

Abstract
Epithelioid glioblastoma (E‐GBM) is a rare aggressive variant of IDH‐wildtype glioblastoma newly recognized in the 2016 World Health Organization classification, composed predominantly of monotonous, patternless sheets of round cells with laterally positioned nuclei and plump eosinophilic cytoplasm. Approximately 50% of E‐GBM harbor BRAF V600E, which is much less frequently found in other types of glioblastomas. Most E‐GBM are recognized as primary/de novo lesions; however, several E‐GBM with co‐ or pre‐existing lower‐grade lesions have been reported. To better understand associations between E‐GBM and the lower‐grade lesions, we undertook a histological and molecular analysis of 14 E‐GBM, 10 of which exhibited lower‐grade glioma‐like components (8 E‐GBM with co‐existing diffuse glioma‐like components, 1 E‐GBM with a co‐existing PXA‐like component and 1 E‐GBM with a pre‐existing PXA). Molecular results demonstrated that the prevalence of BRAF V600E, TERT promoter mutations and CDKN2A/B homozygous deletions in E‐GBM were 13/14 (93%), 10/14 (71%) and 11/14 (79%), respectively, and concurrent BRAF V600E, TERT promoter mutations and CDKN2A/B homozygous deletions were observed in 7/14 (50%) of E‐GBM. These alterations were also frequently seen in the lower‐grade lesions irrespective of the histology. Genetic analysis including array comparative genomic hybridization performed for 5 E‐GBM with co‐ and pre‐existing lower‐grade components revealed that all molecular changes found in the lower‐grade components were also observed in the E‐GBM components, and additional changes were detected in the E‐GBM components. In conclusion, E‐GBM frequently exhibit BRAF V600E, TERT promoter mutations and CDKN2A/B homozygous deletions and these alterations tend to coexist in E‐GBM. Taken together with the facts that only one PXA preceded E‐GBM among these lower‐grade lesions, and that co‐occurrence of BRAF V600E, TERT promoter mutations and CDKN2A/B homozygous deletions have been reported to be rare in conventional lower‐grade diffuse gliomas, the diffuse glioma‐like components may be distinct infiltrative components of E‐GBM, reflecting intratumoral heterogeneity.
Keywords: BRAF, CDKN2A/B, diffuse astrocytoma, epithelioid glioblastoma, TERT
Introduction
Epithelioid glioblastoma (E‐GBM) is a provisional uncommon variant of IDH‐wildtype glioblastoma newly recognized in the 2016 World Health Organization (WHO) classification, characterized by a relatively solid aggregate of monotonous round epithelioid cells with abundant eosinophilic cytoplasm devoid of stellate processes, some eccentrically positioned nuclei, conspicuous nucleoli and distinct cellular membranes 9. E‐GBM tend to occur in young adults and children, and the prognosis is particularly poor, even for glioblastomas, with an approximate median overall survival of 6 months 4, 9, 18, 19. From a genetic standpoint, approximately half of E‐GBM harbor BRAF V600E, which is much less frequently found in other types of glioblastomas 9.
Although E‐GBM are generally recognized as primary/de novo lesions 9, several E‐GBM with a pre‐ or co‐existent lower‐grade components have been reported 1, 11, 18, 21, 22, 24, 28, 34, 35. The fact that most of these lower‐grade lesions documented so far were pleomorphic xanthoastrocytomas (PXA) 1, 24, 34, 35 and that both tumors commonly exhibit BRAF V600E supports the possibility that E‐GBM and PXA are related; the association was reinforced by a recent study identifying heterozygous deletion of ODZ3 (TEMN3) as a shared genetic alteration, found in 7 of 11 E‐GBM and 2 of 5 epithelioid PXA, defined therein as tumors composed predominantly of epithelioid cells with the same features as seen in E‐GBM, but also demonstrating at least a small component of classic PXA 1. In addition, a few E‐GBM with BRAF V600E have been reported to accompany a low‐grade diffuse glioma‐like component 11, 21, 22, 28.
In addition to BRAF V600E as a frequent genetic alteration in PXA, homozygous 9p21.3 deletions involving CDKN2A/B have been identified in 60%–83% of the cases 35, 38. Conversely, only a small number of E‐GBM has been shown to harbor the homozygous deletions 1, 4, 28, 35, and the frequency in E‐GBM is not clear.
Recently, we reported a case of E‐GBM with a diffuse astrocytoma‐like area demonstrating not only BRAF V600E but also TERT promoter mutations in both histologically distinct components 22; the prevalence of TERT promoter mutation in E‐GBM is also not known, and that in PXA and diffuse astrocytoma has been reported to be 4 and 15%–32%, respectively 7, 8, 20.
In this report, we investigated 14 E‐GBM, 10 of which exhibited pre‐ or co‐existing lower‐grade (WHO grade II/III) glioma‐like lesions, and genetic associations between their E‐GBM and lower‐grade components were explored by molecular and cytogenetic analyses performed separately for each component, focusing on the status of BRAF V600E, TERT promoter mutations, CDKN2A/B deletions and ODZ3 deletions.
Materials and Methods
Tumor samples
Fourteen cases of E‐GBM were collected for this study (Table 1). Six cases were previously reported (cases 2, 3, 5, 7, 8, 11) 11, 21, 22, 25, 28, 34. Five cases were from the consultation files of one of the authors (S.N.). Two cases were from the pathology archives of Department of Pathology, Gunma University Hospital. One case was from the pathology archives of Japan Brain Tumor Reference Center. Sections for histological and genetic analyses were prepared from formalin‐fixed paraffin‐embedded (FFPE) tissue specimens. This study was conducted in accordance with the Gunma University Ethical Committee.
Table 1.
Case list with clinical features.
| Case | Age (Yr)/Sex | Location | Histology | Radiation, dose | Chemotherapy | Local recurrence | Dissemination | Extra CNS metastasis | Clinical outcomes |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 21/M | Temporo‐parietal lobe | E‐GBM with DA | Extended focal, 64.8 Gy | MCNU | NA | Yes | Scalp |
Dead, 7months |
| 28 | 12/F | Temporal lobe | PXA | No | No | Yes | No | No | – |
| (Rec) | 25/F | Temporal lobe | E‐GBM | Extended focal, 60 Gy | TMZ | NA | Yes | No |
Dead, 4.3 months |
| 311 | 26/F | Frontal lobe | E‐GBM with DA | Extended focal, 60 Gy | TMZ | No | Yes | Scalp |
Dead, 2 months |
| 4 | 47/M | Parietal lobe | E‐GBM with AA | NA | NA | NA | NA | NA | NA |
| 59 | 22/M | Occipital lobe | E‐GBM with DA | Extended focal | TMZ+IFN | Yes | Yes | Vertebral bodies, lung thoracic wall, liver |
Alive at 24 months |
| 6 | 32/M | Parietal lobe | E‐GBM with PXA | Extended focal, 67 Gy | TMZ | No | No | No |
Alive at 10 months |
| 710 | 18/M | Temporal lobe | E‐GBM with DA | Whole brain, 40 Gy | TMZ+IFN | Yes | Yes | No |
Dead, 2 months |
| 812 | 30/M |
Temporal lobe, basal ganglia |
E‐GBM with OA | Yes | Yes | NA | NA | NA |
Alive at 9 months |
| 9 | 70/M | Parietal lobe | E‐GBM with DA | Extended focal, 40 Gy | TMZ | Yes | Yes | No |
Dead, 7 months |
| 10 | 23/M | Frontal lobe | E‐GBM with DA | Extended focal | PCZ+ACNU+VCR | Yes | Yes | Scalp |
Dead, 3 months |
| 1117 | 18/M | Temporal lobe | E‐GBM | Extended focal | ACNU+IFN | Yes | Yes | No |
Dead, 0.7 months |
| 12 | 32/F | Occipital lobe | E‐GBM | Extended focal, 60 Gy | TMZ | No | No | No |
Alive at 26 months |
| 13 | 47/M | Parietal lobe | E‐GBM |
1st: Extended focal, 60 Gy 2nd: SRT |
1st: TMZ+IFN, 2nd: IFM+CDDP+VP16 |
Yes | Yes | No |
Dead, 25 months |
| 14 | 65/F | Frontal lobe | E‐GBM | Extended focal, 60 Gy | TMZ | NA | NA | NA | Dead |
Abbreviations: Yr = years; M = male; F = female; Rec = recurrence; E‐GBM = epithelioid glioblastoma; DA = diffuse astrocytoma; PXA = pleomorphic xanthoastrocytoma; AA = anaplastic astrocytoma; OA = oligoastrocytoma; NA = not avilable; MCNU = ranimustine; TMZ = temozolomide; IFN = interferon‐beta; PCZ = procarbazine; ACNU = nimustine; VCR = vincristine; SRT = stereotactic radiotherapy; IFM = ifosfamide; CDDP = cisplatin; VP16 = etoposide.
