

SUMMARY
Pancreatic ductal adenocarcinoma (PDAC) is driven by co-existing mutations in KRAS and TP53. However, how these mutations collaborate to promote this cancer is unknown. Here we uncover sequence-specific changes in RNA splicing enforced by mutant p53 which enhance KRAS activity. Mutant p53 increases expression of splicing regulator hnRNPK to promote inclusion of cytosine-rich exons within GTPase Activating Proteins (GAPs), negative regulators of RAS family members. Mutant p53-enforced GAP isoforms lose cell membrane association, leading to heightened KRAS activity. Preventing cytosine-rich exon inclusion in mutant KRAS/p53 PDACs decreases tumor growth. Moreover, mutant p53 PDACs are sensitized to inhibition of splicing via spliceosome inhibitors. These data provide insight into co-enrichment of KRAS and p53 mutations and therapeutics targeting this mechanism in PDAC.
Graphical Abstract
INTRODUCTION
Pancreatic ductal adenocarcinoma (PDAC) is one of the most lethal cancers worldwide (Simoes et al., 2017). It is established that hotspot mutations in KRAS, a guanine nucleotide binding protein and member of the RAS-superfamily of small-GTPases, are present in 95% of PDACs and appear most commonly in codon G12 (Bailey et al., 2016, Collisson et al., 2011, Moffitt et al., 2015). KRAS is active when bound to GTP but inactive when bound to GDP. In physiologic conditions, conversion from the GTP to GDP-bound state occurs when the intrinsic GTPase activity of KRAS is stimulated by GTPase-Activating Proteins (GAPs) (Settleman et al., 1992). Different KRAS mutations have unique intrinsic and GAP-stimulated GTPase activities (Hunter et al., 2015). Among KRAS mutations, G12C has the highest intrinsic GTP hydrolysis rates, while G12D has the highest GAP-stimulated GTP hydrolysis rates (Hunter et al., 2015). These differences have led to development of G12C inhibitors (Janes et al., 2018), however, there is no targeted therapy for other mutant forms of KRAS.
Following mutations in KRAS, mutations in TP53 are the second most common in PDAC. ~40% of PDACs have hotspot missense TP53 mutations, while 30% have truncating mutations (Bailey et al., 2016, Collisson et al., 2011, Moffitt et al., 2015). Hotspots and truncating mutations in TP53 inactivate its tumor suppressor function. However, mice harboring hotspot p53 mutations develop more aggressive PDACs compared to those arising in their p53 heterozygous or null counterparts (Morton et al., 2010, Olive et al., 2004), suggesting that mutant forms of p53 exert gain-of-function activities.
Despite the importance of hotspot mutations in KRAS and TP53 for PDAC formation (Hingorani et al., 2005), the mechanistic basis for the cooperation between these oncoproteins, which might enable the design of therapies targeting PDACs, has been elusive. To this end, it has recently been shown that PDAC is composed of two molecular subtypes based on RNA expression signatures and patient survival (Bailey et al., 2016, Collisson et al., 2011, Moffitt et al., 2015). Of these, the most aggressive subtype is associated with combined mutations in KRAS and TP53 as well as increased expression of genes encoding RNA splicing proteins (Bailey et al., 2016). These data suggest potential altered RNA processing in PDAC. However, connections between TP53 mutations and altered splicing are not known, nor is it clear if adverse forms of PDAC are associated with changes in splicing.
RNA splicing, the enzymatic process of removing segments of premature RNA to produce mature RNA, mediates proteome diversity and gene expression regulation (Escobar-Hoyos et al., 2019). In cancer, RNA splicing is altered by mutations in splicing factors (Dvinge et al., 2016), dysregulated expression of splicing factors (Anczuków et al., 2012), and oncogenic transcription (Hsu et al., 2015), resulting in altered gene expression, the production of functionally aberrant proteins, and/or non-functional mRNAs. Whether and how RNA splicing may be altered in PDAC is not well understood however. To address these questions, we evaluated RNA splicing in PDACs with different TP53 genotypes.
RESULTS
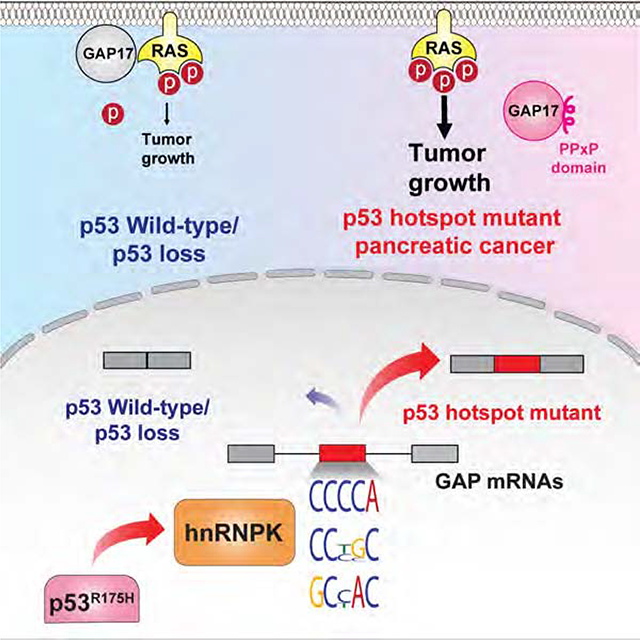
Mutant p53 promotes inclusion of cytosine-rich exons, impacting expression of GAP isoforms
Given an association between TP53 mutations and increased expression of RNA processing machinery, we set out to identify whether changes in splicing are linked to TP53 mutations. We performed supervised analyses of mRNA splicing using RNA-seq data from the ICGC (n = 84)(Bailey et al., 2016) and found that PDACs with TP53 missense hotspot mutations exhibited aberrant use of exons compared to PDACs with WT or truncating mutations in TP53 (Figure S1A–C, Table S1). Mutations at R175 were associated with the greatest change in exon usage (Figure 1A, S1B–C, Table S1).
Figure 1: p53R175H promotes C-rich cassette exons, impacting expression of GAP isoforms in pancreatic cancer.

A. Differentially spliced exons in PDAC patients with missense hotspot mutations in p53 compared to those with WT TP53 and nonsense/frameshift mutations in p53 (“Trunc”). RNA-Seq from the ICGC (Bailey et al., 2016).
B-C. Differentially spliced exons whose inclusion was significantly promoted (red circles) or repressed (blue circles) in p53R175H versus control cells with knockdown of p53R175H. Splicing events quantified using ‘percent spliced in’ (PSI, or Ψ) value. Promoted and repressed exons are defined as those whose Ψ values are increased or decreased ≥10%, respectively (N). Grey circles: exons where ΔΨ is <10%. Statistical significance calculated using Kolmogorov-Smirnov test. PDAC patient organoids (KRASG12D/+; p53R172H/−) and murine KPC cells following transduction with doxycycline-inducible control (shR) or anti-p53 shRNAs (sh1 and 2) (B). RNA-seq and westerns performed after 5 days of doxycycline, n = 3. Murine PanIN organoids (KrasG12D/+; WT p53; Mist1-Cre) bearing p53R172H/+ introduced by CRISPR, compared to WT p53 (C). RNA-seq and westerns performed 20 days after confirmation of genome editing, n= 3.
D. Nucleotide motifs enriched in repressed and promoted exons by p53R175H identified from comparisons in B-C.
E. Spatial distribution and relative frequency of CCC and AAA motifs in exons promoted versus repressed by p53R172H in murine PDAC cells. Shading, 95% confidence interval by bootstrapping.
F. Left, Gene Ontology analysis of mRNAs with polyC exons based on the comparisons in B-C. p values for multiple comparisons. Green bars, biologic processes involving GAPs of RAS GTPases. Right, schema of how GAPs terminate signaling by inducing GTP hydrolysis of GTP-bound RAS.
G. Percentage of GAPs that undergo alternative splicing (AS) in the presence of p53R175H in patient and murine PDAC. GAP mRNAs with exon splicing events in both human and murine PDACs listed.
H. Intersection of GAP mRNAs that gain polyC exons by p53R175H from A-C comparisons. Fisher’s exact test p= 0.0007.
I. Qualitative and quantitative RT-PCRs of GAP17 AS in KPC cells in the presence or absence of mutant p53.
Left, schematic of primers that flank the two exons (exons 16 and 18) surrounding the polyC exon (exon 17). Upper band denotes inclusion of polyC exon, while lower band corresponds to isoform lacking the polyC exon. Right, qRT-PCR quantification of inclusion of exon 17 using primers that flank the exon junctions. Mean ± s.d, n = 3 replicates/condition. Student’s t-test.
J. Western blot and quantifications demonstrating loss of GAP17 isoforms +/− polyC exons under p53R172H knockdown or renilla control in KPC cells. Mean ± s.d, n = 3 replicates/condition. Student’s t-test.
K. RT-PCR (left) and qRT-PCRs (right) of GAP17 AS revealing distinct isoforms in patient PDAC organoids with WT p53, truncated p53 (Trunc), and hotspot mutations (using same primer approach as in (I)). Mean ± s.d, Student’s t-test.
s.d, standard deviation; n, number of repetitions. *p < 0.05, **p < 0.01, ***p < 0.001, **** p < 0.0001. See also Figures S1–S2. Tables S1–S3.
To determine the role of p53R175H in splicing, we silenced mutant p53R175H/− in patient PDAC organoids and murine PDAC cells (KrasG12D/+; Trp53R172H/−; Pdx1-Cre [KPC cells]) and performed RNA-seq. This identified differences in exon utilization in cells expressing p53R175H compared to knockdown of p53R175H (Figures 1B, S1D, S1E, S1F, S1G, Table S2). To test if splicing changes exist in pancreatic intraepithelial neoplasia (PanIN), we introduced p53R172H/+ in the endogenous Trp53 locus of murine PanIN organoids (KrasG12D/+; Mist1-Cre) and compared RNA splicing patterns against isogenic WT p53 PanINs. Similar changes in splicing were observed across p53R172H PanIN and PDAC (Figure 1C, S1H–I, Table S2). Among all splicing alterations observed with p53R175H, most of the changes were in exon usage (Figures 1B, 1C, S1B, S1C, S1E, S1F, S1I; Table S2).
We next characterized the nucleotide sequences in promoted versus repressed exons in the context of mutant p53R175H. Exons repressed in the presence of p53R175H in human and murine pancreas were rich in purines, while exons promoted by p53R175H were enriched in cytosines (polyC exons) (Figure 1D–E, S2A, S2B, Tables S1–2). Protein-coding RNAs displaying retained polyC exons were enriched for transcripts encoding GAPs, the negative regulators of RAS GTPases (Figure 1F, Table S2). RAS GTPases have low intrinsic enzymatic activity, such that efficient GTP hydrolysis requires interaction with GAPs, which accelerate the GTP hydrolysis and decrease small-GTPase signaling (Maertens and Cichowski, 2014, Vigil et al., 2010). We found that 23% (28/120) of all GAPs in humans, and 27% (17/63) in mice were subject to altered splicing in PDACs expressing p53R175H (Figure 1G).
Amongst GAP splicing changes in patient and murine PDAC and PanIN cells, expression of a polyC exon inclusion isoform of GAP17 (+polyC GAP17, gene name ARHGAP17) was the strongest and highly conserved alternative splicing event induced by p53R175H (Figure 1G–J, S2C, S2D, S2E, S2F, S2G, Table S3). The +polyC GAP17 isoform is an annotated, in-frame, protein-coding isoform. In PDAC patient organoids, increased +polyC GAP17 expression was associated with common TP53 hotspot mutations compared to samples with WT or truncating mutations in TP53 (Figure 1K, S2H, Table S3). These findings were confirmed in pancreas, colon, and breast adenocarcinoma cell lines upon silencing of p53R175H (Figure S2I, S2J, S2K). The results demonstrated that adenocarcinoma-derived cells harboring mutant p53 display altered splicing of GAP17 and other GAPs.
