

Abstract
Aluminum is a ubiquitously abundant nonessential element. Aluminum has been associated with neurodegenerative diseases such as Alzheimer's disease (AD), amyotrophic lateral sclerosis, and dialysis encephalopathy. Many continue to regard aluminum as controversial although increasing evidence supports the implications of aluminum in the pathogenesis of AD. Aluminum causes the accumulation of tau protein and Aβ protein in the brain of experimental animals. Aluminum induces neuronal apoptosis in vivo and in vitro, either by endoplasmic stress from the unfolded protein response, by mitochondrial dysfunction, or a combination of them. Some, people who are exposed chronically to aluminum, either from through water and/or food, have not shown any AD pathology, apparently because their gastrointestinal barrier is more effective. This article is written keeping in mind mechanisms of action of aluminum neurotoxicity with respect to AD.
Keywords: aluminum, Alzheimer's disease, amyloid‐β protein, neurofibrillary tangles
Introduction
Aluminum (Al) is the third most abundant element which comprises about 8% of earth's crust, after oxygen and silicon. Al is too reactive chemically to occur as a free metal in nature. Instead, it always occurs in combination with other elements such as hydroxide, silicate, sulfate, and phosphate. The wide distribution of this element ensures the potential for causing human exposure. Aluminum is used in the production of every‐day products such as cookware is made from aluminum, soda cans, aluminum foil, antacids, aspirin, vaccines, and flour 33.
It has been suggested that there is a relationship between chronic routine exposure to Al and increased risk of a number of neurodegenerative disorders including Alzheimer's disease (AD), amyotrophic lateral sclerosis (ALS), and AD‐type dementia in Parkinson patients 9.
Failure of amyloid trials has caused many researchers to be more critical of current dogma. Recent studies have suggested that neuroinflammation and neuronal loss precede amyloid‐β plaque deposition in the hAβPP‐J20 mouse model of AD 158, The earliest damage in humans with AD occurs as neurofibrillary tangles (NFTs) in the entorhinal cortex cells of origin for the performant pathway. Neurofibrillary tangle formation involves aluminum interactions with hyperphosphorylated tau 147. AD is one of the prevalent neurodegenerative diseases affecting more than five million people in the United States and is projected to affect more than quadruple that amount during the next 50 years if the present situation persists 130. AD is the most common cause of dementia in the elderly. The well‐defined lesions in AD pathology are extracellular deposition of the amyloid‐β (Aβ) protein, intracellular NFTs, and selective neuronal loss. (Although the cause of AD remains poorly understood, multiple factors are reported to influence AD onset. Mounting genetic and biochemical data support the hypothesis that amyloid‐β protein (Aβ) accumulation and aggregation in the brain are early and central events in the pathogenesis of AD.) The primary factor, among the multiple factors in the pathogenesis of early onset AD, is mutations in amyloid precursor protein (AβPP) and presenilins 1 and 2, that lead to an increase in the production of Aβ42 14. The ε4 allele of apolipo protein E (Apo E) is the most prevalent risk factor linked to AD. Additionally linked to AD are increased levels of homocysteine, cholesterol, and several minor metal ions such as Al, iron, and copper. Dollken et al (1897) first reported Al neurotoxicity in experimental animals and stated that free radical formation is due to Al. Klatzo et al (1965) reported that injections of Al salts into rabbit brain led to the formation of NFTs, one of the hallmarks of AD. It has been shown that rabbits may provide a unique animal system for producing neurofibrillary pathology because rabbits belong to the mammalian order lagomorpha which more closely resembles primates than rodents 52. Crapper et al (1973) reported neurofibrillary degeneration in cats with Al accumulation. It has also been shown that Al salts injected intracerebrally or peripherally in rabbits, mice, cats, and monkeys induce the formation of neurofibrillary threads 6, 11, 12, 22, 23, 25, 45, 52, 77, 142. Al has been shown to accumulate to different extents in a number of regions of rat brain, in particular the hippocampus, which is the site of memory and learning 24, 26, 66, 71. Becaria et al (2002) (described Al as a toxicant when present in high doses in circulation and) suggested that a prolonged exposure to relatively low levels of dietary Al may be responsible for the observed neurotoxic effects. Al has been detected in both senile plaques and NFT‐bearing neurons in the brains of AD patients (McLachlan et al 1996), which suggests roles for this metal in AD. Earlier studies from our laboratory have also shown that acute as well as subacute exposure of rats to Al causes marked reduction in acetyl choline esterase activity 66, 112 and significant deterioration in cognitive functions of rats 148, which are symptoms of AD. Walton (2006) showed that Al was present in the hippocampal neurons of AD patients and hence Al might be playing a role in the formation of NFTs 145. Miu et al (2003) reported the effects of long term Al exposure in terms of behavioral and brain modifications 98. They observed numerous ghost‐like neurons with cytoplasmic and nuclear vacuolations with Al deposits. Their study also reported hippocampus containing extracellular accumulations of Al and amyloid surrounded by nuclei of degenerating cells. The aforementioned modifications are reminiscent of those observed in AD. Perl and Moalem (2006) also presented evidence that Al, a highly reactive element known to crosslink hyperphosphorylated proteins, may play an active role in the pathogenesis of critical neuropathological lesions in AD and the other related disorders. Al binding can enhance Aβ protein penetration of the blood brain barrier 5. Several studies have demonstrated that Al can induce pathological changes similar to AD including the accumulation of tau and Aβ, neuronal apoptosis, conformational changes of Aβ and disease related proteins, and disruption of iron and calcium homeostasis 16, 96, 155, 162, 163, 166, 167. Kawahara (2005) demonstrated that Al can cause the accumulation of tau protein and Aβ protein in experimental animals leading to induced neural apoptosis in vivo as well as in vitro 72. Studies show increased oxidative damage, neuronal degeneration, and hyperphosphorylated tau in rat model of AD, providing further evidence that Al may play a role AD 145, 146. In this article, we review material in favor of, and against, the concept of Al neurotoxicity in the context of AD.
Al Exposure and Assessment
Al has been used for many years in shipbuilding, petroleum processing, rubber industry, medicine, food processing, agriculture, and treatment of water. Various pathological conditions, such as asthma‐like disorders and pulmonary fibrosis, have been reported among workers involved in the Al industry 103. The principal routes of human exposure to Al are by ingestion of food or water containing Al and the inhalation of Al as fine particles 118, 120, 121, 143, 163. The average human Al consumption through food and beverages (excluding drinking water) ranges from 2.5 to 13 mg/day and varies with composition of the food eaten, country of residence, age, and sex. In the US, the total dietary Al intake estimates for adult men and women are 8–9 and 7 mg/day (These Al values are appropriate for fresh foods but are too low for commercially prepared (processed) foods. Given figures are higher but more realistic considering one piece of sliced individually wrapped cheese can contain), respectively 44, 53, 157. The metal can be taken into the body from food, by both naturally occurring Al and artificial dietary additions from sources such as additives, water, cooking utensils, containers (including Al drink cans), aerosols, and dusts. Although it is well known that less than 1% of ingested Al is absorbed, significant absorption and retention can occur either (i) when long‐term intake exceeds 1000 mg/day or (ii) when chronic aluminum exposures at the high end of the human range for dietary aluminum levels (around 100 mg/day) 149.
Even though bioavailability of Al from water (0.3%) is higher than from food (0.1%), due to differences in daily Al intake, drinking water contributes (considerably less) to the overall human exposure to Al 38, 143. The issue of safe levels of Al in drinking water is of considerable interest to public health officials and regulatory agencies because Al is widely used as a coagulant in water treatment 92.
Al intake from drinking water
The World Health Organization 157 has recommended that Al concentrations in drinking water not to exceed 0.2 mg/L, stating “…the positive relationship between Al in drinking water and AD, which was demonstrated in several epidemiological studies, cannot be totally dismissed. Taken together, the relative risks for AD from exposure to Al in drinking water above 100 µg/liter are low (less than 2.0) 67.” The US Environmental Protection Agency set 0.05 mg/L as a limit in 1985. The European Economic Community has established a recommended guideline of 0.05 mg/L and a maximum permissible level of 0.2 mg/L 38. Numerous epidemiological reports were published on the subject of exposure to Al from drinking water and risk of AD 38.
First attempts to relate the actual levels of Al in drinking water to AD were made in 1986, when two parallel Norwegian studies reported higher mortality from dementia (from all causes) in areas with high concentrations of Al in drinking water 38, 143. A cross‐sectional investigation 90 conducted a few years later in England and Wales found that among all other causes of dementia the risk of AD was 1.5 times higher in districts where the mean Al concentration in drinking water exceeded 0.11 mg/L, compared to the districts where Al concentrations were less than 0.01 mg/L; no relation was found between any other neurological condition and concentrations of Al in water 90. Another ecological study, conducted in Newfoundland, Canada, found an excess of dementia mortality (confirmed by death certificates) from the North shore of Bonavista Bay in 1985 and 1986, a phenomenon that could not be explained by differences in sex, age, or other parameters 43. That specific area of the bay was known to have high Al concentration in the drinking water (165 mg/L) and low pH (5.2). A case‐control study conducted in Ontario, Canada, used hospital discharge data with a diagnosis of dementia or presenile dementia to describe a dose‐response relationship between the Al content of drinking water and risk of AD 106. The relative risks associated with the consumption of drinking water containing Al concentrations of <0.01, 0.01–0.1, 0.1–0.199, and >0.200 mg/L were estimated to be 1.00, 1.13, 1.26, and 1.46, respectively. Subsequent reanalysis of the data confirmed a stronger dose‐response relationship for those over 75 years of age 39, 40, 84, 103. No additional adjustments for confounding factors except for age and sex were made in this study. Another Canadian investigation was done by Forbes et al (1994, 1995), who studied the relationship between Al, fluoride, and other constituents in drinking water and cognitive function. The research project was based on the Ontario Longitudinal Study of Aging, where 2000 men have been followed for about 30 years. Analysis of the data showed that men living in areas with high Al and low fluoride concentrations in drinking water were about three times more likely to have some form of mental impairment than those individuals living in the areas with low Al and high fluoride levels. Further analysis suggested that at neutral pH, relatively low Al concentrations, and relatively high fluoride concentrations in drinking water decreased the odds of cognitive impairment by a factor of five 39, 40. A case‐control study on autopsy‐verified material conducted by McLachlan et al (1996) compared the AD patients and controls without brain pathology on the basis of neuropathologic criteria. Al concentration in drinking water at last residence before death was used as the measure of exposure 96. The authors reported elevated risks for histopathologically verified AD to be associated with higher levels of Al (odds ratio (OR) = 1.7, 95% CI 1.2–2.5, for a level of Al >0.1 mg/L). The authors later obtained even larger estimates (OR of 2.5 or greater) and implemented a 10‐year weighted residential history in the analysis. At an Al concentration of 0.125 mg/L in drinking water, the OR for risk of AD was 3.6 (95% CI 1.4–9.9), at 0.150 mg/L the OR was 4.4 (95% CI 0.98–20), and at 0.175 mg/L it was 7.6 (95% CI 0.98–61). The study had several methodological strengths including the diagnostic quality of the data 96.
In France, Rondeau et al (2000) utilized the data from a large prospective Paquid cohort study, which followed 3777 subjects, aged 65 years and older, for up to 8 years, and recorded all new cases of dementia and AD 121. In each residential area the range and mean exposure to Al (0.001–0.459 mg/L, median 0.009 mg/L) and silica (4.2–22.4 mg/L) from drinking water were recorded. The analysis of data adjusted for age, gender, educational level, place of residence, and wine consumption revealed that the risk of dementia was higher for individuals who lived in areas with high levels of Al in water (mean concentration >0.1 mg/L) compared with people residing in areas with Al levels less than 0.1 mg/L (relative risk 1.99, 95% CI 1.20–3.28, P = 0.007). Higher silica concentrations (>11.25 mg/L), adjusted for age and gender, were associated with a reduced risk of dementia (RR = 0.71, 95% CI 0.56–0.91, P = 0.007). The adjusted relative risk of AD for individuals exposed to drinking water with Al concentration > 0.10 mg/L was 2.14 (95% CI 1.21–3.80, P = 0.007). As with dementia, the risk of AD was reduced in the presence of high concentrations of silica (RR = 0.73, 95% CI 0.55–0.99, P = 0.04). Although no dose‐response effect was found, the authors concluded that a concentration of Al in drinking water above 0.1 mg/L may be a risk factor for dementia and AD.
