

Abstract
Giant cell glioblastoma (gcGBM) is a rare histological variant of GBM, accounting for about 1% of all GBM. The prognosis is poor generally though gcGBM does slightly better than the other IDH‐wild‐type GBM. Because of the rarity of the cases, there has been no comprehensive molecular analysis of gcGBM. Previously, single‐gene study identified genetic changes in TP53, PTEN and TERT promoter mutation in gcGBM. In this report, we performed whole‐exome sequencing (WES) to identify somatically acquired mutations and copy number variations (CNVs) in 10 gcGBM genomes. We also examined TERT promoter mutation and MGMT methylation in our cohort. On top of the reported mutations, WES revealed ATRX, PIK3R1, RB1 and SETD2 as the recurrent mutations in gcGBM. Notably, one tumor harbored a mutation in MutS homolog 6 (MSH6) that is a key mismatch repair (MMR) gene. This tumor demonstrated hypermutation phenotype and showed an increased number of somatic mutations. TERT promoter mutation and MGMT methylation were observed in 20% and 40% of our samples, respectively. In conclusion, we described relevant mutation profiling for developing future targeted therapies in gcGBM.
Keywords: giant cell glioblastoma, hypermutation phenotype, whole‐exome sequencing
Introduction
Giant cell glioblastoma (gcGBM) is a rare histological variant of GBM that constitutes approximately 1% of all GBM 22, 35. Histologically, gcGBM is characterized by numerous bizarre, multinucleated giant cells as large as 0.5 mm in diameter with astrocytic differentiation 22, 37. Compared to regular GBM, gcGBM is more often present in younger patients 35. Despite the poor prognosis for GBM, this variant is associated with improved clinical outcome compared to the regular IDH‐wild‐type GBM 24, 38, 42. At present, the best treatment strategy of gcGBM has not yet been ascertained. Like GBM, complete surgical resection, radiation and chemotherapy are the mainstay of gcGBM management.
The underlying genetic alterations caused gcGBM remain obscure because of scarcity of samples for molecular examination and the low incidence of the disease. TP53 mutation is common in gcGBM and was identified in about 80% of tumors using first‐generation sequencing approach 30, 32. Mutations in PTEN and TERT promoter were also found in gcGBM to a lesser extent 34, 39. Similar to primary GBM, gcGBM often do not carry IDH mutation 25, 56. At chromosomal level, 50% and 42% of gcGBM showed loss of chromosome 10q and 19q, respectively 34. However, frequent EGFR amplification detected in regular GBM is only observed in less than 10% of gcGBM 30, 32.
Advancement in high‐throughput next‐generation sequencing allows oncologists to decipher genomic landscape of tumors. We conducted whole‐exome sequencing (WES) of 10 gcGBM samples and matched blood. The goals of this study were to sequence the exome of gcGBM to identify potentially recurrent somatic mutations and copy number variations (CNVs) in gcGBM.
Materials and Methods
Frozen tissue specimens
A total of 10 primary, treatment‐naïve giant cell GBMs were obtained at the time of neuro‐oncology service between years 2010 and 2015 at Huashan Hospital, Shanghai. Tissues were snap frozen and stored at −80°C until use. Samples were histologically reviewed by two neuro‐pathologists (Ng HK and Chen H) and diagnosed according to WHO 2016. Clinicopathological information of the patients is summarized in Table 1. This study was approved by the Ethics Committee of Huashan Hospital, Fudan University.
Table 1.
Patient characteristics of 10 giant cell glioblastomas in this study.
| Patient | Age | Sex | KPS score at diagnosis | Survival status | Overall survival (months) | Chemotherapy (yes/no) | Radiotherapy (yes/no) | Extent of resection (total vs non‐total) |
|---|---|---|---|---|---|---|---|---|
| 1 | 39 | F | 70 | Dead | 10.37 | Yes | Yes | Total |
| 2 | 17 | M | 90 | Dead | 11.50 | Yes | Yes | Total |
| 3 | 39 | F | 80 | Dead | 12.67 | Yes | Yes | Total |
| 4 | 12 | F | 90 | Dead | 12.93 | Yes | Yes | Total |
| 5 | 42 | M | 80 | Dead | 13.23 | Yes | Yes | Total |
| 6 | 41 | M | 90 | Dead | 27.16 | Yes | Yes | Total |
| 7 | 50 | F | 90 | Dead | 28.00 | Yes | Yes | Total |
| 8 | 30 | F | 80 | Alive | 35.16 | Yes | Yes | Total |
| 9 | 63 | M | 80 | Alive | 36.70 | Yes | Yes | Total |
| 10 | 51 | F | 80 | Alive | 38.00 | Yes | Yes | Total |
Whole‐exome sequencing
DNA was extracted from tissue and blood using Qiagen DNAeasy kits (Qiagen). DNA was quantified using Nanodrop ND‐100 (Thermo Scientific, Waltham, MA) and Qubit 2.0 Fluorometer (Life Technologies, Carlsbad, CA, USA). Library preparation, exome capture and sequencing were done by Genetron Health (Shanghai, China). Genomic DNA were captured and amplified with Agilent Technologies SureSelect Human All Exon version 5 (Agilent Technologies, Santa Clara, CA, USA), followed by paired‐end sequencing on HiSeq2500 platform (Illumina Inc., San Diego, CA, USA). The raw data of WES are available upon request.
Data analysis
Sequencing reads were mapped to the human genome (GRCh37) using Burrows‐Wheeler Alignment tool (BWA). Duplicate reads were then marked using Picard (http://broadinstitute.github.io/picard/), and mapped reads around known deletions were locally realigned using Genome Analysis Toolkit (GATK) to improve the overall quality of alignment.
Single nucleotide variants (SNVs) and short insertions and deletions (indels) were detected using MuTect2 10 and Strelka 43, and their functional effects were annotated using ANNOVAR 52. Potential germline mutations were removed if a mutation had a frequency of greater than 1% in public databases (ExAC, ESP and 1000 Genomes Project). Copy number variations were interrogated with CNVkit 49. GISTIC version 2.0 (http://archive.broadinstitute.org/cancer/cga/gistic) analysis was performed to identify significantly recurrent copy number gains and losses at focal level, defined as regions spanning <50% of a chromosome arm. A log2 ratio above 0.3 was considered as “gain,” and a log2 ratio below −0.3 was considered as “loss.” Amplification and homozygous loss were considered when log2 ratio was >2.0 and <2.0, respectively.