GAP isoforms promoted by mutant p53 increase GTP-hydrolysis of RAS but have impaired membrane localization
We next explored functional differences in +polyC and non-polyC (−polyC) GAP17 isoforms on KRASG12D and WT KRAS and Rho small GTPases. Previous reports suggest that oncogenic KRAS with specific activating mutations remain susceptible to GAP-mediated GTP hydrolysis (Hunter et al., 2015). Based on the findings that p53R175H promoted polyC exon inclusion in 25% of GAPs, we hypothesized that +polyC GAPs would have decreased activity compared to −polyC GAPs, thereby favoring active forms of KRASG12D and other small GTPases.
Supporting this concept, knockdown of p53R175H decreased active, GTP-bound forms of KRASG12D, Rho, and Rap1 (Figure 2A, S3A, S3B, S3C). In addition, expression of human p53R175H in murine p53 null PDAC cells (KrasG12D/+; Trp53Flox/Flox; Pdx1-Cre [KPFLC cells]), increased levels of active KRASG12D and associated Erk and Cofilin signaling compared to WT p53 and p53 null cells (Figure 2B, S3D, S3E, S3F). Moreover, expression of p53R175H in PDACs and other adenocarcinomas was positively associated with activated RAS transduction gene signatures (Figure 2C, S3G).
Figure 2: GAP17 isoforms promote GTP-hydrolysis of RAS small-GTPases and impact membrane localization.

A. Affinity precipitation of GTP-bound KRASG12D in KPC cells following transduction with doxycycline-inducible control (shR) or anti-p53 shRNAs (sh1 and 2). Analysis performed after 5 days of doxycycline. Precipitation using GST-Raf1-RBD fusion protein, blotting using RasG12D antibody. Mean ± s.d, n= 3 replicates/condition. Student’s t-test.
B. Affinity precipitation of GTP-bound KRASG12D in murine PDAC cells with null p53 (KPFLC cells) overexpressing human wild-type p53 (WT), p53R175H (R175H) and empty vector (EV). Precipitation and blotting performed as in A. Mean ± s.d, n= 3 replicates/condition. Student’s t-test.
C. GSEA plots showing p53R175H tumors positively associated with small GTPase and RAS-activated gene signatures in PDAC patients (Bailey et al., 2016).
D. Western demonstrating overexpression of + polyC or −polyC GAP17 isoforms or EV in KPC cells.
E. Left, affinity precipitation of GTP-bound active KRASG12D in KPC cells following overexpression of +polyC and −polyC GAP17 isoforms. Precipitation and blotting performed as in A. Right, relative Erk phosphorylation in KPC cells following overexpression of +polyC or −polyC GAP17 isoforms or EV. n = 3 replicates/condition. Mean ± s.d, Student’s t-test.
F. Fractional GTP remaining (%) after combining purified WT KRAS, WT Rho-A, or KRASG12D mutant proteins with purified full-length GAP17 +polyC and −polyC isoform proteins. Catalytic domain of p120 used as control. Measurements taken after 2 h of reaction. Coomassie stain for purified individual full-length +polyC and −polyC GAP17 proteins. Mean ± s.d, n = 3 replicates/condition. Student’s t-test.
G. Real time measurement of GTP hydrolysis of recombinant full-length WT and mutant G12D KRAS and recombinant GAP17 Rho-GAP domain (residues 245–489). Measurements taken up to 20200 sec. Mean ± s.d, n = 3 replicates/condition.
H. Residue motif enrichment encoded by polyC exons in GAPs promoted by p53R175H/−. Bottom, alignment of representative PPXP motifs gained in GAPs by p53R175H/−.
I. Western for expression of GAP17 in KPFLC cells encoding EV or +polyC GAP17 with WT PPXP motif or mutant PPXP motif (“AAxA”).
J. Affinity precipitation of GTP-bound active KRASG12D (left) or p-Erk (right) in KPFLC cells following overexpression of PPXP or AAXA +polyC GAP17 constructs or EV. Precipitation and blotting performed as in A. n = 3 replicates/condition. Mean ± s.d, Student’s t-test.
K. Western for expression of FLAG-tagged GAP17 isoforms (+polyC and −polyC) and +polyC mutant (AAXA) in p53-null murine PDAC KPFLC cells.
L. Relative membrane localization of each FLAG-tagged GAP17 isoform in p53-null PDACs KPFLC cells after 20% FBS stimulation and obtaining cell-membrane enriched fraction. Anti-FLAG antibody used to measure membrane localization. Pan-cadherin: membrane marker; actin: cytoplasmic fraction; histone H3: nuclear fraction. n = 3 replicates/condition. Mean ± s.d, Student’s t-test.
M. Relative membrane and cytosolic localization of endogenous GAP17 in KPC cells following transduction with doxycycline-inducible control (shR) or anti-p53 shRNAs (sh1 and 2). n = 3 replicates/condition. Mean ± s.d, Student’s t-test.
N. Immunofluorescence of endogenous GAP17 in KPC cells following transduction with shR or anti-p53 shRNAs. Arrowheads: membrane co-localization of GAP17 and cadherin. Scale bars: 50 and 5 μm. n = 10–20 random photos/condition. Mean ± s.d, Student’s t-test.
s.d, standard deviation; n, number of repetitions. *p < 0.05, **p < 0.01, ***p < 0.001, **** p < 0.0001. See also Figure S3.
To test whether +polyC and −polyC Gap17 differentially affected GTP-bound KrasG12D and Rho, we expressed GAP17 or additional GAPs, either lacking or containing polyC exons in p53R172H/− PDAC cells (Figures 2D, S3H, S3I). We found that the −polyC GAP17 dramatically reduced active KRASG12D and Rho, as well as downstream Erk and Cofilin signaling (Figure 2E, S3J, S3K). In contrast, expression of +polyC GAP17 did not alter levels of active KRASG12D and Rho and sustained downstream signaling in p53R172H/−-mutant PDAC cells (Figure 2E, S3J, S3K). Overall these results suggested that +polyC and −polyC GAP17 isoforms differentially influence the activity of RAS GTPases in cells.
To test whether the levels of active, GTP-bound KRASG12D and RHO associated with expression of +polyC GAP17 were due to diminished ability to stimulate GTPase activity, we tested the influence of GAP17 isoforms on GTP hydrolysis of WT KRAS, KRASG12D, and WT RHO in cell-free biochemical assays using recombinant proteins (Figure 2F–G).These experiments demonstrated that inclusion of the polyC exon does not alter the GAP activity of GAP17 towards RHO (Figure 2F). Moreover, both GAP17 isoforms slightly enhanced the intrinsic GTPase activity of WT KRAS (Figure 2F–G), although the activity of GAP17 was not as potent as the cognate RAS GAP p120RasGAP. Interestingly, the GTPase-stimulating activity of both forms of GAP17 was also seen towards mutant KRASG12D, suggesting that intrinsic GAP activity of GAP17 was not directly altered by use of the polyC exon.
The fact that there was no difference in GAP17 isoforms to influence levels of GTP-bound KRAS or Rho in cell-free assays, while +polyC GAP17 expression increased KRASG12D and RHO GTPase signaling within cells, suggested that inclusion of the polyC exon might alter cellular localization and/or protein-protein interactions rather than directly influence GTPase activity. GAPs are cytoplasmatic proteins requiring interactions with other proteins or lipids to translocate to the membrane, where RAS small-GTPases display intrinsic membrane localization (Huang et al., 1993). Remarkably, 40% of coding polyC exons promoted by p53R175H encoded a PPxP amino acid motif-canonical SH3-binding motifs known to mediate protein-protein interactions in GAPs and other proteins (Feng et al., 1995) (Figure 2H). In the case of GAP17, the polyC exon encodes four PPxP motifs at the C-terminus, downstream of the membrane association and GAP domains (Figure S3L). We next exchanged prolines in the +polyC GAP17 PPxP motifs for alanines (AAXA), and overexpressed PPXP and AAXA +polyC GAP17 isoforms in p53 null PDAC cells to determine their relative abilities to regulate KRASG12D (Figure 2I). AAxA +polyC GAP17 decreased GTP-bound KRASG12D and Erk signaling, while PPXP +polyC GAP17 increased active KRASG12D and Rho (Figure 2J, S3M). This suggests that the PPxP motifs impair the ability of +polyC GAP17 to decrease active KRASG12D and Rho within cells.
Given that GAP17 needs to localize to the membrane to effectively influence KRAS activity, we next tested if PPxP motifs prevented +polyC GAP17 from accessing the cell membrane. We expressed N-terminal FLAG-tagged +polyC GAP17, PPxP +polyC GAP17, and AAxA +polyC GAP17 and measured their association with the membrane (Figure 2K). Compared to −polyC GAP17, PPxP +polyC GAP17 had markedly decreased membrane association (Figure 2L). Conversion of PPxP motifs to AAxA rescued this defect in membrane association, suggesting that the PPxP is responsible for altered membrane association of +polyC GAP17 (Figure 2L).
To further evaluate how the PPxP domains influence subcellular localization, we tested the distribution of endogenous GAP17 in KPC cells). Knockdown of p53R172H increased membrane localization and decreased cytosolic localization of endogenous GAP17 (Figure 2M–N). Overall, these data support the concept that mutant p53 enhances KRASG12D and Rho signaling by promoting inclusion of exons that effectively sequester GAP17, and likely other GAPs, away from membrane-associated RAS small-GTPases, thereby maximizing oncogenic signaling.
Mutant p53 regulates expression of hnRNPK to promote polyC exons and GTP-bound KRASG12D
To understand how mutant p53 alters splicing, we evaluated splicing factors that could be transcriptionally regulated by mutant p53. RNA-seq revealed that mutant p53 was positively associated with expression of two RNA processing factors: hnRNPK and DDX42 (Figure S4A–C, Table S4). We focused on hnRNPK, as it is an RNA binding protein known to bind C-rich sequences in RNA and plays key roles in RNA splicing, stability, and translation (Paziewska et al., 2004). Compared to WT p53, knock-in of p53R172H was associated with increased hnRNPK expression, while knockdown of p53R175H decreased hnRNPK expression in murine and human PDAC cells (Figure 3A–B; Figure S4D, S4E; an effect not related to cell cycle changes).
Figure 3: Mutant p53 regulates hnRNPK to promote inclusion of +polyC GAP17.

A. Expression of hnRNPK in PanIN organoids (KrasG12D/+; Mist1-Cre) bearing p53R172H/+ compared to WT p53. qRT-PCR and western performed 20 days after genome editing confirmation. n = 3 replicates/condition. Mean ± s.d, Student’s t-test.
B. Expression of hnRNPK after knockdown of p53R175H in PDAC patient organoids (left, KRASG12D/+; TP53R175H/−) and KPC cells following transduction with doxycycline-inducible control (shR) or anti-p53 shRNAs (sh1 and 2) (right). qRT-PCR and western performed after 5 days of doxycycline. n = 3 replicates/condition. Mean ± s.d, Student’s t-test.
C. Top, nucleotide sequence logo of full and half-site p53 binding sites (Wei et al., 2006). Bottom, predicted p53-DNA binding sites in mouse and human hnRNPK promoters.
D. TdTomato reporter expression in WT hnRNPK promoter (−1429 to +260) and with “CATG”-deleted from p53 binding sites in p53 null PDAC cells (KPFLC cells), engineered to express human p53 mutants, WT p53, or empty vector (EV) control. n = 3 replicates/condition. Mean ± s.d, Student’s t-test.
E. TdTomato expression driven by the hnRNPK promoter using WT and “CATG”-deleted promoter sites in isogenic KPC cells. n = 3 replicates/condition. Mean ± s.d, Student’s t-test.
F. hnRNPK western in KPC cells following transduction with control non-targeting shRNA (NC) or anti-hnRNPK shRNAs (sh1 and 2).
G. Sequences enriched in repressed and promoted exons from RNA-Seq of KPC cells with knockdown of hnRNPK or negative control.
H. Intersection of differentially spliced exons by p53R172H and hnRNPK in KPC cells. Promoted (Fisher’s exact test p= 9.48E-06) and repressed (Fisher’s exact test p= 0.000149) exons shown.
I. Recombinant GST-hnRNPK protein (left). In vitro filter binding assays confirms interaction between recombinant hnRNPK and GAP17 +polyC exon. Known interactor of hnRNPK (polyC sequence) shown as positive control (C16-grey). Negative control in yellow (polyN sequence N16). Mean ± s.d, n = 3 replicates/condition. Student’s t-test.
