or an equivalent volume of PBS (1 μL). Sagittal sections were immunostained for pathologically phosphorylated α synuclein (EP1536Y rabbit monoclonal pSer129 antibody; see Table S1). The nuclear marker Hoechst (pseudocolored blue) was used to denote cytoarchitectonic boundaries. Stitched images of sagittal brain sections from 9 month old male (top) and female (bottom) mice perfused 6 months post infusion are displayed from medial to lateral. All sections were processed in parallel and images were captured at the same camera and exposure settings. Note that you must zoom in on the computer screen to view the pathology. Abbreviations are defined in Table S2. To view the original, higher resolution Adobe Illustrator or EPS files, please link to https://dsc.duq.edu/pharmacology/ or https://www.dropbox.com/sh/a6r5ylg1tco6trm/AAC9Mb2gWuP29ABbmdGuNoaJa?dl=0
Figure S2. Neurons with α synucleinopathic inclusions at the boundary zone between the substantia nigra and the ventral tegmental area are not tyrosine hydroxylase+. Mice were infused in the right rear olfactory bulb with either preformed α synuclein fibrils (5 μg 1 μL) or an equivalent volume of PBS (1 μL). After 6 months, sagittal brain sections were stained with the rabbit monoclonal EP1536Y pSer129 antibody (pseudocolored red) for pathologically phosphorylated α synuclein (see Table S1) and tyrosine hydroxylase (pseudocolored green). Nuclei were counterstained with the Hoechst reagent (pseudocolored blue). This confocal Z stack video is of the same brain section as displayed in Figure 10. No colocalization of the two markers was observed. To view the original, higher resolution Adobe Illustrator or EPS files, please link to https://dsc.duq.edu/pharmacology/ or https://www.dropbox.com/sh/a6r5ylg1tco6trm/AAC9Mb2gWuP29ABbmdGuNoaJa?dl=0
Figure S3. α synucleinopathic inclusions in the ventral tegmental area are not tyrosine hydroxylase+. Mice were infused in the right rear olfactory bulb with either preformed α synuclein fibrils (5 μg 1 μL) or an equivalent volume of PBS (1 μL). After 6 months, sagittal brain sections were stained with the rabbit monoclonal EP1536Y pSer129 antibody (pseudocolored red) for pathologically phosphorylated α synuclein (see Table S1) and tyrosine hydroxylase (TH; pseudocolored green). No robust colocalization of the two markers was observed, and the arrow in the bottom row points to a single pSer129 positive structure that may be lightly TH positive. Abbreviations are defined in Table S2. To view the original, higher resolution Adobe Illustrator or EPS files, please link to https://dsc.duq.edu/pharmacology/ or https://www.dropbox.com/sh/a6r5ylg1tco6trm/AAC9Mb2gWuP29ABbmdGuNoaJa?dl=0
Figure S4. Impact of preformed α synuclein fibril injections in the OB AON on dopaminergic cell numbers in the ventral mesencephalon. Mice were infused in the right olfactory bulb anterior olfactory nucleus with either preformed α synuclein fibrils (5 μg 1 μL) or an equivalent volume of PBS (1 μL). Sagittal brain sections were stained with antibodies against the dopaminergic marker tyrosine hydroxylase (TH) and the nuclear marker Hoechst and 200× images were stitched together to form large, high resolution photomontages of the ventral mesencephalon from every section of a 1 in 5 sagittal series. A blinded observer traced the anatomical boundaries of the ventral tegmental area and the anterior, posterior, and lateral subregions of the substantia nigra, pars compacta in cellSens software. (a) Every Hoechst+ cell (within the TH+ area) and every TH+ cell encompassing a Hoechst+ nucleus was manually counted by a blinded observer. These raw data were then used for the cell density measurements presented in Figure 12. Data are presented as (a) raw, unnormalized TH+ cell counts, and (b) raw, unnormalized Hoechst+ cell counts. Two‐way ANOVAs were followed by the Bonferroni post hoc correction. Shown are the mean and SD from an n of 3 8 mice per group (see Methods and Figure 1 for animal numbers). P ≤ 0.05, P ≤ 0.01 for PBS vs fibrils; P ≤ 0.05 vs 3 9 month male group; P ≤ 0.05 vs 11 17 month male group. Abbreviations are defined in Table S2. To view the original, higher resolution Adobe Illustrator or EPS files, please link to https://dsc.duq.edu/pharmacology/ or https://www.dropbox.com/sh/a6r5ylg1tco6trm/AAC9Mb2gWuP29ABbmdGuNoaJa?dl=0
Acknowledgments
Funded by NINDS/NIA grants 1R15NS093539‐01 (PI RKL), 1R21NS107960‐01 (PI RKL) and NSF 1726368 (Co‐PI RKL). RKL designed and directed the studies and wrote the paper. DMM helped to draft the paper and design the studies, performed all the brain surgeries, perfusions, behavioral analyses and statistical analyses. DMM, YW, TNB, KMM, SAT and JFS conducted microscopy and image analyses. KCL provided the fibrils and helpful discussions. All authors provided feedback on the manuscript, including significant feedback from TNB. We thank Joe Sallmen for generous help with confocal microscopy, Nancy Hosni and Deb Willson for excellent administrative support, and Denise Butler‐Buccilli and Christine Close for outstanding animal care.
None of the authors have any conflicts of interest to declare.
