

Abstract
The secretory sorting receptors carboxypeptidase E (CPE) and secretogranin III (SgIII) critically activate peptidic messengers and targeting them at the regulated secretory pathway. In Alzheimer's disease (AD), the wide range of changes includes impaired function of key secretory peptidic cargos such as brain‐derived neurotrophic factor (BDNF) and neuropeptides. Here, we analyzed CPE and SgIII in the cerebral cortex of AD patients and transgenic mice. In the normal human cortex, a preferential location in dendrites and perikarya was observed for CPE, whereas SgIII was mainly associated with axons and terminal‐like buttons. Interestingly, SgIII and CPE were consistently detected in astroglial cell bodies and thin processes. In AD cortices, a strong wide accumulation of both sorting receptors was detected in dystrophic neurites surrounding amyloid plaques. Occasionally, increased levels of SgIII were also observed in plaque associate‐reactive astrocytes. Of note, the main alterations detected for CPE and SgIII in AD patients were faithfully recapitulated by APPswe/PS1dE9 mice. These results implicate for the first time the sorting receptors for regulated secretion in amyloid β‐associated neural degeneration. Because CPE and SgIII are essential in the process and targeting of neuropeptides and neurotrophins, their participation in the pathological progression of AD may be suggested.
Keywords: Alzheimer's disease, astrocytes, carboxypeptidase E, dystrophic neurites, secretogranin III, transgenic
Introduction
Alzheimer disease (AD) is the most prevalent neurodegenerative disorder, characterized by profound cognitive dysfunction and memory loss. The hallmarks of AD include occurrence of senile plaques and neurofibrillary tangles, synaptic and neural loss, and glia‐mediated inflammation 13, 42. Moreover, aberrant function of classical neurotransmitters, neuropeptides and growth factors such as Ach, somatostatin and brain‐derived neurotrophic factor (BDNF) is also a feature of this disorder 6, 47, 51. Although the pathogenesis of AD has not been established, identification of amyloid‐β (Aβ) as the main component of senile plaques, together with subsequent genetic studies, has sustained the critical role of Aβ in the etiology of AD over the last two decades 45. In fact, considerable effort has been focused on inactivating detrimental effects of Aβ deposits through multiple anti‐Aβ therapeutics 18.
Peptidic transmitters in neurons and endocrine cells are targeted, processed and stored in the so‐called dense‐core vesicles (DCV) and secretory granules, respectively. In response to a physiological signal, secretion of neuropeptides, peptidic hormones and specific growth factors, for example, BDNF, is triggered by regulated exocytosis 7. Aggregates of granin family members with unprocessed peptidic transmitters are critical for the biogenesis of these shuttle organelles 23. Moreover, “secretory sorting receptors” play crucial roles in connecting the core aggregates with the vesicular membrane to target them at the regulated secretory pathway 20, 24. The enzyme carboxypeptidase E (CPE, also known as carboxypeptidase H and enkephalin convertase) proteolytically actives peptidic hormone and neuropeptide precursors 9, 16. In addition to its exopeptidase activity, CPE has been revealed as a key secretory sorting receptor targeting proopiomelanocortin/adrenocorticotropic hormone and pro‐BDNF at the regulated secretory pathway in pituitary cells and hippocampal neurons 11, 27. Secretogranin III (SgIII, originally identified as the 1B1075 gene product) has been identified as another secretory sorting receptor, which targets chromogranin A (CgA) at endocrine secretory granules 19. Furthermore, through a cooperative mechanism, proopiomelanocortin‐derived peptides have been described as being transferred from CPE to SgIII, and subsequently to CgA for the efficient processing, storage and release of endocrine hormones 21.
Although SgIII and CPE are key components of the regulated secretory pathway, study of them in the central nervous system (CNS) have been poor. Moreover, to the best of our knowledge, analyses of these proteins in the human brain have not yet been performed. Recent reports could implicate these proteins in AD. First, SgIII and CPE are downregulated in the cerebrospinal fluid of AD patients 1, 38. Moreover, in vivo CPE elimination leads to neuronal degeneration and memory deficits 49, 50. Here, we analyze the expression of CPE and SgIII in the human cerebral cortex and their changes in AD subjects. Strikingly, aberrant accumulation of these sorting receptors was detected in senile plaques of AD patients. Moreover, the recapitulation of human CPE and SgIII alterations by amyloid‐forming transgenic mice suggests a role for Aβ in impairing secretory sorting receptors in AD.
Materials and Methods
Human brain tissues
Thirteen non‐AD and 11 AD post‐mortem human samples (aged 49–81; post‐mortem delays between 2.15 and 8.5 h) were obtained from the Institute of Neuropathology Brain Bank IDIBELL‐Hospital Universitari de Bellvitge (Hospitalet de Llobregat, Spain) following approval by the local ethics committee. Subjects were selected on the basis of post‐mortem diagnosis of AD according to Consortium to Establish a Registry for Alzheimer's Disease (CERAD) criteria 31. AD cases corresponded to Braak stages V and VI. No neurological symptoms or signs were recorded in control cases.
Transgenic mouse
The experiments were carried out on male APPswe/PS1dE9 mice (n = 4) and wild‐type littermates (n = 4) from The Jackson Laboratory (Bar Harbor, ME, USA) 22. Genotypes were identified by polymerase chain reaction (PCR) amplification of tail DNA. The animal colony was kept under controlled temperature (22 ± 2°C), humidity (40–60%) and light (12‐h cycles) conditions, and treated in accordance with the European Community Council Directive (86/609/ECC). The study was approved by the local ethical committee (University of Barcelona).
Antibodies
Polyclonal antibodies against SgIII were purchased from Sigma‐Aldrich (Diesenhofen, Germany) and Proteintech Group Inc. (Chicago, IL, USA). Monoclonal and polyclonal antibodies against CPE were obtained from BD Transduction Laboratories (San Jose, CA, USA) and Proteintech Group Inc., respectively. Antibodies against glial fibrillary acidic protein (GFAP), β‐actin, Aβ, AT8 and voltage‐dependent anion channel (VDAC) were from Millipore Iberica (Madrid, Spain), DAKO (Glostrup, Denmark), Innogenetics (Gent, Belgium) and Calbiochem (La Jolla, CA, USA).