Pathway and GO enrichment analyses
ConsensusPathDB of the Max Planck Institute for Molecular Genetics was used in pathway and GO enrichment analyses 16. We searched for pathways as defined KEGG, with a minimum of 5% members and a Q‐value cutoff at 0.05. Also, we performed an enrichment analysis based on Gene Ontology Biological Process Level 5 with the same website and analysis tool, and a Q‐value of <0.05 was regarded as significant.
TERT promoter mutation analysis
PCR amplification was conducted on DNA extracted from formalin‐fixed, paraffin‐embedded (FFPE) tissues. The primer sequences were 5′‐GTCCTGCCCCTTCACCTT‐3′ and 5′‐CAGCGCTGCCTGAAACTC‐3′, and amplified a 163‐bp fragment with KAPA2G Robust HotStart ReadyMix (Sigma). The PCR product was purified and sequenced with BigDye Terminator Cycle Sequencing kit v1.1 (Life Technologies). The products were resolved in 3130xl Genetic Analysis.
O6‐methylguanine‐DNA methyltransferase (MGMT) methylation analysis
MGMT methylation status was evaluated by methylation‐specific polymerase chain reaction (MSP) according to our previous report 11.
Results
Clinical characteristic of 10 gcGBM
To explore the mutational profiling in gcGBM, we performed WES on 10 primary, treatment‐naïve gcGBM tumors and matched blood samples. The mean and median ages of the samples were 38.4 and 40 years old, respectively. This is consistent with the literature that gcGBM is present in younger population compared to classical GBM 33, 35. Male and female ratio was 1:1.5. All tumors were found in the hemisphere with majority of them (70%; 7/10) located in the frontal lobe (Table 1). Tumors located in the temporal lobe were found in two cases. All patients had undergone surgery, chemotherapy and radiotherapy. Histology of all cases can be found in Supporting Information Figure S1.
Mutational spectrum in gcGBM
The mean coverage was 75.19 × for gcGBM and 85.97 × for normal samples. A total of 1312 tumor‐specific (somatic) mutations were identified, of which 947 were non‐silent. Interestingly, our cohort has a median of 35 coding mutations per tumor, which is about 2 times lower compared to regular GBM 29. Excluding the hypermutated tumor which will be discussed below, the nine samples had a median rate of 0.62 coding mutations per megabase, which is 3.5‐fold lower compared to regular GBM 4.
We found 28 recurrent mutations (Figure 1; Supporting Information Table S1). TP53 mutation was present in 4 of 10 tumors, representing the most frequent genomic alteration in our sample set. The result is in concordance with previous literature showing high frequency of TP53 mutation in gcGBM 32, 39. Notably, mutation in ATRX, PIK3R1, RB1 and SETD2 was identified in 20%, 20%, 20% and 30% of gcGBM, respectively. These gene mutations have not been reported in gcGBM, albeit in low‐ and high‐grade gliomas 8, 13, 55. We also identified PTEN mutation in 20% of gcGBM, and the incidence is in agreement with the literature 32, 39. We did validate the mutations identified in gcGBMs by Sanger sequencing. Examples are shown in Figures 2, 3, 4.
Figure 1.
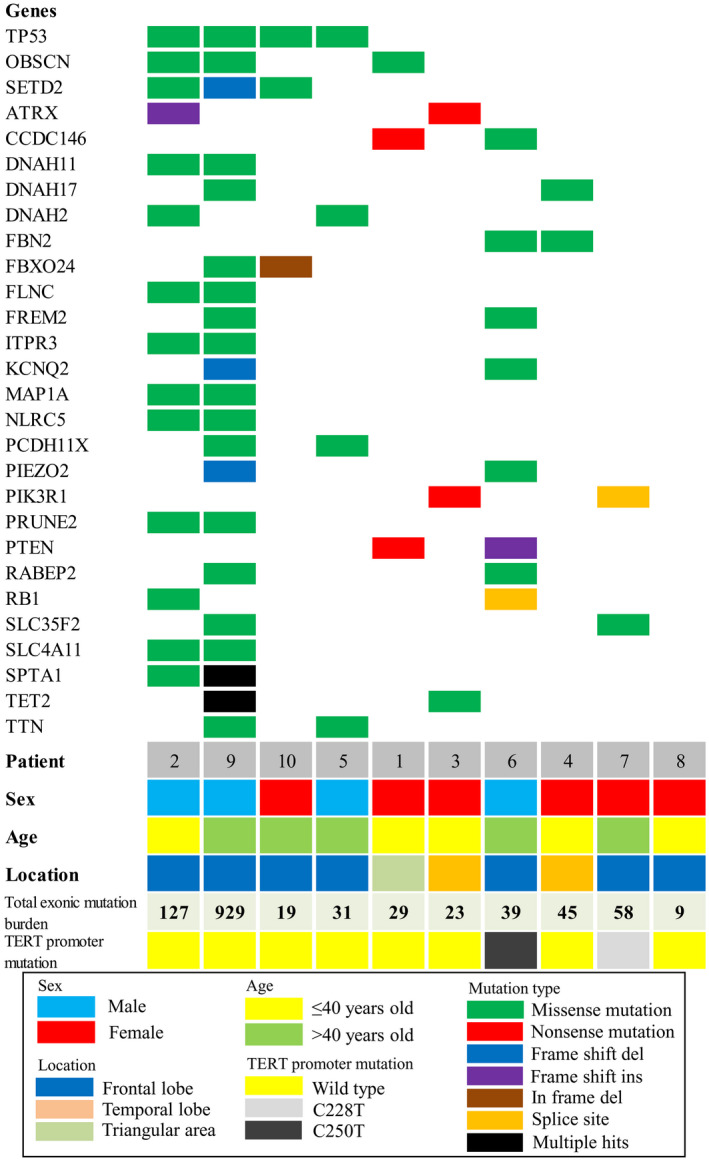
Mutation spectrum of giant cell glioblastomas. The heat map illustrates recurrent mutations found in 10 gcGBM samples. Type of mutation is represented by different colors. The bottom indicates age, sex, tumor location, number of mutations and TERT promoter mutation status of individual samples.