References
- 1. Aarsland D, Perry R, Brown A, Larsen JP, Ballard C (2005) Neuropathology of dementia in Parkinson's disease: a prospective, community‐based study. Ann Neurol 58:773–776. [DOI] [PubMed] [Google Scholar]
- 2. Adamowicz DH, Roy S, Salmon DP, Galasko DR, Hansen LA, Masliah E, Gage FH (2017) Hippocampal alpha‐synuclein in dementia with lewy bodies contributes to memory impairment and is consistent with spread of pathology. J Neurosci 37:1675–1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Adler CH, Beach TG (2016) Neuropathological basis of nonmotor manifestations of Parkinson's disease. Mov Disord 31:1114–1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Alberico SL, Cassell MD, Narayanan NS (2015) The vulnerable ventral tegmental area in Parkinson's disease. Basal Ganglia 5:51–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Alerte TN, Akinfolarin AA, Friedrich EE, Mader SA, Hong CS, Perez RG (2008) Alpha‐synuclein aggregation alters tyrosine hydroxylase phosphorylation and immunoreactivity: lessons from viral transduction of knockout mice. Neurosci Lett 435:24–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Alim MA, Ma QL, Takeda K, Aizawa T, Matsubara M, Nakamura M et al (2004) Demonstration of a role for alpha‐synuclein as a functional microtubule‐associated protein. J Alzheimers Dis 6:435–442; discussion 43–9. [DOI] [PubMed] [Google Scholar]
- 7. Amschl D, Neddens J, Havas D, Flunkert S, Rabl R, Romer H et al (2013) Time course and progression of wild type alpha‐synuclein accumulation in a transgenic mouse model. BMC Neurosci 14:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Anderson JP, Walker DE, Goldstein JM, de Laat R, Banducci K, Caccavello RJ et al (2006) Phosphorylation of Ser‐129 is the dominant pathological modification of alpha‐synuclein in familial and sporadic Lewy body disease. J Biol Chem 281:29739–29752. [DOI] [PubMed] [Google Scholar]
- 9. Attems J, Jellinger KA (2008) The dorsal motor nucleus of the vagus is not an obligatory trigger site of Parkinson's disease. Neuropathol Appl Neurobiol 34:466–467. [DOI] [PubMed] [Google Scholar]
- 10. Attems J, Walker L, Jellinger KA (2014) Olfactory bulb involvement in neurodegenerative diseases. Acta Neuropathologica 127:459–475. [DOI] [PubMed] [Google Scholar]
- 11. Attems J, Walker L, Jellinger KA (2015) Olfaction and aging: a mini‐review. Gerontology 61:485–490. [DOI] [PubMed] [Google Scholar]
- 12. Baba M, Nakajo S, Tu PH, Tomita T, Nakaya K, Lee VM et al (1998) Aggregation of alpha‐synuclein in Lewy bodies of sporadic Parkinson's disease and dementia with Lewy bodies. Am J Pathol 152:879–884. [PMC free article] [PubMed] [Google Scholar]
- 13. Baba T, Kikuchi A, Hirayama K, Nishio Y, Hosokai Y, Kanno S et al (2012) Severe olfactory dysfunction is a prodromal symptom of dementia associated with Parkinson's disease: a 3 year longitudinal study. Brain 135(Pt 1):161–169. [DOI] [PubMed] [Google Scholar]
- 14. Baldereschi M, Di Carlo A, Rocca WA, Vanni P, Maggi S, Perissinotto E, Grigoletto F, Amaducci L, Inzitari D (2000) Parkinson's disease and parkinsonism in a longitudinal study: two‐fold higher incidence in men. ILSA Working Group. Italian Longitudinal Study on Aging. Neurology 55:1358–1363. [DOI] [PubMed] [Google Scholar]
- 15. Beach TG (2017) A review of biomarkers for neurodegenerative disease: will they swing us across the valley? Neurol Ther 6(Suppl 1):5–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Beach TG, Adler CH, Lue L, Sue LI, Bachalakuri J, Henry‐Watson J et al (2009) Unified staging system for Lewy body disorders: correlation with nigrostriatal degeneration, cognitive impairment and motor dysfunction. Acta Neuropathol 117:613–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Beach TG, Adler CH, Sue LI, Peirce JB, Bachalakuri J, Dalsing‐Hernandez JE et al (2008) Reduced striatal tyrosine hydroxylase in incidental Lewy body disease. Acta Neuropathol 115:445–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Beach TG, White CL 3rd, Hladik CL, Sabbagh MN, Connor DJ, Shill HA et al (2009) Olfactory bulb alpha‐synucleinopathy has high specificity and sensitivity for Lewy body disorders. Acta Neuropathol 117:169–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Benedetti MD, Maraganore DM, Bower JH, McDonnell SK, Peterson BJ, Ahlskog JE et al (2001) Hysterectomy, menopause, and estrogen use preceding Parkinson's disease: an exploratory case‐control study. Mov Disord 16:830–837. [DOI] [PubMed] [Google Scholar]
- 20. Bennett DA, Beckett LA, Murray AM, Shannon KM, Goetz CG, Pilgrim DM, Evans DA (1996) Prevalence of parkinsonian signs and associated mortality in a community population of older people. N Engl J Med 334:71–76. [DOI] [PubMed] [Google Scholar]
- 21. Bertrand E, Lechowicz W, Szpak GM, Lewandowska E, Dymecki J, Wierzba‐Bobrowicz T (2004) Limbic neuropathology in idiopathic Parkinson's disease with concomitant dementia. Folia Neuropathol 42:141–150. [PubMed] [Google Scholar]
- 22. Bland ST, Gonzales RA, Schallert T (1999) Movement‐related glutamate levels in rat hippocampus, striatum, and sensorimotor cortex. Neurosci Lett 277:119–222. [DOI] [PubMed] [Google Scholar]
- 23. Bosco A, Longoni AM, Vecchi T (2004) Gender effects in spatial orientation: cognitive profiles and mental strategies. Appl Cogn Psychol 18:519–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Braak H, Braak E, Yilmazer D, de Vos RA, Jansen EN, Bohl J, Jellinger K (1994) Amygdala pathology in Parkinson's disease. Acta Neuropathol 88:493–500. [DOI] [PubMed] [Google Scholar]
- 25. Braak H, Del Tredici K, Bratzke H, Hamm‐Clement J, Sandmann‐Keil D, Rub U (2002) Staging of the intracerebral inclusion body pathology associated with idiopathic Parkinson's disease (preclinical and clinical stages). J Neurol 249(Suppl 3):III/1–5. [DOI] [PubMed] [Google Scholar]
- 26. Braak H, Del Tredici K, Rub U, de Vos RA, Jansen Steur EN, Braak E (2003) Staging of brain pathology related to sporadic Parkinson's disease. Neurobiol Aging 24:197–211. [DOI] [PubMed] [Google Scholar]
- 27. Braak H, Rub U, Del Tredici K (2006) Cognitive decline correlates with neuropathological stage in Parkinson's disease. J Neurol Sci 248:255–258. [DOI] [PubMed] [Google Scholar]