Conventional histological analysis
Three‐micrometer‐thick tissue sections were cut and stained with hematoxylin and eosin. Immunohistochemical staining was performed on FFPE tissue sections. Primary antibodies directed against the following antigens were applied: vimentin (V9; 1:200; Dako, Glostrup, Denmark), glial fibrillary acidic protein (GFAP; 1:5000) 27, Olig2 (1:5000) 40, cytokeratin (CAM5.2; 1:5; BD Bioscience, San Jose, CA, USA), p53 protein (monoclonal, 1:50; Leica Microsystems, Wetzlar, Germany), ATRX (polyclonal, 1:500; Sigma, St. Louis, MO, USA), BAF47/INI1 (BAF47; 1:100; BD Bioscience, San Jose, CA, USA), BRG1 (polyclonal; 1:1000; Millipore, Temecula, CA, USA) and Ki‐67 (MIB‐1; 1:100; Dako). For coloration, a commercially available biotin‐streptavidin immunoperoxidase kit (Histofine, Nichirei, Tokyo, Japan) and diaminobenzidine were employed.
For vimentin, GFAP, Olig2, p53 and CAM5.2 the intensity of the staining was graded as negative, weak, moderate or strong and the extent was scored as follows: −, totally negative; 1+, <10% of tumor cells are positive; 2+, 10%–50% of tumor cells are positive; 3+, >50% of tumor cells are positive.
DNA extraction
DNA was extracted from FFPE tissue sections, as previously described 29. For E‐GBM cases with a lower‐grade area (cases 1, 3–10), the extraction was performed separately from an E‐GBM area and a lower‐grade area.
Array comparative genomic hybridization
Array comparative genomic hybridization (CGH) analysis was carried out using a 4 × 180 K CGH oligonucleotide microarray (Agilent Technologies, Santa Clara, CA, USA), as described previously 29. The sizes of gains and losses were refined by manual inspection of probe intensity plots. The log2 ratio of <–1.0 at the region of interest was considered to represent a homozygous deletion, and a value of −1.0 to −0.2 was considered to represent a heterozygous deletion 33.
Direct DNA sequencing for BRAF, IDH1/2, H3F3A, TP53 and TERT promoter mutations
Genomic DNA extracted from FFPE sections was amplified and sequenced using the primers described previously 2, 12, 26, 31, 36, 37. PCR products were sequenced on a 3130xl Genetic Analyzer (Applied Biosystems, Foster City, CA, USA) with the Big Dye Terminator v.1.1 Cycle Sequencing Kit (Applied Biosystems) following standard procedures.
Multiplex ligation‐dependent probe amplification analysis
Copy number changes in the CDKN2A/B genes, chromosomes 1 and 19 were analyzed by multiplex ligation‐dependent probe amplification (MLPA) analysis. The SALSA MLPA probemix P088‐C2 (MRC‐Holland, Amsterdam, the Netherlands) was used, and electrophoresis data were analyzed using Gene Mapper software (Life Technologies, Carlsbad, CA, USA) and normalized by Coffalyzer.net software (MRC‐Holland). A dosage quotient (probe ratio) of between 0.4 and 0.7 was taken to be indicative of a heterozygous deletion, while a value < 0.4 was taken to represent a homozygous deletion 16.
Fluorescence in situ hybridization analysis
Dual‐probe hybridization using an intermittent microwave irradiation method was applied to 4‐μm‐thick FFPE tissue sections, as described previously 39. To investigate the copy number of ODZ3, a 4q34.3–4q35.1 probe was prepared from a bacterial artificial chromosome (BAC) clone, RP11–713O21, labeled with ENZO Orange‐dUTP (Abbott Molecular Inc., Des Plaines, IL, USA) and a 4p15.1 probe was prepared from a BAC clone RP11–81N11 labeled with ENZO Green‐dUTP (Abbott Molecular Inc.). Metaphase fluorescence in situ hybridization (FISH) to verify clone mapping positions was performed using the peripheral blood cell cultures of a healthy donor.
Results
Clinical data
Relevant clinical data are summarized in Table 1. Fourteen patients were between 18 and 70 years of age, with half of them being less than 30‐year old. The female to male ratio was 10:4. All lesions were supratentorial. Thirteen patients presented with a primary lesion, and one patient had a previous history of PXA 13 years prior to the onset (case 2) 34. All 14 patients underwent surgical resection, and 12 of these cases with available clinical data received subsequent chemoradiotherapy with temozolomide or other alkylating agents. Cerebrospinal fluid dissemination and extra‐CNS metastasis were documented in ten and three cases, respectively. Of 14 cases, 10 had sufficient clinical follow‐up to evaluate outcomes. Of these, seven patients (cases 1–3, 7, 9–11) died of disease within 7 months after the surgery due to the early tumor dissemination, whereas three patients (cases 5, 12, 13) survived over 2 years with or without local recurrence.
Histopathological findings
The histological features are summarized in Table 2 (Figure 1A–L). Nine of 14 E‐GBM exibited co‐existing lower‐grade components: E‐GBM with a diffuse astrocytoma‐like component (cases 1, 3, 5, 7, 9, 10; Figure 1A–C), E‐GBM with an anaplastic astrocytoma‐like component (case 4; Figure 1D), E‐GBM with an oligoastrocytoma‐like component (case 8; Figure 1E,F) or E‐GBM with a PXA‐like component (case 6; Figure 1G). The interface between E‐GBM and lower‐grade components was relatively sharp in cases 3, 5, 6, 8–10 (Figure 1A) and intraparenchymal and perivascular calcifications were frequently observed in these lower‐grade areas (cases 1–4, 8–10; Figure 1A,C,E).
Table 2.