Figure 2.

Histology and radiologic feature of a gcGBM from a 17‐year‐old male with RB1 mutation. A. H&E section of the tumor and B. T1‐enhanced MRI of the tumor mass. C. Integrative Genomics Viewer (IGV) screenshot illustrates RB1 mutation. D. RB1 mutation was confirmed by Sanger sequencing.
Figure 3.
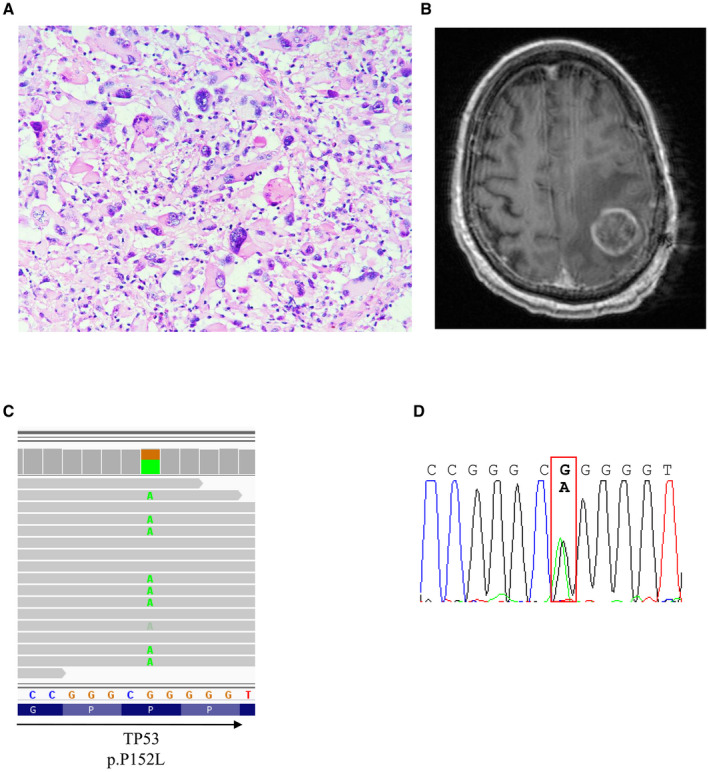
Histology and radiologic feature of a gcGBM from a 42‐year‐old male with TP53 mutation. A. H&E section and B. T1‐enhanced images of the tumor. C. TP53 mutation was visualized with IGV and D. validated by Sanger sequencing.
Figure 4.
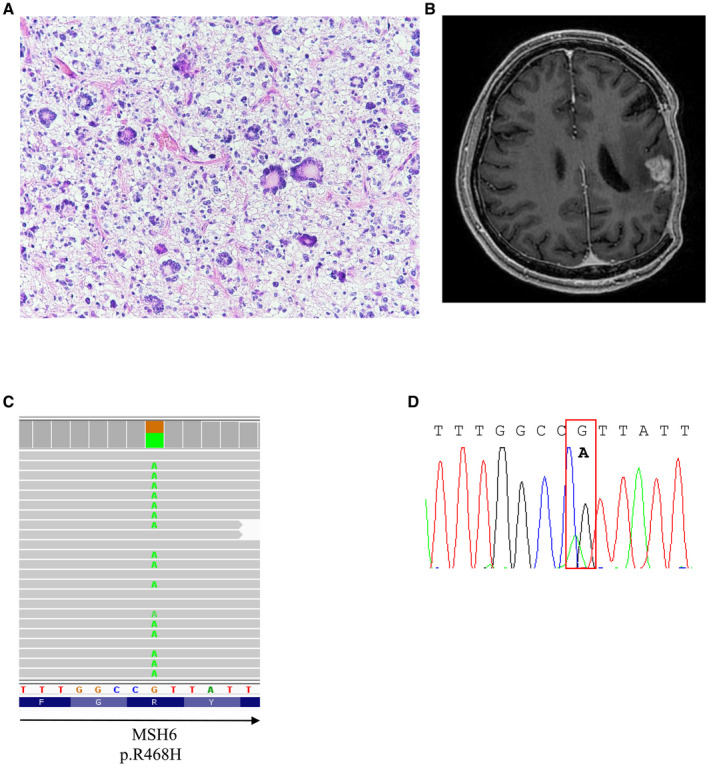
Representative figures of H&E and MRI imaging of a 63‐year‐old gcGBM possessing MSH6 mutation. A. H&E section and B. T1‐enhanced images of the tumor. C. The missense mutation was visualized with IGV and D. confirmed by Sanger sequencing.
Pathway analysis revealed that the 28 recurrent alterations found in our exome study were significantly (i.e., FDR Q‐value < 0.05) enriched with 3 pathways in KEGG. As shown in Table 2, they were “Glioma—Homo sapiens (human)” (FDR Q‐value = 3.15 × 10−5), “Melanoma—Homo sapiens (human)” (FDR Q‐value = 3.15 × 10−5) and “Endometrial cancer—Homo sapiens (human)” (FDR Q‐value = 2.59 × 10−4). The recurrent alterations contained in the last two pathways are essentially subsets of those contained in the glioma pathway. Gene ontology analysis of the 28 recurrent alternations identified “GO:0034349‐ glial cell apoptotic process” as the only significantly enriched term from the GO Biological Process Level 5 (FDR Q‐value = 1.68 × 10−2; Table 3).
Table 2.
Pathway analysis of 28 recurrent mutations in giant cell glioblastoma.
| KEGG pathways | Recurrent alterations | P‐value | FDR Q‐value |
|---|---|---|---|
| Glioma—Homo sapiens (human) | PIK3R1, RB1, PTEN, TP53 | 2.98 × 10−6 | 3.15 × 10−5 |
| Melanoma—Homo sapiens (human) | PIK3R1, RB1, PTEN, TP53 | 3.15 × 10−6 | 3.15 × 10−5 |
| Endometrial cancer—Homo sapiens (human) | PIK3R1, PTEN, TP53 | 7.78 × 10−5 | 2.59 × 10−4 |
Only significant (Q‐value < 0.05) pathways from KEGG with at least 5% members containing recurrent alterations are shown.