In addition, intriguing findings by Exley and Esiri (2006) have provided additional support for the Al‐AD hypothesis 34. The authors reported a rapidly progressive, fatal neurological illness a 58‐year‐old woman who, at autopsy, showed dramatic Aβ deposition of cerebral cortical and leptomeningeal blood vessels, modest numbers of NFTs and Lewy bodies, and evidence of very high Al content in affected brain regions 34. The deceased was amongst the 20,000 people who were accidentally exposed to exceedingly high concentrations of Al (approximately 500–3000 times the acceptable limit of 0.200 mg/L) in their water supplies in July 1988. Although this case alone cannot confirm the causative role of Al in the development of AD, it is certainly the first autopsy‐documented case of AD‐like condition potentially linked to Al exposure from drinking water, which warrants further investigations and surveillance of all affected individuals 37.
Several studies have purported to show lack of significant difference in AD in human populations exposed to high vs. low aluminum levels in their drinking water. Wettstein et al (1991) compared the cognitive skills between two groups of senile long‐term residents of Zurich. One group of subjects lived in an area with high Al concentration in drinking water (mean concentration <0.10 mg/L); the other group lived in the area with relatively low Al levels (mean <0.01 mg/L). The authors did not find any substantial difference in cognitive impairment between the two groups. However, the significance of these negative results might be limited by the fact that the study only tested for cognitive impairment of any kind and relied on only two sources of drinking water 151. Thus the bioavailability of Al was likely to be different in the two water sources. Likewise, Martyn et al (1997) found no association between the prevalence of AD and higher Al concentrations in drinking water when comparing 106 men with early onset AD 91. Also, it should be noted that this study was based on patients with early‐onset AD who are more affected by their genetic constitution than patients with sporadic AD. These results were similar to results of Forster DP et al (1995) whose study was also based on early‐onset AD patients. Early‐onset AD patients are more likely to contain mutations in their AbetaPP and/or presenilin genes, so they are differ from patients with sporadic AD by being more affected by their genetic constitution rather than by environmental influences in AD. The preponderance of these epidemiological studies has shown a positive relationship between risk for sporadic AD and levels of aluminum in public drinking water supplies.
Al intake from diet (epidemiological studies)
Even though most epidemiological studies have focused Al exposure from drinking water, Al exposure from food consumption is typically 10‐fold or greater than exposure from drinking water 120. Al in the food supply comes from natural sources, water used in food preparation, and food additives. Depending on several variables, the Al content of food can vary greatly and, in certain vegetables, Al may be adhering to the vegetable through the soil.
Dietary Al toxicity in healthy individuals in relation to AD has not been reported, primarily because of the difficulties, or even potential impossibility, for accurately assessing chronic dietary Al exposure, particularly where the subjects include AD patients. One pilot study conducted by Rogers and Simon (1999) in a geriatric center, employed 23 case‐control pairs. Close relatives were interviewed about dietary intake of the cases, with special attention to food high in Al such as American cheese, chocolate pudding, doughnuts, pancakes, or muffins. In the same study, resulting ORs were unstable and not statistically significant, except for one food category that contained waffles, pancakes, biscuits, muffins, corn bread or corn tortillas. Although interesting, this pilot study strongly suggested the need for additional confirmatory studies with larger sample size and more rigorous design. Other sources of dietary Al include tea, which contains particularly high levels of Al (300–2677 mg/kg), and may contribute up to 50% of the total daily Al intake in the countries with high tea consumption and small intake of Al from other sources 111, 156. Yet, several studies that explored the link between tea consumption and risk of AD were unsuccessful in providing evidence to support this hypothesis.
In a case‐control study comparing cases of clinically diagnosed presenile dementia of the Alzheimer's type and controls matched for age and sex, exposure to Al from tea was not a significant risk factor for dementia 42. Similarly, no relationship was found for exposure to Al from drinking water or medicinal sources in this investigation. A case control study completed in Canada produced a similar OR for the risk of AD; however, the serving amount of tea served was not specified 42.
Some foods, known as Al accumulators (eg, herbs and tea leaves) may naturally contain more than 5 mg/g of this metal. Intake of dietary Al is higher in the United States than in other countries, due to wide use of Al‐containing food additives 120, 143. A previously mentioned study by Rogers and Simon (1999) also found no significant relation between tea consumption and AD, which the authors explained by a possible loss of Al during the tea processing and binding of Al to organic compounds, possibly lemon juice. Thus, despite relatively high Al content and high dietary consumption, the role of tea in development of AD or similar pathologies remains controversial 120. Overall, the results of epidemiological investigations are promising and suggest an association between chronic exposure to Al and risk of AD. Although, to prove this hypothesis, meta‐analysis studies are needed. The significant contribution of food to Al exposure may mean that an omitted‐variable bias has been present in many previous studies that have only focused on Al in water and, if this is true, these studies might have significantly underestimated the relationship of Al to AD.
Recently Molloy et al (2007), conducted a double‐blind, randomized, controlled study. The authors could not find any significant differences in neuropsychological test scores after brief exposure to high Al ingestion (150 μg/L) in normal volunteers or in patients with cognitive impairment 101. The results did not support the hypothesis that Al ingested at these doses produces acute effects on cognition or adverse effects, nor did they reveal that AD patients are more vulnerable to such outcomes in the short‐term. It is possible that higher doses of Al and citrate over a much longer exposure period might produce subacute neuropsychological effects. The Al doses used resulted in serum Al levels exceeding presumed threshold values for neurotoxicity, and in the absence of measuring data to the contrary, the use of higher doses of Al or longer exposure of humans to neurotoxic serum Al levels in future studies would be limited by ethical considerations about possible long term implications of such exposures.
Al neurotoxicity—in vitro studies and in vivo studies (in experimental animals)
Hewitt et al (1991) explanted cores of subaqueductal midbrain nuclei and maintained on thin cubes of Gelfoam in the central well of an organ culture dish. Cultures were treated with 11, 13, and 15 μM/L concentration of Al maltolate for up to 24 days. Neurofilamentous aggregates were induced which were positive for the high and middle MW neurofilament subunits. No tau immunopositivity was seen but this must be placed in perspective, as many of the present day monoclonal antibodies were not available when this study was carried out 57.
In Griffioen et al (2004), studied whether in human teratocarcinoma (NT2) precursor cells, and found Al maltolate at a concentration of 500 μM caused significant cell death. Nuclear fragmentation suggestive of apoptosis was observed as early as 3 h and increased significantly through 24 h 54. TUNEL positive nuclei were also observed. The release of cytochrome c suggestive of mitochondrial injury was demonstrated by Western blot (WB) analysis. Savory et al (2006) studied the development of a neuronal culture system to evaluate the neurotoxic effects of Al maltolate on fetal rabbit midbrain sections containing the oculomotor nucleus 126.
Studies from our laboratory 64, 65, 67, 70, 71 on chronic Al exposure depicted a distribution of Al in different brain regions and body organs of male albino rats (wistar strain) weighing approximately 100–130 g. All animals received about 10 mg Al/kg bw/day, in the form of aluminum lactate dissolved in distilled water, intragastrically for 12 weeks. Distribution of Al in the different regions of brain (cerebral cortex, hippocampus, corpus striatum, cerebellum, and brain stem) revealed that, following subacute Al exposure, the maximum accumulation of Al occurred in the hippocampus followed by the corpus striatum, brain stem, cerebral cortex, and cerebellum whereas, following acute Al exposure, the maximum accumulation was in the corpus striatum followed by hippocampus, brain stem, cerebral cortex, and cerebellum 67, 71. With chronic 71 and subacute 67 exposure of aluminum to Wistar male rats, the maximum accumulation was in the hippocampus. There was almost a 24‐fold increase in aluminum accumulation in the hippocampus following chronic aluminum exposure and 80‐fold following subacute exposure as compared to control animals.
This result demonstrates the cumulative nature of Al in the hippocampus during subacute and chronic treatment. It can be reasoned, that excessive accumulation of Al in the hippocampus could result in loss of memory which is a characteristic feature of neurodegenerative diseases in which Al has been implicated.
Studies from our laboratory, on the subcellular localization of this metal ion, revealed that Al resides primarily in the nucleus of the cell 67. This finding suggests that Al may be more stably bound to the phosphate groups of DNA in the nuclear fraction rather than to the other cellular constituents. An appreciable amount of Al was also bound avidly to cytosolic, microsomes,and to mitochondria, which may be responsible for the inhibition of some of the mitochondrial functions such as cellular respiration, oxidative phosphorylation, and antioxidant enzyme activity 67.
Another study from our laboratory confirmed that maximum accumulation was in hippocampus, which exhibited more than 24‐fold increase in Al levels, followed by corpus stratum fivefold) and finally cerebral cortex fourfold). In the same study, the accumulation of neurofilaments which are fundamental units of NFTs in cerebral cortex, corpus striatum, and hippocampus was also observed. This high molecular weight neurofilament protein (200 kDa) was identified and confirmed by immunoblotting using the antineurofilament [H] as the primary antibody 71 (Figure 1). These results were consistent with results by Leterrier et al (1992) 83.
Figure 1.

(A) Immunofluorescence of high molecular weight neurofilament in cerebral cortex, (B) immunofluorescence of high molecular weight. neurofilaments in corpus striatum, (C) immunofluorescence of high molecular weight neurofilaments in hippocampus (Male albino rats (Wistar strain) exposed to 10 mg/(kg bw day), aluminum for a period of 12 weeks, smears were prepared from the three neuronal regions studied earlier, that is, cerebral cortex, corpus striatum, and hippocampus. These were initially stained with antineurofilament anti body followed by staining with FITC‐labeled secondary antibody. The slides were then observed under a fluorescence microscope) Toxicology 219 (2006) 1–10.
Savory et al (2003) employed Al maltolate for intravenous administration to adult rabbits for 8–30 weeks. Although no consistent neurological symptoms were observed in the Al‐treated animals, weight loss were noted. A slight increase in brain tissue Al concentration was seen by an electrothermal atomic absorption spectrophotometric technique 125. Bertholf et al (1989) observed neurofibrillary changes in the oculomotor nucleus in about one‐third of the treated rabbits, as demonstrated by a monoclonal antibody to the 200 kDa subunit of neurofilament protein 7. More distinct evidence of neurofibrillary changes has been detected by Forrester and Yokel (1989) in a comparative study of subcutaneous and intraventricular administration of Al lactate to adult New Zealand white rabbits. For the subcutaneous study, rabbits received daily subcutaneous injections of 400 μmol/kg Al lactate over a period of 4 weeks. Although no consistent clinical pattern of neurotoxicity was apparent, two of the animals displayed some evidence of motor abnormalities which included foot spread on landing, loss of the righting reflex and gait abnormalities. Neurofibrillary changes, detected by the Bielschowsky's silver method, were evident in the hippocampus and frontal cortex of the affected animals 41.
In a later report aimed specifically at relating Al neurotoxicity to age, Yokel used the subcutaneous and intraventricular routes of Al exposure in older (2–3.4 years) New Zealand white rabbits. Because the older animals were more susceptible to the higher dose (400 μmol/kg) of Al lactate used in the younger animals, the dose was reduced to 200 μmol/kg. Tissue Al concentrations in the frontal cortex and hippocampus were found to be significantly elevated even at this lower dose of Al 161.
Mawal‐Dewan et al (1996) reported NFTs, one of the histologic hallmarks of AD, using the rabbit model system and Al maltolate intracisternal injections, analyzed by light microscopy 95. Other pathological hallmarks of AD, such as neuritic plaques, are not observed in Al maltolate‐induced encephalopathy. Intriguingly, they found an abundance of NFTs coupled to a relative paucity of neuritic plaques characteristic of the ALS/parkinsonism‐dementia complex of Guam, suggesting that widespread NFTs (without Aβ plaques or significant Aβ deposition) indicate neurodegeneration 95. In AD, neuritic plaques often show a perivascular predilection, suggesting that dystrophic neurites and accompanying amyloidogenic accumulation may be pathogenically linked to vascular or microvascular associated factors. Cerebrovascular pathology may be relevant in the pathogenesis of naturally occurring AD, and may account for increased Al accumulation during central nervous system damage. Conversely, in experimental Al‐induced neurofibrillary degeneration, cerebrovascular pathology is not observed in the nonsenescent laboratory rabbit. In experimental neurodegeneration, the mode of Al delivery in the central nervous system is often direct, as with intracisternal, intraventricular, or intracerebral modes of administration, thereby, effectively bypassing the blood‐brain barrier. This may be related to the type and distribution of neurofibrillary degeneration in rabbits which, in some respects, differs from the more naturally occurring NFTs in AD in humans 94, 95. Furthermore, the formation of neuritic plaques appears to be species dependent, being found in few mammalian species and then, only in the aged 126. As described below, these differences are less compelling when compared to the broad array of immunochemical similarities which exist between experimental Al‐induced neurofibrillary degeneration in rabbits and the various neurofibrillary lesions of neurodegenerative disorders in humans. The relationship of Al‐induced neurofibrillary degeneration in rabbits to the neuritic pathology in human neurodegenerative disorders has been viewed with scepticism, because of the apparent ultrastructural differences between the experimental lesions in rabbits and NFTs in humans. Compared to the tangles of AD, which mostly contain paired helical filaments, those produced by Al are predominantly straight (intermediate‐like) filaments 74, 153, 154, 155.