Immunohistochemistry
Human and mouse samples were fixed in 4% paraformaldehyde in 0.1 M phosphate buffer, pH 7.4, by immersion and intracardiac perfusion, respectively. Animals were perfused under deep anesthesia (ketamine hydrochloride/xylazine hydrochloride). Brain samples were cryoprotected in a 30% sucrose solution, frozen and sectioned with a cryostat. For peroxidase immunohistochemistry, histological sections were soaked for 30 minutes in phosphate buffered saline (PBS) containing 10% methanol and 3% H2O2 and subsequently washed in PBS. Pretreatment with formic acid was used to enhance labeling of plaques. To suppress nonspecific binding, brain sections were incubated in 10% serum‐PBS containing 0.1% Triton X‐100, 0.2% glycine and 0.2% gelatin for 1 h at room temperature. Incubations with the primary antibodies were carried out overnight at 4°C in PBS containing 1% fetal calf serum, 0.1% Triton X‐100 and 0.2% gelatin. SgIII and CPE detection in human and mouse brains was indistinctively performed with antibodies from different sources. Immunoglobulin binding was detected with the avidin‐biotin‐peroxidase method (Vectastain ABC kit, Vector Laboratories Inc., Burlingame, CA, USA). The peroxidase complex was visualized by incubating the sections with 0.05% diaminobenzidine and 0.01% H2O2 in PBS. Some immunoreactions were enhanced with the cobalt‐nickel coprecipitation technique. Sections were mounted, dehydrated and coverslipped in Eukitt® (Sigma‐Aldrich).
For double‐label peroxidase immunohistochemistry, the first immunostaining was performed as above. The second immunolabeling reaction was developed in a medium containing 0.01% benzidine dihydrochloride, 0.025% sodium nitroprusside (Merck, Darmstadt, Germany) and 0.005% H2O2 in PBS, pH 6 37. Double‐label fluorescent immunohistochemistry was performed by incubation with different fluorochrome‐conjugated secondary antibodies (Alexa Fluor 488 and Alexa Fluor 568, Molecular Probes, Eugene, OR, USA), and cell nuclei were stained with 4’,6‐diamidino‐2‐phenylindole (Molecular Probes). Endogenous autofluorescence was quenched by Sudan Black B (Sigma‐Aldrich) treatment. Sections were mounted with Mowiol (Merk Chemicals Ltd., Nottingham, UK) and observed with a Leica TCS SPE scanning confocal microscope. The specificity of the immunostaining was tested by preincubating the primary antibodies with an excess of antigen (Proteintech Group Inc.), replacing the primary antibodies with an equivalent concentration of nonspecific IgG and omitting them. No immunostaining was observed in these conditions.
Electron microscopy
APPswe/PS1dE9 and wild‐type mice aged 9 months were deeply anaesthetized prior to intracardiac perfusion with 4% paraformaldehyde and 0.1% glutaraldehyde. Brains were removed, dissected and then postfixed by immersion in 1% osmium tetraoxide. Tissue specimens were embedded in epon‐812 (Electron Microscopy Sciences, Hatfield, PA, USA) and cut with an ultramicrotome. Semithin sections of the hippocampus, entorhinal cortex and neocortex were stained with toluidine blue, and selected ultrathin sections (70 nm) were subjected to an etching treatment in order to expose hidden antigenic sites. Ultrathin sections were incubated in a blocking and quenching free‐aldehyde solution prior to incubation with the primary antibodies. After washing, the sections were incubated in a gold‐conjugated secondary antibody (Agar, Monocomp, Madrid, Spain) and directly visualized with a Jeol Jem 1011 electron microscope (JEOL GmbH, München, Germany).
Western blotting
Human brain tissues were homogenized in ice‐cold lysis buffer containing 50 mM Tris‐HCl pH 7.4, 150 mM NaCl, 5 mM MgCl2, 1 mM EGTA, 1% Triton X‐100 and protease inhibitor cocktail (Roche Diagnostics GmbH, Mannheim, Germany). Samples of postnuclear lysates were electrophoresed in 12% SDS‐PAGE (Bio‐Rad Laboratories, Hercules, CA, USA) and then transferred to nitrocellulose membranes (Whatman® Schleicher & Schuell, Keene, NH, USA). The membranes were blocked in a solution containing 5% nonfat milk powder in Tris‐buffered saline and Tween 20 (TBST; 140 mM NaCl, 10 mM Tris/HCl, pH 7.4 and 0.1% Tween 20) for 1 h at room temperature and then incubated with primary antibodies in blocking buffer for 2 h at room temperature. SgIII and CPE were detected with antibodies from Sigma‐Aldrich and BD Transduction Laboratories, respectively. After several washes in blocking solution, the membranes were incubated for 1 h with horseradish peroxidase‐conjugated secondary antibodies (DAKO). Bound antibodies were visualized with enhanced chemiluminescence reagents ECL™ (GE Healthcare, Buckinghamshire, UK). Blot images were captured with a scanner.
Results
CPE and SgIII are distributed in specific neuronal and astroglial microdomains in the human cerebral cortex
To study CPE and SgIII in the human cerebral cortex, we performed an immunocytochemical analysis of samples from autopsies with different well‐validated antibodies. We examined cortical areas typically affected in AD, such the neocortex, the entorhinal cortex, the subiculum and the hippocampal formation. In general, we found that both proteins were abundant in all analyzed cortices, although CPE immunostaining was more intense and more extended than that for SgIII. Both proteins were widely detected through the gray and white matter, associated with neuronal and non‐neuronal cell bodies and processes (Figures 1, 2, 3). Similar results were consistently obtained using antibodies from different sources (data not shown).
Figure 1.
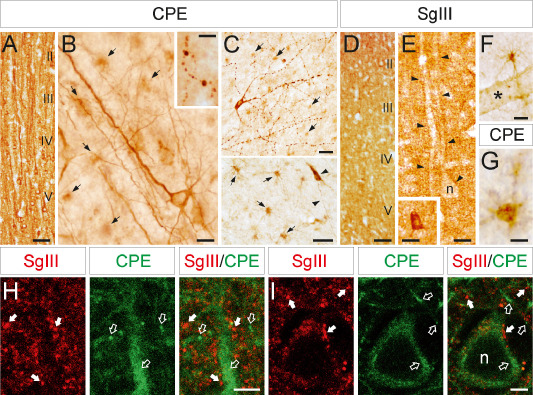
CPE and SgIII protein expression in the neocortex. (A,B) In the gray matter, an intense CPE immunoreaction is abundantly located in dendrites and neuronal perikarya through all cortical layers (A), especially in inner levels (B), whereas varicose axons (inset in B) and glial‐like cells (arrows in B) are occasionally and faintly labeled, respectively. (C) Two different images showing neuronal somata and processes (arrowheads) and numerous fibrous astrocytes (arrows) immunolabeled for CPE in the white matter. (D,E) SgIII is detected in punctuate structures through the neuropil (D), outlining neuronal perykaria and proximal dendrites (arrowheads in E), and in interneuron somata (inset in E). (F,G) A granular immunoreaction for both SgIII (F) and CPE (G) is located in fibrous astroglial cell bodies and processes. (H,I) Confocal double immunofluorescence showing the location of SgIII (arrows) and CPE (open arrows) in the neuropil (H) and a neuronal soma (I). Note how most punctate structures and dendrite shafts are differentially labeled for SgIII and CPE. Scale bar in μm: A and D, 100; B and E–G, 25; C, 50; inset in B, 10; inset in E, 25; H and I, 5. n = nucleus; asterisk = blood vessel.