- 28. Braak H, Rub U, Gai WP, Del Tredici K (2003) Idiopathic Parkinson's disease: possible routes by which vulnerable neuronal types may be subject to neuroinvasion by an unknown pathogen. J Neural Transm 110:517–536. [DOI] [PubMed] [Google Scholar]
- 29. Braak H, Rub U, Jansen Steur EN, Del Tredici K, de Vos RA (2005) Cognitive status correlates with neuropathologic stage in Parkinson disease. Neurology 64:1404–1410. [DOI] [PubMed] [Google Scholar]
- 30. Brown RM, Crane AM, Goldman PS (1979) Regional distribution of monoamines in the cerebral cortex and subcortical structures of the rhesus monkey: concentrations and in vivo synthesis rates. Brain Res 168:133–150. [DOI] [PubMed] [Google Scholar]
- 31. Brunjes PC, Illig KR, Meyer EA (2005) A field guide to the anterior olfactory nucleus (cortex). Brain Res Brain Res Rev 50:305–335. [DOI] [PubMed] [Google Scholar]
- 32. Burke RE, Dauer WT, Vonsattel JP (2008) A critical evaluation of the Braak staging scheme for Parkinson's disease. Ann Neurol 64:485–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Burke RE, O'Malley K (2013) Axon degeneration in Parkinson's disease. Exp Neurol 246:72–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Cai L, Gibbs RB, Johnson DA (2012) Recognition of novel objects and their location in rats with selective cholinergic lesion of the medial septum. Neurosci Lett 506:261–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Candy JM, Perry RH, Perry EK, Irving D, Blessed G, Fairbairn AF, Tomlinson BE (1983) Pathological changes in the nucleus of Meynert in Alzheimer's and Parkinson's diseases. J Neurol Sci 59:277–289. [DOI] [PubMed] [Google Scholar]
- 36. Cantuti‐Castelvetri I, Keller‐McGandy C, Bouzou B, Asteris G, Clark TW, Frosch MP, Standaert DG (2007) Effects of gender on nigral gene expression and parkinson disease. Neurobiol Dis 26:606–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Cartelli D, Aliverti A, Barbiroli A, Santambrogio C, Ragg EM, Casagrande FV et al (2016) alpha‐synuclein is a novel microtubule dynamase. Sci Rep 6:33289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Cersosimo MG (2017) Propagation of alpha‐synuclein pathology from the olfactory bulb: possible role in the pathogenesis of dementia with Lewy bodies. Cell Tissue Res 373:233–243. [DOI] [PubMed] [Google Scholar]
- 39. Cholerton B, Johnson CO, Fish B, Quinn JF, Chung KA, Peterson‐Hiller AL et al (2018) Sex differences in progression to mild cognitive impairment and dementia in Parkinson's disease. Parkinsonism Relat Disord 50:29–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Chu Y, Kordower JH (2007) Age‐associated increases of alpha‐synuclein in monkeys and humans are associated with nigrostriatal dopamine depletion: Is this the target for Parkinson's disease? Neurobiol Dis 25:134–149. [DOI] [PubMed] [Google Scholar]
- 41. Colloby SJ, McParland S, O'Brien JT, Attems J (2012) Neuropathological correlates of dopaminergic imaging in Alzheimer's disease and Lewy body dementias. Brain 135(Pt 9):2798–2808. [DOI] [PubMed] [Google Scholar]
- 42. Damier P, Hirsch EC, Agid Y, Graybiel AM (1999) The substantia nigra of the human brain. II. Patterns of loss of dopamine‐containing neurons in Parkinson's disease. Brain 122(Pt 8):1437–1448. [DOI] [PubMed] [Google Scholar]
- 43. De Wijk RA, Cain WS (1994) Odor identification by name and by edibility: life‐span development and safety. Hum Factors 36:182–187. [DOI] [PubMed] [Google Scholar]
- 44. Del Ser T, Hachinski V, Merskey H, Munoz DG (2001) Clinical and pathologic features of two groups of patients with dementia with Lewy bodies: effect of coexisting Alzheimer‐type lesion load. Alzheimer Dis Assoc Disord 15:31–44. [DOI] [PubMed] [Google Scholar]
- 45. Del Tredici KBH (2000–2013) Idiopathic Parkinson's Disease: Staging an α‐Synucleinopathy with a Predictable Pathoanatomy. Landes Biosci: Madame Curie Biosci Database. https://www.ncbi.nlm.nih.gov/books/NBK6077/#_A25898_ [Google Scholar]
- 46. Del Tredici K, Rub U, De Vos RA, Bohl JR, Braak H (2002) Where does parkinson disease pathology begin in the brain? J Neuropathol Exp Neurol 61:413–426. [DOI] [PubMed] [Google Scholar]
- 47. Delic V, Chandra S, Abdelmotilib H, Maltbie T, Wang S, Kem D et al (2018) Sensitivity and specificity of phospho‐ser129 alpha‐synuclein monoclonal antibodies. J Comp Neurol 526:1978–1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Diaz D, Valero J, Airado C, Baltanas FC, Weruaga E, Alonso JR (2009) Sexual dimorphic stages affect both proliferation and serotonergic innervation in the adult rostral migratory stream. Exp Neurol 216:357–364. [DOI] [PubMed] [Google Scholar]
- 49. Dickson DW, Ruan D, Crystal H, Mark MH, Davies P, Kress Y, Yen SH (1991) Hippocampal degeneration differentiates diffuse Lewy body disease (DLBD) from Alzheimer's disease: light and electron microscopic immunocytochemistry of CA2‐3 neurites specific to DLBD. Neurology 41:1402–1409. [DOI] [PubMed] [Google Scholar]
- 50. Dickson DW, Schmidt ML, Lee VM, Zhao ML, Yen SH, Trojanowski JQ (1994) Immunoreactivity profile of hippocampal CA2/3 neurites in diffuse Lewy body disease. Acta Neuropathol 87:269–276. [DOI] [PubMed] [Google Scholar]
- 51. Dirr ER, Ekhator OR, Blackwood R, Holden JG, Masliah E, Schultheis PJ, Fleming SM (2018) Exacerbation of sensorimotor dysfunction in mice deficient in Atp13a2 and overexpressing human wildtype alpha‐synuclein. Behav Brain Res 343:41–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Dluzen DE, McDermott JL, Liu B (1996) Estrogen alters MPTP‐induced neurotoxicity in female mice: effects on striatal dopamine concentrations and release. J Neurochem 66:658–666. [DOI] [PubMed] [Google Scholar]
- 53. Doty RL (2009) The olfactory system and its disorders. Seminars Neurol 29:74–81. [DOI] [PubMed] [Google Scholar]
- 54. Doty RL (2012) Olfaction in Parkinson's disease and related disorders. Neurobiol Dis 46:527–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Doty RL (2012) Olfactory dysfunction in Parkinson disease. Nat Rev Neurol 8:329–339. [DOI] [PubMed] [Google Scholar]