Interestingly, tau has been observed to form Alzheimer's‐like filaments in vitro 26, 104. Al‐induced neurofibrillary degeneration is characterized predominantly by immunohistochemical staining with antibodies specific for epitopes of hyperphosphorylated neurofilament protein60, 144. The primary constituent of paired helical filaments in human NFTs is tau in association with abnormally phosphorylated neurofilament proteins, ubiquitin, α1‐antichymotrypsin, AβPP, and Aβ. Studies over the past several years 102, 135 have demonstrated that immunochemical similarities between the composition of the Al‐induced lesions and those found in AD are far closer than was originally surmised. Using a variety of mAbs that recognize both nonphosphorylated and phosphorylated tau, namely Tau‐1, Tau‐2, AT8, PHF‐1, and Alz‐50 102, 123, it has been observed that abnormally phosphorylated tau is present in these Al‐induced neurofilamentous aggregates 60. Savory et al (1996) reported the time course of aggregation of these cytoskeletal proteins. The results indicate that the aggregates may become detectable by silver staining as early as 24 h following Al maltolate administration and those neurofilament proteins predominate. Nonphosphorylated/phosphorylation‐independent epitopes, as detected by mAb SMI‐33, are found first, followed by the phosphorylated forms, immunostained by mAb SMI 31, at approximately 72 h. Tau is also detectable by around 72 h, although the characteristic epitopes of AD as recognized by mAbs AT8 and PHF‐1 are most distinct at 6–7 days following Al injection. It has been proposed that phosphorylation of cytoskeletal proteins drives the formation of the neurofilamentous aggregates, particularly in human neurodegenerative disorders 96. As these aggregates are hyperphosphorylated, phosphorylation alone would render these protein accumulations unstable, because of the preponderance of negative charges on the phosphate groups. Thus, it is reasonable to propose that some positively charged species constitute an inherent factor in the formation and stabilization of the neurofibrillary protein aggregates which are rich in phosphate groups and would have an extremely strong affinity for positively charged anions. This could occur in AD and in experimental Al‐induced neurofibrillary degeneration; in the latter, Al3+ is an obvious candidate for this role. Savory et al (1996) have shown some features of Al‐induced changes in rabbits using the intracisternal route of administration. This study demonstrates dramatic neurofibrillary changes using the Bielschowsky's silver impregnation technique on a frozen section of temporal cortex; these changes have many similarities at the light microscopic level to NFTs seen in humans. WB analysis of homogenized tissue is clearly positive for tau protein which is the major constituent of the NFTs seen in AD 123.
A recent report claimed that 100 µM Al maltolate induces tau aggregation in vitro (Mizoroki et al 2007), but lacks that same effect in vivo 100. Their research results provided evidence showing Al does not encourage the etiology of AD. Savory and Ghribi 119, 120, 121 and Exley (2007) criticized the methodology utilized by Mizoroki et al The response from Mizoroki et al has been to indicate that the absence of Al maltolate treatment is a proof for the claim that Al is not a causative agent in the etiology of AD 32. However, brain amyloidosis can be increased in AβPP transgenic mice through the use of dietary Al. Recently Mizoroki et al (2007) concluded that the failure of treatment of transgenic mice with Al maltolate to induce Alzheimer's‐like neuropathological changes provides proof that Al is not a causative agent in the etiology of AD. Dietary Al has been shown to increase brain amyloidosis in AβPP transgenic mice 114. As NFTs and neuritic plaques appear to be seen in only a few animal species including aged bears 23 and, of course, humans, it is not surprising that Al compounds in most species fail to reproduce the neuropathological and biochemical abnormalities seen in AD. Such a lack of an exact match in abnormal features does not rule out this metal ion as a contributing factor in the etiology of AD.
Al should certainly be high on the list of possible candidates for contributing to AD. First, it is highly abundant in the environment, being the most abundant metal in the Earth's crust. Second, it is extremely neurotoxic once it gains access to the central nervous system. Its proven toxicity in dialysis encephalopathy attests to this neurotoxicity, as do many experimental animal studies 126. Studies using rabbits may be particularly relevant to the investigation of human disease as they belong to the mammalian order Lagomorpha, according to an impressive 88 protein sequences 77. The Mizoroki et al (2007) experiments have not tested the hypothesis that a physiologically significant concentration of Al could influence the aggregation of tau in vivo. Therefore, it is unreasonable for the authors to conclude that “these results indicate that Al has no direct link to AD pathology” without conducting further studies to test this hypothesis 100.
Al exposure and neurobehavioral changes
Based on earlier reports from our laboratory 64, 65, 67, it has been shown that following chronic and subacute exposure to Al, male wistar rats showed maximum accumulation of Al in hippocampus. There were decreased number of ligand receptor binding sites (Bmax) observed in the hippocampus, cerebral cortex and corpus striatum of Al treated animals, with the effect being most pronounced in the hippocampus. From these reports, it can be concluded that hippocampus which is the seat of memory and learning is the most affected region of the brain. Decrease in the Bmax value suggests that there is a decrease in the number of receptors in hippocampus, implying that a decreased transmission of nerve impulses may be responsible for neurobehavioral alterations 64, 65, 67. A similar decrease in the number of receptors can be responsible for the state of dementia, as observed in patients with AD 92. Our data also revealed a significant retardation in the retention of the learned task in the Al‐exposed group compared to the control group. Hippocampal damage has been correlated with significantly decreased performance during memory function tests in experimental animals 64, 65, 67, 109, 113. The possibility that altered motor function could influence the changes in the memory tasks could be ruled out, as no significant changes in the spontaneous locomotor activity and rota rod tests were observed. Therefore, these tests can serve as an index of memory function.
A number of studies have been performed to evaluate the neurocognitive functions both in rodents as well as in humans, and many studies conclude that Al is potentially toxic to mammals even though Al neurotoxicity has been documented previously. This might be the case because neurobehavioral studies of Al in rodents have generally not produced consistent results. In one study testing neurocognitive function, rats exposed to Al were evaluated for motor activity in an open‐field apparatus as well as for learning in a passive avoidance test. Al exposure presented no significant effects on behavior of the rats, although, exposure did result in a decrease in the total numbers of synapses with age 30. Golub et al (1992) exposed adult mice to Al, using a diet high in Al content (1000 micrograms Al/g diet) for 90 days. Following exposure, a decrease in motor activity, grip strength, and startle responsiveness in the mice was observed 51. The behavioral and neurochemical effects of Al exposure on mice gestation and offspring have been long documented 3; and now, it is well established that an excess of Al during gestation and lactation, at levels below maternal toxicity, can result in persistent neurobehavioral deficits in the offspring of rats and mice 51, 139. No deleterious effects of Al ingestion were found in cognitive behavior when 22‐day old rats were given Al hydroxide for 60 days 139. However, studies on the neurobehavior effects of oral Al exposure in adult animals have shown contradictory results 11, 50. Similarly, no significant differences between control and Al‐treated rats were found in behavioral tests for short and long‐term memory 18. Although, on the contrary, Connor et al (1988, 1989) observed impaired performance of rats in a passive avoidance task after administration of Al sulfate in drinking water for 1 month. The passive avoidance conditioning test results were similar to previously reported findings 27, 28, with no significant effects of Al on this parameter. Recently, Li et al (2006) have reported cognitive dysfunction in mice exposed to Al chloride and suggested that an increase of dissociated Ca2+ in hippocampus neurons may be a possible mechanism for cognitive impairment 85.
Al induced symptoms mimic AD symptoms
The neurotoxic symptoms resulting from Al exposure include speech disturbances, dyspraxia, tremors, partial paralysis, and marked decline in learning and memory. Additionally, Al is also linked to other pathologies such as dialysis dementia, microcytic anemia, osteomalacia, and epilepsy 30. Our laboratory studies 66, 71 have also shown that Al exposure causes neuropathological (Figure 1), neurobehavioral, and neurochemical changes resulting in impaired learning ability.
Mounting evidence in recent years has suggested that Al has severe toxic manifestations on the central nervous system. Al causes changes in acetylcholinesterase activity from acute and subacute exposure as well as significant deterioration in cognitive functions in rats 21, 66, 78, 108, 166. Observation of the central cholinergic system after Al exposure, of 10 mg/kg of body weight/day for 4 weeks, revealed a deleterious effect on the activities of biosynthetic (choline acetyltransferase) and hydrolytic (acetylcholinesterase) enzymes of the neurotransmitter acetylcholine. Following the dose regimen, the levels of acetylcholine were significantly lowered in different brain regions and there was a significant decrease in high‐affinity choline uptake. Muscarinic acetylcholine receptor binding studies have revealed a decreased number of binding sites, with the maximum effect manifesting in the hippocampus. Exogenous addition of 10 μM desferrioxamine restored the muscarinic receptor binding completely. Impaired cholinergic functioning results in severe effects on cognitive functions, manifesting as neurobehavioral deficits shown by decreased performance in active (52%) and passive (73.30%) avoidance tests. The neurobehavioral deficits observed in these studies suggest that Al is toxic and is altering cholinergic neurotransmission 66.
Recently, Ribes et al (2008) have indicated that chronic low level exposure to Al lactate may lead to disrupted spatial learning and neurogenesis in a transgenic mouse model of AD. In this study, from 5 months age onwards they fed wild type and APP‐Tg 2576 mice with diets supplemented with Al lactate at 0 and 1 mg/g of diet for 120 days. The activity in an open‐field and learning in a water maze were evaluated after 3 months of Al exposure. Transgenic mice presented with impairments to both acquisition and retention of the water maze task. These mice also showed higher amounts of Aβ fragments in the brain. Al exposure impaired learning and memory in mice and increased the total number of proliferating cells in the dentate gyrus of hippocampus. The authors suggested that low Al doses might impair cognition in the general population at doses comparable to current levels of human exposure 115.
Another chronic study with Al gluconate showed behavioral alterations such as disturbed exploratory behavior, catatonic behavior, and emotional reactivity alterations as well as brain modifications such as decrease in neuronal density in the parietal and temporal cortex andinvaginated nuclei with intense eosinophilic cytoplasm in the hippocampus, temporal, and parietal cortex. Experimental adult rats that exhibited significantly lower performance scores in the shuttle‐box task along with lower and fluctuating performance scores indicative of impaired spatial memory in the Morris water maze were found to have hippocampal neurons and cortical neurons with chromatin clumping and vacuolations in the nucleus and cytoplasm 98. The Miu authors have also demonstrated decreased scores of activity and emotionality in Al exposed, aging animals along with cellular and ultrastructural degenerative signs by electron‐microscopic analysis 99. Jing et al (2004) performed similar experiments that corroborated the conclusion that rats exposed to Al present learning and memory impairments 63.
The reports of neurocognitive dysfunctions have also been published on humans. The relationship between an elevated body Al burden and central nervous system function was studied in 25 current mild steel welders and 65 aluminum welders and body burden was estimated. They analyzed Al concentrations both in serum (S‐Al) as well as in urine (U‐Al) with graphite furnace atomic absorption spectrometry with Zeeman background correction. Referents, low‐exposure and high‐exposure groups were defined according to an aggregated measure of aluminum body burden, the group median S‐A1 levels being 0.08, 0.14, and 0.46 µmol/L, respectively, and the corresponding values for U‐A1 being 0.4, 1.8, and 7.1 µmol/L 119. The subjects presented symptoms such as increase in fatigue, mild depression, and memory and concentration problems. Neuropsychological testing revealed adverse effects of Al in attention related tasks and the working memory system. Giorgianni et al (2003) also studied cognitive disorders among welders exposed to Al and concluded that workers exposed to Al presented impairment in memory and concentration 49. Some other epidemiological studies also showed poorer performance in cognitive tests or a higher level of neurological symptoms for workers occupationally exposed to Al 117, 136. In another study, Kiesswetter et al (2009) performed a longitudinal study, over 4 years, on potential neurotoxic effects of Al and assessed the association of Al exposure and neurobehavioral performance of Al welders in the train and truck construction industry 75. The biomonitoring data of the welders showed a high long‐term stability but also presented with sensitivity to acute shift dependent exposure changes. Al welders working in this profession an average of 15 years showed no significantly increased symptom levels compared to the control groupExplorative regression and covariance analyses revealed neither a correlation between biomonitoring and performance variables nor a significant difference between Al exposed and controls in the performance courses during the 4 year period 75. In their first longitudinalstudies, recorded neurobehavioral health and Al exposure of welders in the train and truck construction industry over 4 years, with intermediate results after 2 years 15, that showed that welders in this industrial sector were exposed to considerably higher levels of Al (90–150 lg Al/g creatinine in urine) than welders of a parallel study in the automobile industry (30–40 lg Al/g creatinine in urine) 76.