Figure 2.
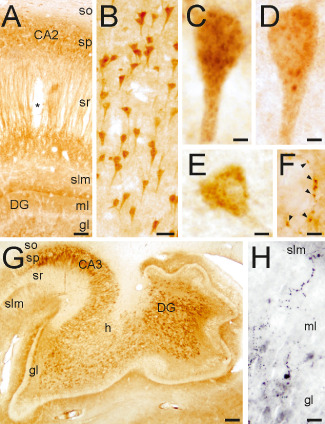
CPE and SgIII protein expression in the hippocampus. (A) CPE distribution in the hippocampus. (B) Pyramidal cell bodies labeled for SgIII. (C,D) Granule‐like compartments stained for SgIII (C) and CPE (D) in CA1 pyramidal neurons. (E) A CPE‐positive interneuron in the stratum oriens. (F) CPE‐labeled terminal‐like buttons on a negative soma in the hilus. (G) Pattern of SgIII immunolabeling in CA3 and the dentate gyrus. (H) SgIII varicose fibers in the inner portion of the molecular layer of the dentate gyrus. Scale bars in μm: A, 150; B, 50; C‐F, 5; G, 1000, H, 25. Abbreviations: so = stratum oriens; sp = stratum pyramidale; sr = stratum radiatum; slm = stratum lacunosum‐moleculare; ml = molecular layer; gl = granular layer; h = hilus; DG = dentate gyrus.
Figure 3.
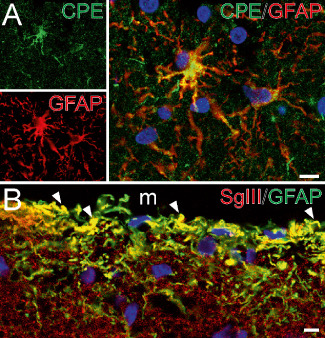
Astroglial‐identified cells express CPE and SgIII in the cerebral cortex. (A, B) Confocal images illustrating double immunofluorescence of CPE and SgIII with GFAP in the white matter (A) and the upper layers of the parietal cortex (B). Note the location of CPE in the perinuclear area and processes of astrocytes. In B, SgIII decorates punctuate structures and thin processes exhibiting GFAP. Arrowheads indicate yellow color in merge images. Nuclei are in blue color. Scale bar in μm: A, 10; B, 5. M = meninge.
In the neocortex, robust CPE immunolabeling was detected filling dendrites and neuronal perikarya. Dendritic staining was found mainly in apical shafts running across the entire thickness of the cortex (Figure 1A). The strongest immunoreactive somata were located in inner cortical layers, including pyramidal and multipolar neurons. CPE immunoreactivity was also detected in some varicose axons and slightly in glial‐like cells (Figure 1B). In the white matter, scattered pyramidal/polymorphic and fusiform neurons displayed high levels of CPE in the superficial and deep regions, respectively (Figure 1C). Neuronal varicose processes and numerous astroglial‐like cells were positive for CPE in different myelinated areas (Figure 1C,G).
Of note, a differential immunolabeling pattern was found in the gray matter of neocortex for SgIII compared with CPE. Faint SgIII labeling was detected within large pyramidal cell bodies (Figure 1D). Somatic SgIII was mainly restricted to perinuclear secretory organelles, whereas labeling in dendritic shaft was weak or absent (Figure 1E). Characteristically, SgIII was found as immunoreactive puncta throughout the neuropil, occasionally outlining neuronal perykaria and proximal dendrites (Figure 1E). Scattered interneurons exhibited intense SgIII immunoreactivity (inset in Figure 1E). Some glial‐like cells and processes in the outer layers also displayed this granin. In the white matter, astrocyte‐like cells and deep fusiform neurons were weakly labeled for SgIII (Figure 1F).
The differential distribution of CPE and SgIII in neuronal microdomains of the isocortex was further substantiated by double confocal immunofluorescence. Numerous SgIII‐containing puncta were detected in the neuropil and frequently opposite CPE‐positive somata and dendrites of pyramidal neurons (Figure 1H,I). Only a few SgIII‐immunofluorescent puncta also exhibited CPE signal. Moreover, a differential location was also detected inside large pyramidal cell bodies. CPE entirely filled the perikaryon, whereas SgIII antibodies faintly labeled structures around the nucleus, apparently not overlapping with CPE (Figure 1I).
In the entorhinal cortex, subicular complex and the hippocampus, the immunolabeling pattern of CPE was similar to that detected in the neocortex. Pyramidal and nonpyramidal neurons displayed intense CPE staining in dendritic shafts and perikarya (Figure 2A,D,E). In dentate gyrus and CA regions CPE was mainly detected in dendrites with a prominent laminar distribution, whereas perikarya and proximal dendrites of subiculum and entorhinal cortex were strongly labeled (Figure 2A). Subcellular structures labeled for CPE included somatic and dendritic granular compartments, varicose fibers and terminal‐like buttons (Figure 2D–F). The pattern of SgIII immunolabeling in the entorhinal cortex was equivalent to that of the isocortex, where neural somata and dendrites lacked this granin. In the hippocampus, mossy fibers and the hilus were strongly labeled for SgIII (Figure 2G). Neuronal perykaria contained abundant SgIII in CA and subicular regions (Figure 2B,C). Marked SgIII immunoreaction was detected in varicose fibers located in the inner portion of the molecular layer of the dentate gyrus and the pyramidal layer of CA2 (Figure 2H). Typically, puncta immunoreactive for SgIII outlined pyramidal dendrites in CA2 (data not shown). Glial‐like cells positive for CPE and SgIII were mainly detected in the white matter.
Finally, we determined whether non‐neuronal cells labeled for CPE and SgIII in the human brain corresponded with astrocytes by using double immunofluorescence. Star‐shaped cells displaying CPE were consistently decorated with the astroglial marker GFAP in several cortical areas (Figure 3A). This colocalization was particularly obvious in fibrous astrocytes of the white matter (Figure 3A). CPE labeling was located in astroglial cell bodies and processes, occasionally detected as a granule‐like structure (Figure 1G). Similar results were obtained for SgIII in GFAP‐identified astrocytes, however, as occurs in neurons, SgIII labeling in astrocytes was weaker than that found for CPE. Characteristically, many glial processes in the outer layers of the cortex, most likely glia limitants and interlaminar astrocytes, were differentially labeled for SgIII (Figure 3B).