- 56. Duda JE, Lee VM, Trojanowski JQ (2000) Neuropathology of synuclein aggregates. J Neurosci Res 61:121–127. [DOI] [PubMed] [Google Scholar]
- 57. Durrett R (2008) Probability Models for DNA Sequence Evolution, 2nd edn. Springer: New York; London. [Google Scholar]
- 58. Elder GJ, Mactier K, Colloby SJ, Watson R, Blamire AM, O'Brien JT, Taylor JP (2017) The influence of hippocampal atrophy on the cognitive phenotype of dementia with Lewy bodies. Int J Geriatric Psychiatry 32:1182–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Fallon JH, Riley JN, Moore RY (1978) Substantia nigra dopamine neurons: separate populations project to neostriatum and allocortex. Neurosci Lett 7:157–162. [DOI] [PubMed] [Google Scholar]
- 60. Fearnley JM, Lees AJ (1991) Ageing and Parkinson's disease: substantia nigra regional selectivity. Brain 114(Pt 5):2283–2301. [DOI] [PubMed] [Google Scholar]
- 61. Fleming SM, Tetreault NA, Mulligan CK, Hutson CB, Masliah E, Chesselet MF (2008) Olfactory deficits in mice overexpressing human wildtype alpha‐synuclein. Eur J Neurosci 28:247–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Fu L, Sztul E (2015) Characterization of intracellular aggresomes by fluorescent microscopy. Methods Mol Biol 1258:307–317. [DOI] [PubMed] [Google Scholar]
- 63. Fujiwara H, Hasegawa M, Dohmae N, Kawashima A, Masliah E, Goldberg MS et al (2002) alpha‐Synuclein is phosphorylated in synucleinopathy lesions. Nat Cell Biol 4:160–164. [DOI] [PubMed] [Google Scholar]
- 64. Georgiev D, Hamberg K, Hariz M, Forsgren L, Hariz GM (2017) Gender differences in Parkinson's disease: A clinical perspective. Acta Neurol Scand 136:570–584. [DOI] [PubMed] [Google Scholar]
- 65. German DC, Manaye KF, White CL 3rd, Woodward DJ, McIntire DD, Smith WK et al (1992) Disease‐specific patterns of locus coeruleus cell loss. Ann Neurol 32:667–676. [DOI] [PubMed] [Google Scholar]
- 66. Gerstenberger J, Bauer A, Helmschrodt C, Richter A, Richter F (2016) The novel adaptive rotating beam test unmasks sensorimotor impairments in a transgenic mouse model of Parkinson's disease. Behav Brain Res 304:102–110. [DOI] [PubMed] [Google Scholar]
- 67. Giraldez‐Perez R, Antolin‐Vallespin M, Munoz M, Sanchez‐Capelo A (2014) Models of alpha‐synuclein aggregation in Parkinson's disease. Acta Neuropathol Commun 2:176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Haber SN, Knutson B (2010) The reward circuit: linking primate anatomy and human imaging. Neuropsychopharmacology 35:4–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Haberly LB, Price JL (1978) Association and commissural fiber systems of the olfactory cortex of the rat. J Comp Neurol 178:711–740. [DOI] [PubMed] [Google Scholar]
- 70. Haberly LB, Price JL (1978) Association and commissural fiber systems of the olfactory cortex of the rat. II. Systems originating in the olfactory peduncle. J Comp Neurol 181:781–807. [DOI] [PubMed] [Google Scholar]
- 71. Hall H, Reyes S, Landeck N, Bye C, Leanza G, Double K et al (2014) Hippocampal Lewy pathology and cholinergic dysfunction are associated with dementia in Parkinson's disease. Brain 137(Pt 9):2493–2508. [DOI] [PubMed] [Google Scholar]
- 72. Halliday GM, Blumbergs PC, Cotton RG, Blessing WW, Geffen LB (1990) Loss of brainstem serotonin‐ and substance P‐containing neurons in Parkinson's disease. Brain Res 510:104–107. [DOI] [PubMed] [Google Scholar]
- 73. Halliday GM, Li YW, Blumbergs PC, Joh TH, Cotton RG, Howe PR et al (1990) Neuropathology of immunohistochemically identified brainstem neurons in Parkinson's disease. Ann Neurol 27:373–385. [DOI] [PubMed] [Google Scholar]
- 74. Hanell A, Marklund N (2014) Structured evaluation of rodent behavioral tests used in drug discovery research. Front Behav Neurosci 8:252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Harding AJ, Halliday GM (2001) Cortical Lewy body pathology in the diagnosis of dementia. Acta Neuropathol 102:355–363. [DOI] [PubMed] [Google Scholar]
- 76. Harding AJ, Stimson E, Henderson JM, Halliday GM (2002) Clinical correlates of selective pathology in the amygdala of patients with Parkinson's disease. Brain 125(Pt 11):2431–2445. [DOI] [PubMed] [Google Scholar]
- 77. Hawkes CH, Del Tredici K, Braak H (2007) Parkinson's disease: a dual‐hit hypothesis. Neuropathol Appl Neurobiol 33:599–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Hawkes CH, Del Tredici K, Braak H (2009) Parkinson's disease: the dual hit theory revisited. Ann N Y Acad Sci 1170:615–622. [DOI] [PubMed] [Google Scholar]
- 79. Heimer L (1978) The Olfactory Cortex and the Ventral Striatum, Plenum Press: New York. [Google Scholar]
- 80. Hindle JV (2010) Ageing, neurodegeneration and Parkinson's disease. Age Ageing 39:156–161. [DOI] [PubMed] [Google Scholar]
- 81. Hirohata M, Ono K, Morinaga A, Ikeda T, Yamada M (2009) Anti‐aggregation and fibril‐destabilizing effects of sex hormones on alpha‐synuclein fibrils in vitro . Exp Neurol 217:434–439. [DOI] [PubMed] [Google Scholar]
- 82. Hirsch EC, Graybiel AM, Duyckaerts C, Javoy‐Agid F (1987) Neuronal loss in the pedunculopontine tegmental nucleus in Parkinson disease and in progressive supranuclear palsy. Proc Natl Acad Sci U S A 84:5976–5980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Hoglinger GU, Alvarez‐Fischer D, Arias‐Carrion O, Djufri M, Windolph A, Keber U et al (2015) A new dopaminergic nigro‐olfactory projection. Acta Neuropathol 130:333–348. [DOI] [PubMed] [Google Scholar]
- 84. Hubbard PS, Esiri MM, Reading M, McShane R, Nagy Z (2007) Alpha‐synuclein pathology in the olfactory pathways of dementia patients. J Anat 211:117–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Iacono D, Geraci‐Erck M, Rabin ML, Adler CH, Serrano G, Beach TG, Kurlan R (2015) Parkinson disease and incidental Lewy body disease: Just a question of time? Neurology 85:1670–1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Ito H, Takano H, Arakawa R, Takahashi H, Kodaka F, Takahata K et al (2012) Effects of dopamine D2 receptor partial agonist antipsychotic aripiprazole on dopamine synthesis in human brain measured by PET with L‐[beta‐11C]DOPA. PLoS One 7:e46488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Jellinger K (1988) The pedunculopontine nucleus in Parkinson's disease, progressive supranuclear palsy and Alzheimer's disease. J Neurol Neurosurg Psychiatry 51:540–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Jellinger KA (2000) Morphological substrates of mental dysfunction in Lewy body disease: an update. J Neural Transm Suppl 59:185–212. [DOI] [PubMed] [Google Scholar]