Histological, immunohistochemical, molecular and cytogenetic features.
| Case | Histology | Vascular wall invation | Calcification | Epithelioid :spindle :lower grade † | Coagulative necrosis/Palisading necrosis | Immunohistochemistry | Genetic analysis | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Mitosis/HPF | vimentin | GFAP | Olig2 | p53 | CAM5.2 | ATRX |
INI1/ BRG1 |
MIB‐1 LI (%) |
Direct DNA sequence | MLPA | FISH | |||||||||
| IDH1/2 | TP53 | BRAF | TERTp | CDKN2A | ODZ3 | |||||||||||||||
| 1 | E‐GBM | Present | – | 6:0:4 | Present/Absent | 3 | 3+ (s) | 1+ (m) | 2+ (m) | 2+ (w) | – | + | +/+ | 17.3 | – | – | + | C228T | Homo del * | LOH(−) * |
| DA | Absent | Present | – | 0 | 2+ (w) | 3+ (m) | 3+ (m) | 2+ (w) | – | + | +/+ | 2.3 | – | – | + | C228T | Homo del * | LOH(−) * | ||
| 234 | E‐GBM | Absent | – | 10:0:0 | Present/Absent | 14 | 3+ (m) | 1+ (s) | 2+ (m) | 1+ (s) | – | + | +/+ | 48.4 | – | – | + | – | Homo del * | LOH(+) * |
| PXA | Absent | Present | 0:0:10 | – | 0 | 3+ (m) | 2+ (s) | 2+ (m) | 1+ (w) | – | + | +/+ | <1 | ND | – | + | ND | Homo del * | LOH(−) * | |
| 321 | E‐GBM | Present | – | 6:0:4 | Present/Absent | 22 | 3+ (s) | 1+ (s) | 1+ (m) | 1+ (w) | – | + | +/+ | 62.6 | – | – | + | C228T | Homo del * | LOH(−) * |
| DA | Absent | Present | – | 0 | 1+ (m) | 3+ (m) | 3+ (s) | – | – | + | +/+ | <1 | – | – | – | – | No CNA * | LOH(−) * | ||
| 4 | E‐GBM | Present | – | 7:0:3 | Present/Absent | 15 | 3+ (s) | 3+ (s) | 2+ (m) | 1+ (m) | – | – | +/+ | 24.1 | – | ND | + | – | Homo del * | LOH(−) * |
| AA | Absent | Present | – | 4 | 3+ (s) | 3+ (m) | 3+ (s) | 1+ (m) | – | – | +/+ | 5.1 | – | ND | + | – | Homo del * | LOH(−) * | ||
| 528 | E‐GBM | Present | – | 18:1:1 | Present/Absent | 15 | 3+ (s) | 1+ (m) | 1+ (w) | 1+ (w) | – | + | +/+ | 23.8 | – | – | + | C228T | Homo del * | LOH(+) * |
| DA | Absent | Absent | – | 0 | 1+ (w) | 3+ (m) | 2+ (w) | 1+ (w) | – | + | +/+ | 2.9 | – | – | + | C228T | Homo del * | LOH(−) * | ||
| 6 | E‐GBM | Present | – | 9:0:1 | Present/Absent | 4 | 3+ (m) | 2+ (s) | 2+ (s) | 2+ (m) | 1+ (w) | + | +/+ | 16.3 | – | – | + | C228T | Homo del | LOH(−) |
| PXA | Absent | Absent | – | 0 | 3+ (s) | 3+ (s) | 3+ (m) | 1+ (m) | – | + | +/+ | 2.3 | – | – | + | C228T | Homo del | LOH(−) | ||
| 722 | E‐GBM | Present | – | 7:1:2 | Present/Absent | 32 | 3+ (s) | 1+ (s) | 2+ (m) | 2+ (m) | 2+ (s) | + | +/+ | 45.8 | – | – | + | C228T | Homo del | LOH(−) |
| DA | Absent | Absent | – | 1 | 2+ (m) | 3+ (m) | 3+ (s) | 1+ (w) | – | + | +/+ | 1.6 | – | – | + | C228T | Homo del | LOH(−) | ||
| 811 | E‐GBM | Present | – | 6:3:1 | Present/Absent | 5 | 3+ (s) | 1+ (s) | 2+ (m) | 1+ (w) | 2+ (m) | + | +/+ | 17.7 | – | – | + | C228T | Homo del | LOH(−) |
| OA | Absent | Present | – | 0 | 3+ (s) | 3+ (s) | 3+ (s) | 1+ (w) | – | + | +/+ | 3.7 | – | – | + | C228T | Homo del | LOH(−) | ||
| 9 | E‐GBM | Present | – | 7:0:3 | Present/Absent | 9 | 3+ (s) | 1+ (s) | 1+ (w) | 1+ (m) | 1+ (w) | + | +/+ | 35.5 | – | – | + | C250T | Homo del | LOH(−) |
| DA | Absent | Present | – | 0 | 1+ (m) | 3+ (s) | 3+ (s) | 1+ (w) | – | + | +/+ | 3.3 | – | – | + | C250T | No CNA | LOH(−) | ||
| 10 | E‐GBM | Present | – | 6:3:1 | Present/Present | 15 | 3+ (s) | 1+ (s) | 1+ (m) | 2+ (s) | – | + | +/+ | 17.7 | – |
c.633delT p.R213fs * 34 |
+ | C228T | No CNA | LOH(−) |
| DA | Absent | Present | – | 0 | 1+ (w) | 3+ (m) | 2+ (s) | 1+ (m) | – | + | +/+ | <1 | – | – | + | – | No CNA | LOH(−) | ||
| 1125 | E‐GBM | Present | – | 10:0:0 | Present/Present | 9 | 3+ (s) | 1+ (s) | 1+ (w) | 1+ (w) | – | ND | +/+ | 27.1 | – | – | + | C228T | No CNA | LOH(−) |
| 12 | E‐GBM | Present | – | 7:3:0 | Present/Absent | 4 | 3+ (m) | 1+ (m) | 3+ (s) | 2+ (m) | 2+ (m) | + | +/+ | 18.0 | – | – | + | – | Homo del | LOH(−) |
| 13 | E‐GBM | Present | – | 9:1:0 | Present/Present | 19 | 1+ (s) | 1+ (s) | – | 2+ (s) | 1+ (s) | + | +/+ | 30.4 | – |
c.742C>A p.R248R |
– | C228T | No CNA | LOH(−) |
| 14 | E‐GBM | Present | – | 8:2:0 | Present/Absent | 18 | 3+ (m) | 1+ (s) | 3+ (m) | 1+ (m) | 1+ (s) | + | +/+ | 20.7 | – | – | + | – | Homo del | LOH(−) |
The instensity and extent of immunopositive tumor cells were scored as follows: w = weak; m = moderate; s = strong; −, totally negative; 1+, <10%; 2+, 10–50%; 3+, >50%.
*The results of FISH and MLPA analyses were compatible with array CGH data.
†The areas of epithelioid, anaplastic spindle and lower grade glioma components were estimated.
Abbreviations: HPF = high power field; GFAP = glial fibrillary acidimic protein; Olig2 = oligodendrocyte transcription factor 2; MIB‐1 LI = MIB‐1 labeling index; FISH = fluorescence in situ hybridization; dPCR = digital polymerase chain reaction; IDH = Isocitrate dehydrogenase; TERTp = Telomerase reverse transcriptase promoter; E‐GBM = epithelioid glioblastoma; DA = diffuse astrocytoma; PXA = pleomorphic xanthoastrocytoma; AA = anaplastic astrocytoma; OA = oligoastrocytoma; LOH = loss of heterozygosity; Homo del = homozygous deletion; CNA = copy number aberration; ND = not detected.