Taken together, these results show that CPE and SgIII are widely expressed by neuronal and astroglial cell populations in the human cerebral cortex. Moreover, a differential targeting of these sorting receptors is strikingly evidenced in microdomains of specific neuronal subsets.
CPE and SgIII are aberrantly accumulated in cortical plaques of AD patients
To investigate alterations in CPE and SgIII in AD, we performed immunological analyses of the cerebral cortex of patients and age‐matched controls. With Western blotting, a robust band around 55 kDa for CPE was detected in cortical tissues, whereas SgIII mainly displayed two greater bands corresponding to the precursor and mature forms (Figure 4A). No differences were found in the intensity or the electrophoretical mobility of the bands in the hippocampus (Figure 4A) and neocortex (data not shown) of AD patients (n = 6) compared to controls (n = 6).
Figure 4.
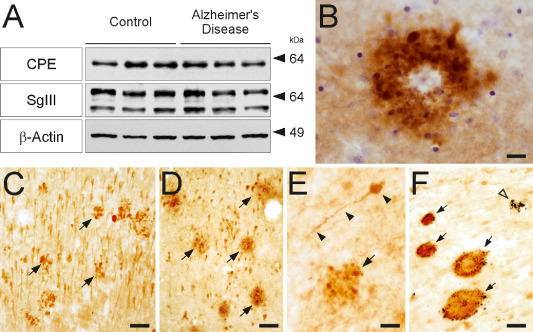
CPE and SgIII are accumulated in senile plaques of AD patients. (A) Western blots showing protein levels of CPE and SgIII in homogenates of hippocampus from AD patients and age‐matched controls. Densitometric analyses revealed no significant differences among groups in protein expression levels, normalized to β‐actin (P > 0.05). The mobility of molecular mass markers (in kDa) is indicated. (B–F) Aberrant accumulation of SgIII (B,D,F) and CPE (C,E) are consistently detected in AD plaques of different areas. (B) SgIII‐labeled corona plaque in upper layers of the neocortex, Nissl counterstaining in blue. (C,D) Arrows indicate numerous plaques in the CA1 region of the hippocampus. (E) CPE‐positive dystrophic neurite (arrowheads) and a plaque (arrow) in the white matter. (F) Double immunostaining against SgIII (brown) and Aβ (blue) in the hippocampus showing four double‐labeled senile plaques (arrows) and one lacking SgIII. Scale bar in μm: B, 20; C,D and F, 50; E, 15.
Although total levels of CPE and SgIII were preserved in the AD cortices, dramatic changes in these two proteins were revealed in the AD cerebral cortex (n = 6) compared with controls (n = 7) with immunocytochemistry (Figure 4B–F). In general, distribution patterns of CPE and SgIII appeared normal in nonplaque areas of AD brains. Some sample‐to‐sample variability in the labeling intensity was detected in pyramidal cell bodies, likely related to differences in specimen processing. A striking accumulation of sorting receptors was widely found in senile plaques (Figure 4C,D). CPE‐ and SgIII‐positive plaques were detected in the neocortex, mainly in superficial layers. In decreasing order, the sorting receptors were also observed in the entorhinal cortex, subiculum, CA1 hippocampal region, molecular layer of the dentate gyrus and myelinated tracts. Double immunocytochemistry showed that more than 90% the Aβ‐labeled plaques were positive for CPE and SgIII (Figure 4F). We observed different degrees of colocalization of CPE and SgIII in senile plaques. Major overlapping between these proteins was frequently detected in the same plaque (Figure 5), although low colocalization and even single‐labeled plaques were also observed (data not shown).
Figure 5.
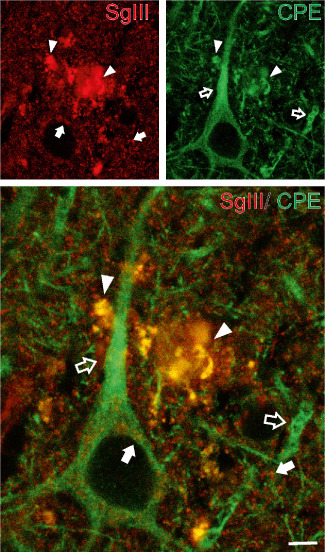
Colocalization of SgIII with CPE in AD senile plaques. Confocal images showing double immunofluorescence of SgIII and CPE in layer V of the parietal cortex. Aberrant plaque structures display a high grade of overlapping (yellow color in merge image, arrowheads). Note the differential distribution of SgIII (arrows) and CPE (open arrows) in the neuropil and subcelullar locations around the nucleus. Scale bar: 5 μm.
To identify structures accumulating secretory sorting receptors in AD plaques, different markers were used (Figures 6, 7). Only a small amount of CPE and SgIII labeling colocalized with Aβ deposits (Figure 6). Because an array of proteins destined for secretion has been shown to co‐deposit with Aβ in senile plaques 46, extracellular accumulation of secreted CPE and SgIII is conceivable. Most of the aberrant CPE and SgIII signal in plaques was associated with cellular structures in the vicinity of both diffuse and focal Aβ deposits (Figure 6). Typically, focal Aβ deposits were surrounded by a CPE‐ and SgIII‐immunoreactive corona (Figure 4B). Neuritic identity of CPE‐ and SgIII‐containing structures was determined by AT8 and VDAC immunolabeling. Dystrophic neurites identified by the pathological phosphorylated form of tau, recognized by AT8 antibodies, were also decorated for CPE and SgIII (Figure 6). Moreover, both sorting receptors colocalized with the mitochondrial porin VDAC (Figure 6). Interestingly, CPE and SgIII colocalizations with dystrophic neurite markers were partial. This observation suggests that a subpopulation, or certain microdomains, of degenerating neurites specifically accumulates these sorting receptors. Because CPE and SgIII are also expressed by astrocytes, we performed double labeling with GFAP to determine alterations in plaque‐surrounding activated glia. Detailed inspection of immunofluorescence revealed that levels of sorting receptor were occasionally increased in reactive astrocytes, mainly for SgIII (Figure 7).
Figure 6.
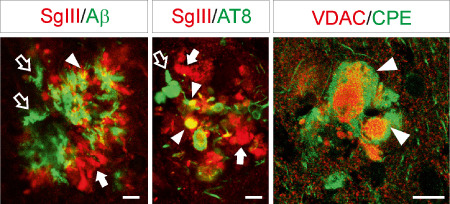
Dystrophic neurites aberrantly accumulate SgIII and CPE. Confocal double immunofluorescence of SgIII with Aβ and the dystrophic neurite markers AT8 and VDAC in senile plaques of the parietal cortex. SgIII poorly and moderately colocalize with Aβ and AT8, respectively (arrowheads). Arrows and open arrows indicate single labeling structures. Image on the right shows pathological enlarged neurites accumulating CPE, the core of which exhibits VDAC. Scale bar: 10 μm.