- 89. Jellinger KA (2008) A critical reappraisal of current staging of Lewy‐related pathology in human brain. Acta Neuropathol 116:1–16. [DOI] [PubMed] [Google Scholar]
- 90. Jellinger KA (2009) A critical evaluation of current staging of alpha‐synuclein pathology in Lewy body disorders. Biochim Biophys Acta 1792:730–740. [DOI] [PubMed] [Google Scholar]
- 91. Jellinger KA (2009) Formation and development of Lewy pathology: a critical update. J Neurol 256(Suppl 3):270–279. [DOI] [PubMed] [Google Scholar]
- 92. Jiao Y, Sun Z, Lee T, Fusco FR, Kimble TD, Meade CA et al (1999) A simple and sensitive antigen retrieval method for free‐floating and slide‐mounted tissue sections. J Neurosci Methods 93:149–162. [DOI] [PubMed] [Google Scholar]
- 93. Juraska JM, Wilson CJ, Groves PM (1977) The substantia nigra of the rat: a Golgi study. J Comp Neurol 172:585–600. [DOI] [PubMed] [Google Scholar]
- 94. Keogh MJ, Wei W, Aryaman J, Walker L, van den Ameele J, Coxhead J et al (2018) High prevalence of focal and multi‐focal somatic genetic variants in the human brain. Nat Commun 9:4257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Kim C, Lv G, Lee JS, Jung BC, Masuda‐Suzukake M, Hong CS et al (2016) Exposure to bacterial endotoxin generates a distinct strain of alpha‐synuclein fibril. Sci Rep 6:30891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Kopito RR (2000) Aggresomes, inclusion bodies and protein aggregation. Trends Cell Biol 10:524–530. [DOI] [PubMed] [Google Scholar]
- 97. Kordower JH, Olanow CW, Dodiya HB, Chu Y, Beach TG, Adler CH et al (2013) Disease duration and the integrity of the nigrostriatal system in Parkinson's disease. Brain 136(Pt 8):2419–2431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Kothawala A, Kilpatrick K, Novoa JA, Segatori L (2012) Quantitative analysis of alpha‐synuclein solubility in living cells using split GFP complementation. PLoS One 7:e43505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Lebow MA, Chen A (2016) Overshadowed by the amygdala: the bed nucleus of the stria terminalis emerges as key to psychiatric disorders. Mol Psychiatry 21:450–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Leverenz JB, Hamilton R, Tsuang DW, Schantz A, Vavrek D, Larson EB et al (2008) Empiric refinement of the pathologic assessment of Lewy‐related pathology in the dementia patient. Brain Pathol 18:220–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Leverenz JB, Umar I, Wang Q, Montine TJ, McMillan PJ, Tsuang DW et al (2007) Proteomic identification of novel proteins in cortical lewy bodies. Brain Pathol 17:139–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Litim N, Morissette M, Di Paolo T (2016) Neuroactive gonadal drugs for neuroprotection in male and female models of Parkinson's disease. Neurosci Biobehavioral Rev 67:79–88. [DOI] [PubMed] [Google Scholar]
- 103. Loughlin SE, Fallon JH (1983) Dopaminergic and non‐dopaminergic projections to amygdala from substantia nigra and ventral tegmental area. Brain Res 262:334–338. [DOI] [PubMed] [Google Scholar]
- 104. Lucas BA, Lavi E, Shiue L, Cho H, Katzman S, Miyoshi K et al (2018) Evidence for convergent evolution of SINE‐directed Staufen‐mediated mRNA decay. Proc Natl Acad Sci U S A 115:968–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Luk KC, Kehm VM, Zhang B, O'Brien P, Trojanowski JQ, Lee VM (2012) Intracerebral inoculation of pathological alpha‐synuclein initiates a rapidly progressive neurodegenerative alpha‐synucleinopathy in mice. J Exp Med 209:975–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Mak SK, McCormack AL, Langston JW, Kordower JH, Di Monte DA (2009) Decreased alpha‐synuclein expression in the aging mouse substantia nigra. Exp Neurol 220:359–365. [DOI] [PubMed] [Google Scholar]
- 107. Marder K, Tang MX, Alfaro B, Mejia H, Cote L, Jacobs D et al (1998) Postmenopausal estrogen use and Parkinson's disease with and without dementia. Neurology 50:1141–1143. [DOI] [PubMed] [Google Scholar]
- 108. Marui W, Iseki E, Kato M, Akatsu H, Kosaka K (2004) Pathological entity of dementia with Lewy bodies and its differentiation from Alzheimer's disease. Acta Neuropathol 108:121–128. [DOI] [PubMed] [Google Scholar]
- 109. Mason DM, Nouraei N, Pant DB, Miner KM, Hutchison DF, Luk KC et al (2016) Transmission of alpha‐synucleinopathy from olfactory structures deep into the temporal lobe. Mol Neurodegener 11:49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. McKeith IG, Dickson DW, Lowe J, Emre M, O'Brien JT, Feldman H et al (2005) Diagnosis and management of dementia with Lewy bodies: third report of the DLB Consortium. Neurology 65:1863–1872. [DOI] [PubMed] [Google Scholar]
- 111. McKeith IG, Galasko D, Kosaka K, Perry EK, Dickson DW, Hansen LA et al (1996) Consensus guidelines for the clinical and pathologic diagnosis of dementia with Lewy bodies (DLB): report of the consortium on DLB international workshop. Neurology 47:1113–1124. [DOI] [PubMed] [Google Scholar]
- 112. McNaught KS, Shashidharan P, Perl DP, Jenner P, Olanow CW (2002) Aggresome‐related biogenesis of Lewy bodies. Eur J Neurosci 16:2136–2148. [DOI] [PubMed] [Google Scholar]
- 113. McRitchie DA, Cartwright HR, Halliday GM (1997) Specific A10 dopaminergic nuclei in the midbrain degenerate in Parkinson's disease. Exp Neurol 144:202–213. [DOI] [PubMed] [Google Scholar]
- 114. Mestas J, Hughes CC (2004) Of mice and not men: differences between mouse and human immunology. J Immunol 172:2731–2738. [DOI] [PubMed] [Google Scholar]
- 115. Milber JM, Noorigian JV, Morley JF, Petrovitch H, White L, Ross GW, Duda JE (2012) Lewy pathology is not the first sign of degeneration in vulnerable neurons in Parkinson disease. Neurology 79:2307–2314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Miller DB, Ali SF, O'Callaghan JP, Laws SC (1998) The impact of gender and estrogen on striatal dopaminergic neurotoxicity. Ann N Y Acad Sci 844:153–165. [PubMed] [Google Scholar]