Recently, Lu et al (2014) studied Tau‐protein expression and cognitive disorders in sixty six 88 retired Al smelting workers. The cognitive functions were assessed with the Mini Mental State Examination. The tau‐protein expression in peripheral blood lymphocytes was analyzed with western blots. The cognitive functions of the exposed group were significantly decreased. Twelve mild cognitive impairment cases in the exposed group and four mild cognitive impairment cases in the control group were diagnosed. Significantly higher p‐tau181 and p‐tau231 levels were detected in the Al‐exposed workers than in the control. They suggested, that long‐term exposure to Al may cause cognitive disorders 88.
Many, but not all, publications to date have shown cognitive impairments and/or neurobehavioral changes following Al exposure. While many studies reported neurobehavioral changes, other studies not showing lack of Al exposure‐related deterioration of performance has led some to question the diversity of results 2, 15, 61, 75, 76. Taken together, these conflicting findings on learning impairments may be attributed to the differences in duration of dose and route of exposure. More studies of occupational Al exposure need to be conducted with a broader range of concentrations (doses), different routes, and varying durations of exposure to Al to corroborate the conditions needed to demonstrate Al neurotoxicity in humans. Studies that show a positive effect of Al on cognitive performances should be explained by studies that promote a negative findings, just as positive studies attempts to explain the occasional study with negative results.
Mechanism of Al Toxicity in Relation to AD
The exact mechanism by which Al crosses the blood brain barrier (BBB) is unknown. Most of the studies that have examined the effects of acute injections of Al have reported that it does not disrupt the BBB or alter cerebral blood flow. However, because Al and iron possess similar properties, Al is thought to cross the BBB using the same mechanism as iron, namely in transferrin receptor 68.
Additionally, Al has been shown to increase the rate of transmembrane diffusion through the BBB and to selectively change saturable transport systems without disrupting the integrity of the membranes or altering the hemodynamics of the central nervous system and causing pathological features of AD. Hence, to understand the toxicity of Al, identification of an appropriate Al compound which can easily diffuse through BBB is immensely important. The electroneutral Al‐maltolate appears ideal as this compound can deliver a significant amount of free aqueous Al at physiological pH. Unlike most other Al salts, such as AlCl3, it produces insoluble complexes at neutral pH. Al‐maltolate increases the soluble Al concentration from 4–6 mM compared to other organic Al salts like Al‐lactate or Al‐aspartate (soluble Al concentration is ∼55–330 μM). Al‐maltolate is soluble and stable from pH 3.0 to 10.0, while hydrolytically stable at **pH 7.0, and has no speciation chemistry problems. Al‐maltolate is suitable over other Al compounds because of its very high metal solubility at pH 7.0 and prominent kinetic restrictions to ligand exchange reactions in neutral solution. Based on our own contributions, Bharathi et al (2003), Liang et al (2012), Kaneko et al (2006), Wang et al (2014), and those of Savory et al (1996, 1999, 2003, 2006), it is understood that Al‐maltolate is a suitable compound for toxicological and neuropathological studies in relevance to AD 8, 69, 87, 123, 124, 125, 126, 127, 150.
Al is a neurotoxic element and there are various proposed mechanisms for its neurotoxicity including: impaired glucose metabolism, free radical damage via increased lipid peroxidation, protein modification, consequences on signal transduction, disturbances in the axonal transport, and altered phosphorylation of neurofilaments 71. Some studies have reported that Al kills individual neurons secondary to intracellular accumulation and that cell death is not secondary to the generation of reactive oxygen species nor the accumulation of intracellular calcium even though Al causes an elevation in both 66, 71. Several studies have explored the role of Al induced oxidative stress via free radical generation in the early onset or evolution of AD in transgenic models. The most notable feature in the oxidative stress hypothesis for AD and other neurodegenerative diseases is that cumulative oxidative damage over time could account for the late life onset and slowly progressive nature of AD and Parkinson's disease (PD) 24.
Evidence for increased oxidative stress in AD includes: increased levels of elements capable of initiating free radical formation such as Al, mercury, and iron in brain 17; increased protein oxidation; increased DNA oxidation; increased lipid peroxidation and decreased polyunsaturated fatty acids in AD brain; and the ability of Aβ to generate free radicals 17. Pratico et al (2002) reported that chronic dietary administration of Al can increase Aβ levels and accelerate plaque deposition in an animal model of AD‐like amyloidosis. Increase in the levels of specific and sensitive markers of oxidative stress in vivo correlated with increased levels of Aβ and was consistent with an exacerbation of brain lipid peroxidation 114.
Al promotes phosphorylation of the tau protein 133, which results in it being less soluble, increasing the likelihood that the protein will aggregate. Tau protein is the microtubular associated protein that accumulates in NFTs. By targeting tau, Al may act to promote the development of NFTs. This result has led some to suggest that AD is a result of hypoactive phosphatases and overactive kinases 47, 122. There is also evidence that Al may alter the dynamics of Aβ 4. The core center of amyloid plaques is known to contain an overabundance of Aβ42, which is less soluble than the more abundant Aβ40. This alone suggests the possibility that abnormal cleavage of AβPP may play a role in the development of AD. However, there is now clear evidence that when Al complexes with Aβ42, it reduces solubility, increases precipitation of β‐sheets, and facilitates Aβ flux across the BBB 31.
In spite of all of the evidence for a central role of Aβ (especially from genetic studies) and tau protein in AD, there remains some reason to question whether the abnormalities in these proteins are causative of the disease or are only reflections of the true underlying etiologic factor 107. Bancher et al (1997) showed that while neurons in physical contact with an amyloid deposit or containing a NFT were more likely to die than those neurons which did not, most dying cells were not located within amyloid deposits and most dying cells did not bear a tangle, nor were they near to a neuron with a tangle. Although the precipitated forms of Aβ and tau protein may not be physiologically active, there certainly remains the possibility that circulating or intracellular forms of these proteins, perhaps forming complexes with Al, are toxic, but other primary causes of neuronal cell death cannot be excluded 4.
Role of Al on AβPP processing in the formation of Aβ
Another finding in Al‐induced neurodegeneration has been the observation of increased levels of AβPP and Aβ in neurons of Al treated rats 132 and rabbits 60. NFTs in AD also exhibit this Aβ staining but the most prevalent pattern is the presence of Aβ in the neuritic plaques. It is logical to hypothesize that in AD, increased Aβ initially appears intracellularly, followed by extracellular deposition after cell death. Other works 59, 110, 164 have described an Al‐induced secondary structural transition in the non‐Aβ component of AD amyloid (NACP), generating approximately a 33% α‐helix, which renders NACP resistant to proteases. Based on this finding, it is suggested that Al may influence NACP protein turnover and induce aggregation via structural modifications thus leading to Aβ deposition 113. A microprobe method for detecting Al within plaques would have to be extremely sensitive. This required sensitivity is because Aβ has a molecular weight of 5000 kDa and Al(III) is trivalent (Al3+) with an atomic weight of 27. This means it is possible that the ratio of Aβ to Al on a weight to weight basis could be 550:1.
Candy et al (1986) reported the presence of Al within plaques in the brain of an AD patient 16. Landsberg et al (1992) failed to confirm this finding with the alternative microanalytical technique of particle‐induced X‐ray emission (PIXE) using a nuclear microscopy; however, this nuclear microscopic technique is relatively insensitive below 15 μg/g of Al, making the significance of the report questionable 82. The nuclear microscope has yet to be validated for Al detection in biological tissue.
While the formation of NFTs is most likely a product and not a cause of neuronal death, Aβ, the principal component of the senile plaques (SP) characteristic of AD, has been shown to be neurotoxic, possibly because it traps excess toxic metal ions. Al is a constituent of SP and influences the aggregation and toxicity of Aβ 36. If Aβ is involved in neuronal cell death in AD, then the presence of Al in brain interstitial fluid (BIF) could mediate its action. This mediation may even involve Al‐ATP which has been shown to promote amyloidosis of Aβ. It is not clear whether Al is also involved in the secretion into BIF of superphysiological concentrations of Aβ following cleavage of its precursor protein (AβPP) by a β‐secretase. However, Al has been shown to promote aggregation of Aβ in vitro and accumulation of AβPP in neurons and glia 36.
The function of AβPP is not fully understood, and it has been shown to be neuroprotective. When cleaved by α‐secretase AβPP releases a soluble fragment, sAβPP, which suppresses elevation of intracellular Ca2+ by reducing glutamate‐induced NMDA currents 36. The secretion of sAβPP is regulated glutamaterigically through phosphatidyl‐inositol‐linked receptors. The potentiation of neuronal excitotoxicity by Al‐ATP would be expected to result in the increased secretion of sAβPP. However, the co‐localization of Al and AβPP in the Golgi complex and irreversible accumulation of AβPP induced by Al in neurons and glia 36 could suggest that AβPP binding of intraneuronal Al may prevent the release of sAβPP and, therefore, the amelioration of excitotoxicity. This could also be indirect evidence of an influence of Al on AβPP processing 84, 89.
The presence of Al in the cores of senile plaques has been controversial for a long time 17, 31, 62, 140. One of the reasons in these studies may be that Al in amyloid fibers, in the core, could not be stained histochemically while Al in the NFTs and nuclei of nerve cells in the AD brain is stained clearly by histochemistry 104, 134, 145. If Al is lightly dispersed in a tissue or is present in small amounts, the conditions are less than favorable for Al staining at the level of light microscopy. It seems likely that Al does not attach to the surface of amyloid fibers in the core after the aggregation of Aβ peptides but instead, that Al binds just before, and during, the course of Aβ peptide aggregation. It is widely believed that amyloid‐Al complex are more toxic than Al or amyloid on their own and consequently play a key role in the etiology of AD 29. However, Aβ plaques are not necessarily toxic, given that Wujek JR et al (1996) reported that cells can readily grow on amyloid plaque removed from AD brain tissue 159. Amyloid fibers in the core have been reported to consist mainly of the aggregates of Aβ peptides that have a beta‐sheet structure 129, 141. Recently Yumoto et al (2009) examined the presence of Al at autopsy of five AD patients using energy‐dispersive X‐ray spectroscopy combined with transmission electron microscopy (TEM‐EDX). TEM‐EDX analysis allows simultaneous imaging of subcellular structures with high spatial resolution and analysis of small quantities of elements contained in the same subcellular structures. The Yumoto study, demonstrated colocalization of Al and Aβ peptides in amyloid fibers in the cores of senile plaques 165.
The results support the following possibilities in the brains of patients with AD: there is evidence for Al could be involved in the aggregation of Aβ peptides to form toxic fibrils 31. Al induction of Aβ peptides into the beta‐sheet structure 35; and Al might facilitate iron‐mediated oxidative reactions, which cause severe damage to brain tissues 10, 55. First, Al aggregation of Aβ peptides in vivo is probable because Al (Al3+) is a trivalent cation, Al can interact with acidic groups of the peptides and bind these peptides to one another. It has been reported that Al ions promote aggregation of physiological concentrations of Aβ peptides in vitro 89. House et al (2004) and Khan et al (2005) reported that Al ions accelerate the formation of amyloid fibrils in vitro 58, 73. Second, Al causes the conformational change of Aβ peptides into the beta‐sheet structure in vivo. Ricchelli et al (2005) reported that Al induces structural modification of Aβ peptides enriched in beta‐sheet conformations in vitro 116. Exley (2006) pointed out that only Al and Iron (Fe), but not copper (Cu) or zinc (Zn), are involved in the formation of the beta‐sheet structure of Aβ peptides 31. It has been proposed that Aβ peptides containing the beta‐sheet structure are directly incorporated into membranes, forming Ca‐permeable ion channels, and causing elevation of intracellular calcium levels that lead to nerve cell death. Third, Al promotes oxidative injury to brain tissues both on its own and by facilitating Fe‐mediated oxidation reactions in the AD brain 31. Fe is involved in the formation of free hydroxyl radicals via Fenton chemistry, which may cause deleterious effects in brain tissues 31. It has been reported that Fe accumulates in the cores of senile plaques and that deposition of Aβ peptides facilitates the generation of reactive oxygen species in the presence of Fe in vitro 19, 20, 22, 137. Xie et al (1996) reported that Al can facilitate Fe‐mediated oxidative injury in cultured neurons 160. In addition, Khan et al (2005) reported that Al potentiates the Fe(II)/Fe(III) redox cycle in favor of Fe(II) in vitro 73. These authors suggested that oxidative damage in the vicinity of senile plaques may be the result of Fenton reactions catalyzed by the co‐deposition of Aβ peptides with Fe and Al. Based on these studies, it can be concluded that Al is linked to the generation of Aβ neurotoxic species.