Figure 1.
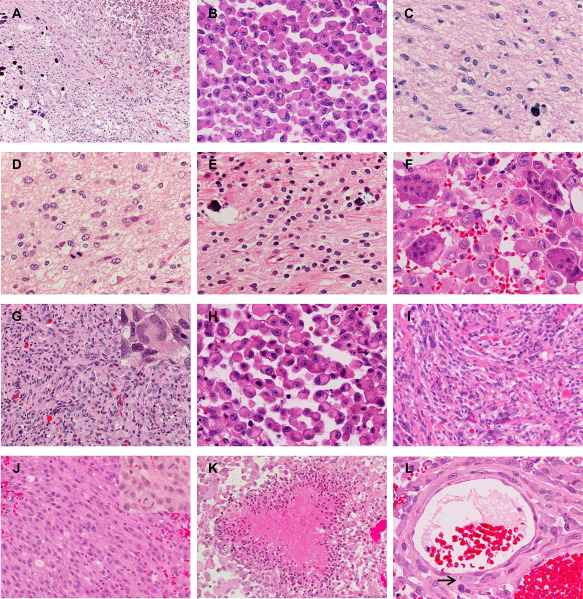
Microscopic appearance. A–C. Epithelioid glioblastoma (E‐GBM) and diffuse astrocytoma (DA)‐like components in case 9. The interface between the E‐GBM (top right) and DA‐like (bottom left) components is relatively sharp (A). Note the calcification in the DA‐like component (A,C). The E‐GBM area is composed of monotonous, discohesive round cells with laterally positioned nuclei and eosinophilic cytoplasm (B). The DA‐like component exhibits mild cellular proliferation of well‐differentiated neoplastic fibrillary astrocytes (C). D. The anaplastic astrocytoma‐like component in case 4, presenting 4 mitoses per 10 high‐power fields. E,F. The oligoastrocytoma‐like component with calcification (E) and osteoclast‐like giant cells intermingled with epithelioid tumor cells (F) in case 8. G. The PXA‐like component in case 6 shows a fascicular arrangement of spindle‐shaped cells with some multinucleated pleomorphic cells (inset). H,I. E‐GBM (H) and its precursor PXA lesion (I) in case 2. The PXA shows numerous eosinophilic granular bodies (I). J. A component with anaplastic spindle‐shaped cells with monotonous nuclei and thick processes (case 12). Frequent mitosis is seen (inset). K. Palisading necrosis in the E‐GBM component (case 10). L. Epithelioid tumor cells invade the vascular wall in the subarachnoid space (case 6). The arrow indicates a tumor cell right beneath the endothelium. Original magnification: A, x100; G, I–K, x200; B–F, H, L inset in G and J, x400.
All E‐GBM in this study were mainly composed of monotonous, discohesive sheets of epithelioid cells with eosinophilic, rounded cytoplasm lacking processes (Figure 1B,H). The nuclei were large, laterally positioned, and variable in shape; round, ovoid, reniform or crescent nuclei with distinct nucleoli were observed. The cytoplasm often contained filamentous‐like or hyalinized inclusions with or without fine basophilic granules, and small cytoplasmic vacuoles were also found in some tumor cells. Unlike conventional GBMs, an interspersed fibrillary matrix was not observed between these tumor cells. Mitotic figures were frequent. In case 8, there were numerous osteoclast‐like giant cells intermingling with epithelioid cells (Figure 1F) 11.
In diffuse astrocytoma‐like components, a mildly cellular proliferation of glial tumor cells with oval to irregular nuclei and cytoplasmic processes, in the background of a loosely structured matrix were observed (cases 1, 3–5, 7, 9, 10; Figure 1C). Subpial and perineuronal accumulation of tumor cells, indicating the infiltrative nature of the tumor cells, were observed in some cases. Mitotic figures were scant (case 7) or absent (cases 1, 3, 5, 9, 10). A lower‐grade component of case 4 showed similar histology but exhibited 4 mitotic figures per 10 high‐power fields, meeting the criteria for “anaplastic” astrocytoma (Figure 1D). In a oligoastrocytoma‐like component in case 8, fibrillary astrocytic tumor cells and oligodendroglial tumor cells with a perinuclear halo were intimately mixed (Figure 1E), where no mitotic figures were observed.
In case 6, a PXA‐like component co‐existing with E‐GBM demonstrated a fascicular arrangement of spindle‐shaped cells with some mononucleated or multinucleated pleomorphic cells (Figure 1G). Perivascular lymphocytic cuffings and intercellular reticlin meshwork were noted, although eosinophilic granular bodies (EGBs) could not be identified. Mitotic figures were not observed.
The pre‐existing PXA lesion in case 2 was composed of a combination of spindle‐shaped, xanthic and pleomorphic, multinucleated giant astrocytes, with numerous EGBs, perivascular lymphocytic cuffings and abundant reticulin meshwork (Figure 1I).
In addition to the typical E‐GBM areas, seven cases contained another anaplastic area composed of spindle‐shaped cells with relatively monotonous nuclei and thick processes (cases 5, 7, 8, 10, 12–14; Figure 1J). Characteristics of localized astrocytomas (PXA and pilocytic astrocytoma) as follows were not observed: piloid cytoplasmic processes, xanthomatous change, Rosenthal fibers or EGBs. These areas were not regarded as lower‐grade components because of frequent mitosis, albeit less than in the E‐GBM components and necrosis seen in these components.
In areas of these epithelioid and anaplastic spindle‐shaped cells, coagulative necrosis was observed in all cases, whereas palisading necrosis was identified in three cases (cases 10, 11, 13; Figure 1K), and microvascular proliferation was absent in all cases. These tumor cells tended to extend within the subarachnoid space, and in most cases, characteristic vascular wall invasion (Figure 1L) and intratumoral hemorrhage were observed.
The immunohistochemical findings are summarized in Table 2 (Figure 2A–L). E‐GBM exhibited diffuse and strong staining with vimentin (Figure 2A). GFAP immunoreactivity was identified in a limited number of epithelioid cells with a few cytoplasmic processes (Figure 2B), but was diffusely observed in lower‐grade glioma cells (Figure 2C). Anaplastic spindle‐shaped cells were generally sparsely immunostained for GFAP (Figure 2D). Nuclear Olig2 staining was variable in extent in epithelioid cells; mostly negative in cases 5, 9, 11 and 13 (Figure 2E), whereas diffusely positive in cases 12 and 14 (Figure 2F). Most E‐GBM showed weak to moderate p53 immunoreactivity (Figure 2G), but in cases 10 and 13, strong immunostaining was observed (Figure 2H). CAM5.2 staining was focally observed in epithelioid cells in case 6–9, 12–14. The loss of ATRX nuclear expression was detected only in case 4 (Figure 2I). The retention of nuclear staining of INI1 and BRG1 was observed throughout the specimen in all cases (Figure 2J). MIB‐1 staining depicted markedly higher proliferation indexes in the E‐GBM components (Figure 2K) than those in the lower‐grade components (Figure 2L).
Figure 2.