Figure 7.
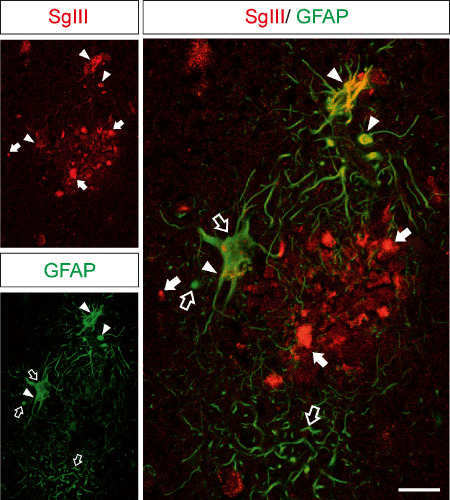
Increased levels of SgIII in plaque‐surrounding reactive astrocytes. Confocal double immunofluorescence of SgIII and GFAP in the AD hippocampus. Hypertrophied GFAP‐labeled astrocytes that surround an amyloid plaque aberrantly contain high levels of SgIII. Arrows and open arrows indicate single labeling structures. Yellow color in merge image indicates colocalization (arrowheads). Scale bar: 25 μm.
Finally, we evaluated whether CPE and SgIII accumulate in the other hallmark of AD, the neurofibrillary tangles. As AT8 labeling evidenced, no changes in the level of sorting receptors were found in tangle‐bearing neuronal somata (data not shown).
We conclude that CPE and SgIII are aberrantly accumulated in degenerating neurites and activated astroglia of AD plaques.
APPswe/PS1dE9 transgenic mice mimic the CPE and SgIII alterations found in AD subjects
Because Aβ plays a key role in contributing to the main alterations of AD, we next investigated the Aβ involvement in CPE and SgIII changes in vivo. We used amyloid‐forming transgenic mice APPswe/PS1dE9. In these mice, the APPswe mutation causes Aβ deposits, whereas the dE9 variant of PS1 accelerates the amyloid pathology as early as 6 months of age 22. CPE and SgIII protein expression were analyzed in 12‐month‐old transgenic and wild‐type animals. In general, the distribution pattern for both proteins in the mouse brain was similar to that in the human brain (data not shown). In control mice, strong CPE labeling was frequently detected in neuronal dendrites and perikarya, whereas a weaker signal for SgIII was observed mainly associated with punctate structures and fibers. Moreover, both SgIII and CPE were detected in astroglial cells. Of note, intense labeling for CPE and SgIII was found in numerous plaques through all the CNS of transgenic mice. These accumulations were evident in regions such as the cerebral cortex, striatum, thalamus and cerebellum (Figure 8). As in AD patients, CPE and SgIII were aberrantly accumulated in the same plaques, where a high degree of colocalization was frequently observed (Figure 9A,B). Double labeling of CPE and SgIII with AT8, VDAC and Aβ showed that sorting receptors were mainly accumulated in dystrophic neurites surrounding Aβ deposits (data not shown). To identify subcellular structures containing CPE and SgIII in degenerating neurites we performed immunogold labeling. At the ultrastructural level, dystrophic neurites were packed with heterogeneous collections of vesicular and vacuolar structures, including single‐ and double‐membrane organelles, and they were commonly filled with dense or multilamellar contents (Figure 9C,D). CPE and SgIII labeling in dystrophic neurites was associated with enlarged and autophagic‐like vesicles, indicating abnormal forms of vesicular compartments.
Figure 8.
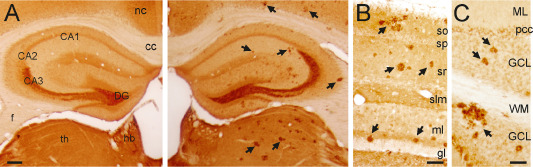
SgIII and CPE accumulation in APPswe/PS1dE9 mice. (A) SgIII distribution in wild‐type (left) and transgenic (right) mice. Strong SgIII accumulation is associated in plaques through hippocampus, neocortex and thalamus of the APPswe/PS1dE9 mouse. In both genotypes, mossy fibers are intensely labeled. (B) Higher magnification of the transgenic hippocampus immunolabeled for SgIII. (C) CPE‐positive accumulations in the APPswe/PS1dE9 cerebellar cortex. Scale bars: A, 250 μm; B and C, 100 μm. Abbreviations: nc = neocortex; cc = corpus callosum; CA = regions of the hippocampus; f = fimbria; hb = habenula; th = thalamus; DG = dentate gyrus; so = stratum oriens; sp = stratum pyramidale; sr = stratum radiatum; slm = stratum lacunosum‐moleculare; ml = molecular layer; gl = granular layer.
Figure 9.
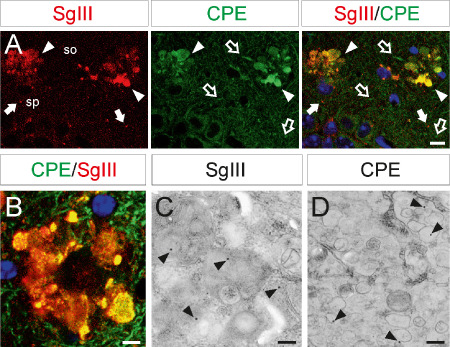
CPE and SgIII accumulation in dystrophic neurites of transgenic mice. CPE and SgIII colocalization in degenerating neurites in the CA1 region of the hippocampus (upper images) and somatosensorial cortex (lower image) by double confocal immunofluorescence. Blue color labels cell nuclei. Ultrastructural images show immunogold staining of SgIII and CPE in autophagic‐like (left) and enlarged (right) vesicles in cortical dystrophic neurites. Scale bars in μm: A, 10; B, 5; C and D, 0.5. Abbreviations: so = stratum oriens; sp = stratum pyramidale.
These results show that amyloid‐forming transgenic mice accurately recapitulate the aberrant CPE and SgIII accumulation detected in AD dystrophic neurites.
Discussion
In the present study, we show new and compelling evidence implicating the sorting receptors of the regulated secretory pathway in the Aβ‐induced neurodegeneration of AD. We report for the first time the specific distribution of CPE and SgIII in neuronal and glial cells in the healthy human brain. Moreover, a dramatic accumulation of these sorting receptors in senile plaques of AD patients and Aβ‐forming transgenic mice is demonstrated.