Table 3.
Gene Ontology (GO) terms associated with recurrent mutations in giant cell glioblastoma.
| GO term ID | Biological process description | Recurrent alterations | P‐value | FDR Q‐value |
|---|---|---|---|---|
| GO:0034349 | Glial cell apoptotic process | RB1, TP53 | 1.84 × 10−4 | 1.68 × 10−2 |
Only significant (Q‐value < 0.05) terms from the GO Biological Process Level 5 with at least 5% members containing recurrent alternations are shown.
Copy number variations
CNVs were determined using software package CNVKit, which analyzed copy number based on read depth in the on‐target and off‐target reads 49. As shown in Figure 5, arm‐level copy number variation was identified in all patients except patients 8 and 10. Recurrent arm‐level gains were found at chromosomes 7 (n = 6) and 20 (n = 2). Recurrent arm‐level losses were occurred at chromosomes 10q (n = 2) and 22q (n = 2). One case displayed loss of the long arm of chromosome 10 (10q) also carried a PTEN mutation.
Figure 5.
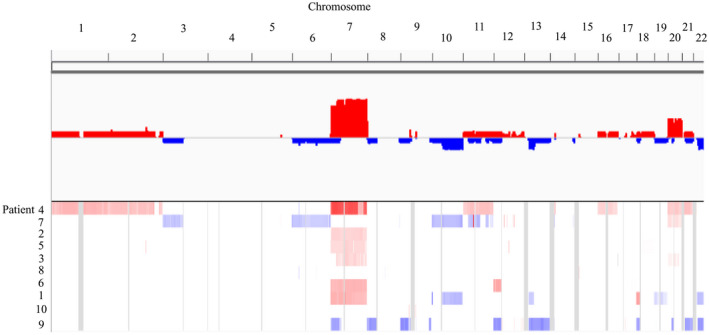
Arm‐level copy number variations in gcGBM. Chromosome gains (shown in red) and losses (shown in blue) were identified by WES. The numbers on top of the plot represent chromosome number (1–22), and the numbers on the left indicate patient number.
We then conducted GISTIC analysis to determine statistically significant recurrent focal gains and losses. As shown in Figure 6 and Table 4, we found in the gcGBM genome eight significantly focal loss regions including 6p21.32, 7q34, 8p11.22, 14q32.33, 19q13.42, 22q11.23 and 22q13.1. Genes located in these regions include ADAM3A, APOBEC3A, GSTT1, HLA‐DRB6, JAG2, KIR2DL1, KIR2DL3, KIR2DL4, LINC00226, LINC00221 and PRSS3P2. No recurrent focal gains were found in this cohort. We did detect a gcGBM from a 50‐year‐old female patient harboring amplifications for EGFR, MDM2 and CDK4. Neither EGFR, MDM2 nor CDK4 amplification was detected in the other nine samples. Thus, these aberrations did not reach significance in GICTIC analysis.
Figure 6.
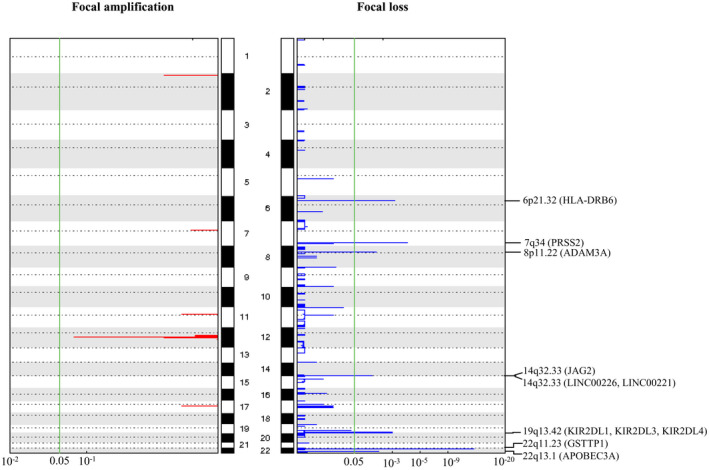
Significant focal copy number variations in gcGBM as revealed by GISTIC 2.0. The plot illustrates the statistical significance of aberrations displayed as FDR Q‐values (x‐axes). The human chromosomes 1 to 22 (hg19) are indicated along the y axis. Eight focal regions surpass the significance threshold (green line). Chromosomal locations of GSTT1, PRSS2, HLA‐DRB6, KIR2DL1, KIR2DL3, KIR2DL4, APOBEC3A, ADAM3A, JAG2, LINC00226 and LINC00221 Q‐value are indicated on the plot.
Table 4.
Focal copy number variations in giant cell glioblastoma.
| Cytoband | FDR Q‐value | Peak boundaries | Genes |
|---|---|---|---|
| 22q11.23 | 5.27 × 10−13 | chr22:24325343‐24373954 | GSTTP1 |
| 7q34 | 9.48 × 10−5 | chr7:142470509‐142482020 | PRSS2 |
| 6p21.32 | 7.04 × 10−4 | chr6:32497787‐32546606 | HLA‐DRB6 |
| 19q13.42 | 1.02 × 10−3 | chr19:55241149‐55331354 | KIR2DL1, KIR2DL3, KIR2DL4 |
| 22q13.1 | 5.18 × 10−3 | chr22:39338026‐39369501 | APOBEC3A |
| 8p11.22 | 6.59 × 10−3 | chr8:39254268‐39427923 | ADAM3A |
| 14q32.33 | 1.07 × 10−2 | chr14:105525907‐105644252 | JAG2 |
| 14q32.33 | 1.15 × 10−2 | chr14:106436558‐107349540 | LINC00226,LINC00221 |
A hypermutation phenotype and somatic MSH6 mutation in gcGBM
We found one gcGBM harbored high number of somatic exonic mutations (Figures 1 and 4). This tumor had 929 somatic mutations which is equivalent to 18.46 exonic mutation/Mb, representing a rate 22 times greater, on average, than the rate found in the other 9 cases.