Al inducesapoptosis, mitochondrial dysfunction, and endoplasmic reticulum stress
The hallmark of most neurodegenerative diseases is loss of neurons and there is mounting evidence that indicates apoptosis may be a significant mechanism for neuronal death in neurodegenerative diseases. Recently, our laboratory results have shown that chronic Al exposure increases ROS generation which is implicated in the impairment of mitochondrial functions in various regions of rat brain 79. All animals received Al about 10 mg/kg bw day, in the form of aluminum lactate dissolved in distilled water, intragastrically for 12 weeks. Following chronic Al exposure, there was significant decrease in the activity of mitochondrial electron transport chain complexes, resulting in reduced ATP synthesis. The levels of various mitochondrial cytochromes were also decreased. Additionally, there was increased oxidative damage to mitochondrial proteins, revealed by increased protein carbonylation and decrease in the activity of mitochondrial aconitase 79. This oxidative damage to mitochondria was reflected morphologically in electron microscopic studies which revealed changes in mitochondrial shape and loss of cristae 80, 81. Savory et al (1999) reported that aging is an important factor in the susceptibility of neurons to oxidative stress and subsequent apoptosis; both processes are observed in AD brain 127.
Mitochondrial dysfunction has been implicated in many neurodegenerative diseases; namely, ischemia‐reperfusion injury, aging, and inflammatory diseases 152. Mitochondria are more prone to reactive oxygen species (ROS) induced cellular injury 86 as they lack protective proteins. Earlier reports from our lab have shown that chronic Al exposure results in decreased activity of cytochrome oxidase (COX) complexes I, II, and IV of the electron transport chain and to excessive production of ROS 79. Electron transport chain (ETC) dysfunction may ensue from mitochondrial damage including oxidation of mitochondrial DNA, proteins, and lipids, in turn leading to the opening of the mitochondrial permeability transition pore (MTP), an event associated with neuronal cell death 56. Enhanced ROS production and oxidative injury have been shown to play a cardinal role in the onset and progression of neurodegenerative disorders 46, 48, 128, 146. Defects in mitochondrial respiratory enzymes have been reported in several neurodegenerative diseases. Decreased complex I activity is reported in the substantia nigra of post‐mortem samples obtained from neurodegenerative disease patients 128. Decreased mRNA levels of the mitochondrial‐encoded COX subunits I, II, and III have also been known to occur in brains of AD patients 131. Reduced expression of nuclear encoded subunits of mitochondrial enzymes of oxidative phosphorylation (OXPHOS), including subunit IV of COX and the β‐subunit of the F1F0‐ATP synthase is also observed in vulnerable areas of AD brains 13.
Mitochondrial changes following cytotoxic stimuli represent a primary event in apoptotic cell death. Al has been demonstrated to accumulate in neurons following cell depolarization. Al inhibits Na+/Ca2+ exchange and thereby induces an excessive accumulation of mitochondrial Ca2+ 138 which leads to opening of the mitochondrial permeability transition pore (MTP), with cytochrome c release and, subsequently, apoptosis by activation of the caspase family of proteases. Release of cytochrome c from the mitochondria into the cytoplasm has been shown to involve three distinct pathways. One implicates the opening of the MTP, the second involves translocation of the proapoptogenic Bax to mitochondria which can form a channel by itself, and the third apparently results from the interaction of Bax with the voltage‐dependent anion channel (VDAC) to form a larger channel which is permeable to cytochrome c. On the contrary, the antiapoptotic regulator, Bcl‐2, has the ability to block the release of cytochrome c from mitochondria by mechanisms such as a direct blockade of the MTP opening or by functioning as a docking protein 1. Al induced cytochrome c release is markedly reduced by cyclosporin A, a specific inhibitor of the MTP opening, and implicates opening of the MTP as the process by which cytochrome c translocates from mitochondria to the cytoplasm 1.
The endoplasmic reticulum (ER) is a multifaceted organelle that regulates protein synthesis, protein folding and trafficking, cellular responses to stress, and intracellular Ca2+ levels 93. Disruption of Ca2+ homeostasis, inhibition of protein N‐linked glycosylation, expression of mutant proteins, and various other types of cellular stresses cause accumulation of misfolded proteins in the ER lumen, resulting in ER stress 97. Al maltolate and Aβ42 induce stress in the ER of rabbit brain. In response to the induced stress, the ER‐resident protein, gadd 153, and the inducible transcriptional factor, NF‐kB, are both translocated into the nucleus. Furthermore, this ER stress elicits an adaptive response triggering a signaling pathway called the unfolded protein response (UPR), which alleviates stress by induction of ER‐localized chaperones, initiation of a degradation system, and attenuation of protein synthesis. However, if the stress is prolonged, it may lead to processing of the ER‐resident protease, procaspase‐12, as well as activation of calpain, caspase‐3, caspase‐6, and apoptosis. Although mitochondrial apoptotic signals are important in regulating apoptosis, the ER and nuclear organelles may also participate in the molecular mechanisms of apoptosis following neurotoxic stimuli. Recent evidence suggests that ER stress‐mediated apoptotic signal pathways may contribute to the pathogenesis of AD 105. However, the precise molecular mechanisms underlying mitochondrial dysfunction and ER stress in neuronal cell death and neurodegeneration remain unknown.
Al appears to have a significant role in the induction of apoptosis, mitochondrial dysfunction, and ER stress in neurons and this induction is an important step to understand while moving toward AD neurodegeneration and pathogenesis.
Concluding Remarks
Plausible explanations for chronic Al intake as the cause of AD have been presented.
Many studies have revealed the neurotoxic role of Al in spatial learning, long‐term potentiation and unaltered neurogenesis mechanisms, cellular respiration mechanisms through ETC chain and TCA cycle, cell death mechanisms through apoptosis, inflammatory response through NF‐κB mechanism, antioxidant activity through SOD, and, of particular note in this review, in the formation of the hallmarks of AD such as senile plaques and NFT formation (Figures 2 and 3). In most of the studies reviewed here, the routes of Al exposure were through intragastric, oral, intracisternal, intraventricular, or intracerebral modes of administration and are effectively bypassed the blood‐brain barrier (BBB). Oral studies are very good because they mimic human Al exposure risk, but because only a limited amount of Al is absorbed, it necessary to have a much longer exposure periods for Al to accumulate in neurons to toxic amounts. AD has a long prodromal phase, with pathologic changes often preceding the onset of clinical symptoms by more than a decade. High Al absorption may someday be recognized as a risk factor for AD. Humans usually are exposed to Al neurotoxicity either through the skin or through food additives. For this reason, it is crucial to do more research on the bio‐availability of this metal and its effectiveness in crossing the gastrointestinal and the blood brain barriers, which could show prime cellular changes in the pathogenesis of AD.
Figure 2.
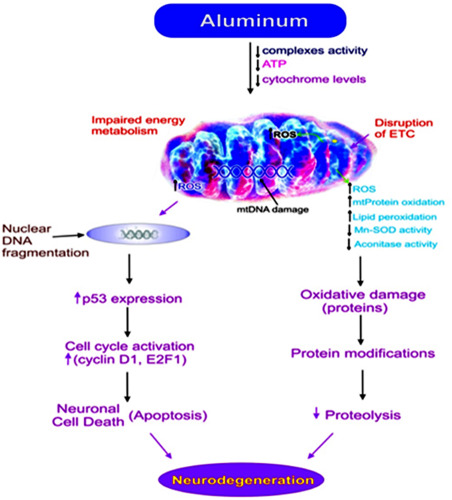
Possible Mechanisms for the role of Aluminum in the pathogenesis of Neurodegeneration via mitochondria (mt).
Figure 3.

Hypothetical model for aluminum neurotoxicity in neurodegeneration—depicts a general overview of damage and disrupted regulatory pathways leading to AD.
Acknowledgment
Work in the authors' laboratories was supported in part by Indian Council of Medical Research (ICMR), New Delhi.
References
- 1. Adams JM, Cory S (1998) The Bcl‐2 protein family: arbiters of cell survival. Science (New York, NY) 281:1322–1326. [DOI] [PubMed] [Google Scholar]
- 2. Akila R, Stollery BT, Riihimaki V (1999) Decrements in cognitive performance in metal inert gas welders exposed to aluminium. Occup Environ Med 56:632–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Alleva E, Rankin J, Santucci D (1998) Neurobehavioral alteration in rodents following developmental exposure to aluminum. Toxicol Ind Health 14:209–221. [DOI] [PubMed] [Google Scholar]
- 4. Bancher C, Lassmann H, Breitschopf H, Jellinger KA (1997) Mechanisms of cell death in Alzheimer's disease. J Neural Transm Suppl 50:141–152. [DOI] [PubMed] [Google Scholar]
- 5. Banks WA, Jaeger LB, Urayama A, Kumar VB, Hileman SM, Gaskin FS et al (2006) Preproenkephalin targeted antisenses cross the blood‐brain barrier to reduce brain methionine enkephalin levels and increase voluntary ethanol drinking. Peptides 27:784–796. [DOI] [PubMed] [Google Scholar]
- 6. Becaria A, Campbell A, Bondy SC (2002) Aluminum as a toxicant. Toxicol Ind Health 18:309–320. [DOI] [PubMed] [Google Scholar]
- 7. Bertholf RL, Herman MM, Savory J, Carpenter RM, Sturgill BC, Katsetos CD et al (1989) A long‐term intravenous model of aluminum maltol toxicity in rabbits: tissue distribution, hepatic, renal, and neuronal cytoskeletal changes associated with systemic exposure. Toxicol Appl Pharmacol 98:58–74. [DOI] [PubMed] [Google Scholar]
- 8. Bharathi, Jagannatha Rao KS, Stein R (2003) First evidence on induced topological changes in supercoiled DNA by an aluminium D‐aspartate complex. J Biol Inorg Chem 8:823–830. [DOI] [PubMed] [Google Scholar]
- 9. Bondy SC (2014) Prolonged exposure to low levels of aluminum leads to changes associated with brain aging and neurodegeneration. Toxicology 315:1–7. [DOI] [PubMed] [Google Scholar]
- 10. Bondy SC, Guo‐Ross SX, Pien J (1998) Mechanisms underlying the aluminum‐induced potentiation of the pro‐oxidant properties of transition metals. Neurotoxicology 19:65–71. [PubMed] [Google Scholar]
- 11. Brining SK, Jones CR, Chang MC (1996) Effects of chronic beta‐amyloid treatment on fatty acid incorporation into rat brain. Neurobiol Aging 17:301–309. [DOI] [PubMed] [Google Scholar]
- 12. Brooks WM, Lynch PJ, Ingle CC, Hatton A, Emson PC, Faull RL et al (2007) Gene expression profiles of metabolic enzyme transcripts in Alzheimer's disease. Brain Res 1127:127–135. [DOI] [PubMed] [Google Scholar]
- 13. Bu G (2009) Apolipoprotein E and its receptors in Alzheimer's disease: pathways, pathogenesis and therapy. Nat Rev Neurosci 10:333–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Buchta M, Kiesswetter E, Otto A, Schaller KH, Seeber A, Hilla W, Windorfer K et al (2003) Longitudinal study examining the neurotoxicity of occupational exposure to aluminium‐containing welding fumes. Int Arch Occup Environ Health 76:539–548. [DOI] [PubMed] [Google Scholar]