CPE and SgIII in the human cerebral cortex
In general, the distribution patterns of CPE and SgIII shown in this study are in agreement with those formerly found in rodents 29, 34. However, some differential features were observed for the human brain. First, we found immunolabeling for these two proteins, robustly for CPE, in neuronal populations in both the superficial and the deep white matter, whereas no information of this kind has been reported for rodents. Moreover, human fibrous and interlaminar‐like astrocytes widely display CPE and SgIII. In rodents, SgIII, but not CPE, was recently reported in astrocytes in situ, mainly protoplasmic cells 37. Although differences related to antibody cross‐reactivity cannot be ruled out, species‐specific features in CPE and SgIII expressions are suggested. In fact, several distinctions have been shown between human white matter neurons and astrocytes and those of rodents 17, 33.
One of the most striking observations of this study is the differential localization of CPE and SgIII in neuronal subsets. A nice study performed by Hosaka et al 21 demonstrated colocalization and functional interaction between SgIII and CPE to facilitate prohormone sorting in endocrine secretory granules. Therefore, an overlapping localization for these sorting receptors would be expected in subcellular neuronal domains. However, although both proteins were present in axons, a predominant location in dendrites was detected for CPE but not for SgIII. In contrast to what is found in endocrine cells, these observations suggest a differential targeting of DCV sorting receptors in neurons, at least for distinctive subpopulations. Although a lack of immunolabeling could reflect protein levels below the detection threshold, the present differences between CPE and SgIII argue for differential targeting. Previous studies have lent support to the idea of differential routing of DCV cargo in neurons. For instance, in Aplysia bag cells and hypothalamic neurons differential packaging and targeting toward dendrites and nerve endings has been shown for neuropeptides and their processing endopeptidases 10, 26, 28, 35, 43. Therefore, a potential role for CPE and SgIII in specialized sorting and processing of DCV cargo in neuronal microdomains is proposed.
The regulated secretory pathway in astrocytes
In recent years, a growing body of evidence has shown that astrocytes can release non‐peptidic transmitters such as adenosine triphosphate (ATP), glutamate and d‐serine in a regulated fashion, to influence excitability and plasticity of neuronal networks 2. Regulated secretion for peptidic gliotransmitters has also been reported in cultured astrocytes. For instance, in vitro release of the hormones and growth factors atrial natriuretic peptide, neuropeptide Y and BDNF is triggered from astrocytes by different stimuli 4, 25, 40. Moreover, signaling mechanisms, such as the cyclic adenosine monophosphate (cAMP) pathway and the RE1‐silencing transcription factor/neuron‐restrictive silencer factor transcription factor, have been revealed to control regulated peptide secretion from astroglial cells 36, 39. However, the identity and function of molecular components of this stimuli‐dependent secretory pathway in astrocytes are largely unknown. The present results showing that CPE and SgIII are expressed by astrocytes in human brain sections support the competence of these glial cells for regulated secretion of peptidic messengers in vivo. Therefore, as what we might call “professional” secretory cells, we propose an important role for these two sorting receptors in the trafficking and secretion of peptidic transmitters in astroglial cells in vivo.
Involvement of CPE and SgIII in AD
Here, we show for the first time that CPE and SgIII are dramatically accumulated in senile plaques of AD patients and transgenic mice. Aberrant levels of secretory sorting receptors are occasionally located in plaque‐surrounding astrocytes, whereas the main accumulation corresponds within dystrophic neurites. Because activated astrocytes overexpress SgIII in rodents 37, increased levels of secretory sorting receptors in reactive astrocytes around human senile plaques were expected. Moreover, the BDNF overexpression previously found in plaque‐associated astrocytes of AD transgenic mice suggests that Aβ deposits coordinately alter the expression of trophic factors and their secretory components in glial cells 5.
Typically, CPE and SgIII were markedly accumulated in dystrophic neurites close to Aβ deposits in both AD patients and transgenic mice. The lack of overlapping observed in some aberrant neurites may be related to their differential distribution in neurons, mentioned above. The accumulation of sorting receptors in dystrophic neurites likely indicates retention of DCV. This suggestion is supported by previous reports showing accumulation of classical granins and DCV cargos in AD dystrophic neurites, such as CgA, Secretogranin II and BDNF 5, 15, 48. Because typical markers of synaptic vesicles, small clear vesicles, are defective in dystrophic neurites (ie, synapsin and synaptophysin) 14, a predominant accumulation of peptide‐containing vesicles in plaque‐associate neuronal projections is plausible. Interestingly, the aberrant intracellular increase of CPE and SgIII agrees with the extracellular downregulation reported in cerebrospinal fluid of AD patients 1, 38, which suggests impairment in trafficking and secretion of these proteins. Furthermore, as proposed for some classical granins 3, SgIII and CPE could serve as diagnostic biomarkers of AD.
Of note, the aberrant accumulation of CPE and SgIII detected in dystrophic neurites of AD patients was faithfully recapitulated by APPswe/PS1dE9 mice. Because transgenic animals produce Aβ and develop amyloid plaques, a critical role for Aβ leading to neuritic CPE and SgIII alterations in AD is suggested. In agreement, previous studies performed on cultured cells have shown that Aβ disrupts trafficking of BDNF‐containing DCV, likely impairing neurite transport 12, 44. Recent in vivo observations, showing that amyloid deposition precedes neuritic pathology in APPswe/PS1dE9 mice, support a mediation of plaque‐associated Aβ changing secretory sorting receptors in AD 30. Although CPE and SgIII accumulation may represent a late‐stage effect of neuritic dystrophy in AD, a contribution of these proteins in the neuropathological progression of AD may be suggested. It has been shown that improving levels of growth factors and neuropeptides, typically decreased in AD and sorted to the regulated secretory pathway (ie, BDNF and somatostatin), partially recover AD‐altered neural circuitries 32, 41. Therefore, beyond transcriptional mechanisms, we propose that aberrant accumulation of CPE and SgIII may participate in AD progression by altering sorting, processing and secretion of DCV cargos critical in this neurodegenerative disease. In this view, the elimination of CPE leads to abnormal dendritic pattering, neuronal degeneration and memory deficits 8, 49, 50.
In conclusion, we show here for the first time the differential distribution of CPE and SgIII in neuronal and astroglial microdomains in the human cerebral cortex. Moreover, Aβ‐associated accumulation of CPE and SgIII in AD dystrophic neurites and activated astroglia is uncovered. These data implicate the processing enzymes and sorting receptors for the regulated secretion of neuropeptides and neurotrophins in AD neural degeneration.
Competing Interests
The authors declare that they have no competing interests.