The tumor with high number of somatic mutations carried a mutation in MutS homolog 6 (MSH6), resulting a change of amino acid from arginine (R) to histidine (H) at residue 468 at location inside the MutS domain I involved in DNA binding. Mutation at this position has been identified in prostate and stomach cancers 17, 41. In The Cancer Genome Atlas (TCGA) GBM study, MSH6 mutation was identified in 1.1% of tumors, and 2/3 of the mutations were located outside of MutS domain I.
TERT promoter mutation in gcGBM
TERT promoter mutation was identified in 2/10 (20%) samples. One sample carried C228T mutation, and another sample had C250T. The ages of these tumors were 41 and 50 years old, and the overall survival of them was 27–28 months. The mutation frequency in this study was similar to that in Oh et al study showing TERT promoter mutation in one‐quarter of gcGBM 34.
MGMT methylation status in gcGBM
Four gcGBM samples (40%) were positive for MGMT methylation as revealed by MSP. Similar prevalence of MGMT methylation was reported by Lohkamp et al 28. MGMT methylation was not associated with age, sex or overall survival.
Discussion
Giant cell glioblastoma (gcGBM) is a very rare disease and occurs in approximately 1% of all GBM 24, 38, 39, 44. Because of uncommon nature of this tumor entity, very few studies have reported the genomic and genetic alterations underlying this disease. We here employed next‐generation sequencing approach to reveal genomic‐wide mutational landscape of this entity, and identified potential‐mutated genes for target therapy.
First, we described the tumor mutation rate in gcGBM is much lower than the rates observed in regular GBM and other cancers 4, 29. Then, we found TP53 is the most frequent mutated gene in gcGBM, accounting for 40% of our cohort. The result is concordant with previous report showing the high prevalence of p53 mutation in gcGBM 30, 31, 32. TP53 is a well‐known tumor suppressor that is involved in cell cycle regulation and is dysregulated in 30% of primary de novo GBM and 65% of secondary GBM 36. Previous studies indicated nearly 80% of GBM had dysregulated p14ARF/MDM2/p53 pathway either by p53 mutation, amplification of MDM2 or deletion/mutation of p14ARF 19. In our cohort, amplification of MDM2 was found in one case, and this sample carried the wild‐type TP53 (see discussion). Deletion of p14ARF was identified in one case and the tumor had a mutation in TP53 gene. Mutation of p14ARF was not found in our cohort. Thus, 50% of our cohort (case #2, #5, #7, #9 and #10) showed p53 mutation, MDM2 amplification or p14ARF deletion. Besides TP53, RB1 (chromosome 13q14) is another well‐known tumor suppressor that also controls progression through G1 into the S‐phase of the cell cycle. Aberration of RB1 is found in around 10% of GBM according to TCGA 4. In this study, two gcGBM samples (20%) harbored RB1 mutation and one of them also carried a mutation in TP53 gene. Although we found RB1 mutation at a rate higher than in regular GBM, we have to be cautious in interpreting the data with our small sample size.
PTEN and PIK3R1 are members of RTK/RAS/PI(3)K signaling pathway, that plays key roles in proliferation, differentiation and survival of cancer cells. Nearly 90% of the regular GBMs showed various alterations leading to an aberrant activation of RTK/RAS/PI(3)K signaling cascade 8. In this study, we found 20% of gcGBM‐harbored PTEN mutation. The prevalence is consistent with the literature 39. PIK3R1 encodes a regulatory protein p85α, that forms the PI(3)K complex with a catalytically active protein p110α, and it is mutated in ~10% of the regular GBMs according to TCGA 8. We found two samples (20%) of gcGBM‐harbored PIK3R1 mutation, and one of them also had copy number gain in PIK3R1. PTEN and PIK3R1 mutations were mutually exclusive in this study and they did not overlap with TP53 mutation. Collectively, they made up of 40% of our cohort (case #1, #3, #6 and #7; Figure 1). We speculated that these tumors showed activation of RTK/RAS/PI(3)K signaling pathway. Further studies are required to verify the role of this pathway in the pathogenesis of gcGBM.
Furthermore, we reported for the first time recurrent mutation in ATRX and SETD2 in gcGBM. Mutation in ATRX (alpha thalassemia/mental retardation syndrome X‐linked), a SWI2/SNF2 family of DNA helicases functioned in chromatin modulation and maintenance of telomeres, is known in pediatric and adult gliomas 23, but has not been reported in gcGBM. Mutation in ATRX leads to epigenetic alterations, and epigenetic alteration is known to play a role in tumorigenesis 14, 20. In astrocytic tumors, DNA methylation profile was different between tumors with low‐ and high‐ATRX expression 6. Thus, it is possible that ATRX mutation could lead to alterations in global genomic methylation and gene expression in gcGBM. The gene SETD2 (KMT3A) is located on chromosome 3p21.31, and it encodes a methyltransferase that mediates trimethylation of H3K36 (H3K36me3). SETD2 is also involved in chromatin organization 48. Mutation of SETD2 is often described in pediatric high‐grade gliomas (PHGG) and many other tumors, including kidney, lung and liver cancers 9, 13, 26. In PHGG, SETD2 mutation leads to disruption of trimethyltransferase activity and distinct global DNA methylation signature 9, 13, 48. Recent study further demonstrated that SETD2 mutation is present in low‐ and high‐grade gliomas of children and adults and of hemisphere and midline 51. SETD2 mutation in gliomas was also associated with decreased H3K36me3 expression 13, 51. We further examined SETD2 and H3K36me3 levels by immunohistochemistry in our cohort. We adopted a semiquantitative scoring system in which signal intensity and percentage of positive cells were considered as parameters in grading IHC staining result 27. We found SETD2 expression level was not significantly different between SETD2‐wild‐type and SETD2‐mutant tumors. Yet, H3K36me3 level (determined by antibody Abcam ab9050) was significantly lower in the three SETD2‐mutant gcGBMs [0.33 ± 0.577 (mean ± SD)] compared to the wild‐type gcGBMs [4.43 ± 2.936 (mean ± SD); P = 0.049; Supporting Information Figure S2]. Our results suggested SETD2 mutations lead to a decrease in H3K36me3 expression in gcGBM.