J. RT-PCR and qRT-PCRs of GAP17 splicing in KPC cells with knockdown of hnRNPK compared to cells expressing control shRNAs (NC). RT-PCR using primers that flank the junctions of polyC exon (exon 17). Upper band: inclusion of exon 17; lower band: isoform lacking exon 17. qRT-PCRs for quantification of +polyC GAP17 retention using primers that flank junctions between exon 17 and adjacent exons. Mean ± s.d, n = 3 replicates/condition. Student’s t-test.
K. RT-PCR and qRT-PCRs of GAP17 splicing in KPFLC cells with overexpression of hnRNPK compared to cells expressing EV. RT-PCRs and qRT-PCRs as described in J. Mean ± s.d, n = 3 replicates/condition. Student’s t-test.
L. Affinity precipitation of GTP-bound KRASG12D in KPC cells following transduction with control shRNA (NC) or anti-hnRNPK shRNAs (sh1 and 2). Precipitation using GST-Raf1-RBD fusion protein, blotting using RasG12D antibody. Mean ± s.d, n = 3 replicates/condition. Student’s t-test.
M. Kaplan-Meier survival (log-rank Mantel-Cox test) of patients with PDACs expressing high- or low-hnRNPK mRNA (defined by the Akaike Information Criterion (Kletting and Glatting, 2009)). RNA-Seq from TCGA (n= 179) and ICGC (Bailey et al., 2016) (n= 88).
N. Statistical enrichment of p53 mutations (y axis) across PDACs expressing low (n=232) or high (n= 29) hnRNPK (y axis). Student’s t-test.
O. hnRNPK expression in ICGC PDACs (n= 88) with p53 hotspot mutations, WT p53, nonsense or frameshift mutations (“truncating mutations”), and other mutations. Student’s t-test.
P. T umor volume by 3D-ultrasound of mutant p53 KPC cells expressing control or anti-hnRNPK sh1 and 2. Arrow, tumor regression time point. Representative ultrasound images of tumors (T). Mean ± s.d, n = 5–6 animals/condition, Student’s t-test.
Q. Tumor volume by 3D-ultrasound of p53 null KPFLC cells expressing control or anti-hnRNPK. Representative ultrasound images of PDAC tumors (T). Mean ± s.d, n = 5–6 animals/condition, Student’s t-test.
s.d, standard deviation; n, number of repetitions. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. See also Figure S4 and Tables S4–5.
To test whether mutated p53 promotes hnRNPK expression, we first identified p53 DNA binding consensus sites in the hnRNPK promoter (Figure 3C) and performed an hnRNPK promoter-activity reporter assay in p53 null PDAC cells (KPFLC cells) with expression of human WT p53 and mutant p53 (Figure S4F). hnRNPK promoter activity was significantly upregulated by p53 mutants compared to WT p53 or EV control (Figure 3D). However, deleting p53-consensus sites resulted in loss of hnRNPK reporter activation by p53 mutant forms (Figure 3D–E). These results were validated in murine pancreatic duct epithelial cells (PDEC, KrasG12D; Trp53+/+) (Agbunag et al., 2006) with expression of murine p53R172H or p53R270H (Figure S4G). These results suggest that hnRNPK expression is regulated by mutant p53 and depends upon p53 DNA binding sites its promoter.
To determine whether hnRNPK mediated the splicing changes promoted by p53R175H, we silenced hnRNPK in p53R172H-mutant PDAC cells (Figure 3F, S4H) and performed RNA-seq. As in the case of p53R175H, knockdown of hnRNPK significantly altered exon splicing (Figure S4I, Tables S5). Importantly, exons promoted by hnRNPK were enriched in C-rich sequences while exons repressed by hnRNPK knockdown were enriched in purines (Figure 3G), mirroring the sequence-specific splicing changes induced by p53R175H. Indeed, splicing changes induced by p53R175H in these same cells were also induced by hnRNPK (Fisher’s exact test p< 0.0001, Figure 3H, Tables S5). Transcripts whose splicing was similarly regulated by p53R175H and hnRNPK included GAP17 and GAP12, both of which underwent hnRNPK-promoted inclusion of polyC exons. We confirmed direct binding of recombinant hnRNPK to GAP17 exon 17, with similar affinity to a previously established hnRNPK target (Figure 3I, S4J). Silencing or overexpressing hnRNPK in p53-mutant (Figure 3J, S4K) and p53-null PDAC cells (Figure 3K, S4L), respectively, mimicked the effects of mutant p53 in these same cells. We next tested if hnRNPK promoted active, GTP-bound KRASG12D. Suppression of hnRNPK in KPC cells decreased cell proliferation and levels of active KRASG12D by ~50% (Figure 3L, S4M). Overall, these results suggest that p53R175H mediates changes in splicing of GAP17 and enforces KRASG12D activity by regulating the expression of hnRNPK.
Previous studies reveal that increased hnRNPK expression is associated with shorter survival in several cancers (Carpenter et al., 2006, Ciarlo et al., 2012, Wen et al., 2010). Similarly, we found that PDAC patients with higher hnRNPK expression have shortened survival (Figure 3M). In addition, high hnRNPK expression in PDACs was associated with increased p53 hotspot mutations compared to PDACs with low hnRNPK expression (Figure 3N–O). To determine whether hnRNPK is required for tumor maintenance, we tested the effects of inhibiting hnRNPK in orthotopic syngeneic p53 mutant and p53-null PDAC models. Knockdown of hnRNPK in p53 mutant tumors increased survival and decreased tumor volume and cell proliferation (Figure 3P, S4N, S4O, S4P). In contrast, hnRNPK knockdown did not impede the growth of p53-null PDACs (Figure 3Q). These data demonstrate that hnRNPK expression contributes to tumor maintenance in p53 mutant PDAC.
Targeting mutant p53 GAP17 isoforms decreases KRASG12D activation and increases survival
We next sought to evaluate the requirement of mutant p53-mediated splicing changes in GAP17 by manipulating GAP17 isoform expression in p53-mutant PDACs. We first altered the proportion of +polyC and −polyC GAP isoforms, without impacting total levels of GAP17, by targeting the polyC exons with isoform-specific shRNAs (Figure 4A, S5A). Isoform-specific knockdown of +polyC GAP17 decreased cell proliferation and active KRASG12D and Erk1/2 by 50% (Figure 4B–C), and 75% decrease in active Rho and downstream mediators (Figure 4D), mimicking the effects seen with suppression of p53R172H (Figure 2A–B, S3A). In a similar manner, knockdown of the +polyC GAP4 isoform also decreased proliferation and downstream signaling from KRASG12D and Rho (Figure S5B, S5C, S5D). These results suggested that manipulation of GAP splicing can attenuate the activity of RAS GTPases and downstream signaling.
Figure 4: Targeting GAP17 isoforms decreases KRASG12D activation and increases survival in pancreatic cancer models.

A. Top left, schematic of isoform specific shRNAs targeting +polyC exon of GAP17 promoted by p53R172H. Bottom left, RT-PCR demonstrating isoform-specific knockdown of +polyC GAP17 in KPC cells using shRNAs against polyC exons (PolyC sh1 and sh2), compared to non-targeting control. Upper band: +polyC isoform; lower band: −polyC isoform. Right, qRT-PCRs for quantification of +polyC GAP17 or −polyC GAP17 isoform using primers that flank junctions between polyC exon and adjacent exons (exons 16 and 18) or primers that flank the junction between exons 16 and 18. Mean ± s.d, n = 3 replicates/condition. Student’s t-test.
B. Proliferation of KPC cells expressing control or +polyC GAP17 sh1 and sh2. Mean ± s.d, n = 3 repetitions/condition, Student’s t-test.
C. Affinity precipitation of GTP-bound active KRASG12D (left) or p-ERK (right) in KPC cells with knockdown of polyC GAP17 isoforms or control. Precipitation using GST-RAF1-RBD fusion protein, blotting using RASG12D antibody. Mean ± s.d, n = 3 replicates/condition. Student’s t-test.
D. Affinity precipitation of GTP-bound active RHO (left) and phosphorylated COFILIN (inactive form, bottom left) in KPC cells with knockdown of + polyC GAP17 isoform and control. Mean ± s.d, n = 3 replicates, Student’s t-test.
E. Kaplan-Meier survival (log-rank Mantel-Cox test) following orthotopic syngeneic transplantation of KPC cells expressing control or shRNAs against +polyC GAP17. n = 5–6 animals/condition.
F. Tumor volume by 3D-ultrasound 30 days post-orthotopic injection of cells expressing control or +polyC GAP17 sh1 or 2. Ultrasound images of tumors (T). S: Skin. Mean ± s.d, n = 5–6 animals/condition, Student’s t-test.
G. Number of hepatic and splenic metastases per animal bearing PDACs with expression of control or +polyC GAP17 sh1 and 2. Mean ± s.d, n = 5 animals/condition, Student’s t-test.
H. Kaplan-Meier survival (log-rank Mantel-Cox test) following orthotopic syngeneic transplantation of KPC cells with overexpression of +polyC or −polyC GAP17 isoforms or empty vector (EV). n = 5–6 animals/condition.
I. Tumor volume by 3D-ultrasound 30 days post-orthotopic injection of KPC cells expressing + polyC GAP17, −polyC GAP17, or EV. Representative ultrasound images of tumors (T). S: Skin. Mean ± s.d, n= 5 animals/condition, one-way ANOVA with Tukey multiple comparison.
J. Number of hepatic and splenic metastases per animal bearing PDACs with overexpression of +polyC or −polyC GAP17 or EV. Mean ± s.d, n = 3 repetitions/condition, Student’s t-test.
s.d, standard deviation; n, number of repetitions. *p < 0.05, **p < 0.01, ***p < 0.001, **** p < 0.0001. See also Figure S5.
To determine whether p53R175H-driven +polyC GAP17 isoforms enhance tumor burden, we next tested the effects of inhibiting polyC GAP17 in vivo. In an orthotopic, syngeneic p53 mutant PDAC model, knockdown of +polyC GAP17 extended animal survival by >40% compared to control mice with +polyC GAP17-expressing tumors (Figure 4E, S5E). In addition, tumor volume and metastases were markedly decreased by +polyC GAP17 depletion (Figure 4F–G, S5F). Further, overexpression of +polyC GAP17 decreased survival and increased tumor size and metastatic burden (Figure 4H–J, S5G, S5H). These data indicate that the changes in GAP17 mRNA splicing induced by mutant p53R172H significantly increase aggressiveness of p53 mutant PDAC.
The spliceosome and +polyC GAP isoforms are therapeutic vulnerabilities in p53R172H PDAC
Recently, a number of small-molecule compounds that bind the SF3b complex and impair its interaction with RNA have been developed (Seiler et al., 2018, Yoshimi and Abdel-Wahab, 2017). Given the ability of such compounds to impede exon splicing globally, we tested the dependency of p53R175H tumors on inclusion of C-rich exons by SF3b inhibitor treatment of PDACs with or without p53R172H.
Daily treatment with H3B-8800 in mice bearing established orthotopic p53R172H and p53 null PDACs increased survival and decreased tumor volume compared to isogenic PDACs treated with vehicle (Figure 5A, S5I, S5J, S5K, S5L). However, the largest differences in survival were among the mutant p53R172H mice treated with vehicle and H3B-8800 (Figure 5A, S5L). These changes were associated with marked PDAC apoptosis and occurred in a cell-autonomous manner, as these cells were also more sensitive to spliceosome modulation in vitro using either H3B-8800 or E7107 (Figure 5B, S5M, S5N). In addition, p53-null murine PDAC cells engineered to express human p53 mutants showed greater sensitivity to H3B-8800 than isogenic cells expressing WT or empty vector control (Figure 5C).
Figure 5: The spliceosome and polyC GAP isoforms are therapeutic vulnerabilities in p53R172H-pancreatic cancers.

A. Kaplan-Meier survival (log-rank Mantel–Cox test) following orthotopic syngeneic transplantation of KPC cells with either control renilla shRNA (shR) or shRNAs against p53 (sh1) and treated with either vehicle or H3B-8800 (at 8 mg/kg). Data are mean ± s.d, n = 5–11 injected animals/condition. For p53R172H shR vehicle versus H3B-8800, hazard ratio (HR) = 156, 95%CI= 17.16–1431; for p53R172H sh1 vehicle versus H3B-8800, HR= 76.5, 95%CI 9.47–617.9; for p53R172H sh1 versus shR (both with H3B-8800), HR= 17.25, 95%CI 4.92–60.4.