- 117. Miller DB, Ali SF, O'Callaghan JP, Laws SC (1998) The impact of gender and estrogen on striatal dopaminergic neurotoxicity. Ann N Y Acad Sci 844:153–165. [PubMed] [Google Scholar]
- 118. Mrak RE, Griffin WS (2007) Dementia with Lewy bodies: Definition, diagnosis, and pathogenic relationship to Alzheimer's disease. Neuropsychiatr Dis Treat 3:619–625. [PMC free article] [PubMed] [Google Scholar]
- 119. Murray HE, Pillai AV, McArthur SR, Razvi N, Datla KP, Dexter DT, Gillies GE (2003) Dose‐ and sex‐dependent effects of the neurotoxin 6‐hydroxydopamine on the nigrostriatal dopaminergic pathway of adult rats: differential actions of estrogen in males and females. Neuroscience 116:213–222. [DOI] [PubMed] [Google Scholar]
- 120. Nakano I, Hirano A (1984) Parkinson's disease: neuron loss in the nucleus basalis without concomitant Alzheimer's disease. Ann Neurol 15:415–418. [DOI] [PubMed] [Google Scholar]
- 121. Nelson PT, Schmitt FA, Jicha GA, Kryscio RJ, Abner EL, Smith CD et al (2010) Association between male gender and cortical Lewy body pathology in large autopsy series. J Neurol 257:1875–1881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Nitkowska M, Czyzyk M, Friedman A (2014) Reproductive life characteristics in females affected with Parkinson's disease and in healthy control subjects—a comparative study on Polish population. Neurol Neurochir Pol 48:322–327. [DOI] [PubMed] [Google Scholar]
- 123. Niu H, Shen L, Li T, Ren C, Ding S, Wang L et al (2018) Alpha‐synuclein overexpression in the olfactory bulb initiates prodromal symptoms and pathology of Parkinson's disease. Transl Neurodegener 7:25. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 124. Nouraei N, Mason DM, Miner KM, Carcella MA, Bhatia TN, Dumm BK et al (2018) Critical appraisal of pathology transmission in the alpha‐synuclein fibril model of Lewy body disorders. Exp Neurol 299(Pt A):172–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Oades RD, Halliday GM (1987) Ventral tegmental (A10) system: neurobiology. 1. Anatomy and connectivity. Brain Res 434:117–165. [DOI] [PubMed] [Google Scholar]
- 126. Olanow CW, Perl DP, DeMartino GN, McNaught KS (2004) Lewy‐body formation is an aggresome‐related process: a hypothesis. Lancet Neurol 3:496–503. [DOI] [PubMed] [Google Scholar]
- 127. Osterberg VR, Spinelli KJ, Weston LJ, Luk KC, Woltjer RL, Unni VK (2015) Progressive aggregation of alpha‐synuclein and selective degeneration of lewy inclusion‐bearing neurons in a mouse model of parkinsonism. Cell Rep 10:1252–1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Pardo‐Bellver C, Cadiz‐Moretti B, Novejarque A, Martinez‐Garcia F, Lanuza E (2012) Differential efferent projections of the anterior, posteroventral, and posterodorsal subdivisions of the medial amygdala in mice. Front Neuroanat 6:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Parkkinen L (2005) Impact of α‐synuclein pathology on aging. In: University of Kuopio Department of Neurology Series of Reports, Uo K. (ed.), p. 43. [Google Scholar]
- 130. Parkkinen L, Pirttila T, Alafuzoff I (2008) Applicability of current staging/categorization of alpha‐synuclein pathology and their clinical relevance. Acta Neuropathol 115:399–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Parkkinen L, Pirttila T, Tervahauta M, Alafuzoff I (2005) Widespread and abundant alpha‐synuclein pathology in a neurologically unimpaired subject. Neuropathology 25:304–314. [DOI] [PubMed] [Google Scholar]
- 132. Paulus W, Jellinger K (1991) The neuropathologic basis of different clinical subgroups of Parkinson's disease. J Neuropathol Exp Neurol 50:743–755. [DOI] [PubMed] [Google Scholar]
- 133. Paumier KL, Luk KC, Manfredsson FP, Kanaan NM, Lipton JW, Collier TJ et al (2015) Intrastriatal injection of pre‐formed mouse alpha‐synuclein fibrils into rats triggers alpha‐synuclein pathology and bilateral nigrostriatal degeneration. Neurobiol Dis 82:185–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Pearce RK, Hawkes CH, Daniel SE (1995) The anterior olfactory nucleus in Parkinson's disease. Move Disorders: Off J Move Disorder Soc 10:283–287. [DOI] [PubMed] [Google Scholar]
- 135. Pellegrini L, Wetzel A, Granno S, Heaton G, Harvey K (2017) Back to the tubule: microtubule dynamics in Parkinson's disease. Cell Mol Life Sci 74:409–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Pencea V, Bingaman KD, Freedman LJ, Luskin MB (2001) Neurogenesis in the subventricular zone and rostral migratory stream of the neonatal and adult primate forebrain. Exp Neurol 172:1–16. [DOI] [PubMed] [Google Scholar]
- 137. Piggott MA, Marshall EF, Thomas N, Lloyd S, Court JA, Jaros E et al (1999) Striatal dopaminergic markers in dementia with Lewy bodies, Alzheimer's and Parkinson's diseases: rostrocaudal distribution. Brain 122(Pt 8):1449–1468. [DOI] [PubMed] [Google Scholar]
- 138. Pinto JM, Wroblewski KE, Kern DW, Schumm LP, McClintock MK (2014) Olfactory dysfunction predicts 5‐year mortality in older adults. PLoS One 9:e10754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Polinski NK, Volpicelli‐Daley LA, Sortwell CE, Luk KC, Cremades N, Gottler LM et al (2018) Best practices for generating and using alpha‐synuclein pre‐formed fibrils to model Parkinson's disease in rodents. J Parkinsons Dis 8:303–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Rentz DM, Weiss BK, Jacobs EG, Cherkerzian S, Klibanski A, Remington A et al (2018) Sex differences in episodic memory in early midlife: impact of reproductive aging. Menopause 24:400–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Rey NL, George S, Steiner JA, Madaj Z, Luk KC, Trojanowski JQ et al (2018) Spread of aggregates after olfactory bulb injection of alpha‐synuclein fibrils is associated with early neuronal loss and is reduced long term. Acta Neuropathol 135:65–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Rey NL, George S, Steiner JA, Madaj Z, Luk KC, Trojanowski JQ et al (2018) Spread of aggregates after olfactory bulb injection of alpha‐synuclein fibrils is associated with early neuronal loss and is reduced long term. Acta Neuropathol 135:65–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Rey NL, Steiner JA, Maroof N, Luk KC, Madaj Z, Trojanowski JQ, Lee VM, Brundin P (2016) Widespread transneuronal propagation of alpha‐synucleinopathy triggered in olfactory bulb mimics prodromal Parkinson's disease. J Exp Med 213:1759–1778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Rey NL, Steiner JA, Maroof N, Luk KC, Madaj Z, Trojanowski JQ, Lee VM. Brundin P (2016) Widespread transneuronal propagation of alpha‐synucleinopathy triggered in olfactory bulb mimics prodromal Parkinson's disease. J Exp Med. [DOI] [PMC free article] [PubMed]