Acknowledgments
We are grateful to Dr. Javier DeFelipe (Cajal Institute, Spain) for helpful discussions, Drs José Antonio del Rio and Vanessa Gil (Catalonian Institute for Bioengineering, Spain) for providing some human brain samples, Margarita Carmona for technical assistance and Tom Yohannan for editorial assistance. This work was supported by grants from the Spanish Ministry of Science and Innovation (BFU2007‐67889 and BFU2010‐22132) and MAPFRE Foundation to FA and Seventh Framework Programme of the European Commission, grant agreement 278486: DEVELAGE to IF. EP is a researcher of the Ramón y Cajal program (MICINN‐Spain).
References
- 1. Abdi F, Quinn JF, Jankovic J, McIntosh M, Leverenz JB, Peskind E et al (2006) Detection of biomarkers with a multiplex quantitative proteomic platform in cerebrospinal fluid of patients with neurodegenerative disorders. J Alzheimers Dis 9:293–348. [DOI] [PubMed] [Google Scholar]
- 2. Araque A, Navarrete M (2010) Glial cells in neuronal network function. Philos Trans R Soc Lond B Biol Sci 365:2375–2381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bartolomucci A, Possenti R, Mahata SK, Fischer‐Colbrie R, Loh YP, Salton SR (2011) The extended granin family: structure, function, and biomedical implications. Endocr Rev 32:755–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bergami M, Santi S, Formaggio E, Cagnoli C, Verderio C, Blum R et al (2008) Uptake and recycling of pro‐BDNF for transmitter‐induced secretion by cortical astrocytes. J Cell Biol 183:213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Burbach GJ, Hellweg R, Haas CA, Del Turco D, Deicke U, Abramowski D et al (2004) Induction of brain‐derived neurotrophic factor in plaque‐associated glial cells of aged APP23 transgenic mice. J Neurosci 24:2421–2430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Burgos‐Ramos E, Hervás‐Aguilar A, Aguado‐Llera D, Puebla‐Jiménez L, Hernández‐Pinto AM, Barrios V, Arilla‐Ferreiro E (2008) Somatostatin and Alzheimer's disease. Mol Cell Endocrinol 286:104–111. [DOI] [PubMed] [Google Scholar]
- 7. Burgoyne RD, Morgan A (2003) Secretory granule exocytosis. Physiol Rev 83:581–632. [DOI] [PubMed] [Google Scholar]
- 8. Carrel D, Du Y, Komlos D, Hadzimichalis NM, Kwon M, Wang B et al (2009) NOS1AP regulates dendrite patterning of hippocampal neurons through a carboxypeptidase E‐mediated pathway. J Neurosci 29:8248–8258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cawley NX, Wetsel WC, Murthy SR, Park JJ, Pacak K, Loh YP (2012) New roles of carboxypeptidase e in endocrine and neural function and cancer. Endocr Rev 33:216–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chun JY, Korner J, Kreiner T, Scheller RH, Axel R (1994) The function and differential sorting of a family of aplysia prohormone processing enzymes. Neuron 12:831–844. [DOI] [PubMed] [Google Scholar]
- 11. Cool DR, Normant E, Shen F, Chen HC, Pannell L, Zhang Y, Loh YP (1997) Carboxypeptidase E is a regulated secretory pathway sorting receptor: genetic obliteration leads to endocrine disorders in Cpe(fat) mice. Cell 88:73–83. [DOI] [PubMed] [Google Scholar]
- 12. Decker H, Lo KY, Unger SM, Ferreira ST, Silverman MA (2010) Amyloid‐beta peptide oligomers disrupt axonal transport through an NMDA receptor‐dependent mechanism that is mediated by glycogen synthase kinase 3beta in primary cultured hippocampal neurons. J Neurosci 30:9166–9171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ferrer I (2012) Defining Alzheimer as a common age‐related neurodegenerative process not inevitably leading to dementia. Prog Neurobiol 97:38–51. [DOI] [PubMed] [Google Scholar]
- 14. Ferrer I, Martí E, Tortosa A, Blasi J (1998) Dystrophic neurites of senile plaques are defective in proteins involved in exocytosis and neurotransmission. J Neuropathol Exp Neurol 57:218–225. [DOI] [PubMed] [Google Scholar]
- 15. Ferrer I, Marín C, Rey MJ, Ribalta T, Goutan E, Blanco R et al (1999) BDNF and full‐length and truncated TrkB expression in Alzheimer disease. Implications in therapeutic strategies. J Neuropathol Exp Neurol 58:729–739. [DOI] [PubMed] [Google Scholar]
- 16. Fricker LD (1988) Carboxypeptidase E. Annu Rev Physiol 50:309–321. [DOI] [PubMed] [Google Scholar]
- 17. García‐Marín V, Blazquez‐Llorca L, Rodriguez JR, Gonzalez‐Soriano J, DeFelipe J (2010) Differential distribution of neurons in the gyral white matter of the human cerebral cortex. J Comp Neurol 518:4740–4759. [DOI] [PubMed] [Google Scholar]
- 18. Golde TE, Petrucelli L, Lewis J (2010) Targeting Abeta and tau in Alzheimer's disease, an early interim report. Exp Neurol 223:252–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hosaka M, Watanabe T, Sakai Y, Uchiyama Y, Takeuchi T (2002) Identification of a chromogranin A domain that mediates binding to secretogranin III and targeting to secretory granules in pituitary cells and pancreatic beta‐cells. Mol Biol Cell 13:3388–3399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hosaka M, Watanabe T (2010) Secretogranin III: a bridge between core hormone aggregates and the secretory granule membrane. Endocr J 57:275–286. [DOI] [PubMed] [Google Scholar]
- 21. Hosaka M, Watanabe T, Sakai Y, Kato T, Takeuchi T (2005) Interaction between secretogranin III and carboxypeptidase E facilitates prohormone sorting within secretory granules. J Cell Sci 118:4785–4795. [DOI] [PubMed] [Google Scholar]
- 22. Jankowsky JL, Fadale DJ, Anderson J, Xu GM, Gonzales V, Jenkins NA et al (2004) Mutant presenilins specifically elevate the levels of the 42 residue beta‐amyloid peptide in vivo: evidence for augmentation of a 42‐specific gamma secretase. Hum Mol Genet 13:159–170. [DOI] [PubMed] [Google Scholar]
- 23. Kim T, Tao‐Cheng JH, Eiden LE, Loh YP (2001) Chromogranin A, an “on/off” switch controlling dense‐core secretory granule biogenesis. Cell 106:499–509. [DOI] [PubMed] [Google Scholar]
- 24. Kim T, Gondré‐Lewis MC, Arnaoutova I, Loh YP (2006) Dense‐core secretory granule biogenesis. Physiology (Bethesda) 21:124–133. [DOI] [PubMed] [Google Scholar]