B. Dose-response curves in p53R172H expressing cells (shR) compared to sh1 cells treated with H3B-8800 for 48h. Relative proliferation index established by comparing cells treated with DMSO. Inset indicates inhibitor concentration that reduced proliferation by half (IC50) and that killed half of the cells (LD50). NA-Not applicable. Data represent mean ± s.d; n = 3 repetitions/condition, Student’s t-test. RNA-seq performed across a range of non-cytotoxic concentrations (grey box) to compare differences in exon splicing in cells with or without p53R172H.
C. IC50s to H3B-8800 in p53-null PDAC cells (KPFLC cells) overexpressing human wild-type p53 (WT), missense hotspot mutants R175H; R248Q; R273H, or empty vector (EV). Treatment for 48 h was done 30 days after confirmed overexpression. Data represent mean ± s.d; n = 3 repetitions/condition, Student’s t-test.
D. Sashimi plots illustrating C-rich exon (exon 17; E17) in GAP17 in murine PDAC cells without (sh1) or with mutant p53R172H (shR), untreated with H3B-8800 (top two sashimi plots). Bottom three sashimi plots refer to cells with mutant p53R172H (shR) treated with three non-cytotoxic H3B-8800 concentrations. PSI value is provided for each condition.
E. RT-PCR of loss of polyC exons in representative GAPs (Asap1, Rasa4 and Gap4) in KPC cells expressing p53R172H treated with non-cytotoxic H3B-8800 concentrations or vehicle, using primers that flank polyC exons. Upper bands in RT-PCRs denote the +polyC isoform, while lower bands correspond −polyC exon to isoforms.
F. Left, westerns of cDNAs encoding polyC and no polyC isoforms of GAPs (GAP17 and Asap1) or EV in KPC cells. Molecular weight difference between +polyC and −polyC ASAP1 isoforms is not large enough to distinguish by western. Right, dose-response curves in p53R172H-expressing cells overexpressing polyC and non-polyC isoforms of GAPs GAP17 and ASAP1, or EV treated with H3B-8800 for 48 h. Mean ± s.d; n = 3–5 repetitions/condition, Student’s t-test.
G. Schematic of antisense oligonucleotides (ASOs) (top left) and corresponding RT-PCRs (bottom left) and qRT-PCRs (right) for quantification of +polyC GAP17 or −polyC GAP17 isoforms in KPC cells 48 h after treatment with 10 μM ASOs. Mean ± s.d, n = 3 repetitions, Student’s t-test.
H. Left, tumor volume after in vivo ASO treatment of KPC sub-cutaneous xenografts. Tumor volume measured twice/week during treatment with non-targeting ASO control (NC) or +polyC GAP17-targeting ASOs. ASOs given at 12.5 mg/kg, every other day intratumorally. Right, representative PDAC images. Mean ± s.d, One-way ANOVA, with Tukey multiple comparison.
I. Immunohistochemistry quantification for p-ERK1/2 in PDACs treated with non-targeting ASO control or +polyC GAP17-targeting ASOs. Mean ± s.d, n = 5 PDACs, One-way ANOVA, with Tukey multiple comparison.
J. Relative p-ERK1/2 (left) and p-COFILIN (right) in non-targeting ASO control (NC) or +polyC GAP17 targeting ASOs (ASO1 and 2). Lysates generated from viable tumor areas. Data represent mean ± s.d, n = 5 PDAC, One-way ANOVA, with Tukey multiple comparison.
K. Percentage necrotic tumor area following in vivo ASO treatment of KPC allografts. Data represent mean ± s.d, n = 3–5 PDACs, One-way ANOVA, with Tukey multiple comparison.
s.d, standard deviation; n, number of repetitions; # p< 0.1*p < 0.05, **p < 0.01, ***p < 0.001, ****p< 0.0001. See also Figure S5.
We hypothesized that increased sensitivity of p53R172H PDACs to H3B-8800 was due to requirement for the splicing changes promoted by mutant p53. To test this, we compared exon usage in cells with or without p53R172H at increasing non-cytotoxic concentrations of H3B-8800. At these concentrations, H3B-8800 reduced expression of p53R172H-associated isoforms, including GAP17 (Figure 5D–E, Figure S5O). Given that H3B-8800 induces splicing changes unrelated to those associated with p53R172H, we next tested whether expression of +polyC isoforms in GAPs could rescue the lethal effects of H3B-8800. The lethal effects were partially rescued by expressing cDNAs encoding +polyC isoforms of GAPs. In contrast, expressing corresponding cDNAs for −polyC isoforms did not rescue cell growth (Figure 5F, S5P). These data suggest that p53-mutant PDACs depend on sustained expression of +polyC GAPs.
To test if +polyC Gap17 is a therapeutic target for mutant p53 PDACs and extend the experiments using isoform specific shRNAs (Figure 4A–E), we designed two morpholino antisense oligonucleotides (ASOs) to block GAP17 polyC exon inclusion by blocking 3’ or 5’ splice sites (Figure S5Q). Both ASOs decreased +polyC GAP17 and increased −polyC GAP17 protein levels while maintaining constant GAP17 mRNA levels relative to control ASO (Figure 5G, S5Q). We then tested ASO in vivo. We established subcutaneous PDAC allografts in syngeneic immunocompetent mice and treated these with ASO via intratumoral injection for 12 days. ASOs targeting polyC exon of GAP17, but not control ASO, significantly reduced tumor growth and phospho-Erk while increasing phospho-cofilin and tumor necrosis (Figure 5H–K). We conclude that blocking p53-mutant associated GAP17 isoforms restores GAP17 GAP activity in murine PDAC, thereby decreasing tumor growth.
DISCUSSION
Here we identify that p53R175H, as well as other p53 hotspot mutations, rewire the splicing of mRNAs encoding GAPs to promote maximal activation of Kras signaling in PDACs. Despite decades of studies on the role of p53R175H, effects of mutant forms of p53 on RNA splicing or KRASG12D activity have not been recognized. In PDAC mouse models, mutations in Kras are necessary for tumor initiation but not sufficient to maintain GTP-bound levels of KRASG12D, suggesting that KRAS activity and tumorigenicity is subject to additional regulatory mechanisms (Ardito et al., 2012, di Magliano and Logsdon, 2013, Logsdon and Lu, 2016). We found that mutant p53, hnRNPK, and +polyC GAP isoforms maintain GTP-bound levels of mutant KRAS and enhance its downstream signaling, thereby promoting tumor growth.
These results also highlight oncogenic mechanisms of GAP regulation. Although GAPs are tumor suppressors inactivated either by deletion or mutations in different cancers (Vigil et al., 2010), they are mutated in <1% of PDAC cases. Here, we demonstrate a non-genetic and plastic mechanism for GAP inactivation by mutant p53, in a manner that promotes the activity of RAS small-GTPases. Importantly, the PPXP motifs encoded by GAP isoforms promoted by mutant p53 are classical SH3-binding motifs known to mediate protein-protein interactions (Feng et al., 1995). Further studies are needed to elucidate the precise biochemical role of SH3 domains in mediating GAP regulation. Overall, these findings implicate mutant p53 in deregulated RAS signaling in PDAC, and potentially in other malignancies harboring TP53 mutations.
Given that mutant KRAS and p53 have proven “undruggable”, there have been intense efforts to identify druggable critical downstream targets and nodes required by these oncoproteins (Aguirre and Hahn, 2018). Here, we show that PDACs harboring mutant KRASG12D and p53R172H are selectively dependent on hnRNPK, +polyC GAPs, and intact spliceosome function. These findings suggest specific downstream targets that can be exploited to target tumors with both oncogenic KRAS and mutant p53. The observation that PDACs expressing KRAS and p53 are preferentially sensitive to ASOs targeting +polyC GAP isoforms, as well as splicing inhibitors, suggest that this dependency may be leveraged for therapeutic benefit in a biomarker-driven manner. Given the recent clinical success and FDA approval of ASOs for spinal muscular atrophy (Stein and Castanotto, 2017), the correction of GAP mis-splicing using such agents may prove similarly effective in cancers with p53R175H mutations.
Collectively, these findings uncover a biologically critical and therapeutically exploitable mechanism of cooperation between the two most frequently mutated and co-existing oncogenes in human cancers: KRAS and TP53.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Luisa Escobar-Hoyos (luisa.escobar-hoyos@vale.edu). This study did not generate new unique reagents.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
PDAC patient derived organoids:
Studies were approved by the Institutional Review Boards of Memorial Sloan Kettering Cancer Center (MSK) and Dana-Farber Cancer Center in accordance to the Declaration of Helsinki protocol. Patients provided samples after their informed consent and primary human de-identified PDAC samples derived from resection or fine needle biopsies. Genomic were analyzed using MSK IMPACT (Cheng et al., 2015) assay or FoundationOne (Leeksma et al., 2018) assay, both as previously described.
Animals.
All animals were housed at Memorial Sloan Kettering Cancer Center (MSKCC). All animal procedures were completed in accordance with the Guidelines for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committees at MSKCC. All mouse experiments were performed in accordance with a protocol approved by the MSKCC Institutional Animal Care and Use Committee (11–12-029). 6–10 week old female albino mutant coisogenic C57BL/6 strain were used for all xenografts. Power analysis was used to determine appropriate sample size to detect significant changes in animal median survival, which were based on previous median survival analyses in our laboratory.
Cell culture.
Primary patient derived and murine PanIN organoids were culture following the protocols previously described (Boj et al., 2015). Briefly, cells were embedded in matrigel 3D cultures and media included several growth factors and enzyme inhibitors to support the growth of the cells. Cultures were split at 80% confluence. Murine PDAC cells (KrasG12D/+; p53R172H/−; Pdx1-Cre [KPC cells] and KrasG12D/+; p53 Flox; Pdx1-Cre [KPFLC cells]) and 293T cells, were cultured in DMEM (Gibco) supplemented with 10% fetal bovine serum (FBS) and 1% Pen Strep (Gibco). Murine pancreatic duct epithelial cells (PDEC, KrasG12D; p53+/+), were cultured based on previous reports (Agbunag et al., 2006, Lee and Bar-Sagi, 2010). All cell lines were incubated at 37°C and 5% CO2. Primary patient derived organoids were generated at Memorial Sloan Kettering Cancer Center (MSKCC) using primary patient samples from tumor resection under Institutional Review Boards approval and in accordance to the Declaration of Helsinki protocol. Informed consents were obtained from all human subjects. Murine PanIN organoids were generated using the Mist1:CreERT2 (CiMist1) Lox-Stop-Lox (LSL)-KrasG12D/+ mice (Tuveson et al., 2006) with td Tomato tracer and following the protocol from Boj et al., 2015 (Boj et al., 2015). Murine PDAC cells were obtained from Dr. Robert Vonderheide, generated from the PDX-1-Cre; LSL-KrasG12D/+; LSL-Trp53R172H/+ (Hingorani et al., 2005). All cells are tested yearly for mycoplasma contamination. Stable cell lines expressing shRNAs or cDNAs were generated by lentiviral or retroviral transduction in the presence of appropriate antibiotic resistance markers or by Fluorescence Activated cell sorting (FACS) sorting of cells with fluorescent marker.
METHOD DETAILS
Patient derived RNASeq data from ICGC.
FastQ files derived from the Bailey et al.,(2016) were used to identify the isoform expression measurements from PDACs of patients with different mutation status for TP53.
RNA-seq quantification.
For a robust analysis of subtle expression and splicing effects, alignment-based and alignment-free methods were combined. Reads were aligned to Gencode annotation v25 for human or vM12 for mouse using STAR v2.4.1d and quantified by QoRTs v1.1.8(Hartley and Mullikin, 2015), while alignment-free quantification was performed by Kallisto v0.43.0(Bray et al., 2016) accounting for hexamer bias and using 100 bootstrap iterations to estimate the uncertainty due to the finite depth of coverage.
Differential gene expression and splicing defect estimation.