- 15. Campbell A (2002) The potential role of aluminium in Alzheimer's disease. Nephrol, Dial, Transplant 17(Suppl. 2):17–20. [DOI] [PubMed] [Google Scholar]
- 16. Candy JM, Oakley AE, Klinowski J, Carpenter TA, Perry RH, Atack JR et al (1986) Aluminosilicates and senile plaque formation in Alzheimer's disease. Lancet (London, England) 1:354–357. [DOI] [PubMed] [Google Scholar]
- 17. Castellani RJ, Moreira PI, Liu G, Dobson J, Perry G, Smith MA et al (2007) Iron: the Redox‐active center of oxidative stress in Alzheimer disease. Neurochem Res 3:1640–1645. [DOI] [PubMed] [Google Scholar]
- 18. Clauberg M, Joshi JG (1992) Effects of prolonged chronic dietary treatment with AlCl3 on selected metabolisms and memory in rats. Ann New York Acad Sci 648:289–290. [DOI] [PubMed] [Google Scholar]
- 19. Collingwood JF, Chong RK, Kasama T, Cervera‐Gontard L, Dunin‐Borkowski RE, Perry G et al (2008) Three‐dimensional tomographic imaging and characterization of iron compounds within Alzheimer's plaque core material. J Alzheimer's Dis 14:235–245. [DOI] [PubMed] [Google Scholar]
- 20. Connor DJ, Harrell LE, Jope RS (1989) Reversal of an aluminum‐induced behavioral deficit by administration of deferoxamine. Behav Neurosci 103:779–783. [DOI] [PubMed] [Google Scholar]
- 21. Connor DJ, Jope RS, Harrell LE (1988) Chronic, oral aluminum administration to rats: cognition and cholinergic parameters. Pharmacol, Biochem, Behav 31:467–474. [DOI] [PubMed] [Google Scholar]
- 22. Cork LC, Powers RE, Selkoe DJ, Davies P, Geyer JJ, Price DL (1988) Neurofibrillary tangles and senile plaques in aged bears. J Neuropathol Exp Neurol 47:629–641. [DOI] [PubMed] [Google Scholar]
- 23. Coyle JT, Puttfarcken P (1993) Oxidative stress, glutamate, and neurodegenerative disorders. Science (New York, NY) 262:689–695. [DOI] [PubMed] [Google Scholar]
- 24. Crapper DR, Krishnan SS, Dalton AJ (1973) Brain aluminum distribution in Alzheimer's disease and experimental neurofibrillary degeneration. Science (New York, NY) 180:511–513. [DOI] [PubMed] [Google Scholar]
- 25. Crowther RA, Olesen OF, Smith MJ, Jakes R, Goedert M (1994) Assembly of Alzheimer‐like filaments from full‐length tau protein. FEBS Lett 337:135–138. [DOI] [PubMed] [Google Scholar]
- 26. Dollken (1897) Ueber die Wirkung des Al mit besonderer Besucksichtigung der durch das Al verursachten Lasionen im Zentralenervensystem. Arch Exp Pathol Pharmacol 40:98–120. [Google Scholar]
- 27. Domingo JL (1995) Adverse effects of potential agents for the treatment of Alzheimer's disease: a review. Adverse Drug Reactions Toxicol Rev 14:101–115. [PubMed] [Google Scholar]
- 28. Domingo JL (1995) Reproductive and developmental toxicity of aluminum: a review. Neurotoxicol Teratol 17:515–521. [DOI] [PubMed] [Google Scholar]
- 29. Drago D, Bettella M, Bolognin S, Cendron L, Scancar J, Milacic R et al (2008) Potential pathogenic role of beta‐amyloid(1‐42)‐aluminum complex in Alzheimer's disease. Int J Biochem Cell Biol 40:731–746. [DOI] [PubMed] [Google Scholar]
- 30. Edwardson JA, Klinowski J, Oakley AE, Perry RH, Candy JM (1986) Aluminosilicates and the ageing brain: implications for the pathogenesis of Alzheimer's disease. Ciba Found Symp 121:160–179. [DOI] [PubMed] [Google Scholar]
- 31. Exley C (2006) Aluminium and iron, but neither copper nor zinc, are key to the precipitation of beta‐sheets of Abeta_{42} in senile plaque cores in Alzheimer's disease. J Alzheimer's Dis 10:173–177. [DOI] [PubMed] [Google Scholar]
- 32. Exley C (2007) Aluminium, tau and Alzheimer's disease. J Alzheimer's Dis 12:313–315; author reply 7–8. [DOI] [PubMed] [Google Scholar]
- 33. Exley C (2013) Human exposure to aluminium. Environ Sci Process Impacts 15:1807–1816. [DOI] [PubMed] [Google Scholar]
- 34. Exley C, Esiri MM (2006) Severe cerebral congophilic angiopathy coincident with increased brain aluminium in a resident of Camelford, Cornwall, UK. J Neurol, Neurosurg, Psychiatr 77:877–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Exley C, Price NC, Kelly SM, Birchall JD (1993) An interaction of beta‐amyloid with aluminium in vitro. FEBS Lett 324:293–295. [DOI] [PubMed] [Google Scholar]
- 36. Exley C (1999) A molecular mechanism of aluminium‐induced Alzheimer's disease? J Inorg Biochem 76:133–140. [DOI] [PubMed] [Google Scholar]
- 37. Flaten TP (2001) Aluminium as a risk factor in Alzheimer's disease, with emphasis on drinking water. Brain Res Bull 55:187–196. [DOI] [PubMed] [Google Scholar]
- 38. Forbes WF, Gentleman JF, Park E (1995) Dementia and age at death. Exp Gerontol 30:445–453. [DOI] [PubMed] [Google Scholar]
- 39. Forbes WF LS, Gentleman JF (1995) Geochemical risk factors for mental functioning, based on the Ontario Longitudinal Study of Agigng (LSA) V. Comparisons of the results, relavant to Al water concentrations, obtained from the LSA and from death certificates mentioning dementia. Can J Aging 14:642–656. [Google Scholar]
- 40. Forbes WF MC, Hayward LM, Agwani (1994) Geochemical risk factors for mental functioning, based on the Ontario Longitudinal Study of Agigng (LSA) II. The role of pH. Can J Aging 13:249–267. [Google Scholar]
- 41. Forrester TM, Yokel RA (1985) Comparative toxicity of intracerebroventricular and subcutaneous aluminum in the rabbit. Neurotoxicology 6:71–80. [PubMed] [Google Scholar]
- 42. Forster DP, Newens AJ, Kay DW, Edwardson JA (1995) Risk factors in clinically diagnosed presenile dementia of the Alzheimer type: a case‐control study in northern England. J Epidemiol Community Health 49:253–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Frecker MF (1991) Dementia in Newfoundland: identification of a geographical isolate? J Epidemiol Community Health 45:307–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Frisardi V, Solfrizzi V, Capurso C, Kehoe PG, Imbimbo BP, Santamato A et al (2010) Aluminum in the diet and Alzheimer's disease: from current epidemiology to possible disease‐modifying treatment. J Alzheimer's Dis 20:17–30. [DOI] [PubMed] [Google Scholar]
- 45. Games D, Adams D, Alessandrini R, Barbour R, Berthelette P, Blackwell C et al (1995) Alzheimer‐type neuropathology in transgenic mice overexpressing V717F beta‐amyloid precursor protein. Nature 373:523–527. [DOI] [PubMed] [Google Scholar]
- 46. Garcia T, Esparza JL, Nogues MR, Romeu M, Domingo JL, Gomez M (2010) Oxidative stress status and RNA expression in hippocampus of an animal model of Alzheimer's disease after chronic exposure to aluminum. Hippocampus 20:218–225. [DOI] [PubMed] [Google Scholar]
- 47. Garruto RM, Brown P (1994) Tau protein, aluminium, and Alzheimer's disease. Lancet (London, England) 343:989. [DOI] [PubMed] [Google Scholar]
- 48. Gibson GE, Starkov A, Blass JP, Ratan RR, Beal MF (2010) Cause and consequence: mitochondrial dysfunction initiates and propagates neuronal dysfunction, neuronal death and behavioral abnormalities in age‐associated neurodegenerative diseases. Biochim Biophys Acta 1802:122–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Giorgianni C, Faranda M, Brecciaroli R, Beninato G, Saffioti G, Muraca G et al (2003) Cognitive disorders among welders exposed to aluminum. Giornale italiano Medicina Lavoro Ergonomia 25(Suppl. 3):102–103. [PubMed] [Google Scholar]
- 50. Golub MS, Han B, Keen CL (1996) Developmental patterns of aluminum and five essential mineral elements in the central nervous system of the fetal and infant guinea pig. Biol Trace Element Res 55:241–251. [DOI] [PubMed] [Google Scholar]
- 51. Golub MS, Han B, Keen CL, Gershwin ME (1992) Effects of dietary aluminum excess and manganese deficiency on neurobehavioral endpoints in adult mice. Toxicol Appl Pharmacol 112:154–160. [DOI] [PubMed] [Google Scholar]
- 52. Graur D, Duret L, Gouy M (1996) Phylogenetic position of the order Lagomorpha (rabbits, hares and allies). Nature 379:333–335. [DOI] [PubMed] [Google Scholar]
- 53. Greger JL (1993) Aluminum metabolism. Annu Rev Nutr 13:43–63. [DOI] [PubMed] [Google Scholar]
- 54. Griffioen KJ, Ghribi O, Fox N, Savory J, DeWitt DA (2004) Aluminum maltolate‐induced toxicity in NT2 cells occurs through apoptosis and includes cytochrome c release. Neurotoxicology 25:859–867. [DOI] [PubMed] [Google Scholar]
- 55. Gutteridge JM, Quinlan GJ, Clark I, Halliwell B (1985) Aluminium salts accelerate peroxidation of membrane lipids stimulated by iron salts. Biochim Biophys Acta 835:441–447. [DOI] [PubMed] [Google Scholar]
- 56. Halliwell B (1992) Reactive oxygen species and the central nervous system. J Neurochem 59:1609–1623. [DOI] [PubMed] [Google Scholar]
- 57. Hewitt CD, Herman MM, Lopes MB, Savory J, Wills MR (1991) Aluminium maltol‐induced neurocytoskeletal changes in fetal rabbit midbrain in matrix culture. Neuropathol Appl Neurobiol 17:47–60. [DOI] [PubMed] [Google Scholar]
- 58. House E, Collingwood J, Khan A, Korchazkina O, Berthon G, Exley C (2004) Aluminium, iron, zinc and copper influence the in vitro formation of amyloid fibrils of Abeta42 in a manner which may have consequences for metal chelation therapy in Alzheimer's disease. J Alzheimer's Dis 6:291–301. [DOI] [PubMed] [Google Scholar]
- 59. Howlett DR (2003) Protein misfolding in disease: cause or response? Curr Med Chem—Immun, Endoc Metab Agents 3, 371–383. [Google Scholar]
- 60. Huang Y, Herman MM, Liu J, Katsetos CD, Wills MR, Savory J (1997) Neurofibrillary lesions in experimental aluminum‐induced encephalopathy and Alzheimer's disease share immunoreactivity for amyloid precursor protein, A beta, alpha 1‐antichymotrypsin and ubiquitin‐protein conjugates. Brain Res 771:213–220. [DOI] [PubMed] [Google Scholar]
- 61. Iregren A, Sjogren B, Gustafsson K, Hagman M, Nylen L, Frech W et al (2001) Effects on the nervous system in different groups of workers exposed to aluminium. Occup Environ Med 58:453–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Jacobs RW, Duong T, Jones RE, Trapp GA, Scheibel AB (1989) A reexamination of aluminum in Alzheimer's disease: analysis by energy dispersive X‐ray microprobe and flameless atomic absorption spectrophotometry. Can J Neurol Sci J Can Sci Neurol 16(Suppl. 4):498–503. [DOI] [PubMed] [Google Scholar]
- 63. Jing Y, Wang Z, Song Y (2004) Quantitative study of aluminum‐induced changes in synaptic ultrastructure in rats. Synapse (New York, NY) 52:292–298. [DOI] [PubMed] [Google Scholar]
- 64. Julka D, Gill KD (1996) Altered calcium homeostasis: a possible mechanisms of aluminium‐induced neurotoxicity. Biochim Biophys Acta 1315:47–54. [DOI] [PubMed] [Google Scholar]
- 65. Julka D, Gill KD (1996) Involvement of altered cytoskeletal protein phosphorylation in aluminum‐induced CNS dysfunction. J Biochem Toxicol 11:227–233. [DOI] [PubMed] [Google Scholar]
- 66. Julka D, Sandhir R, Gill KD (1995) Altered cholinergic metabolism in rat CNS following aluminum exposure: implications on learning performance. J Neurochem 65:2157–2164. [DOI] [PubMed] [Google Scholar]