Aside from ATRX, PIK3R1, RB1 and SETD2, we detected OBSCN mutation in 30% (3/10) of gcGBM. The patients were aged 17–63. Copy number variation in MYCN was seen in one patient (#1) and arm‐level copy number variation in chromosome 22q was observed in two patients (#1 and #9; Figure 5). None of these variations was found in non‐OBSCN mutant tumors. The gene OBSCN is located on chromosome 1q42.13. It encodes obscurins which are large cytoskeletal proteins with structural and regulatory roles 40. OBSCN is highly mutated in cancers and the mutation has been found in melanoma, salivary gland carcinoma, pancreatic, breast and colorectal cancers 2, 21, 47. Reduced obscurins expression level has been reported in breast cancer and depletion of obscurins leads to disruption of cell‐cell contacts and acquisition of a mesenchymal phenotype that leads to enhanced tumorigenesis, migration and invasiveness 46. In colorectal carcinoma, OBSCN mutation was found early in carcinogenesis and was considered an early driver of carcinogenesis 54. In contrast, OBSCN was not considered as a cancer driver gene in another study 50.
We then did a pathway analysis with ConsensusPathDB 16 and found that the glioma pathway in KEGG (involving TP53, PTEN, RB1 and PIK3R1 among the 28 recurrent alternations) was altered in 70% of cases (FDR Q‐value = 3.15 × 10−5). Gene ontology analysis suggested that 7.1% of these recurrent mutations (i.e., RB1, TP53) were related to glial cell apoptotic process (FDR Q‐value = 1.68 × 10−2).
Furthermore, we explored CNVs in our cohort. At arm‐level, we showed that gain of chromosome 7 was the most frequent CNV, followed by gain of chromosome 20 and losses of 10q and 22q. EGFR is located on chromosome 7, and is amplified in >40% of regular GBM. However, such alteration is less frequently detected (<10%) in gcGBM 38. We detected EGFR amplification in only one gcGBM (10%) diagnosed at the age of 50. Interesting, the same patient had MDM2 and CDK4 amplifications, and no other patient in this cohort showed EGFR, MDM2 or CDK4 amplification.
At focal level, we identified eight highly recurrent loss regions in gcGBM. All of them had at least one gene located within the focal loss regions. For instance, the focal loss region on chromosome 14q32.33 contains the gene JAG2, which encodes one of the four transmembrane ligands that bind to NOTCH receptors 12. The binding of ligand to NOTCH receptors induces cleavage of receptor, releases NOTCH intracellular domain and activates NOTCH target genes. Another focal loss region located on chromosome 8p11.22 involves the gene ADAM3A, which is deleted in 16% of pediatric high‐grade gliomas 3. We also identified a focal loss at chromosome 14q32 which contains a long noncoding RNA LINC00226 that was downregulated in pancreatic ductal adenocarcinoma 58.
Here, we also reported a single case of primary gcGBM exhibited hypermutated phenotype. This tumor carried a missense mutation at amino acid residue 468 of the MSH6 gene. High frequency of hypermutation has been identified in melanoma, bladder and lung cancer 1, 15, 53. Hypermutation has also been identified in pediatric and adult brain tumors 7, 45. The gene MSH6 is located on chromosome 2p16.3, and it is a main player in DNA mismatch repair system (MMR). Hypermutation phenotype has been linked to MSH6 mutation in GBM after alkylating agent or temozolomide treatment 5, 18. In fact, all four MSH6 mutations found in GBM by TCGA were posttreatment samples, and subsequent study showed absent of MSH6 mutation in the matched treatment‐naïve biopsies 57.
In conclusion, by next‐generation sequencing, we have identified potential clinically relevant mutations for targeted therapy in gcGBM.
Conflict of Interest
The authors have no conflict of interest.
Supporting information
Figure S1. H&E stained sections of 10 gcGBM samples at (A‐J) low and (K‐T) high powers.
Figure S2. Representative images of immunohistochemistry staining for H3K36me3 in gcGBM.
Table S1. List of the detected mutations in gcGBM samples.
Acknowledgments
This study was supported by National Natural Science Foundation of China (NSFC, grant numbers 81472373 and 81702471), and Shenzhen Science Technology and Innovation Commission (reference number JCYJ20170307165432612).
Contributor Information
Hong Chen, Email: cherrychen30@126.com.
Ho‐Keung Ng, Email: hkng@cuhk.edu.hk.
References
- 1. Akbani R, Akdemir KC, Aksoy BA, Albert M, Ally A, Amin SB et al (2015) Cancer Genome Atlas Network. Genomic classification classification of cutaneous melanoma. Cell 161:1681–1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Balakrishnan A, Bleeker FE, Lamba S, Rodolfo M, Daniotti M, Scarpa A et al (2007) Novel somatic and germline mutations in cancer candidate genes in glioblastoma, melanoma, and pancreatic carcinoma. Cancer Res 67:3545–3550. [DOI] [PubMed] [Google Scholar]
- 3. Barrow J, Adamowicz‐Brice M, Cartmill M, MacArthur D, Lowe J, Robson K et al (2011) Homozygous loss of ADAM3A revealed by genome‐wide analysis of pediatric high‐grade glioma and diffuse intrinsic pontine gliomas. Neuro Oncol 13:212–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Brennan CW, Verhaak RG, McKenna A, Campos B, Noushmehr H, Salama SR et al (2013) The somatic genomic landscape of glioblastoma. Cell 155:462–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cahill DP, Levine KK, Betensky RA, Codd PJ, Romany CA, Reavie LB et al (2007) Loss of the mismatch repair protein MSH6 in human glioblastomas is associated with tumor progression during temozolomide treatment. Clin Cancer Res 13:2038–2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cai J, Chen J, Zhang W, Yang P, Zhang C, Li M et al (2015) Loss of ATRX, associated with DNA methylation pattern of chromosome end, impacted biological behaviors of astrocytic tumors. Oncotarget 6:18105–18115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Campbell BB, Light N, Fabrizio D, Zatzman M, Fuligni F, de Borja R et al (2017) Comprehensive Analysis of Hypermutation in Human Cancer. Cell 171:1042–1056.