Differentially expressed genes were then identified from aligned data using DESeq2 v1.10.1(Love et al., 2014) and from alignment-free data using Sleuth v0.28.1(Pimentel et al., 2017). Splicing defects at known loci (exons etc.) were identified using SUPPA v1 (Trincado et al., 2018). SUPPA extracts the PSI value for each event by transformation of alignment-free transcript quantification. Statistical significance is then calculated using a Kolmogorov-Smirnov test to compare the PSI distributions of two conditions as a function of the expression of the transcripts defining the events. To further validate the significance of the splicing defects, the empirical version of the SUPPA algorithm was applied to samples permuted in such a way as to eliminate any biological effect.
Tumorigenicity and Metastasis Assays.
KPC cells expressing (i) inducible shRNAs against R or p53, (ii) constitutive shRNAs against polyC exons or control (iii) constitutive cDNAs of +polyC, −polyC isoforms and empty vector (EV) control were mixed in 1:1 matrigel (BD Biosciences) and simple media to a final of 150,000 cells in a 30 μl. Cells were orthotopically implanted into the tail of the pancreas of B6 albino mice (Charles river). 5–11 mice were implanted for each-individual genetically-engineered stable cell line. For H3B-8800 studies, after 10 days post-implantation with KPC cells, animals were placed under a doxy food (0.625g/Kg Doxycycline Hyclate) to induce the expression of shRNAs against renilla or p53. At the same time, animals were randomized to receive oral gavage doses infusions of vehicle (5% Ethanol, 95% methyl cellulose (0.5% in water) or 8 mg/kg of H3B-8800 daily until the time of death. Tumor growth was measured via 3D-ultrasound imaging starting 10 days post-implantation weekly. For median survival assessment, animals were sacrificed following the endpoints approved by IACUC: (i) Animals showing signs of significant discomfort, (ii) ascites or overt signs of tumor metastasis or gastrointestinal bleeding (blood in stool), (iii) animals losing >15% of their body, and (iv) animals with tumors >2cm in diameter. Animals with tumors expressing inducible shRNAs, were fed with doxycycline food (0.625 Doxycycline Hyclate). Investigators responsible for monitoring and measuring the xenografts of individual tumors were not blinded.
In vitro anti-sense oligonucleotide (ASO) transfection.
To deliver morpholinos into cultured cell lines, we followed the manufacture’s instruction (GeneTools LLC). Briefly, we used 6uM Endo-Porter after adding morpholinos (final concentration: 10uM). RNA and proteins are collected 48 h after delivery. ASO target sequence - #1 ACACCAAAGCTACACAGAGAGAAGA, #2 CAGCAGAAGTGGGCT CCTT ACCT GC.
In vivo anti-sense oligonucleotide (ASO) transfection.
Treatment with ASOs was started when the sub-cutaneous tumor volume in mice reached 100–200 mm3. Cohorts were treated i.t. with 12.5 mg/kg scrambled or poison exon–targeting Vivo-Morpholinos (#1 ACACCAAAGCTACACAGAGAGAAGA, #2 CAGCAGAAGTGGGCTCCTTACCTGC, GeneTools LLC) dissolved in 50 μl PBS, every 2 days for 8 doses total. The mice were dissected 24 h after the final treatment.
Patient survival calculations based on hnRNPK expression.
To determine the best low/high cutoff for hnRNPK mRNA from TCGA data set, Cox regression model was run repeatedly from the lowest level of hnRNPK expression (6612.71 RSEM reads) to the highest (16155.8 RSEM reads) and the best model was chosen based on the Hazard Ratio (HR), p value, and the lowest Akaike information criterion (indicator of the model fit). Once the best model was chosen, that hnRNPK mRNA expression (9697.7 RSEM reads) was used to stratify low verse high hnRNPK mRNA. Overall survival analyses were performed to test the relationship between hnRNPK mRNA expression and clinical outcomes. The association between hnRNPK mRNA expression and overall survival was estimated through univariate Cox proportional hazard models using log rank test.
Retroviral Constructs and virus production.
Inducible shRNAs against TP53 and Trp53 (human and murine p53) or renilla (R) were cloned into the TRMPV-Neo vector (pSIN-TRE-dsRed-miR30-PGK-Venus-IRES-NeoR) as previously described (Weissmueller et al., 2014, Zuber et al., 2011). Constitutive expression of shRNAs against polyC exons for the isoforms were cloned into the pSicoR vector (Addgene, 11579) as previously described (Ventura et al., 2004). Constitutive expression of shRNAs against hnRNPK were cloned into the GIPZ Lentiviral shRNA vector (Dharmacon). shRNA sequences are described in Key Resource Table.Constitutive expression of cDNAs encoding for +polyC and −polyC GAPs and indirect regulators of GTPase signaling shRNAs were cloned into the pLVX-IRES-mCherry vector (Clontech 631237). Constitutive expression of cDNAs encoding for human TP53 wild-type and mutants (R175H; R248Q and R273H) were cloned into the pLVX-IRES-PURO vector (Modified from Clontech 631237). All constructs were verified by sequencing. Lentiviruses and retroviruses were produced by transiently transfecting shRNAs or cDNA constructs using the Dharmacon Trans-Lentiviral shRNA Packaging Kit with Calcium Phosphate Transfection Reagent protocols into 293T cells and harvesting viral supernatants 48 h after transfection.
KEY RESOURCE TABLES.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal murine p53 | Leica | p53-CM5P-L |
| Mouse monoclonal anti-Human p53 | Santa Cruz | sc-126 |
| Mouse monoclonal Arhgap17 (Gap17) | Santa Cruz | sc-514438 |
| Rabbit polyclonal Arhgap17 (Gap17) | Invitrogen | PA5-50113 |
| Mouse monoclonal Total Ras | Cell Signaling | 8832 |
| Mouse monoclonal Total Rho | Cell Signaling | 8789 |
| Rabbit monoclonal Ras G12D | Cell Signaling | 14429 |
| Rabbit monoclonal Rap1 | Cell Signaling | 8825 |
| Mouse monoclonal Erk 1/2 | Cell Signaling | 4696 |
| Rabbit monoclonal Phospho Erk 1/2 | Cell Signaling | 4370 |
| Rabbit monoclonal Cofilin | Cell Signaling | 8503 |
| Rabbit monoclonal phospho-cofilin | Cell Signaling | 3313 |
| Rabbit polyclonal Asap1 | Abcam | 208170 |
| Rabbit monoclonal Flag | Cell Signaling | 14793S |
| Rabbit polyclonal hnRNPK | Cell Signaling | 4675 |
| Mouse monoclonal B-actin | Cell Signaling | 3700 |
| Rabbit monoclonal Gapdh | Santa Cruz | sc-5174 |
| Rabbit monoclonal Gapdh | Cell Signaling | 5174 |
| Rabbit polyclonal Pan-Cadherin | Cell Signaling | 4068 |
| Rabbit polyclonal GFP | Cell Signaling | 2555 |
| Rabbit polyclonal Histone 3 | Cell Signaling | 9715 |
| Bacterial and Virus Strains | ||
| E.coli BL21DE3 | New England BioLabs | C2527I |
| Biological Samples | ||
| Human: Passage 40 HT-42, HT-42 Patient Derived Organoids cells | MSKCC PDAC organoid biobank. See Table S3. | N/A |
| Human: Patient derived organoids, isolated RNA | Dana-Farber PDAC organoid biobank. | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| H3B-8800 | H3 Biomedicine | H3B-8800 |
| Recombinant human hnRNPK | Custom plasmid with cDNA | P61978 |
| Recombinant human GAP17 | Custom plasmid with cDNA | Q68EM7 |
| Recombinant human KRAS | Custom plasmid with cDNA | Q68EM7 |
| Recombinant human RHO | Custom plasmid with cDNA | P61586 |
| Doxycycline Hyclate, Doxy food (0.625g/Kg) | SAFE complete care competence | E8200 Version 0115 |
| Critical Commercial Assays | ||
| Trans-Lentiviral shRNA Packaging Kit with Calcium Phosphate Transfection Reagent | Dharmacon | TLP5915 |
| Active Ras detection kit | Cell Signaling | 8821 |
| Active Rho detection kit | Cell Signaling | 8820 |
| Active Rap1 detection kit | Cell Signaling | 8818 |
| Subcellular Protein Fractionation Kit | Thermo | 78840 |
| Verso cDNA Synthesis Kit | Thermo | AB1453A |
| RNeasy Mini kit | Qiagen | 74104 |
| GoTaq® Green Master Mix | Promega | M7122 |
| SYBR Green Master Mix | Thermo | 4367659 |
| Deposited Data | ||
| Raw and analyzed data | This paper | GSE114502. |
| Experimental Models: Cell Lines and Patient Derived Organoids | ||
| Mouse: Murine PDAC cells (KrasG12D/+; p53R172H/−; Pdx1-Cre [KPC cells] and KrasG12D/+; p53 Flox; Pdx1-Cre [KPFLC cells]) | Provided by Robert Vonderheide, generated from the PDX-1-Cre; LSL-KrasG12D/+; LSL-Trp53R172H/+(Hingorani et al., 2005). | N/A. |
| Mouse: Murine pancreatic duct epithelial cells (PDEC, KrasG12D; p53+/+), | (Agbunag et al., 2006). | N/A |
| Mouse: Murine PanIN organoids were generated using the Mist1:CreERT2 (CiMist1) Lox-Stop-Lox (LSL)-KrasG12D/+ mice. | This paper | N/A |
| Breast cancer cell line SK-BR-3 (KRAS+/+; p53 R175H) | ATCC | ATCC® HTB-30™ |
| Colon cancer cell line LS-123 (KRASG12S/+; p53R175H) | ATCC | ATCC® CCL-255™ |
| Experimental Models: Organisms/Strains | ||
| Mouse: Albino mutant coisogenic C57BL/6 | Strain code:562 | Charles River Frederick Research Models & Services NCI. |
| Oligonucleotides | ||
| Anti-sense Oligos: Anti-polyC Gap17 #1 ACACCAAAGCTACACAGAGAGAAGA, #2 CAGCAGAAGTGGGCTCCTTACCTGC. | Gene Tools | Custom design. N/A. |
| +PolyC isoforms shRNAs sequences | ||
| GAP17 1F shRNA1 TGGGTGGCACTCTAAATAGATTCAAGAGATCTATTTAGAGTGCCACCCTTTTTTC GAP17 1R shRNA1 TCGAGAAAAAAGGGTGGCACTCTAAATAGATCTCTTGAATCTATTTAGAGTGCCACCCA |
Thermo | Custom design. N/A. |
| GAP17 2F shRNA2 TGGGCCTGTCTTCTCTTCTATTCAAGAGATAGAAGAGAAGACAGGCCCTTTTTTC GAP17 2R shRNA2 TCGAGAAAAAAGGGCCTGTCTTCTCTTCTATCTCTTGAATAGAAGAGAAGACAGGCCCA |
Thermo | Custom design. N/A. |
| GAP4 1F shRNA1 TGATCCGAGATGCCTTTGAATTCAAGAGATTCAAAGGCATCTCGGATCTTTTTTC GAP4 1R shRNA1 TCGAGAAAAAAGATCCGAGATGCCTTTGAATCTCTTGAATTCAAAGGCATCTCGGATCA |
Thermo | Custom design. N/A. |
| GAP4 2F shRNA2 TGGATCTCAGAGATCCGAGATTCAAGAGATCTCGGATCTCTGAGATCCTTTTTTC GAP4 2R shRNA2 TCGAGAAAAAAGGATCTCAGAGATCCGAGATCTCTTGAATCTCGGATCTCTGAGATCCA |
Thermo | Custom design. N/A. |
| hnRNPK 1 shRNA1 CTGGATCTATTATTGGCAA hnRNPK 1 shRNA1 GAGTTGAGACTGTTGATTC |
Thermo | Custom design. N/A. |
| TP53 primers F: GAAGACCCAGGTCCAGATGA R: TGTTTCCTGACTCAGAGGGG |
Thermo | Custom design. N/A. |
| Trp53 primers F: CATGGAGGAGTCACAGTCG R: CACTCGGAGGGCTTCACTT |
Thermo | Custom design. N/A. |
| GAP17 primers R: TTAGAACTCGGGGTGGTGAG F: AGCACCAGGGAGAAACAACA |
Thermo | Custom design. N/A. |
| GAP17 primers R: GACTCTGGAGAGGAAGCGAC F: TGTGAGGTGTGCTTACTGAGAG |
Thermo | Custom design. N/A. |
| RASA4 primers F: CACAGACCGAAGCTGAAGTG R: GTGGTTCCATCCACGACTCT |
Thermo | Custom design. N/A. |
| ASAP1 primers R: ATCCATCTCGTCCTGTCGAA F: CTGTCTCAGCAAGCAAGCAC |
Thermo | Custom design. N/A. |
| GAP4 primers R: TTTTCTCTCTGTGCACTGGG F: GACTCAGTGGCTGGGGTACT |
Thermo | Custom design. N/A. |
| GAP10 primers R: AGAGGGCTGGGACTGTCAC F: AGGAGCCATTTTTGAGGATG |
Thermo | Custom design. N/A. |
| GAPDH primers F: AGGTCGGTGTGAACGGATTTG R: TGTAGACCATGTAGTTGAGGTCA |
Thermo | Custom design. N/A. |
| GAPDH primers F: GCAAATTCCATGGCACCGTC R: TCGCCCCACTTGATTTTGG |
Thermo | Custom design. N/A. |
| +polyC ASAP1 primers F: ACGCCGGGAAAGGTCCAAC R: CCCCAAGGAATTGCGCCT |
Thermo | Custom design. N/A. |
| +polyC GAP17 primers F: GGAGAGCTTTGGTGTGAAGC R: TGGGTTTTGGAGGACTGCTG |
Thermo | Custom design. N/A. |
| −polyC ASAP1 primers F: GATGACAAGCCAAGCCCGAT R: ATCATTACCTTTCCCGGCGT |
Thermo | Custom design. N/A. |
| −polyC GAP17 primers F: CTGCCACATCGGTCCACG R: GGAGGACTCTCCTTCTTCACC |
Thermo | Custom design. N/A. |
| Recombinant DNA | ||
| Plasmid: TRMPV-Neo vector (pSIN-TRE-dsRed-miR30-PGK-Venus-IRES-NeoR) | Weissmueller et al., 2014; Zuber et al., 2011 | N/A |
| Plasmid: pLVX-IRES-mCherry vector | Clontech | 631237 |
| Plasmid: pLVX-IRES-PURO | Clontech | Modified from 631237 |
| Plasmid: pSicoR vector | Addgene | 11579 (Described in Ventura et al., 2004). |
| Plasmid: GIPZ Lentiviral shRNA vector | Dharmacon | RHS4531 |
| Plasmid: pQlinkH | Addgene | 13667. Scheich C, et al., Nucleic Acids Res. 2007. Addgene |
| Software and Algorithms | ||
| ImageJ | Schneider et al., 2012 | https://imagej.nih.gov/ij/ |
| QoRTs v1.1.8 | Hartley and Mullikin, 2015 | N/A |
| Kallisto v0.43.0 | Bray et al., 2016 | N/A |
| DESeq2 v1.10.1 | Love et al., 2014 | N/A |
| Sleuth v0.28.1 | Pimentel et al., 2017 | N/A |
| SUPPA v1 | Trincado et al., 2018 | N/A |
| MEME v4.11.2 | Bailey et al., 2009 | N/A |
| Prism | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| Primer3 | Howard Hughes Medical Institute and by the National Institutes of Health | http://bioinfo.ut.ee/primer3/ |
| Other | ||
| Sequence data, (FastQ files) for alternative splicing analyses in patient derived PDACs. | This paper | Bailey et al., 2016 |
mRNA sequencing.