- 145. Rinaman L, Card JP, Enquist LW (1993) Spatiotemporal responses of astrocytes, ramified microglia, and brain macrophages to central neuronal infection with pseudorabies virus. J Neurosci: Off J Soc Neurosci 13:685–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Rinaman L, Levitt P, Card JP (2000) Progressive postnatal assembly of limbic‐autonomic circuits revealed by central transneuronal transport of pseudorabies virus. J Neurosci: Off J Soc Neurosci 20:2731–2741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Rinaman L, Schwartz G (2004) Anterograde transneuronal viral tracing of central viscerosensory pathways in rats. J Neurosci: Off J Soc Neurosci 24:2782–2786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Rocca WA, Bower JH, Maraganore DM, Ahlskog JE, Grossardt BR, de Andrade M, Melton LJ 3rd (2008) Increased risk of parkinsonism in women who underwent oophorectomy before menopause. Neurology 70:200–209. [DOI] [PubMed] [Google Scholar]
- 149. Rodriguez‐Lopez C, Clasca F, Prensa L (2017) The mesoaccumbens pathway: a retrograde labeling and single‐cell axon tracing analysis in the mouse. Front Neuroanat 11:25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Sacino AN, Brooks M, Thomas MA, McKinney AB, McGarvey NH, Rutherford NJ et al (2014) Amyloidogenic alpha‐synuclein seeds do not invariably induce rapid, widespread pathology in mice. Acta Neuropathol 127:645–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Sandyk R (1989) Estrogens and the pathophysiology of Parkinson's disease. Int J Neurosci 45:119–122. [DOI] [PubMed] [Google Scholar]
- 152. Saunders‐Pullman R, Gordon‐Elliott J, Parides M, Fahn S, Saunders HR, Bressman S (1999) The effect of estrogen replacement on early Parkinson's disease. Neurology 52:1417–1421. [DOI] [PubMed] [Google Scholar]
- 153. Savica R, Grossardt BR, Bower JH, Boeve BF, Ahlskog JE, Rocca WA (2013) Incidence of dementia with Lewy bodies and Parkinson disease dementia. JAMA Neurol 70:1396–1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Schafer DA, Welch MD, Machesky LM, Bridgman PC, Meyer SM, Cooper JA (1998) Visualization and molecular analysis of actin assembly in living cells. J Cell Biol 143:1919–1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Seidel K, Mahlke J, Siswanto S, Kruger R, Heinsen H, Auburger G et al (2015) The brainstem pathologies of Parkinson's disease and dementia with Lewy bodies. Brain Pathol 25:121–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Sengoku R, Saito Y, Ikemura M, Hatsuta H, Sakiyama Y, Kanemaru K et al (2008) Incidence and extent of Lewy body‐related alpha‐synucleinopathy in aging human olfactory bulb. J Neuropathol Exp Neurol 67:1072–1083. [DOI] [PubMed] [Google Scholar]
- 157. Shen D, Coleman J, Chan E, Nicholson TP, Dai L, Sheppard PW, Patton WF (2011) Novel cell‐ and tissue‐based assays for detecting misfolded and aggregated protein accumulation within aggresomes and inclusion bodies. Cell Biochem Biophys 60:173–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Sorge RE, Martin LJ, Isbester KA, Sotocinal SG, Rosen S, Tuttle AH et al (2014) Olfactory exposure to males, including men, causes stress and related analgesia in rodents. Nat Methods 11:629–632. [DOI] [PubMed] [Google Scholar]
- 159. Spillantini MG, Crowther RA, Jakes R, Hasegawa M, Goedert M (1998) Alpha‐Synuclein in filamentous inclusions of Lewy bodies from Parkinson's disease and dementia with lewy bodies. Proc Natl Acad Sci U S A 95:6469–6473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Stern MB, Doty RL, Dotti M, Corcoran P, Crawford D, McKeown DA et al (1994) Olfactory function in Parkinson's disease subtypes. Neurology 44:266–268. [DOI] [PubMed] [Google Scholar]
- 161. Tanaka M, Kim YM, Lee G, Junn E, Iwatsubo T, Mouradian MM (2004) Aggresomes formed by alpha‐synuclein and synphilin‐1 are cytoprotective. J Biol Chem 279:4625–4631. [DOI] [PubMed] [Google Scholar]
- 162. Taylor JP, Tanaka F, Robitschek J, Sandoval CM, Taye A, Markovic‐Plese S, Fischbeck KH (2003) Aggresomes protect cells by enhancing the degradation of toxic polyglutamine‐containing protein. Hum Mol Genet 12:749–757. [DOI] [PubMed] [Google Scholar]
- 163. Thomas ML, Kaufmann CN, Palmer BW, Depp CA, Martin AS, Glorioso DK et al (2016) Paradoxical trend for improvement in mental health with aging: a community‐based study of 1,546 adults aged 21–100 years. J Clin Psychiatry 77:e1019–e1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Toledo JB, Gopal P, Raible K, Irwin DJ, Brettschneider J, Sedor S et al (2016) Pathological alpha‐synuclein distribution in subjects with coincident Alzheimer's and Lewy body pathology. Acta Neuropathol 131:393–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Ubeda‐Banon I, Saiz‐Sanchez D, de la Rosa‐Prieto C, Argandona‐Palacios L, Garcia‐Munozguren S, Martinez‐Marcos A (2010) Alpha‐Synucleinopathy in the human olfactory system in Parkinson's disease: involvement of calcium‐binding protein‐ and substance P‐positive cells. Acta Neuropathol 119:723–735. [DOI] [PubMed] [Google Scholar]
- 166. Van Den Eeden SK, Tanner CM, Bernstein AL, Fross RD, Leimpeter A, Bloch DA, Nelson LM (2003) Incidence of Parkinson's disease: variation by age, gender, and race/ethnicity. Am J Epidemiol 157:1015–1022. [DOI] [PubMed] [Google Scholar]