- 67. Julka D, Vasishta RK, Gill KD (1996) Distribution of aluminum in different brain regions and body organs of rat. Biol Trace Element Res 52:181–192. [DOI] [PubMed] [Google Scholar]
- 68. Kalaria R (2002) Similarities between Alzheimer's disease and vascular dementia. 203–204:29–34. [DOI] [PubMed]
- 69. Kaneko N, Takada J, Yasui H, Sakurai H (2006) Memory deficit in mice administered aluminum‐maltolate complex. Biometals 19:83–89. [DOI] [PubMed] [Google Scholar]
- 70. Kaur A, Gill KD (2005) Disruption of neuronal calcium homeostasis after chronic aluminium toxicity in rats. Basic Clin Pharmacol Toxicol 96:118–122. [DOI] [PubMed] [Google Scholar]
- 71. Kaur A, Joshi K, Minz RW, Gill KD (2006) Neurofilament phosphorylation and disruption: a possible mechanism of chronic aluminium toxicity in Wistar rats. Toxicology 219:1–10. [DOI] [PubMed] [Google Scholar]
- 72. Kawahara M (2005) Effects of aluminum on the nervous system and its possible link with neurodegenerative diseases. J Alzheimer's Dis 8:171–182; discussion 209–215. [DOI] [PubMed] [Google Scholar]
- 73. Khan A, Ashcroft AE, Higenell V, Korchazhkina OV, Exley C (2005) Metals accelerate the formation and direct the structure of amyloid fibrils of NAC. J Inorg Biochem 99:1920–1927. [DOI] [PubMed] [Google Scholar]
- 74. Kidd M (1964) Alzheimer's disease—an electron microscopical study. Brain 87:307–320. [DOI] [PubMed] [Google Scholar]
- 75. Kiesswetter E, Schaper M, Buchta M, Schaller KH, Rossbach B, Kraus T et al (2009) Longitudinal study on potential neurotoxic effects of aluminium: II. Assessment of exposure and neurobehavioral performance of Al welders in the automobile industry over 4 years. Int Arch Occup Environ Health 82:1191–1210. [DOI] [PubMed] [Google Scholar]
- 76. Kiesswetter E, Schaper M, Buchta M, Schaller KH, Rossbach B, Scherhag H et al (2007) Longitudinal study on potential neurotoxic effects of aluminium: I. Assessment of exposure and neurobehavioural performance of Al welders in the train and truck construction industry over 4 years. Int Arch Occup Environ Health 81:41–67. [DOI] [PubMed] [Google Scholar]
- 77. Klatzo I, Wisniewski H, Streicher E (1965) Experimental production of neurofibrillary degeneration. I. Light microscopic observations. J Neuropathol Exp Neurol 24:187–199. [DOI] [PubMed] [Google Scholar]
- 78. Kumar A, Prakash A, Dogra S (2011) Neuroprotective effect of carvedilol against aluminium induced toxicity: possible behavioral and biochemical alterations in rats. Pharmacol Rep 63:915–923. [DOI] [PubMed] [Google Scholar]
- 79. Kumar V, Bal A, Gill KD (2008) Impairment of mitochondrial energy metabolism in different regions of rat brain following chronic exposure to aluminium. Brain Res 1232:94–103. [DOI] [PubMed] [Google Scholar]
- 80. Kumar V, Bal A, Gill KD (2009) Aluminium‐induced oxidative DNA damage recognition and cell‐cycle disruption in different regions of rat brain. Toxicology 264:137–144. [DOI] [PubMed] [Google Scholar]
- 81. Kumar V, Bal A, Gill KD (2009) Susceptibility of mitochondrial superoxide dismutase to aluminium induced oxidative damage. Toxicology 255:117–123. [DOI] [PubMed] [Google Scholar]
- 82. Landsberg JP, McDonald B, Watt F (1992) Absence of aluminium in neuritic plaque cores in Alzheimer's disease. Nature 60:65–68. [DOI] [PubMed] [Google Scholar]
- 83. Leterrier JF, Langui D, Probst A, Ulrich J (1992) A molecular mechanism for the induction of neurofilament bundling by aluminum ions. J Neurochem 58:2060–2070. [DOI] [PubMed] [Google Scholar]
- 84. Levallois P (1995) Aluminum and Alzheimer's disease. Can Med Assoc J = J l'Assoc Med Can 152:467–468. [PMC free article] [PubMed] [Google Scholar]
- 85. Li XP, Yang YJ, Hu H, Wang QN (2006) Effect of aluminum trichloride on dissociated Ca2+ in Hippocampus neuron cell as well as learning and memory. Zhonghua lao dong wei sheng zhi ye bing za zhi = Zhonghua laodong weisheng zhiyebing zazhi = Chin J Ind Hygiene Occup Dis 24:161–163. [PubMed] [Google Scholar]
- 86. Liang FQ, Godley BF (2003) Oxidative stress‐induced mitochondrial DNA damage in human retinal pigment epithelial cells: a possible mechanism for RPE aging and age‐related macular degeneration. Exp Eye Res 76:397–403. [DOI] [PubMed] [Google Scholar]
- 87. Liang RF, Li WQ, Wang XH, Zhang HF, Wang H, Wang JX et al (2012) Aluminium‐maltolate‐induced impairment of learning, memory and hippocampal long‐term potentiation in rats. Ind Health 50:428–436. [DOI] [PubMed] [Google Scholar]
- 88. Lu X, Liang R, Jia Z, Wang H, Pan B, Zhang Q et al (2014) Cognitive disorders and tau‐protein expression among retired aluminum smelting workers. J Occup Environ Med/Am Coll Occup Environ Med 56:155–160. [DOI] [PubMed] [Google Scholar]
- 89. Mantyh PW, Ghilardi JR, Rogers S, DeMaster E, Allen CJ, Stimson ER et al (1993) Aluminum, iron, and zinc ions promote aggregation of physiological concentrations of beta‐amyloid peptide. J Neurochem 61:1171–1174. [DOI] [PubMed] [Google Scholar]
- 90. Martyn CN, Barker DJ, Osmond C, Harris EC, Edwardson JA, Lacey RF (1989) Geographical relation between Alzheimer's disease and aluminum in drinking water. Lancet (London, England) 1:59–62. [PubMed] [Google Scholar]
- 91. Martyn CN, Coggon DN, Inskip H, Lacey RF, Young WF (1997) Aluminum concentrations in drinking water and risk of Alzheimer's disease. Epidemiology (Cambridge, MA) 8:281–286. [DOI] [PubMed] [Google Scholar]
- 92. Mash DC, Flynn DD, Potter LT (1985) Loss of M2 muscarine receptors in the cerebral cortex in Alzheimer's disease and experimental cholinergic denervation. Science (New York, NY) 228:1115–1117. [DOI] [PubMed] [Google Scholar]
- 93. Mattson MP, Culmsee C, Yu Z, Camandola S (2000) Roles of nuclear factor kappaB in neuronal survival and plasticity. J Neurochem 74:443–456. [DOI] [PubMed] [Google Scholar]
- 94. Mawal‐Dewan M, Henley J, Van de Voorde A, Trojanowski JQ, Lee VM (1994) The phosphorylation state of tau in the developing rat brain is regulated by phosphoprotein phosphatases. J Biol Chem 269:30981–30987. [PubMed] [Google Scholar]
- 95. Mawal‐Dewan M, Schmidt ML, Balin B, Perl DP, Lee VM, Trojanowski JQ (1996) Identification of phosphorylation sites in PHF‐TAU from patients with Guam amyotrophic lateral sclerosis/parkinsonism‐dementia complex. J Neuropathol Exp Neurol 55:1051–1059. [PubMed] [Google Scholar]
- 96. McLachlan DR, Bergeron C, Smith JE, Boomer D, Rifat SL (1996) Risk for neuropathologically confirmed Alzheimer's disease and residual aluminum in municipal drinking water employing weighted residential histories. Neurology 46:401–405. [DOI] [PubMed] [Google Scholar]
- 97. Migheli A, Piva R, Casolino S, Atzori C, Dlouhy SR, Ghetti B (1999) A cell cycle alteration precedes apoptosis of granule cell precursors in the weaver mouse cerebellum. Am J Pathol 155:365–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Miu AC, Andreescu CE, Vasiu R, Olteanu AI (2003) A behavioral and histological study of the effects of long‐term exposure of adult rats to aluminum. Int J Neurosci 113:1197–1211. [DOI] [PubMed] [Google Scholar]
- 99. Miu AC, Olteanu AI, Miclea M (2004) A behavioral and ultrastructural dissection of the interference of aluminum with aging. J Alzheimer's Dis 6:315–328. [DOI] [PubMed] [Google Scholar]
- 100. Mizoroki T, Meshitsuka S, Maeda S, Murayama M, Sahara N, Takashima A (2007) Aluminum induces tau aggregation in vitro but not in vivo. J Alzheimer's Dis 11:419–427. [DOI] [PubMed] [Google Scholar]
- 101. Molloy DW, Standish TI, Nieboer E, Turnbull JD, Smith SD, Dubois S (2007) Effects of acute exposure to aluminum on cognition in humans. J Toxicol Environ Health Part A 70:2011–2019. [DOI] [PubMed] [Google Scholar]
- 102. Muma NA, Singer SM (1996) Aluminum‐induced neuropathology: transient changes in microtubule‐associated proteins. Neurotoxicol Teratol 18:679–690. [DOI] [PubMed] [Google Scholar]
- 103. Munoz DG (1995) Aluminum and Alzheimer's disease. Can Med Assoc J = J l'Assoc Med Can 152:468–469. [PMC free article] [PubMed] [Google Scholar]
- 104. Murayama H, Shin RW, Higuchi J, Shibuya S, Muramoto T, Kitamoto T (1999) Interaction of aluminum with PHFtau in Alzheimer's disease neurofibrillary degeneration evidenced by desferrioxamine‐assisted chelating autoclave method. Am J Pathol 155:877–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Nakagawa T, Yuan J (2000) Cross‐talk between two cysteine protease families. Activation of caspase‐12 by calpain in apoptosis. J Cell Biol 150:887–894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Neri LC, Hewitt D (1991) Aluminium, Alzheimer's disease, and drinking water. Lancet (London, England) 338:390. [DOI] [PubMed] [Google Scholar]
- 107. Neve RL, Robakis NK (1998) Alzheimer's disease: a re‐examination of the amyloid hypothesis. Trends Neurosci 21:15–19. [DOI] [PubMed] [Google Scholar]
- 108. Noremberg S, Bohrer D, Schetinger MR, Bairros AV, Gutierres J, Goncalves JF et al () Silicon reverses lipid peroxidation but not acetylcholinesterase activity induced by long‐term exposure to low aluminum levels in rat brain regions. Biol Trace Elem Res. 2015 Jun 7. [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 109. Olton DS, Markowska A, Breckler SJ, Wenk GL, Pang KC, Koliatsos V (1991) Individual differences in aging: behavioral and neural analyses. Biomed Environ Sci 4:166–172. [PubMed] [Google Scholar]
- 110. Paik SR, Lee JH, Kim DH, Chang CS, Kim J (1997) Aluminum‐induced structural alterations of the precursor of the non‐A beta component of Alzheimer's disease amyloid. Arch Biochem Biophys 344:325–334. [DOI] [PubMed] [Google Scholar]
- 111. Pennington JA, Gunderson EL (1987) History of the Food and Drug Administration's total diet study–1961 to 1987. J Assoc Off Anal Chem 70:772–782. [PubMed] [Google Scholar]
- 112. Sharma DR, Wani WY, Sunkaria A, Kandimalla RJ, Verma D, Cameotra SS et al (2013) Quercetin protects against chronic aluminum‐induced oxidative stress and ensuing biochemical, cholinergic, and neurobehavioral impairments in rats. Neurotox Res 23:336–357. [DOI] [PubMed] [Google Scholar]
- 113. Plassman BL, Breitner JC (1996) Recent advances in the genetics of Alzheimer's disease and vascular dementia with an emphasis on gene‐environment interactions. J Am Geriatr Soc 44:1242–1250. [DOI] [PubMed] [Google Scholar]
- 114. Pratico D, Uryu K, Sung S, Tang S, Trojanowski JQ, Lee VM (2002) Aluminum modulates brain amyloidosis through oxidative stress in APP transgenic mice. FASEB J 16:1138–1140. [DOI] [PubMed] [Google Scholar]
- 115. Ribes D, Colomina MT, Vicens P, Domingo JL (2008) Effects of oral aluminum exposure on behavior and neurogenesis in a transgenic mouse model of Alzheimer's disease. Exp Neurol 214:293–300. [DOI] [PubMed] [Google Scholar]