For murine KPC and PanIN cells and primary patient derived organoids, RNA was extracted using Qiagen RNeasy columns. poly(A)-selected, unstranded Illumina libraries were prepared with a modified TruSeq protocol. 0.5× AMPure XP beads were added to the sample library to select for fragments <400 bp, followed by 1× beads to select for fragments >100 bp. These fragments were then amplified with PCR (15 cycles) and separated by gel electrophoresis (2% agarose). 300 bp DNA fragments were isolated and sequenced on the Illumina HiSeq 2000 (~100M 2×49 bp reads per sample).
Nucleotide motif, protein motif and enrichment analyses.
Statistically enriched nucleotide motifs were identified as previously described (Kim et al., 2015). Specifically, an ab initio approach by querying all k-mers of length 4, 5, 6 or 7. An enrichment statistic was defined for each k-mer as the occurrence of each k-mer in all cassette exons that were promoted or excluded in the presence of p53R175H in the different comparison studied systems. This method is equivalent to identifying k-mers that maximally distinguish between promoted and repressed cassette exons. Statistically significant enrichment was identified with the nonparametric Wilcoxon test, performed using the distributions of occurrence for each k-mer in promoted versus repressed exons. Differentially enriched motifs with an adjusted p value < 25% were considered significant. Consensus motifs were then identified by performing k-means clustering on significantly enriched or depleted motifs with n = 5 centers. An identical ab initio analysis was performed using the intronic regions flanking differentially spliced cassette exons (taking 100 nt upstream or downstream of the exons), but no significantly enriched motifs were identified. Enriched protein motifs were identified using MEME v4.11.2 (Bailey et al., 2009). The search considered motifs of length between 4 and 6 appearing in between exon sequences. To validate this approach, all k-mers of length 4, 5, or 6 were tested for enrichment, using a Wilcox test applied to the number of occurrences of each k-mer per kilobase to test for significance. Pathway enrichment analysis was performed using Enrichr (Kuleshov et al., 2016).
Immunoblotting.
Cells were lysed in 1× RIPA buffer (Sigma) with protease and phosphatase inhibitors. After lysis, samples were sonicated and mixed with (2% SDS, 2.5% β-mercaptoethanol) and heated at 95°C for 15 minutes. The antibodies used for western blotting are in Key Resource Table.
Cell proliferation and drug response assays.
KPC cells expressing (i) inducible shRNAs against R or p53 (cells were treated with 0.5 μM of doxycycline [dox, Sigma] five days in advance of experiments to knockdown p53), (ii) constitutive shRNAs against polyC exons or control (iii) constitutive cDNAs of +polyC, −polyC isoforms and empty vector (EV) control were seeded at a density of 700 onto 96-well black plates (Corning). For drug-response curves, the day after seeding, treatments using small-molecule compounds that impede the ability of the Sf3b complex to interact with pre-mRNAs (H3B-8800 and E7107)(Lee and Abdel-Wahab, 2016, Seiler et al., 2018, Yoshimi and Abdel-Wahab, 2017) were performed at different nM concentrations or DMSO (Sigma) vehicle control for 48 h. At each time-point of proliferation or drug treatment, cell viability was assessed using the CellTiter-Glo® Luminescent Cell Viability Assay (Promega).
Detection of Active RAS small-GTPases in cells.
KPC cells expressing shRNAs or cDNAs were lysed and processed using the immunoprecipitation kits for detection of active RAS (Cell signaling, 8821), active Rho (Cell signaling, 8820), active Rap1 (Cell signaling, 8818) following the manufacturer’s instructions. Levels of active RAS small-GTPases were determined by western blotting. To detect the levels of active KrasG12D, after pull-down, we blotted with the Ras G12D Mutant Specific antibody (Cell Signaling,14429).
Expression and Purification of full length or catalytic domain of GAP proteins:
Full length GAP17−polyC and GAP17+polyC were expressed in Expi293F cells (ThermoFisher Scientific) according to manufacturer’s instructions. Briefly, 7.5×106 cells in 25.5 ml growth medium were transfected using 80 μl ExpiFectamine293 Reagent and 30 μg plasmid DNA. Cells were grown at 37°C/8% CO2 shaking at 125 rpm, treated with enhancer solutions 1 and 2 after 18 h and harvested 72h after transfection. Cells were lysed in buffer containing 50 mM Tris pH 7.4, 150 mM NaCl, 1 mM EDTA, 1% Triton, 1 mM DTT and 1x protease inhibitor mix. GAP17 proteins were isolated via FLAG m2 affinity purification and elution with 100 pg/ml FLAG peptide in TBS/1 mM DTT.
GAP17 (residues 245–489), GAP17R288A and p120RASGAP (residues 714–1047) were cloned into an engineered pQlinkH vector with an N-terminal GST tag inserted between the 6x His and TEV cleavage sequence. All proteins were expressed in BL21 (DE3) Rosetta2 cells by overnight expression at 16°C following isopropyl β-D-1-thiogalactopyranoside induction and purified following the Qiagen Nickel NTA purification protocol. Cells were lysed by sonication in 50 mM HEPES pH 7.5, 150 mM NaCl, 5 mM MgCl2, 10 μM GDP (for KRAS) and 5 mM β-mercaptoethanol. Following overnight cleavage with TEV protease, all proteins were Nickel NTA purified again to remove the tag and protease. All proteins were further purified by size exclusion chromatography (Superdex-75 10/300 GL column; GE Life Sciences) and judged greater than 99% pure by SDS-PAGE analysis.
Expression of full length small-GTPases:
GST-tagged or 6x-His KRAS 4B and RHO proteins were expressed in E.coli BL21DE3 for 16 h at 16°C after IPTG induction or using the pQlinkH bacterial expression vector (gift from Konrad Buessow; Addgene plasmid # 13667) containing an N-terminal 6x-His purification tag followed by a Tobacco Etch Virus (TEV) protease cleavage site (Scheich et al., 2007). For GST-tagged proteins, cells were lysed in 50mM Tris pH 7.4, 250mM NaCl, 1mM DTT supplemented with 1x protease inhibitor mix and lysozyme (0.3mg/ ml) using a High-Pressure Homogenizer (Emulsiflex-C5; Avestin). Proteins were purified via affinity purification (Glutathione Sepharose) and size exclusion chromatography (ENrich SEC 650 10×300 Column, Bio-Rad equilibrated with 10mM HEPES, 250mM NaCl, 1mM TCEP, pH 7.4. For 6x-His-tagged proteins, purified following the Qiagen Nickel NTA purification protocol. Cells were lysed by sonication in 50 mM HEPES pH 7.5, 150 mM NaCl, 5 mM MgCl2, 10 μM GDP (for KRAS) and 5 mM β-mercaptoethanol. Following overnight cleavage with TEV protease, all proteins were Nickel NTA purified again to remove the tag and protease. All proteins were further purified by size exclusion chromatography (Superdex-75 10/300 GL column; GE Life Sciences) and judged greater than 99% pure by SDS-PAGE analysis.
GTPase activity assay:
In vitro GTPase assays were performed using the GTPase-Glo Assay (Promega) in white 384 well-plates. Reactions contained 1mM DTT, 5μM GTP, 0.7μM RHOA or 1.5μM KRAS wt/KRASG12D plus recombinant GAP proteins at a 2.5 fold molar access in a final volume of 10 μl. Samples were incubated for 2 h at room temperature prior to further processing following manufacturer’s instructions. Bioluminescence readings were performed on a Tecan infinite M1000Pro plate reader.
GTP hydrolysis:
KRAS GTP hydrolysis was measured using a modification of a previously published method (Shutes and Der, 2005). Briefly, excess GDP was removed by buffer exchange, and a 40-fold molar excess of GTP was added to 400 μl of 250 μM KRAS with 30 mM EDTA. After 1–4 hr at RT, MgCl2 was added to a final concentration of 50 mM and the reaction was placed on ice for 5 min. The mixture was applied to a NAP-5 column (GE) equilibrated with 25 mM HEPES and 50 mM NaCl and fractions were collected. Protein containing fractions were pooled and the protein concentration was measured by A280. Using an xx fluorimeter, KRAS (4 μM), phosphate sensor (5 μM), and GAP protein (10 μM for GAP17, 4 nM for p120RASGAP, when indicated) was added to phosphate-free buffer and fluorescence was measured every 10 s for 20000 s.