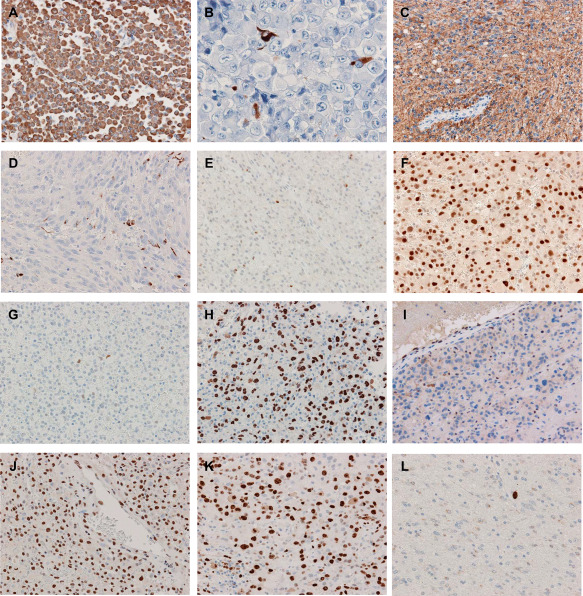
Immunohistochemistry. A. Vimentin is diffusely and strongly positive in E‐GBM (case 9). B,C. A limited number of epithelioid cells with a few cytoplasmic processes are positive for GFAP (B), and the DA‐like area is diffusely positive (C) in case 7. D. Anaplastic spindle‐shaped cells are sparsely immunostained with GFAP (case 5). E,F. The extent and intensity of nuclear Olig2 staining is variable in the E‐GBM components; few and moderate in case 3 (E), whereas diffuse and strong in case 12 (F). G,H. The E‐GBM area has sparse and weak p53 immunoreactivity in case 3 (G), but diffuse and strong in case 13 (H). I. The loss of ATRX nuclear expression in case 4 (E‐GBM area). J. The nuclear staining of INI1 is retained (E‐GBM area of case 3). K,L. The MIB‐1 labeling index is approximately 60% in the E‐GBM component (K) and less than 1% in the DA‐like component (L) in case 3. Original magnification: A, C–L, x200; B, x400.
Genetic analysis
The results of genetic analysis are summarized in Table 2. The prevalence of BRAF V600E, TERT promoter mutations and CDKN2A/B homozygous deletions (Supporting Information Figure S1A) detected by direct DNA sequencing or MLPA analysis in our cohort of E‐GBM were 13/14 (93%), 10/14 (71%) and 11/14 (79%), respectively. Concurrent BRAF V600E, TERT promoter mutations and CDKN2A/B homozygous deletions were observed in 7 of 14 E‐GBM (50%; cases 1, 3, 5–9). These 7 E‐GBM were with co‐existing lower‐grade lesions, of which five cases demonstrated the same combination of alterations in their lower‐grade lesions, whereas none of these alterations were detected in the diffuse astrocytoma‐like area of case 3, and a CDKN2A/B homozygous deletion could not be detected in the diffuse astrocytoma‐like area of case 9.
FISH analysis detected loss of heterozygosity (LOH) of ODZ3 in the E‐GBM components of 2/14 cases (cases 2, 5), but not in their lower‐grade components (Supporting Information Figure S1B).
The results of array CGH revealed that all copy number alterations (CNA) observed in the lower‐grade lesions were also detected in the E‐GBM components in all five cases analyzed (Figure 3, Supporting Information Table S1). CNA recurrently found only in the E‐GBM components but not in the lower‐grade lesions were a gain on 3q24 ‐ q26.2 (cases 1, 2), a heterozygous deletion on 4q34.3 ‐ q35.1 involving ODZ3 (cases 2, 5; Figure 3C), a heterozygous deletion on 6q11.1 ‐ q22.33 (cases 2, 3), a heterozygous deletion on 6q23.3 ‐ q24.1 (cases 2, 3), a gain on whole chromosome 7 (cases 1, 3) and a gain on 7q33 ‐ q36.3 (cases 1–3). A homozygous deletion on 3q13.31 involving LSAMP detected only in the E‐GBM component of case 5, which we previously reported 28, was not observed in any other cases. Small homozygous deletions on 4q13.2 (cases 1, 5), 8p11.22 involving ADAM3A and ADAM5 (cases 3, 4), and 22q11.23 involving GSTT1 and GSTTP2 (cases 3, 5), and a small gain on 6p22.1 (cases 1, 4) recurrently found both in the E‐GBM and lower‐grade components were in regions of known benign copy number variants (polymorphisms) reported in the Database of Genomic Variants (DGV; http://dgv.tcag.ca/dgv/app/home). The results of CNA involving CDKN2A/B and ODZ3 were compatible with the results of MLPA and FISH analyses, respectively (Table 2, Figure 3, Supporting Information Figure S1). As for loss of other important tumor suppressor genes, a heterozygous deletion on 13q involving RB1 was found only in the E‐GBM component of case 2, a heterozygous deletion on 10q involving PTEN was found both in the E‐GBM and diffuse astrocytoma‐like components of case 1 and only in the E‐GBM component of case 4, and a heterozygous deletion on 17p involving TP53 was found only in the E‐GBM component of case 2 (Figure 3, Supporting Information Figure S1).
Figure 3.
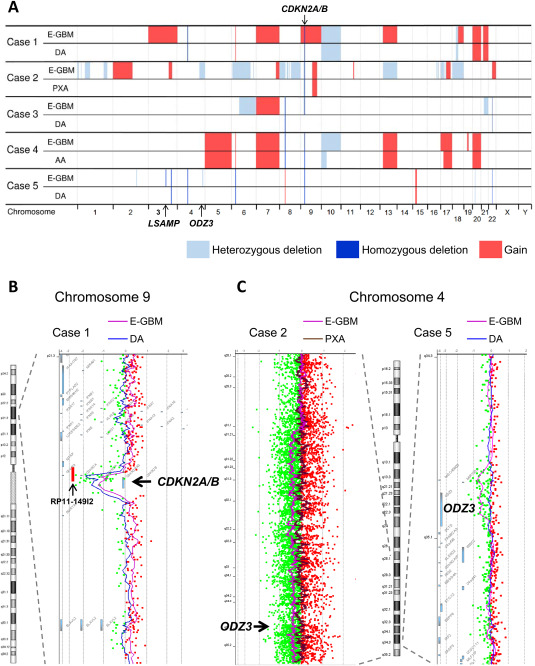
Results of array comparative genomic hybridization (CGH). A. Copy number alterations (CNA) detected by array CGH performed separately for the E‐GBM components and lower‐grade lesions in cases 1–5 are visualized. All CNA observed in the lower‐grade lesions were also detected in the E‐GBM components in all cases. Homozygous deletions involving CDKN2A/B were found both in the E‐GBM components and lower‐grade lesions in cases 1, 2, 4 and 5, but only in the E‐GBM component in case 3. Several CNA were recurrently found only in the E‐GBM components, including a heterozygous deletion on 4q34.3 ‐ q35.1 involving ODZ3 (cases 2, 5). B. A homozygous deletion involving CDKN2A/B in case 1. Chromosome 9 except the deleted region demonstrated a low‐level gain in the E‐GBM component. The positions of CDKN2A/B and a bacterial artificial chromosome clone RP11–149I2, which is commonly used for fluorescence in situ hybridization analysis for investigating the copy number of CDKN2A/B, are indicated. C. Heterozygous deletion involving ODZ3 observed only in the E‐GBM components of cases 2 and 5. The position of ODZ3 is indicated.