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cancer Genome Atlas Research Network (2008) Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 455:1061–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chamdine O, Gajjar A (2014) Molecular characteristics of pediatric high‐grade gliomas. CNS Oncol 3:433–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C et al (2013) Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol 31:213–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dong SM, Pang JC, Poon WS, Hu J, To KF, Chang AR et al (2001) Concurrent hypermethylation of multiple genes is associated with grade of oligodendroglial tumors. J Neuropathol Exp Neurol 60:808–816. [DOI] [PubMed] [Google Scholar]
- 12. D'Souza B, Miyamoto A, Weinmaster G (2008) The many facets of Notch ligands. Oncogene 27:5148–5167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Fontebasso AM, Schwartzentruber J, Khuong‐Quang DA, Liu XY, Sturm D, Korshunov A et al (2013) Mutations in SETD2 and genes affecting histone H3K36 methylation target hemispheric high‐grade gliomas. Acta Neuropathol 125:659–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gibbons RJ, McDowell TL, Raman S, O'Rourke DM, Garrick D, Ayyub H et al (2000) Mutations in ATRX, encoding a SWI/SNF‐like protein, cause diverse changes in the pattern of DNA methylation. Nat Genet 24:368–371. [DOI] [PubMed] [Google Scholar]
- 15. Govindan R, Ding L, Griffith M, Subramanian J, Dees ND, Kanchi KL et al (2012) Genomic landscape of non‐small cell lung cancer in smokers and never‐smokers. Cell 150:1121–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Herwig R, Hardt C, Lienhard M, Kamburov A (2016) Analyzing and interpreting genome data at the network level with ConsensusPathDB. Nat Protoc 11:1889–1907. [DOI] [PubMed] [Google Scholar]
- 17. Hoadley KA, Yau C, Hinoue T, Wolf DM, Lazar AJ, Drill E et al (2018) Cell‐of‐origin patterns dominate the molecular classification of 10,000 tumors from 33 types of cancer. Cell 173:291–304.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hunter C, Smith R, Cahill DP, Stephens P, Stevens C, Teague J et al (2006) A hypermutation phenotype and somatic MSH6 mutations in recurrent human malignant gliomas after alkylator chemotherapy. Cancer Res 66:3987–3991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ichimura K, Bolin MB, Goike HM, Schmidt EE, Moshref A, Collins VP (2000) Deregulation of the p14ARF/MDM2/p53 pathway is a prerequisite for human astrocytic gliomas with G1‐S transition control gene abnormalities. Cancer Res 60:417–424. [PubMed] [Google Scholar]
- 20. Jones PA, Baylin SB (2007) The epigenomics of cancer. Cell 128:683–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kang H, Tan M, Bishop JA, Jones S, Sausen M, Ha PK et al (2017) Whole‐Exome Sequencing of Salivary Gland Mucoepidermoid Carcinoma. Clin Cancer Res 23:283–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Karsy M, Gelbman M, Shah P, Balumbu O, Moy F, Arslan E (2012) Established and emerging variants of glioblastoma multiforme: review of morphological and molecular features. Folia Neuropathol 50:301–321. [DOI] [PubMed] [Google Scholar]
- 23. Karsy M, Guan J, Cohen AL, Jensen RL, Colman H (2017) New molecular considerations for Glioma: IDH, ATRX, BRAF, TERT, H3 K27M. Curr Neurol Neurosci Rep 17:19. [DOI] [PubMed] [Google Scholar]
- 24. Kozak KR, Moody JS (2009) Giant cell glioblastoma: a glioblastoma subtype with distinct epidemiology and superior prognosis. Neuro Oncol 11:833–841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Leske H, Brandal P, Rushing EJ, Niehusmann P (2017) IDH‐mutant giant cell glioblastoma: A neglected tumor variant? Clin Neuropathol 36:293–295. [DOI] [PubMed] [Google Scholar]
- 26. Li J, Duns G, Westers H, Sijmons R, van den Berg A, Kok K (2016) SETD2: an epigenetic modifier with tumor suppressor functionality. Oncotarget 7:50719–50734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Li KK, Qi Y, Xia T, Chan AK, Zhang ZY, Aibaidula A et al (2017) The kinesin KIF14 is overexpressed in medulloblastoma and downregulation of KIF14 suppressed tumor proliferation and induced apoptosis. Lab Invest 97:946–961. [DOI] [PubMed] [Google Scholar]
- 28. Lohkamp LN, Schinz M, Gehlhaar C, Guse K, Thomale UW, Vajkoczy P et al (2016) MGMT promoter methylation and BRAF V600E mutations are helpful markers to discriminate pleomorphic xanthoastrocytoma from giant cell glioblastoma. PLoS One 11:e0156422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Martincorena I, Campbell PJ (2015) Somatic mutation in cancer and normal cells. Science 349:1483–1489. [DOI] [PubMed] [Google Scholar]
- 30. Martinez R, Roggendorf W, Baretton G, Klein R, Toedt G, Lichter P et al (2007) Cytogenetic and molecular genetic analyses of giant cell glioblastoma multiforme reveal distinct profiles in giant cell and non‐giant cell subpopulations. Cancer Genet Cytogenet 175:26–34. [DOI] [PubMed] [Google Scholar]
- 31. Martinez‐Diaz H, Kleinschmidt‐DeMasters BK, Powell SZ, Yachnis AT (2003) Giant cell glioblastoma and pleomorphic xanthoastrocytoma show different immunohistochemical profiles for neuronal antigens and p53 but share reactivity for class III beta‐tubulin. Arch Pathol Lab Med 127:1187–1191. [DOI] [PubMed] [Google Scholar]
- 32. Meyer‐Puttlitz B, Hayashi Y, Waha A, Rollbrocker B, Boström J, Wiestler OD et al (1997) Molecular genetic analysis of giant cell glioblastomas. Am J Pathol 151:853–857. [PMC free article] [PubMed] [Google Scholar]