- 116. Ricchelli F, Drago D, Filippi B, Tognon G, Zatta P (2005) Aluminum‐triggered structural modifications and aggregation of beta‐amyloids. Cell Mol Life Sci 62:1724–1733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Rifat SL, Eastwood MR, McLachlan DR, Corey PN (1990) Effect of exposure of miners to aluminium powder. Lancet (London, England) 336:1162–1165. [DOI] [PubMed] [Google Scholar]
- 118. Riihimaki V, Aitio A (2012) Occupational exposure to aluminum and its biomonitoring in perspective. Crit Rev Toxicol 42:827–853. [DOI] [PubMed] [Google Scholar]
- 119. Riihimaki V, Hanninen H, Akila R, Kovala T, Kuosma E, Paakkulainen H et al (2000) Body burden of aluminum in relation to central nervous system function among metal inert‐gas welders. Scand J Work, Environ Health 26:118–130. [DOI] [PubMed] [Google Scholar]
- 120. Rogers MA, Simon DG (1999) A preliminary study of dietary aluminium intake and risk of Alzheimer's disease. Age Ageing 28:205–209. [DOI] [PubMed] [Google Scholar]
- 121. Rondeau V, Commenges D, Jacqmin‐Gadda H, Dartigues JF (2000) Relation between aluminum concentrations in drinking water and Alzheimer's disease: an 8‐year follow‐up study. Am J Epidemiol 152:59–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Roush W (1995) Protein studies try to puzzle out Alzheimer's tangles. Science (New York, NY) 267:793–794. [DOI] [PubMed] [Google Scholar]
- 123. Savory J, Exley C, Forbes WF, Huang Y, Joshi JG, Kruck T et al (1996) Can the controversy of the role of aluminum in Alzheimer's disease be resolved? What are the suggested approaches to this controversy and methodological issues to be considered? J Toxicol Environ Health 48:615–635. [DOI] [PubMed] [Google Scholar]
- 124. Savory J, Ghribi O (2007) Can studies of aluminum toxicity in vivo and in vitro provide relevant information on the pathogenesis and etiology of Alzheimer's disease? J Alzheimer's Dis 11:429–30; discussion 31–32. [DOI] [PubMed] [Google Scholar]
- 125. Savory J, Herman MM, Ghribi O (2003) Intracellular mechanisms underlying aluminum‐induced apoptosis in rabbit brain. J Inorg Biochem 97:151–154. [DOI] [PubMed] [Google Scholar]
- 126. Savory J, Herman MM, Ghribi O (2006) Mechanisms of aluminum‐induced neurodegeneration in animals: implications for Alzheimer's disease. J Alzheimer's Dis 10:135–144. [DOI] [PubMed] [Google Scholar]
- 127. Savory J, Rao JK, Huang Y, Letada PR, Herman MM (1999) Age‐related hippocampal changes in Bcl‐2:Bax ratio, oxidative stress, redox‐active iron and apoptosis associated with aluminum‐induced neurodegeneration: increased susceptibility with aging. Neurotoxicology 20:805–817. [PubMed] [Google Scholar]
- 128. Schapira AH (1999) Mitochondrial involvement in Parkinson's disease, Huntington's disease, hereditary spastic paraplegia and Friedreich's ataxia. Biochim Biophys Acta 1410:159–170. [DOI] [PubMed] [Google Scholar]
- 129. Schmidt M, Sachse C, Richter W, Xu C, Fandrich M, Grigorieff N (2009) Comparison of Alzheimer Abeta(1‐40) and Abeta(1‐42) amyloid fibrils reveals similar protofilament structures. Proc Natl Acad Sci USA 106:19813–19818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Shcherbatykh I, Carpenter DO (2007) The role of metals in the etiology of Alzheimer's disease. J Alzheimer's Dis 11:191–205. [DOI] [PubMed] [Google Scholar]
- 131. Shi Q, Gibson GE (2007) Oxidative stress and transcriptional regulation in Alzheimer disease. Alzheimer Dis Assoc Disorders 21:276–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Shigematsu K, McGeer PL (1992) Accumulation of amyloid precursor protein in damaged neuronal processes and microglia following intracerebral administration of aluminum salts. Brain Res 593:117–123. [DOI] [PubMed] [Google Scholar]
- 133. Shin RW, Lee VM, Trojanowski JQ (1994) Aluminum modifies the properties of Alzheimer's disease PHF tau proteins in vivo and in vitro. J Neurosci 14(Part 2):7221–7233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Shin RW, Lee VM, Trojanowski JQ (1995) Neurofibrillary pathology and aluminum in Alzheimer's disease. Histol Histopathol 10:969–978. [PubMed] [Google Scholar]
- 135. Singer SM, Chambers CB, Newfry GA, Norlund MA, Muma NA (1997) Tau in aluminum‐induced neurofibrillary tangles. Neurotoxicology 18:63–76. [PubMed] [Google Scholar]
- 136. Sjogren B, Iregren A, Frech W, Hagman M, Johansson L, Tesarz M et al (1996) Effects on the nervous system among welders exposed to aluminium and manganese. Occup Environ Med 53:32–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Smith MA, Harris PL, Sayre LM, Perry G (1997) Iron accumulation in Alzheimer disease is a source of redox‐generated free radicals. Proc Natl Acad Sci USA 94:9866–9868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Szutowicz A, Bielarczyk H, Kisielevski Y, Jankowska A, Madziar B, Tomaszewicz M (1998) Effects of aluminum and calcium on acetyl‐CoA metabolism in rat brain mitochondria. J Neurochem 71:2447–2453. [DOI] [PubMed] [Google Scholar]
- 139. Thorne BM, Donohoe T, Lin KN, Lyon S, Medeiros DM, Weaver ML (1986) Aluminum ingestion and behavior in the Long‐Evans rat. Physiol Behav 36:63–67. [DOI] [PubMed] [Google Scholar]
- 140. Tokutake S, Nagase H, Morisaki S, Oyanagi S (1995) Aluminium detected in senile plaques and neurofibrillary tangles is contained in lipofuscin granules with silicon, probably as aluminosilicate. Neurosci Lett 185:99–102. [DOI] [PubMed] [Google Scholar]
- 141. Toyama BH, Weissman JS (2011) Amyloid structure: conformational diversity and consequences. Annu Rev Biochem 80:557–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Uno H, Alsum PB, Dong S, Richardson R, Zimbric ML, Thieme CS et al (1996) Cerebral amyloid angiopathy and plaques, and visceral amyloidosis in aged macaques. Neurobiol Aging 17:275–281. [DOI] [PubMed] [Google Scholar]
- 143. Vogt (1986) Water Quality and Health: Study of a Possible Relation Between Al in Drinking Water and Dementia. Sosiale og Okonomiske Studier. Oslo: Statistics Norway.61. [Google Scholar]
- 144. Wakayama I, Nerurkar VR, Strong MJ, Garruto RM (1996) Comparative study of chronic aluminum‐induced neurofilamentous aggregates with intracytoplasmic inclusions of amyotrophic lateral sclerosis. Acta Neuropathol 92:545–554. [DOI] [PubMed] [Google Scholar]
- 145. Walton JR (2006) Aluminum in hippocampal neurons from humans with Alzheimer's disease. Neurotoxicology 27:385–394. [DOI] [PubMed] [Google Scholar]
- 146. Walton JR (2007) An aluminum‐based rat model for Alzheimer's disease exhibits oxidative damage, inhibition of PP2A activity, hyperphosphorylated tau, and granulovacuolar degeneration. J Inorg Biochem 101:1275–1284. [DOI] [PubMed] [Google Scholar]
- 147. Walton JR (2010) Evidence for participation of aluminum in neurofibrillary tangle formation and growth in Alzheimer's disease. J Alzheimer's Dis 22:65–72. [DOI] [PubMed] [Google Scholar]
- 148. Walton JR (2012) Cognitive deterioration and associated pathology induced by chronic low‐level aluminum ingestion in a translational rat model provides an explanation of Alzheimer's disease, tests for susceptibility and avenues for treatment. Int J Alzheimer's Dis 2012:914947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Walton JR (2014) Chronic aluminum intake causes Alzheimer's disease: applying Sir Austin Bradford Hill's causality criteria. J Alzheimer's Dis 40:765–838. [DOI] [PubMed] [Google Scholar]
- 150. Wang L, Hu J, Zhao Y, Lu X, Zhang Q, Niu Q (2014) Effects of aluminium on beta‐amyloid (1‐42) and secretases (APP‐cleaving enzymes) in rat brain. Neurochem Res 39:1338–1345. [DOI] [PubMed] [Google Scholar]
- 151. Wettstein A, Aeppli J, Gautschi K, Peters M (1991) Failure to find a relationship between mnestic skills of octogenarians and aluminum in drinking water. Int Arch Occup Environ Health 63:97–103. [DOI] [PubMed] [Google Scholar]
- 152. Williams MD, Van Remmen H, Conrad CC, Huang TT, Epstein CJ, Richardson A (1998) Increased oxidative damage is correlated to altered mitochondrial function in heterozygous manganese superoxide dismutase knockout mice. J Biol Chem 273:28510–28515. [DOI] [PubMed] [Google Scholar]
- 153. Wisniewski HM, Narang HK, Terry RD (1976) Neurofibrillary tangles of paired helical filaments. J Neurol Sci 27:173–181. [DOI] [PubMed] [Google Scholar]
- 154. Wisniewski HM, Shek JW, Gruca S, Sturman JA (1984) Aluminum‐induced neurofibrillary changes in axons and dendrites. Acta Neuropathol 63:190–197. [DOI] [PubMed] [Google Scholar]
- 155. Wisniewski HM, Sturman JA, Shek JW (1980) Aluminum chloride induced neurofibrillary changes in the developing rabbit a chronic animal model. Ann Neurol 8:479–490. [DOI] [PubMed] [Google Scholar]
- 156. Wong MH, Fung KF, Carr HP (2003) Aluminium and fluoride contents of tea, with emphasis on brick tea and their health implications. Toxicol Lett 137:111–120. [DOI] [PubMed] [Google Scholar]
- 157. World_Health_Organization (1997) Aluminium. Geneva: WHO. Environmental Health Criteria.194. [Google Scholar]
- 158. Wright AL, Zinn R, Hohensinn B, Konen LM, Beynon SB, Tan RP et al (2013) Neuroinflammation and neuronal loss precede Abeta plaque deposition in the hAPP‐J20 mouse model of Alzheimer's disease. PLoS One 8:e59586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Wujek JR DM, Frederickson RC, Brunden KR (1996) Deposits of Aβ beta fibrils are not toxic to cortical and hippocampal neurons in vitro. Neurobiol Aging 17:107–113. [DOI] [PubMed] [Google Scholar]
- 160. Xie CX, Mattson MP, Lovell MA, Yokel RA (1996) Intraneuronal aluminum potentiates iron‐induced oxidative stress in cultured rat hippocampal neurons. Brain Res 743:271–277. [DOI] [PubMed] [Google Scholar]
- 161. Yokel RA (1989) Aluminum produces age related behavioral toxicity in the rabbit. Neurotoxicol Teratol 11:237–242. [DOI] [PubMed] [Google Scholar]
- 162. Yokel RA (2000) The toxicology of aluminum in the brain: a review. Neurotoxicology 21:813–828. [PubMed] [Google Scholar]
- 163. Yokel RA, Florence RL (2006) Aluminum bioavailability from the approved food additive leavening agent acidic sodium aluminum phosphate, incorporated into a baked good, is lower than from water. Toxicology 227:86–93. [DOI] [PubMed] [Google Scholar]
- 164. Yoshimoto M, Iwai A, Kang D, Otero DA, Xia Y, Saitoh T (1995) NACP, the precursor protein of the non‐amyloid beta/A4 protein (A beta) component of Alzheimer disease amyloid, binds A beta and stimulates A beta aggregation. Proc Natl Acad Sci USA 92:9141–9145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Yumoto S, Kakimi S, Ohsaki A, Ishikawa A (2009) Demonstration of aluminum in amyloid fibers in the cores of senile plaques in the brains of patients with Alzheimer's disease. J Inorg Biochem 103:1579–1584. [DOI] [PubMed] [Google Scholar]
- 166. Zatta P, Ibn‐Lkhayat‐Idrissi M, Zambenedetti P, Kilyen M, Kiss T (2002) In vivo and in vitro effects of aluminum on the activity of mouse brain acetylcholinesterase. Brain Res Bull 59:41–45. [DOI] [PubMed] [Google Scholar]
- 167. Zatta PF (1995) Aluminum binds to the hyperphosphorylated tau in Alzheimer's disease: a hypothesis. Med Hypotheses 44:169–172. [DOI] [PubMed] [Google Scholar]