Abbreviations: E‐GBM = epithelioid glioblastoma; DA = diffuse astrocytoma‐like component; PXA = pleomorphic xanthoastrocytoma.
No IDH1/2 or H3F3A mutation or 1p/19q loss was observed in any cases analyzed (Supporting Information Figure S1A). TP53 mutations were found in two cases: a frameshift mutation (c.633delT, pR213fs*34) only in the E‐GBM component of case 10, and a silent mutation in codon 248 hot spot (c.742C > A, p.R248R) in case 13, both of which are reported in the Catalogue of Somatic Mutations in Cancer (COSMIC) database (http://cancer.sanger.ac.uk/cosmic; Table 2).
Discussion
Histologically and molecularly, PXA, particularly anaplastic PXA, has been known to be related with E‐GBM 9, 13. PXA and E‐GBM often share such features as a relatively solid growth pattern, dural attachment and reticulin‐rich areas, and it has been noted that a subset of anaplastic PXA show similar histologic features to E‐GBM, but E‐GBM usually possess more cytologically uniform cells and lack eosinophilic granular bodies unlike anaplastic PXA 1, 17, 18, 19. From a genetic stand point, our results suggest that, although BRAF mutations and CDKN2A/B homozygous deletions are common in E‐GBM and PXA, the frequency of TERT promoter mutation is a definite difference between these tumors compared with the reported frequency in PXA. Investigating 14 E‐GBM, the prevalence of BRAF V600E, TERT promoter mutations and CDKN2A/B homozygous deletions in our cohort were 13/14 (93%), 10/14 (71%) and 11/14 (79%), respectively, and concurrent BRAF V600E, TERT promoter mutations and CDKN2A/B homozygous deletions were observed in 7/14 (50%) of E‐GBM (Table 2). BRAF mutations and CDKN2A/B homozygous deletions are common in PXA (50%–78% and 60%–83%, respectively) 14, 35, 38, whereas, according to a single study, TERT promoter mutations were found in 1/25 (4%) and 3/13 (23%) of PXA and anaplastic PXA, respectively 20. It is of note that, although frequent BRAF V600E, TERT promoter mutations and CDKN2A/B deletions have not been reported so far in any other CNS tumor, this combination of alterations was observed in a subset of melanomas (approximately 11%) 15, and morphological similarities between E‐GBM and melanomas have been pointed out 4, 18, 19.
Although most of the previously reported pre‐ or co‐existent lower‐grade lesions with E‐GBM have been PXA, 8 out of 10 in our present cohort were diffuse gliomas: diffuse astrocytoma‐like components in six cases, an anaplastic astrocytoma‐like component in one case and an oligoastrocytoma‐like component in one case. BRAF V600E, TERT promoter mutations and CDKN2A/B homozygous deletions were also frequently observed in these lower‐grade lesions; however, IDH1/2 mutations, genetic characteristics of lower‐grade diffuse gliomas, were not observed in any lesion (Table 2). H3 K27M mutation has been reported in two cases of E‐GBM in the thalamus and spine, the former also harboring BRAF V600E, but H3F3A K27M mutation was not detected in our present cohort 1, 4.
In the current study, we compared the epithelioid cell and infiltrative lower‐grade areas of E‐GBM in regards to their histology, and they were distinguished in terms of cytological features, growth patterns (infiltrative or relatively solid), proliferative ability and immunohistochemical findings (GFAP in particular). Calcification was seen not only in a oligoastrocytoma‐like area (case 8) but also even in five diffusely infiltrating astrocytoma‐like areas of 7 E‐GBM cases (cases 1, 3, 4, 9, 10); calcification, a feature associated with slow growth, is a common histological finding in oligodendroglial tumors, but is uncommon in diffuse astrocytomas 5.
Co‐occurrence of BRAF V600E, TERT promoter mutations and CDKN2A/B homozygous deletions are rare in diffuse gliomas. Among 491 grades II–III diffuse gliomas in recent reports mostly composed of adult cases 3, 6, 41, no cases shared BRAF V600E, TERT promoter mutations and CDKN2A/B homozygous deletions. As for glioblastomas (302 cases), seven cases had both BRAF and TERT promoter mutations, of which three cases also had CDKN2A/B homozygous deletions; however, epithelioid cell morphology was not mentioned in these series. Meanwhile, concurrent BRAF mutation and CDKN2A homozygous deletion have been found in a subset of diffuse astrocytomas in children and young adults 10, 30. Mistry et al reported that BRAF mutations and CDKN2A deletions (heterozygous or homozygous) constitute a clinically distinct subtype of secondary high‐grade gliomas (sHGG) transforming from pediatric low‐grade gliomas (PLGG) 23. These transforming PLGG included “low‐grade astrocytomas,” pilocytic astrocytomas, PXA and gangliogliomas, and 2 of them (a low grade astrocytoma and a ganglioglioma) had TERT promoter mutations in addition to BRAF mutations and CDKN2A deletions; however, epithelioid cell morphology was not mentioned in relation to sHGG 23. Together with the fact that no E‐GBM was preceded by a lower‐grade diffuse glioma in the current series, the lower‐grade diffuse glioma‐like areas may be distinct infiltrative components of E‐GBM, reflecting intratumoral heterogeneity.
The exception is case 3, where BRAF V600E, TERT promoter mutations and CDKN2A/B homozygous deletions were found in the E‐GBM component, but none of these alterations were detected in the diffuse astrocytoma‐like component. Moreover, although 2 CNA found in the diffuse astrocytoma‐like component were also detected in the E‐GBM component using array CGH (Figure 3A, Supporting Information Table S1), these CNA may be benign copy number variants (polymorphisms); therefore, we could not detect any possible somatic genetic changes in the diffuse astrocytoma‐like component, and could not reveal a genetic association between these 2 components. Unrelated E‐GBM and diffuse astrocytoma may present as a collision tumor in this case; however, if not, underlying more pathogenetically important alterations may be shared both in these components, which could not be detected by the methods used in this study.
In the current study, areas of anaplastic spindle‐shaped cells with relatively monotonous nuclei, a solid growth pattern, decreased GFAP staining and devoid of characteristics of localized astrocytomas (PXA and pilocytic astrocytoma) were found in 7 E‐GBM (Figure 2D, Table 2). This finding has not been featured in relation to E‐GBM, but can be a frequent element in E‐GBM. Another histological characteristic of E‐GBM is tumor cell invasion into the wall of vessels in the subarachnoid space, which was observed in 13 cases in the present study. This characteristic, together with the discohesiveness of tumor cells, may be associated with the extra‐CNS metastasis, leptomeningeal dissemination and extensive intratumoral hemorrhage 21, 28.
Case 4 is the first E‐GBM with loss of nuclear ATRX expression ever reported (Figure 2I, Table 2). ATRX expression is almost invariably lost in the setting of ATRX mutations, and the mutations are frequently observed in IDH‐mutant diffuse astrocytomas and anaplastic astrocytomas 8. A small portion of E‐GBM may exhibit ATRX mutations mutually exclusively with TERT promoter mutations as in diffuse gliomas 8.