- 25. Krzan M, Stenovec M, Kreft M, Pangrsic T, Grilc S, Haydon PG, Zorec R (2003) Calcium‐dependent exocytosis of atrial natriuretic peptide from astrocytes. J Neurosci 23:1580–1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Landry M, Vila‐Porcile E, Hökfelt T, Calas A (2003) Differential routing of coexisting neuropeptides in vasopressin neurons. Eur J Neurosci 17:579–589. [PubMed] [Google Scholar]
- 27. Lou H, Kim SK, Zaitsev E, Snell CR, Lu B, Loh YP (2005) Sorting and activity‐dependent secretion of BDNF require interaction of a specific motif with the sorting receptor carboxypeptidase E. Neuron 45:245–255. [DOI] [PubMed] [Google Scholar]
- 28. Ludwig M, Leng G (2006) Dendritic peptide release and peptide‐dependent behaviours. Nat Rev Neurosci 7:126–136. [DOI] [PubMed] [Google Scholar]
- 29. Lynch DR, Braas KM, Hutton JC, Snyder SH (1990) Carboxypeptidase E (CPE): immunocytochemical localization in the rat central nervous system and pituitary gland. J Neurosci 10:1592–1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Meyer‐Luehmann M, Spires‐Jones TL, Prada C, de Garcia‐Alloza M, Calignon A, Rozkalne A et al (2008) Rapid appearance and local toxicity of amyloid‐beta plaques in a mouse model of Alzheimer's disease. Nature 451:720–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Mirra SS, Gearing M, McKeel DW Jr, Crain BJ, van Hughes JP, Belle G, Heyman A (1994) Interlaboratory comparison of neuropathology assessments in Alzheimer's disease: a study of the Consortium to Establish a Registry for Alzheimer's Disease (CERAD). J Neuropathol Exp Neurol 53:303–3015. [DOI] [PubMed] [Google Scholar]
- 32. Nagahara AH, Merrill DA, Coppola G, Tsukada S, Schroeder BE, Shaked GM et al (2009) Neuroprotective effects of brain‐derived neurotrophic factor in rodent and primate models of Alzheimer's disease. Nat Med 15:331–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Oberheim NA, Takano T, Han X, He W, Lin JH, Wang F et al (2009) Uniquely hominid features of adult human astrocytes. J Neurosci 29:3276–3287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ottiger HP, Battenberg EF, Tsou AP, Bloom FE, Sutcliffe JG (1990) 1B1075: a brain‐ and pituitary‐specific mRNA that encodes a novel chromogranin/secretogranin‐like component of intracellular vesicles. J Neurosci 10:3135–3147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ovsepian SV, Dolly JO (2011) Dendritic SNAREs add a new twist to the old neuron theory. Proc Natl Acad Sci U S A 108:19113–19120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Paco S, Margelí MA, Olkkonen VM, Imai A, Blasi J, Fischer‐Colbrie R, Aguado F (2009) Regulation of exocytotic protein expression and Ca2+‐dependent peptide secretion in astrocytes. J Neurochem 110:143–156. [DOI] [PubMed] [Google Scholar]
- 37. Paco S, Pozas E, Aguado F (2010) Secretogranin III is an astrocyte granin that is overexpressed in reactive glia. Cereb Cortex 20:1386–1397. [DOI] [PubMed] [Google Scholar]
- 38. Perrin RJ, Craig‐Schapiro R, Malone JP, Shah AR, Gilmore P, Davis AE et al (2011) Identification and validation of novel cerebrospinal fluid biomarkers for staging early Alzheimer's disease. Plos ONE 6:e16032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Prada I, Marchaland J, Podini P, Magrassi L, D'Alessandro R, Bezzi P, Meldolesi J (2011) REST/NRSF governs the expression of dense‐core vesicle gliosecretion in astrocytes. J Cell Biol 193:537–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ramamoorthy P, Whim MD (2008) Trafficking and fusion of neuropeptide Y‐containing dense‐core granules in astrocytes. J Neurosci 28:13815–13827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Saito T, Iwata N, Tsubuki S, Takaki Y, Takano J, Huang SM et al (2005) Somatostatin regulates brain amyloid beta peptide Abeta42 through modulation of proteolytic degradation. Nat Med 11:434–439. [DOI] [PubMed] [Google Scholar]
- 42. Serrano‐Pozo A, Frosch MP, Masliah E, Hyman BT (2011) Neuropathological alterations in Alzheimer disease. Cold Spring Harb Perspect Med 1:a006189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sossin WS, Sweet‐Cordero A, Scheller RH (1990) Dale's hypothesis revisited: different neuropeptides derived from a common prohormone are targeted to different processes. Proc Natl Acad Sci U S A 87:4845–4848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Stokin GB, Goldstein LS (2006) Axonal transport and Alzheimer's disease. Annu Rev Biochem 75:607–627. [DOI] [PubMed] [Google Scholar]
- 45. Tanzi RE, Bertram L (2005) Twenty years of the Alzheimer's review disease amyloid hypothesis: a genetic perspective. Cell 120:545–555. [DOI] [PubMed] [Google Scholar]
- 46. Timmer NM, de Kuiperij HB, Waal RM, Verbeek MM (2010) Do amyloid β‐associated factors co‐deposit with Aβ in mouse models for Alzheimer's disease? Alzheimers Dis 22:345–355. [DOI] [PubMed] [Google Scholar]
- 47. Wenk GL (2006) Neuropathologic changes in Alzheimer's disease: potential targets for treatment. J Clin Psychiatry 67:3–7. [PubMed] [Google Scholar]
- 48. Willis M, Leitner I, Jellinger KA, Marksteiner J (2011) Chromogranin peptides in brain diseases. J Neural Transm 118:727–735. [DOI] [PubMed] [Google Scholar]
- 49. Woronowicz A, Koshimizu H, Chang SY, Cawley NX, Hill JM, Rodriguiz RM et al (2008) Absence of carboxypeptidase E leads to adult hippocampal neuronal degeneration and memory deficits. Hippocampus 18:1051–1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Woronowicz A, Cawley NX, Chang SY, Koshimizu H, Phillips AW, Xiong ZG, Loh YP (2010) Carboxypeptidase E knockout mice exhibit abnormal dendritic arborization and spine morphology in central nervous system neurons. J Neurosci Res 88:64–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Zuccato C, Cattaneo E (2009) Brain‐derived neurotrophic factor in neurodegenerative diseases. Nat Rev Neurol 5:311–322. [DOI] [PubMed] [Google Scholar]