- 33. Müller W, Slowik F, Firsching R, Afra D, Sanker P (1987) Contribution to the problem of giant cell astrocytomas. Neurosurg Rev 10:213–219. [DOI] [PubMed] [Google Scholar]
- 34. Oh JE, Ohta T, Nonoguchi N, Satomi K, Capper D, Pierscianek D et al (2016) Genetic Alterations in Gliosarcoma and Giant Cell Glioblastoma. Brain Pathol 26:517–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Oh T, Rutkowski MJ, Safaee M, Sun MZ, Sayegh ET, Bloch O et al (2014) Survival outcomes of giant cell glioblastoma: institutional experience in the management of 20 patients. J Clin Neurosci 21:2129–2134. [DOI] [PubMed] [Google Scholar]
- 36. Ohgaki H (2005) Genetic pathways to glioblastomas. Neuropathology 25:1–7. [DOI] [PubMed] [Google Scholar]
- 37. Ohgaki H, Kleihues P, Plate KH, Nakazato Y, Bignes DD (2016) Giant cell glioblastoma. In: WHO Classification of Tumours of the Central Nervous System, 4th edn.Louis DN,Ohgaki H,Wiestler OD,Cavenee WK (eds), pp. 46–47. IARC Press: Lyon. [Google Scholar]
- 38. Ortega A, Nuño M, Walia S, Mukherjee D, Black KL, Patil CG (2014) Treatment and survival of patients harboring histological variants of glioblastoma. J Clin Neurosci 21:1709–1713. [DOI] [PubMed] [Google Scholar]
- 39. Peraud A, Watanabe K, Schwechheimer K, Yonekawa Y, Kleihues P, Ohgaki H (1999) Genetic profile of the giant cell glioblastoma. Lab Invest 79:123–129. [PubMed] [Google Scholar]
- 40. Perry NA, Ackermann MA, Shriver M, Hu LY, Kontrogianni‐Konstantopoulos A (2013) Obscurins: unassuming giants enter the spotlight. IUBMB Life. 65:479–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Robinson D, Van Allen EM, Wu YM, Schultz N, Lonigro RJ, Mosquera JM et al (2015) Integrative clinical genomics of advanced prostate cancer. Cell 161:1215–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Sabel M, Reifenberger J, Weber RG, Reifenberger G, Schmitt HP (2001) Long‐term survival of a patient with giant cell glioblastoma. Case report. J Neurosurg 94:605–611. [DOI] [PubMed] [Google Scholar]
- 43. Saunders CT, Wong WS, Swamy S, Becq J, Murray LJ, Cheetham RK (2012) Strelka: accurate somatic small‐variant calling from sequenced tumor‐normal sample pairs. Bioinformatics 28:1811–1817. [DOI] [PubMed] [Google Scholar]
- 44. Shabihkhani M, Telesca D, Movassaghi M, Naeini YB, Naeini KM, Hojat SA et al (2017) Incidence, survival, pathology, and genetics of adult Latino Americans with glioblastoma. J Neurooncol 132:351–358. [DOI] [PubMed] [Google Scholar]
- 45. Shlien A, Campbell BB, de Borja R, Alexandrov LB, Merico D, Wedge D et al (2015) Combined hereditary and somatic mutations of replication error repair genes result in rapid onset of ultra‐hypermutated cancers. Nat Genet 47:257–262. [DOI] [PubMed] [Google Scholar]
- 46. Shriver M, Stroka KM, Vitolo MI, Martin S, Huso DL, Konstantopoulos K et al (2015) Loss of giant obscurins from breast epithelium promotes epithelial‐to‐mesenchymal transition, tumorigenicity and metastasis. Oncogene. 34:4248–4259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Sjoblom T, Jones S, Wood LD, Parsons DW, Lin J, Barber TD et al (2006) The consensus coding sequences of human breast and colorectal cancers. Science. 314:268–274. [DOI] [PubMed] [Google Scholar]
- 48. Sun X, Wei J, Wu X, Hu M, Wang L, Wang H et al (2005) Identification and characterization of a novel human histone H3 lysine 36‐specific methyltransferase. J Biol Chem 280:35261–35271. [DOI] [PubMed] [Google Scholar]
- 49. Talevich E, Shain AH, Botton T, Bastian BC (2016) CNVkit: Genome‐Wide Copy Number Detection and Visualization from Targeted DNA Sequencing. PLoS Comput Biol 12:e1004873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Tamborero D, Gonzalez‐Perez A, Perez‐Llamas C, Deu‐Pons J, Kandoth C, Reimand J et al (2013) Comprehensive identification of mutational cancer driver genes across 12 tumor types. Sci Rep 3:2650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Viaene AN, Santi M, Rosenbaum J, Li MM, Surrey LF, Nasrallah MP (2018) SETD2 mutations in primary central nervous system tumors. Acta Neuropathol Commun 6:123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Wang K, Li M, Hakonarson H (2010) ANNOVAR: functional annotation of genetic variants from high‐throughput sequencing data. Nucleic Acids Res 38:e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Weinstein JN, Akbani R, Broom BM, Wang W, Verhaak RG, McConkey D et al (2014) Comprehensive molecular characterization of urothelial bladder carcinoma. Nature 507:315–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Wolff RK, Hoffman MD, Wolff EC, Herrick JS, Sakoda LC, Samowitz WS et al (2018) Mutation analysis of adenomas and carcinomas of the colon: Early and late drivers. Genes Chromosomes Cancer 57:366–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Wu G, Diaz AK, Paugh BS, Rankin SL, Ju B, Li Y et al (2014) The genomic landscape of diffuse intrinsic pontine glioma and pediatric non‐brainstem high‐grade glioma. Nat Genet 46:444–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Yan H, Parsons DW, Jin G, McLendon R, Rasheed BA, Yuan W et al (2009) IDH1 and IDH2 mutations in gliomas. N Engl J Med 360:765–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Yip S, Miao J, Cahill DP, Iafrate AJ, Aldape K, Nutt CL et al (2009) MSH6 mutations arise in glioblastomas during temozolomide therapy and mediate temozolomide resistance. Clin Cancer Res 15:4622–4629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Yu X, Lin Y, Sui W, Zou Y, Lv Z (2017) Analysis of distinct long noncoding RNA transcriptional fingerprints in pancreatic ductal adenocarcinoma. Cancer Med 6:673–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. H&E stained sections of 10 gcGBM samples at (A‐J) low and (K‐T) high powers.
Figure S2. Representative images of immunohistochemistry staining for H3K36me3 in gcGBM.
Table S1. List of the detected mutations in gcGBM samples.