For investigating the copy number of CDKN2A/B by FISH analysis, the BAC clone RP11–149I2 is commonly used; however, the position of the BAC clone does not fit into the homozygously deleted regions in 3 of 5 E‐GBM (cases 1, 4, 5) detected by array CGH (Figure 3B, Supporting Information Table S1). We tested FISH analysis using the BAC clone for the five cases analyzed by array CGH, and validated the deletions in cases 2 and 3, but could not in the cases 1, 4, 5 (data not shown). Deleted regions involving CDKN2A/B in E‐GBM may be narrower, as seen in other tumors 32; therefore, other methods such as MLPA and array‐based analysis may be suitable, the former being more appropriate for routine use and screening large cohorts.
In conclusion, our results show that E‐GBM frequently exhibit BRAF V600E, TERT promoter mutations and CDKN2A/B homozygous deletions, these alterations tend to coexist in E‐GBM, and that diffuse glioma‐like components as well as PXA‐like components are commonly observed in E‐GBM. Although PXA can rarely secondarily progress to E‐GBM, the diffuse glioma‐like components may reflect intratumoral heterogeneity. Further studies with large cohorts are needed to better understand the pathogenesis of E‐GBM.
Supporting information
Additional supporting information may be found online in the Supporting Information section at the end of the article.
Table S1. Copy number abberations (CNA) detected by array comparative genomic hybridization.
Figure S1. MLPA and FISH analysis. A. MLPA analysis shows CDKN2A/B homozygous deletion (dosage quotient < 0.4) both in oligoastrocytoma‐like and E‐GBM components of case 8. IDH1/2 mutation or 1p/19q loss is not detected in either component. B. LOH of ODZ3 is detected in recurrent E‐GBM but not in primary PXA of case 2, using a red probe targeting ODZ3 and a reference probe labeled in green. E‐GBM; epithelioid glioblastoma, PXA; pleomorphic xanthoastrocytoma, MLPA; multiplex ligation‐dependent probe amplification, FISH; fluorescence in situ hybridization, LOH; loss of heterozygosity.
The authors have no conflict of interest.
References
- 1. Alexandrescu S, Korshunov A, Lai SH, Dabiri S, Patil S, Li R et al (2016) Epithelioid glioblastomas and anaplastic epithelioid pleomorphic xanthoastrocytomas–same entity or first cousins? Brain Pathol 26:215–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Arai M, Nobusawa S, Ikota H, Takemura S, Nakazato Y (2012) Frequent IDH1/2 mutations in intracranial chondrosarcoma: a possible diagnostic clue for its differentiation from chordoma. Brain Tumor Pathol 29:201–206. [DOI] [PubMed] [Google Scholar]
- 3. Arita H, Yamasaki K, Matsushita Y, Nakamura T, Shimokawa A, Takami H et al (2016) A combination of TERT promoter mutation and MGMT methylation status predicts clinically relevant subgroups of newly diagnosed glioblastomas. Acta Neuropathol Commun 4:79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Broniscer A, Tatevossian RG, Sabin ND, Klimo P, Jr , Dalton J, Lee R et al (2014) Clinical, radiological, histological and molecular characteristics of paediatric epithelioid glioblastoma. Neuropathol Appl Neurobiol 40:327–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Burger PC, Scheithauer BW (2007) Diffuse astrocytoma. In: AFIP Atlas of Tumor Pathology Fourth Series Fascicle 7, Tumors of the Central Nervous System. Silverberg SG, Sobin LH (eds), p. 48. Armed Forces Institute of pathology: Washington, DC. [Google Scholar]
- 6. Ceccarelli M, Barthel FP, Malta TM, Sabedot TS, Salama SR, Murray BA et al (2016) Molecular profiling reveals biologically discrete subsets and pathways of progression in diffuse glioma. Cell 164:550–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chan AK, Yao Y, Zhang Z, Chung NY, Liu JS, Li KK et al (2015) TERT promoter mutations contribute to subset prognostication of lower‐grade gliomas. Mod Pathol 28:177–186. [DOI] [PubMed] [Google Scholar]
- 8. Eckel‐Passow JE, Lachance DH, Molinaro AM, Walsh KM, Decker PA, Sicotte H et al (2015) Glioma groups based on 1p/19q, IDH, and TERT promoter mutations in tumors. N Engl J Med 372:2499–2508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ellison DW, Kleinschmidt‐DeMasters BK, Park SH (2016) Epithelioid glioblastoma. In: WHO Classification of Tumors of the Central Nervous System, 4th edn. Louis DN, Ohgaki H, Wiestler OD, Cavenee WK (eds), pp. 50–51. IARC Press: Lyon. [Google Scholar]
- 10. Forshew T, Tatevossian RG, Lawson AR, Ma J, Neale G, Ogunkolade BW et al (2009) Activation of the ERK/MAPK pathway: a signature genetic defect in posterior fossa pilocytic astrocytomas. J Pathol 218:172–181. [DOI] [PubMed] [Google Scholar]
- 11. Funata N, Nobusawa S, Yamada R, Shinoura N (2016) A case of osteoclast‐like giant cell‐rich epithelioid glioblastoma with BRAF V600E mutation. Brain Tumor Pathol 33:57–62. [DOI] [PubMed] [Google Scholar]
- 12. Gessi M, van de Nes J, Griewank K, Barresi V, Buckland ME, Kirfel J et al (2014) Absence of TERT promoter mutations in primary melanocytic tumours of the central nervous system. Neuropathol Appl Neurobiol 40:794–797. [DOI] [PubMed] [Google Scholar]
- 13. Giannini C, Paulus W, Louis DN, Liberski PP, Figarella‐Branger D, Capper D (2016) Anaplastic pleomorphic xanthoastrocytoma. In: WHO Classification of Tumors of the Central Nervous System, 4th edn. Louis DN, Ohgaki H, Wiestler OD, Cavenee WK (eds), pp. 98–99. IARC Press: Lyon. [Google Scholar]
- 14. Giannini C, Paulus W, Louis DN, Liberski PP, Figarella‐Branger D, Capper D (2016) Pleomorphic xanthoastrocytoma. In: WHO Classification of Tumors of the Central Nervous System, 4th edn. Louis DN, Ohgaki H, Wiestler OD, Cavenee WK (eds), pp. 94–97. IARC Press: Lyon. [Google Scholar]
- 15. Hayward NK, Wilmott JS, Waddell N, Johansson PA, Field MA, Nones K et al (2017) Whole‐genome landscapes of major melanoma subtypes. Nature 545:175–180. [DOI] [PubMed] [Google Scholar]
- 16. Jeuken J, Sijben A, Alenda C, Rijntjes J, Dekkers M, Boots‐Sprenger S et al (2009) Robust detection of EGFR copy number changes and EGFR variant III: technical aspects and relevance for glioma diagnostics. Brain Pathol 19:661–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kepes JJ (1993) Pleomorphic xanthoastrocytoma: the birth of a diagnosis and a concept. Brain Pathol 3:269–274. [DOI] [PubMed] [Google Scholar]
- 18. Kleinschmidt‐DeMasters BK, Aisner DL, Birks DK, Foreman NK (2013) Epithelioid GBMs show a high percentage of BRAF V600E mutation. Am J Surg Pathol 37:685–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kleinschmidt‐DeMasters BK, Alassiri AH, Birks DK, Newell KL, Moore W, Lillehei KO (2010) Epithelioid versus rhabdoid glioblastomas are distinguished by monosomy 22 and immunohistochemical expression of INI‐1 but not claudin 6. Am J Surg Pathol 34:341–354. [DOI] [PubMed] [Google Scholar]