Membrane and cytoplasmic fractionation method:
The translocation of Gap17 from the cytosol to the plasma membrane is stimulated by active EGFR and other growth-factor signaling and downstream Gap17 tyrosine phosphorylations in other cell types (Beck et al., 2014, Beck et al., 2013). To track plasma membrane localization experiments of Gap17 we first depleted FBS from the media (24h) to eliminate growth factor signaling and tyrosine phosphorylation of Gap17. Then we performed an acute growth factor stimulation (20% FBS/20min), to challenge and measure the transient membrane localization of Gap17 isoforms under synchronized stimulation of RAS small-GTPases. These protocols have been previously used to track subcellular localization of GAP17 (Beck et al., 2014, Beck et al., 2013) and GAPs (Molloy et al., 1989). We used the Subcellular Protein Fractionation Kit for Cultured Cells (Thermo Cat # 78840), according to manufacturer and previous studies (Cascio et al., 2011, Draker et al., 2011, Loo et al., 2009, Roger et al., 2010). Briefly, the cells were incubated with a buffer that causes selective membrane permeabilization, releasing soluble cytoplasmic contents. After, high sucrose buffer (1.25M) was used for plasma membrane fraction and collected after centrifugation. Importantly, this method does not solubilize the nuclear membranes.
Immunofluorescence stains:
Briefly, cells were fixed with 100% methanol and blocked using 1X PBS/5% normal horse serum. Permeabilization was performed using 1X PBS/0.5% Triton™ X-100. Primary antibody against Gap 17 (Invitrogen PA5–50113, 1:100) was left overnight. Secondary immunofluorescent antibody was used to detect Gap17 (Cell Signaling Cat # A-11034) and DAPI to recognize the nuclei. Cells were mounted and visualized under confocal microscope at 40X.
RNA isolation, RT-PCR and qRT-PCR.
RNA isolation was performed with the RNeasy Mini kit (Qiagen). Reverse transcription was performed using the Verso cDNA Synthesis Kit (Thermo Scientific). RT-PCRs were performed using the GoTaq® Green Master Mix (Promega) and with an initial incubation at 95°C for 10 minutes followed by 35–40 cycles of 15 seconds at 95°C and 1 minute at 58–60°C. Reaction products were analyzed on 1–2 % agarose gels with ethidium bromide staining. qPCR was performed using SYBR Green Master Mix (Applied Biosystems) according to the manufacturer’s instructions. Reactions were performed using a StepOnePlus Real Time PCR System (Applied Biosystems) with an initial incubation at 95°C for 10 minutes followed by 40 cycles of 15 seconds at 95°C and 1 minute at 60°C. All samples including the template controls were assayed in triplicate. The relative number of target transcripts was normalized to the number of GAPDH transcripts found in the same sample. The relative quantification of target gene expression was performed with the standard curve or comparative cycle threshold (CT) method (Schmittgen and Livak, 2008). Primers were designed using Primer3 (available at http://bioinfo.ut.ee/primer3/). Primers used for each reaction are described in Key Resource Table.
Immunohistochemistry.
Immunohistochemistry was performed on formalin-fixed paraffin-embedded (FFPE) tissue blocks from pancreata from mice using the indirect immunoperoxidase method to identify the presence of GFP (Cell Signaling, 2956) and phospho-ERK 1/2 (Cell Signaling, 4370). Antigen retrieval was performed in citrate buffer at 120°C for 10 minutes in a decloaking chamber. Endogenous peroxidase was blocked by 3% hydrogen peroxide and sections were incubated overnight at 4°C. After primary antibody, biotinylated-horse secondary antibodies (R.T.U. Vectastain ABC kit; Vector Laboratories, Burlingame, CA) were added. Development was done with 3, 3’ diaminobenzidine (DAB) (Dako, Carpinteria, CA), and counter-stain was done with hematoxylin. Negative controls were performed on all runs using an equivalent concentration of a subclass-matched immunoglobulin.
HNRNPK bacterial Expression and Protein Purification.
Transformed Rosetta Cells (Novagen) were cultured in LB medium, (5g Tryptone, 2.5g Yeast Extract, and 5g NaCl) until optical density reached 0.6, cultures were transferred to 4°C and allowed to cool. Protein expression was induced for 14–20 hr with IPTG at 15°C. Cells were pelleted, lysed in lysis buffer (25 mM Tris-HCl, 150 mM NaCl, 3 mM MgCl2 1% Triton 100X, 40 mg/mL Lysozyme (Thermo Scientific)) for 30 min in the presence of protease inhibitor cocktail (Roche), sonicated and clarified by centrifuging at 17,500 rpm, passed through a 0.45 pM filter (GE) and purified using GST-Sepharose in column format (GST-trap FF, GE). A 4 L bacterial culture was used for column purification and eluted protein was concentrated by centrifugation (Amicon Ultra-4 Centrifugal Filter Units) and buffer exchanged into final buffer (100 mM KCl, 1 mM DTT, 20 mM HEPES). Proteins were quantified using Peirce Reagent (Thermo Scientific) and purity and quality of protein was assessed by PAGE followed by Coomassie staining.
Filter Binding Assay.
Custom RNA oligonucleotides were synthesized by IDT (Integrated DNA Technologies) and RBPs were purified as described (Dominguez et al., 2018). polyN16: NNNNNNNNNNNNNNNN (where N is any base), polyC16: CCCCCCCCCCCCCCCC, and GAP17 exon: UGCCCGCUGGCCCAGAGCCCCCUCCCCAGAG. RNA was end-labeled with 32P by incubating with Polynucleotide Kinase (NEB) according to manufacturer protocol. Filter assay was performed following the protocol described previously (Dominguez et al., 2018) for use with a 96-well dot-blot apparatus (Biorad). RBP and radio-labeled RNA were incubated in 100 μL binding buffer (100 mM KCl, 1 mM DTT, 10% glycerol, 20 mM HEPES) for 60 minutes at room temperature. Final concentration of RNA was 330 pM and protein concentration ranged from 2.5 nM-1 μM. Radioactivity signal was captured on a storage phosphor screen and subsequently measured on Typhoon instrument.
QUANTIFICATION AND STATISTICAL ANALYSIS
All experiments were performed on biological replicates unless otherwise specified. Sample size for each experimental group/condition is reported in the appropriate figure legends and methods. For significance testing, analyses were chosen if data met the assumptions of the tests. Data was checked for comparable variance prior to statistical analysis. Statistically significant differences between control and experimental groups were determined using two-tailed unpaired Student’s t-test, one-way ANOVA with Tukey multiple comparison difference test, Mann-Whitney test, Kolmogorov-Smirnov test, Wilcoxon test, and log-rank test as indicated in the appropriate figure legend and methods text.
DATA AND SOFTWARE AVAILABILITY
RNA-seq data deposition
RNA-seq data from this study are deposited in the Gene Expression Omnibus (GEO) under accession number: GSE114502.
Supplementary Material
Table S1: Percent spliced in (Psi) values for differentially spliced events in PDAC patient samples with p53R282G/W versus p53 truncating mutations. Related to Figure 1.
Table S2: Percent spliced in (Psi) values for differentially spliced events and pathways enrichment analysis in the three systems with manipulation of the p53 status. Related to Figure 1.
Table S3: Venn Diagrams of commonly spliced events in the three systems with manipulation of the p53 status. Related to Figure 1.
Table S4: Differential gene expression by p53R175H in the three systems with manipulation of the p53 status. Related to Figure 3.
Table S5: Percent spliced in (Psi) values for differentially spliced events by hnRNPK and comparison against differentially spliced events by p53R175H. Related to Figure 3.
SIGNIFICANCE.
To date, mutant KRAS and p53 have proven to be very difficult to target, preventing successful treatment of PDAC and other cancers. Here we identified that p53 hotspot mutations promote expression of RNA binding proteins which modify the splicing of GAPs in a manner that promotes maximal activation of KRAS. Altering GAP isoform ratios using antisense oligonucleotides or spliceosome inhibitors significantly decreased oncogenic KRAS signaling, decreased tumor growth, and increased survival in PDAC preclinical models. These results uncover a link between mutations in p53 and RNA splicing, a mechanism regulating the activity of oncogenic KRAS via alternative splicing, and the basis for biomarker-driven treatment for PDAC based on modulation of RNA splicing.
ACKNOWLEDGEMENTS
We thank APGI for RNA-seq data and H3 Biomedicine for H3B-8800 and E7107. This work was supported by NIH R01 CA204228-01 and P30 CA023108 (S.D.L.), PanCAN-AACR grants to S.D.L. and L.F.E-H, Suzanne Cohn Simon Pancreatic Cancer Research Fund (S.D.L.), NCI K99 CA226342 01 (L.F.E-H), National Pancreas Foundation grant (L.F.E-H), Hirshberg Foundation (L.F.E-H), and NCI Cancer Center Support Grant (P30 CA08748). R.K.B. and O.A.-W. are supported by NIH/NHLBI (R01 HL128239). O.A.-W. is supported by the Henry & Marilyn Taub Foundation, the Edward P. Evans Foundation, and the Leukemia and Lymphoma Society. We thank the support from the DFG (OE531/2-2 to AO and KU2531/2 to DK) and the IMF Münster (OE121701 to AO).