- 167. Volpicelli‐Daley LA, Luk KC, Patel TP, Tanik SA, Riddle DM, Stieber A et al (2011) Exogenous alpha‐synuclein fibrils induce Lewy body pathology leading to synaptic dysfunction and neuron death. Neuron 72:57–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Watson RE Jr, Wiegand SJ, Clough RW, Hoffman GE (1986) Use of cryoprotectant to maintain long‐term peptide immunoreactivity and tissue morphology. Peptides 7:155–159. [DOI] [PubMed] [Google Scholar]
- 169. Waxman EA, Duda JE, Giasson BI (2008) Characterization of antibodies that selectively detect alpha‐synuclein in pathological inclusions. Acta Neuropathol 116:37–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Wilson RS, Yu L, Schneider JA, Arnold SE, Buchman AS, Bennett DA (2011) Lewy bodies and olfactory dysfunction in old age. Chem Sens 36:367–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Yacoubian TA, Cantuti‐Castelvetri I, Bouzou B, Asteris G, McLean PJ, Hyman BT, Standaert DG (2008) Transcriptional dysregulation in a transgenic model of Parkinson disease. Neurobiol Dis 29:515–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Yoneyama N, Watanabe H, Kawabata K, Bagarinao E, Hara K, Tsuboi T et al (2018) Severe hyposmia and aberrant functional connectivity in cognitively normal Parkinson's disease. PLoS One 13:e0190072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Zarow C, Lyness SA, Mortimer JA, Chui HC (2003) Neuronal loss is greater in the locus coeruleus than nucleus basalis and substantia nigra in Alzheimer and Parkinson diseases. Arch Neurol 60:337–341. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Preformed α synuclein fibril injections in the olfactory peduncle induce a similar topographic extent of α synucleinopathy in males and females. Mice were infused in the right olfactory bulb anterior olfactory nucleus with either preformed α synuclein fibrils (5 μg 1 μL) or an equivalent volume of PBS (1 μL). Sagittal sections were immunostained for pathologically phosphorylated α synuclein (EP1536Y rabbit monoclonal pSer129 antibody; see Table S1). The nuclear marker Hoechst (pseudocolored blue) was used to denote cytoarchitectonic boundaries. Stitched images of sagittal brain sections from 9 month old male (top) and female (bottom) mice perfused 6 months post infusion are displayed from medial to lateral. All sections were processed in parallel and images were captured at the same camera and exposure settings. Note that you must zoom in on the computer screen to view the pathology. Abbreviations are defined in Table S2. To view the original, higher resolution Adobe Illustrator or EPS files, please link to https://dsc.duq.edu/pharmacology/ or https://www.dropbox.com/sh/a6r5ylg1tco6trm/AAC9Mb2gWuP29ABbmdGuNoaJa?dl=0
Figure S2. Neurons with α synucleinopathic inclusions at the boundary zone between the substantia nigra and the ventral tegmental area are not tyrosine hydroxylase+. Mice were infused in the right rear olfactory bulb with either preformed α synuclein fibrils (5 μg 1 μL) or an equivalent volume of PBS (1 μL). After 6 months, sagittal brain sections were stained with the rabbit monoclonal EP1536Y pSer129 antibody (pseudocolored red) for pathologically phosphorylated α synuclein (see Table S1) and tyrosine hydroxylase (pseudocolored green). Nuclei were counterstained with the Hoechst reagent (pseudocolored blue). This confocal Z stack video is of the same brain section as displayed in Figure 10. No colocalization of the two markers was observed. To view the original, higher resolution Adobe Illustrator or EPS files, please link to https://dsc.duq.edu/pharmacology/ or https://www.dropbox.com/sh/a6r5ylg1tco6trm/AAC9Mb2gWuP29ABbmdGuNoaJa?dl=0
Figure S3. α synucleinopathic inclusions in the ventral tegmental area are not tyrosine hydroxylase+. Mice were infused in the right rear olfactory bulb with either preformed α synuclein fibrils (5 μg 1 μL) or an equivalent volume of PBS (1 μL). After 6 months, sagittal brain sections were stained with the rabbit monoclonal EP1536Y pSer129 antibody (pseudocolored red) for pathologically phosphorylated α synuclein (see Table S1) and tyrosine hydroxylase (TH; pseudocolored green). No robust colocalization of the two markers was observed, and the arrow in the bottom row points to a single pSer129 positive structure that may be lightly TH positive. Abbreviations are defined in Table S2. To view the original, higher resolution Adobe Illustrator or EPS files, please link to https://dsc.duq.edu/pharmacology/ or https://www.dropbox.com/sh/a6r5ylg1tco6trm/AAC9Mb2gWuP29ABbmdGuNoaJa?dl=0
Figure S4. Impact of preformed α synuclein fibril injections in the OB AON on dopaminergic cell numbers in the ventral mesencephalon. Mice were infused in the right olfactory bulb anterior olfactory nucleus with either preformed α synuclein fibrils (5 μg 1 μL) or an equivalent volume of PBS (1 μL). Sagittal brain sections were stained with antibodies against the dopaminergic marker tyrosine hydroxylase (TH) and the nuclear marker Hoechst and 200× images were stitched together to form large, high resolution photomontages of the ventral mesencephalon from every section of a 1 in 5 sagittal series. A blinded observer traced the anatomical boundaries of the ventral tegmental area and the anterior, posterior, and lateral subregions of the substantia nigra, pars compacta in cellSens software. (a) Every Hoechst+ cell (within the TH+ area) and every TH+ cell encompassing a Hoechst+ nucleus was manually counted by a blinded observer. These raw data were then used for the cell density measurements presented in Figure 12. Data are presented as (a) raw, unnormalized TH+ cell counts, and (b) raw, unnormalized Hoechst+ cell counts. Two‐way ANOVAs were followed by the Bonferroni post hoc correction. Shown are the mean and SD from an n of 3 8 mice per group (see Methods and Figure 1 for animal numbers). P ≤ 0.05, P ≤ 0.01 for PBS vs fibrils; P ≤ 0.05 vs 3 9 month male group; P ≤ 0.05 vs 11 17 month male group. Abbreviations are defined in Table S2. To view the original, higher resolution Adobe Illustrator or EPS files, please link to https://dsc.duq.edu/pharmacology/ or https://www.dropbox.com/sh/a6r5ylg1tco6trm/AAC9Mb2gWuP29ABbmdGuNoaJa?dl=0