- 20. Koelsche C, Sahm F, Capper D, Reuss D, Sturm D, Jones DT et al (2013) Distribution of TERT promoter mutations in pediatric and adult tumors of the nervous system. Acta Neuropathol 126:907–915. [DOI] [PubMed] [Google Scholar]
- 21. Kuroda J, Nobusawa S, Nakamura H, Yokoo H, Ueda R, Makino K et al (2016) A case of an epithelioid glioblastoma with the BRAF V600E mutation colocalized with BRAF intact low‐grade diffuse astrocytoma. Neuropathology 36:181–186. [DOI] [PubMed] [Google Scholar]
- 22. Matsumura N, Nakajima N, Yamazaki T, Nagano T, Kagoshima K, Nobusawa S et al (2017) Concurrent TERT promoter and BRAF V600E mutation in epithelioid glioblastoma and concomitant low‐grade astrocytoma. Neuropathology 37:58–63. [DOI] [PubMed] [Google Scholar]
- 23. Mistry M, Zhukova N, Merico D, Rakopoulos P, Krishnatry R, Shago M et al (2015) BRAF mutation and CDKN2A deletion define a clinically distinct subgroup of childhood secondary high‐grade glioma. J Clin Oncol 33:1015–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Miyahara M, Nobusawa S, Inoue M, Okamoto K, Mochizuki M, Hara T (2016) Glioblastoma with rhabdoid features: report of two young adult cases and review of the literature. World Neurosurg 86:515.e1–515.e9. [DOI] [PubMed] [Google Scholar]
- 25. Nagai S, Kurimoto M, Ishizawa S, Hayashi N, Hamada H, Kamiyama H, Endo S (2009) A rare astrocytic tumor with rhabdoid features. Brain Tumor Pathol 26:19–24. [DOI] [PubMed] [Google Scholar]
- 26. Nagaishi M, Nobusawa S, Yokoo H, Sugiura Y, Tsuda K, Tanaka Y et al (2016) Genetic mutations in high grade gliomas of the adult spinal cord. Brain Tumor Pathol 33:267–269. [DOI] [PubMed] [Google Scholar]
- 27. Nakazato Y, Ishizeki J, Takahashi K, Yamaguchi H, Kamei T, Mori T (1982) Localization of S‐100 protein and glial fibrillary acidic protein‐related antigen in pleomorphic adenoma of the salivary glands. Lab Invest 46:621–626. [PubMed] [Google Scholar]
- 28. Nobusawa S, Hirato J, Kurihara H, Ogawa A, Okura N, Nagaishi M et al (2014) Intratumoral heterogeneity of genomic imbalance in a case of epithelioid glioblastoma with BRAF V600E mutation. Brain Pathol 24:239–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Nobusawa S, Lachuer J, Wierinckx A, Kim YH, Huang J, Legras C et al (2010) Intratumoral patterns of genomic imbalance in glioblastomas. Brain Pathol 20:936–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Schiffman JD, Hodgson JG, VandenBerg SR, Flaherty P, Polley MY, Yu M et al (2010) Oncogenic BRAF mutation with CDKN2A inactivation is characteristic of a subset of pediatric astrocytomas. Cancer Res 70:512–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Schindler G, Capper D, Meyer J, Janzarik W, Omran H, Herold‐Mende C et al (2011) Analysis of BRAF V600E mutation in 1,320 nervous system tumors reveals high mutation frequencies in pleomorphic xanthoastrocytoma, ganglioglioma and extra‐cerebellar pilocytic astrocytoma. Acta Neuropathol 121:397–405. [DOI] [PubMed] [Google Scholar]
- 32. Schwab CJ, Jones LR, Morrison H, Ryan SL, Yigittop H, Schouten JP, Harrison CJ (2010) Evaluation of multiplex ligation‐dependent probe amplification as a method for the detection of copy number abnormalities in B‐cell precursor acute lymphoblastic leukemia. Genes Chromosomes Cancer 49:1104–1113. [DOI] [PubMed] [Google Scholar]
- 33. Tagawa H, Karnan S, Suzuki R, Matsuo K, Zhang X, Ota A et al (2005) Genome‐wide array‐based CGH for mantle cell lymphoma: identification of homozygous deletions of the proapoptotic gene BIM. Oncogene 24:1348–1358. [DOI] [PubMed] [Google Scholar]
- 34. Tanaka S, Nakada M, Nobusawa S, Suzuki SO, Sabit H, Miyashita K, Hayashi Y (2014) Epithelioid glioblastoma arising from pleomorphic xanthoastrocytoma with the BRAF V600E mutation. Brain Tumor Pathol 31:172–176. [DOI] [PubMed] [Google Scholar]
- 35. Vaubel RA, Caron AA, Yamada S, Decker PA, Eckel Passow JE, Rodriguez FJ et al (2017) Recurrent copy number alterations in low‐grade and anaplastic pleomorphic xanthoastrocytoma with and without BRAF V600E mutation. Brain Pathol [Epub ahead of print; doi: 10.1111/bpa.12495]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Watanabe K, Tachibana O, Sato K, Yonekawa Y, Kleihues P, Ohgaki H (1996) Overexpression of the EGF receptor and p53 mutations are mutually exclusive in the evolution of primary and secondary glioblastomas. Brain Pathol 6:217–224. [DOI] [PubMed] [Google Scholar]
- 37. Watanabe T, Nobusawa S, Kleihues P, Ohgaki H (2009) IDH1 mutations are early events in the development of astrocytomas and oligodendrogliomas. Am J Pathol 174:1149–1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Weber RG, Hoischen A, Ehrler M, Zipper P, Kaulich K, Blaschke B et al (2007) Frequent loss of chromosome 9, homozygous CDKN2A/p14(ARF)/CDKN2B deletion and low TSC1 mRNA expression in pleomorphic xanthoastrocytomas. Oncogene 26:1088–1097. [DOI] [PubMed] [Google Scholar]
- 39. Yokoo H, Kinjo S, Hirato J, Nakazato Y (2006) Fluorescence in situ hybridization targeted for chromosome 1p of oligodendrogliomas (in Japanese). Rinsho Kensa 50:761–766. [Google Scholar]
- 40. Yokoo H, Nobusawa S, Takebayashi H, Ikenaka K, Isoda K, Kamiya M et al (2004) Anti‐human Olig2 antibody as a useful immunohistochemical marker of normal oligodendrocytes and gliomas. Am J Pathol 164:1717–1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zacher A, Kaulich K, Stepanow S, Wolter M, Köhrer K, Felsberg J et al (2017) Molecular diagnostics of gliomas using next generation sequencing of a glioma‐tailored gene panel. Brain Pathol 27:146–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional supporting information may be found online in the Supporting Information section at the end of the article.
Table S1. Copy number abberations (CNA) detected by array comparative genomic hybridization.
Figure S1. MLPA and FISH analysis. A. MLPA analysis shows CDKN2A/B homozygous deletion (dosage quotient < 0.4) both in oligoastrocytoma‐like and E‐GBM components of case 8. IDH1/2 mutation or 1p/19q loss is not detected in either component. B. LOH of ODZ3 is detected in recurrent E‐GBM but not in primary PXA of case 2, using a red probe targeting ODZ3 and a reference probe labeled in green. E‐GBM; epithelioid glioblastoma, PXA; pleomorphic xanthoastrocytoma, MLPA; multiplex ligation‐dependent probe amplification, FISH; fluorescence in situ hybridization, LOH; loss of heterozygosity.