DECLARATION OF INTERESTS
L.F.E. is a consultant for KDx Diagnostics Inc and OncoGenesis Inc. O.A.-W. served as a consultant for H3B Biomedicine, Foundation Medicine Inc, Merck, and Janssen and is on the scientific advisory board of Envisagenics; O.A.-W. received prior research funding from H3B Biomedicine unrelated to the current manuscript and serves on the Scientific Advisory Board of Envisagenics Inc. S.D.L. is a member of the Scientific Advisory Board for Nybo Pharmaceuticals and co-founder of Episteme Prognostics.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Agbunag C, Lee KE, Buontempo S, and Bar-Sagi D (2006). Pancreatic duct epithelial cell isolation and cultivation in two-dimensional and three-dimensional culture systems. Methods Enzymol 407, 703–710. [DOI] [PubMed] [Google Scholar]
- Aguirre AJ, and Hahn WC (2018). Synthetic Lethal Vulnerabilities in KRAS-Mutant Cancers. Cold Spring Harb Perspect Med 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anczuków O, Rosenberg AZ, Akerman M, Das S, Zhan L, Karni R, Muthuswamy SK, and Krainer AR (2012). The splicing factor SRSF1 regulates apoptosis and proliferation to promote mammary epithelial cell transformation. Nature structural & molecular biology 19, 220–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ardito CM, Gruner BM, Takeuchi KK, Lubeseder-Martellato C, Teichmann N, Mazur PK, Delgiorno KE, Carpenter ES, Halbrook CJ, Hall JC, et al. (2012). EGF receptor is required for KRAS-induced pancreatic tumorigenesis. Cancer Cell 22, 304–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey P, Chang DK, Nones K, Johns AL, Patch AM, Gingras MC, Miller DK, Christ AN, Bruxner TJ, Quinn MC, et al. (2016). Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 531, 47–52. [DOI] [PubMed] [Google Scholar]
- Bailey TL, Boden M, Buske FA, Frith M, Grant CE, Clementi L, Ren J, Li WW, and Noble WS (2009). MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res 37, W202–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck S, Fotinos A, Gawaz M, and Elvers M (2014). Nadrin GAP activity is isoform- and target-specific regulated by tyrosine phosphorylation. Cell Signal 26, 1975–1984. [DOI] [PubMed] [Google Scholar]
- Beck S, Fotinos A, Lang F, Gawaz M, and Elvers M (2013). Isoform-specific roles of the GTPase activating protein Nadrin in cytoskeletal reorganization of platelets. Cell Signal 25, 236–246. [DOI] [PubMed] [Google Scholar]
- Boj SF, Hwang CI, Baker LA, Chio II, Engle DD, Corbo V, Jager M, Ponz-Sarvise M, Tiriac H, Spector MS, et al. (2015). Organoid models of human and mouse ductal pancreatic cancer. Cell 160, 324–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bray NL, Pimentel H, Melsted P, and Pachter L (2016). Near-optimal probabilistic RNA-seq quantification. Nat Biotechnol 34, 525–527. [DOI] [PubMed] [Google Scholar]
- Carpenter B, McKay M, Dundas SR, Lawrie LC, Telfer C, and Murray GI (2006). Heterogeneous nuclear ribonucleoprotein K is over expressed, aberrantly localised and is associated with poor prognosis in colorectal cancer. Br J Cancer 95, 921–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cascio S, Zhang L, and Finn OJ (2011). MUC1 protein expression in tumor cells regulates transcription of proinflammatory cytokines by forming a complex with nuclear factor-kappaB p65 and binding to cytokine promoters: importance of extracellular domain. J Biol Chem 286, 42248–42256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng DT, Mitchell TN, Zehir A, Shah RH, Benayed R, Syed A, Chandramohan R, Liu ZY, Won HH, Scott SN, et al. (2015). Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT): A Hybridization Capture-Based Next-Generation Sequencing Clinical Assay for Solid Tumor Molecular Oncology. The Journal of molecular diagnostics : JMD 17, 251–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciarlo M, Benelli R, Barbieri O, Minghelli S, Barboro P, Balbi C, and Ferrari N (2012). Regulation of neuroendocrine differentiation by AKT/hnRNPK/AR/beta-catenin signaling in prostate cancer cells. Int J Cancer 131, 582–590. [DOI] [PubMed] [Google Scholar]
- Collisson EA, Sadanandam A, Olson P, Gibb WJ, Truitt M, Gu S, Cooc J, Weinkle J, Kim GE, Jakkula L, et al. (2011). Subtypes of pancreatic ductal adenocarcinoma and their differing responses to therapy. Nat Med 17, 500–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- di Magliano MP, and Logsdon CD (2013). Roles for KRAS in pancreatic tumor development and progression. Gastroenterology 144, 1220–1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominguez D, Freese P, Alexis MS, Su A, Hochman M, Palden T, Bazile C, Lambert NJ, Van Nostrand EL, Pratt GA, et al. (2018). Sequence, Structure, and Context Preferences of Human RNA Binding Proteins. Mol Cell 70, 854–867 e859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Draker R, Sarcinella E, and Cheung P (2011). USP10 deubiquitylates the histone variant H2A.Z and both are required for androgen receptor-mediated gene activation. Nucleic Acids Res 39, 3529–3542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dvinge H, Kim E, Abdel-Wahab O, and Bradley RK (2016). RNA splicing factors as oncoproteins and tumour suppressors. Nat Rev Cancer 16, 413–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escobar-Hoyos LF, Knorr K, and Abdel-Wahab O (2019). Aberrant RNA Splicing in Cancer. Annual Reviews in Cancer Biology 3, 167–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng S, Kasahara C, Rickles RJ, and Schreiber SL (1995). Specific interactions outside the proline-rich core of two classes of Src homology 3 ligands. Proc Natl Acad Sci U S A 92, 12408–12415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartley SW, and Mullikin JC (2015). QoRTs: a comprehensive toolset for quality control and data processing of RNA-Seq experiments. bMc Bioinformatics 16, 224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hingorani SR, Wang L, Multani AS, Combs C, Deramaudt TB, Hruban RH, Rustgi AK, Chang S, and Tuveson DA (2005). Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell 7, 469–483. [DOI] [PubMed] [Google Scholar]
- Hsu TY, Simon LM, Neill NJ, Marcotte R, Sayad A, Bland CS, Echeverria GV, Sun T, Kurley SJ, Tyagi S, et al. (2015). The spliceosome is a therapeutic vulnerability in MYC-driven cancer. Nature 525, 384–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang DC, Marshall CJ, and Hancock JF (1993). Plasma membrane-targeted ras GTPase-activating protein is a potent suppressor of p21ras function. Mol Cell Biol 13, 2420–2431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter JC, Manandhar A, Carrasco MA, Gurbani D, Gondi S, and Westover KD (2015). Biochemical and Structural Analysis of Common Cancer-Associated KRAS Mutations. Mol Cancer Res 13, 1325–1335. [DOI] [PubMed] [Google Scholar]
- Janes MR, Zhang J, Li LS, Hansen R, Peters U, Guo X, Chen Y, Babbar A, Firdaus SJ, Darjania L, et al. (2018). Targeting KRAS Mutant Cancers with a Covalent G12C-Specific Inhibitor. Cell 172, 578–589 e517. [DOI] [PubMed] [Google Scholar]
- Kim E, Ilagan JO, Liang Y, Daubner GM, Lee SC, Ramakrishnan A, Li Y, Chung YR, Micol JB, Murphy ME, et al. (2015). SRSF2 Mutations Contribute to Myelodysplasia by Mutant-Specific Effects on Exon Recognition. Cancer cell 27, 617–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kletting P, and Glatting G (2009). Model selection for time-activity curves: the corrected Akaike information criterion and the F-test. Z Med Phys 19, 200–206. [DOI] [PubMed] [Google Scholar]
- Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z, Koplev S, Jenkins SL, Jagodnik KM, Lachmann A, et al. (2016). Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res 44, W90–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KE, and Bar-Sagi D (2010). Oncogenic KRas suppresses inflammation-associated senescence of pancreatic ductal cells. Cancer Cell 18, 448–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SC, and Abdel-Wahab O (2016). Therapeutic targeting of splicing in cancer. Nature medicine 22, 976–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leeksma AC, Taylor J, Wu B, Gardner JR, He J, Nahas M, Gonen M, Alemayehu WG, Te Raa D, Walther T, et al. (2018). Clonal diversity predicts adverse outcome in chronic lymphocytic leukemia. Leukemia. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logsdon CD, and Lu W (2016). The Significance of Ras Activity in Pancreatic Cancer Initiation. Int J Biol Sci 12, 338–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loo LH, Lin HJ, Singh DK, Lyons KM, Altschuler SJ, and Wu LF (2009). Heterogeneity in the physiological states and pharmacological responses of differentiating 3T3-L1 preadipocytes. J Cell Biol 187, 375–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maertens O, and Cichowski K (2014). An expanding role for RAS GTPase activating proteins (RAS GAPs) in cancer. Adv Biol Regul 55, 1–14. [DOI] [PubMed] [Google Scholar]
- Moffitt RA, Marayati R, Flate EL, Volmar KE, Loeza SG, Hoadley KA, Rashid NU, Williams LA, Eaton SC, Chung AH, et al. (2015). Virtual microdissection identifies distinct tumor- and stroma-specific subtypes of pancreatic ductal adenocarcinoma. Nat Genet 47, 1168–1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molloy CJ, Bottaro DP, Fleming TP, Marshall MS, Gibbs JB, and Aaronson SA (1989). PDGF induction of tyrosine phosphorylation of GTPase activating protein. Nature 342, 711–714. [DOI] [PubMed] [Google Scholar]
- Morton JP, Timpson P, Karim SA, Ridgway RA, Athineos D, Doyle B, Jamieson NB, Oien KA, Lowy AM, Brunton VG, et al. (2010). Mutant p53 drives metastasis and overcomes growth arrest/senescence in pancreatic cancer. Proc Natl Acad Sci U S A 107, 246–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olive KP, Tuveson DA, Ruhe ZC, Yin B, Willis NA, Bronson RT, Crowley D, and Jacks T (2004). Mutant p53 gain of function in two mouse models of Li-Fraumeni syndrome. Cell 119, 847–860. [DOI] [PubMed] [Google Scholar]
- Paziewska A, Wyrwicz LS, Bujnicki JM, Bomsztyk K, and Ostrowski J (2004). Cooperative binding of the hnRNP K three KH domains to mRNA targets. FEBS Lett 577, 134–140. [DOI] [PubMed] [Google Scholar]
- Pimentel H, Bray NL, Puente S, Melsted P, and Pachter L (2017). Differential analysis of RNA-seq incorporating quantification uncertainty. Nat Methods 14, 687–690. [DOI] [PubMed] [Google Scholar]
- Roger L, Jullien L, Gire V, and Roux P (2010). Gain of oncogenic function of p53 mutants regulates E-cadherin expression uncoupled from cell invasion in colon cancer cells. J Cell Sci 123, 1295–1305. [DOI] [PubMed] [Google Scholar]
- Scheich C, Kummel D, Soumailakakis D, Heinemann U, and Bussow K (2007). Vectors for co-expression of an unrestricted number of proteins. Nucleic Acids Res 35, e43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmittgen TD, and Livak KJ (2008). Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc 3, 1101–1108. [DOI] [PubMed] [Google Scholar]
- Seiler M, Yoshimi A, Darman R, Chan B, Keaney G, Thomas M, Agrawal AA, Caleb B, Csibi A, Sean E, et al. (2018). H3B-8800, an orally available small-molecule splicing modulator, induces lethality in spliceosome-mutant cancers. Nature medicine. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Settleman J, Albright CF, Foster LC, and Weinberg RA (1992). Association between GTPase activators for Rho and Ras families. Nature 359, 153–154. [DOI] [PubMed] [Google Scholar]
- Shutes A, and Der CJ (2005). Real-time in vitro measurement of GTP hydrolysis. Methods 37, 183–189. [DOI] [PubMed] [Google Scholar]
- Simoes PK, Olson SH, Saldia A, and Kurtz RC (2017). Epidemiology of pancreatic adenocarcinoma. Chin Clin Oncol 6, 24. [DOI] [PubMed] [Google Scholar]
- Stein CA, and Castanotto D (2017). FDA-Approved Oligonucleotide Therapies in 2017. Mol Ther 25, 1069–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trincado JL, Entizne JC, Hysenaj G, Singh B, Skalic M, Elliott DJ, and Eyras E (2018). SUPPA2: fast, accurate, and uncertainty-aware differential splicing analysis across multiple conditions. Genome Biol 19, 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuveson DA, Zhu L, Gopinathan A, Willis NA, Kachatrian L, Grochow R, Pin CL, Mitin NY, Taparowsky EJ, Gimotty PA, et al. (2006). Mist1-KrasG12D knock-in mice develop mixed differentiation metastatic exocrine pancreatic carcinoma and hepatocellular carcinoma. Cancer Res 66, 242–247. [DOI] [PubMed] [Google Scholar]
- Ventura A, Meissner A, Dillon CP, McManus M, Sharp PA, Van Parijs L, Jaenisch R, and Jacks T (2004). Cre-lox-regulated conditional RNA interference from transgenes. Proc Natl Acad Sci U S A 101, 10380–10385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vigil D, Cherfils J, Rossman KL, and Der CJ (2010). Ras superfamily GEFs and GAPs: validated and tractable targets for cancer therapy? Nat Rev Cancer 10, 842–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei CL, Wu Q, Vega VB, Chiu KP, Ng P, Zhang T, Shahab A, Yong HC, Fu Y, Weng Z, et al. (2006). A global map of p53 transcription-factor binding sites in the human genome. Cell 124, 207–219. [DOI] [PubMed] [Google Scholar]
- Weissmueller S, Manchado E, Saborowski M, Morris J. P. t., Wagenblast E, Davis CA, Moon SH, Pfister NT, Tschaharganeh DF, Kitzing T, et al. (2014). Mutant p53 drives pancreatic cancer metastasis through cell-autonomous PDGF receptor beta signaling. Cell 157, 382–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen F, Shen A, Shanas R, Bhattacharyya A, Lian F, Hostetter G, and Shi J (2010). Higher expression of the heterogeneous nuclear ribonucleoprotein k in melanoma. Ann Surg Oncol 17, 2619–2627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshimi A, and Abdel-Wahab O (2017). Molecular Pathways: Understanding and Targeting Mutant Spliceosomal Proteins. Clin Cancer Res 23, 336–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuber J, McJunkin K, Fellmann C, Dow LE, Taylor MJ, Hannon GJ, and Lowe SW (2011). Toolkit for evaluating genes required for proliferation and survival using tetracycline-regulated RNAi. Nat Biotechnol 29, 79–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1: Percent spliced in (Psi) values for differentially spliced events in PDAC patient samples with p53R282G/W versus p53 truncating mutations. Related to Figure 1.
Table S2: Percent spliced in (Psi) values for differentially spliced events and pathways enrichment analysis in the three systems with manipulation of the p53 status. Related to Figure 1.
Table S3: Venn Diagrams of commonly spliced events in the three systems with manipulation of the p53 status. Related to Figure 1.
Table S4: Differential gene expression by p53R175H in the three systems with manipulation of the p53 status. Related to Figure 3.
Table S5: Percent spliced in (Psi) values for differentially spliced events by hnRNPK and comparison against differentially spliced events by p53R175H. Related to Figure 3.
Data Availability Statement
RNA-seq data deposition
RNA-seq data from this study are deposited in the Gene Expression Omnibus (GEO) under accession number: GSE114502.