

Abstract
Stroke is one of the leading causes of death and disability worldwide. Inflammation plays a key role across the time course of stroke, from onset to the post‐injury reparative phase days to months later. Several regulatory molecules are implicated in inflammation, but the most established inflammatory mediator of acute brain injury is the cytokine interleukin‐1. Interleukin‐1 is regulated by large, macromolecular complexes called inflammasomes, which play a central role in cytokine release and cell death. In this review we highlight recent advances in inflammasome research and propose key roles for inflammasome components in the progression of stroke damage.
Keywords: inflammation, inflammasome, interleukin‐1, pyroptosis, NLRP3, ischemic stroke, hemorrhagic stroke
Introduction
Stroke is the second leading cause of death worldwide, affecting approximately 17 million people per annum 26. Of those affected by stroke, around two thirds either die or are left disabled 17. Two major subtypes of stroke exist and are defined by their etiology. Ischemic stroke occurs through embolic or thrombotic occlusion of a cerebral vessel, resulting in insufficient oxygen and glucose delivery required for cellular respiration. Hemorrhagic stroke occurs when a diseased or abnormal vessel ruptures, leading to the development of a neurotoxic hematoma within the brain. Ischemic strokes are most prevalent, accounting for 87% of all cases, with spontaneous non‐traumatic intracerebral hemorrhage (ICH) being the second most common subtype of stroke, accounting for around 12% of all stroke cases worldwide 25. Roughly 40% of ICH cases prove fatal and, of those that survive, less than 40% live independently at 12 months 4. It is becoming increasingly clear that inflammatory processes are inherent across the time course of stroke, from endothelial activation shortly after stroke onset (in both ischemic and hemorrhagic stroke) through to the post‐injury reparative phase days to months after stroke 41, 71. During the acute phase of stroke, sterile inflammation (ie, inflammation processes that occurs in the absence of pathogens) leads to the production of pro‐inflammatory cytokines, such as interleukin‐1 (IL‐1), which, in turn, propagate the neuroinflammatory response through activation of resident cells (ie, microglia) and recruitment of inflammatory cells. Neutrophils are the first cells to migrate into the brain after stroke, followed by macrophages and lymphocytes a few days after injury 34, 46. Moreover, inflammatory cell activation is not contained within the necrotic core of ischemic brain tissue but, instead, is observed within the peri‐infarct region. The size of this “inflammatory penumbra” positively correlates with the extent of brain damage and, consequently, is a key target for therapeutic intervention 33.
Minutes to hours after stroke, cell death leads to the induction of an inflammatory response through the release of danger signals termed damage associated molecular patterns (DAMPs). DAMPS are endogenous components which, under normal conditions, are either inert or sequestered intracellularly and thus not detected by immune cells. Examples of DAMPs released from dying cells include the chromatin‐associated protein HMGB1, purine metabolites such as ATP, heat shock proteins and uric acid. In addition to DAMPs, some intracellular stores of pro‐inflammatory cytokines can be released by necrotic cells such as IL‐1α 14. DAMPs trigger inflammatory responses through the activation of pattern recognition receptors (PRRs) present on or within innate immune cells. The activation of some cytosolic PRRs leads to the formation of large multiprotein complexes called inflammasomes. Inflammasomes are a type of intracellular PRR that combine pathogen associated molecular patterns (PAMPs) or DAMPs recognition to the activation of caspase‐1. The presence of oligomerization domains allows the inflammasome sensor molecules to seed large quaternary structures, which ultimately concentrate caspase‐1 peptides leading to auto‐cleavage and activation 87. The majority of inflammasomes contain an N‐terminal pyrin domain, which recruits the adaptor molecule apoptosis‐associated speck‐like protein containing a CARD (ASC) through homotypic pyrin domain interactions 87. In turn, ASC forms a large speck structure consisting of multimeric ASC dimers and recruits caspase‐1 to the assembling inflammasome complex via a C‐terminal CARD 28. Caspase‐1 then processes pro forms of the cytokines IL‐1β and IL‐18, and facilitates their release through a caspase‐1 dependent form of cell death termed pyroptosis 18. Each inflammasome senses PAMPs or DAMPs through unique, direct or indirect, interactions, allowing a multitude of molecular patterns to be sensed and acted on, some of which are implicated in chronic and acute brain injury 59, 90.
As a role for IL‐1 in stroke damage is discussed at length elsewhere 12, here we focus on the role of inflammasomes in the inflammatory and cell death processes seen in stroke.
Evidence for The Involvement of Inflammasomes in Stroke
Of the five known inflammasome forming proteins, to date, four have been cited in stroke literature, namely NOD‐, LRR‐ and PYD‐containing 1 (NLRP1), NLRP3, absent in melanoma 2 (AIM2), and NOD‐, LRR‐ and CARD‐containing 4 (NLRC4). It is important to note NLRP3 is unique amongst inflammasomes in that, at basal levels, its expression is insufficient to respond to activating stimuli 6. Thus, similar to IL‐1β the NLRP3 inflammasome has a two‐step processing system and must receive an initial priming signal which instigates NFkB signaling. The second signal is provided by stimuli that are unique to each inflammasome (discussed later) and cause the assembly of the inflammasome structure. Therefore, when critically analyzing inflammasome activity within tissues it is essential to have a true readout of inflammasome complex formation.
There are a number of studies highlighting upregulation of inflammasome components in various animal models of stroke (see Table 1). At the mRNA and protein level NLRP3 is significantly upregulated 12 hours after transient middle cerebral artery occlusion (tMCAo) in mice, peaking at 24 hours and remains elevated beyond 48 hours 95. Using the same model of stroke another group blotted for protein NLRP3, NLRP1 and the adaptor ASC, and found all to become elevated within one hour and remain above sham levels for 24 hours 97. Within the ischemic penumbra of rats subjected to tMCAo there is a steady rise in NLRP3, NLRP1, NLRC4, AIM2 and ASC mRNA over time, which remained at higher levels than shams up to 72 hours post‐surgery 58. In the permanent embolic MCAo (eMCAo) mouse model of stroke NLRP3 is expressed within the ischemic penumbra within 24 hours 43 and in a thromboembolic occlusion mouse model, ASC protein levels are significantly raised in cortical lysates 7 days post‐stroke 1. Data from both ICH and subarachnoid hemorrhage (SAH) stroke models indicate that the NLRP3 protein is upregulated at 12 hours and remains elevated beyond 24 hours 15, 20, 27, 70. Clearly, NLRP3 is the most cited inflammasome sensor molecule in pre‐clinical stroke research and has also been shown in the brain tissue of stroke patients 97. Whilst the expression of inflammasomes within the stroke brain does not indicate causative effects, it does highlight the presence of sensor molecules in a pathology heavily reliant on IL‐1 signalling. Some studies have taken inflammasome biology further by identifying the cellular distribution of inflammasomes in the brain post‐stroke.
Table 1.
Evidence for inflammasomes in stroke pathology.
| Inflammasome | Evidence of involvement in stroke | Cell type | Stroke model |
|---|---|---|---|
| NLRP3 | Increased mRNA and/or protein expression in stroke brains | Neurons 58, 97, microglia 54, 66 | Clinical ischemic brain tissue 97 |
| Rat and mouse tMCAo 23, 54, 58, 61, 66, 91, 97, 100, 101 | |||
| Mouse eMCAo 43 | |||
| Mouse autologous blood injection 70, 96, 98 | |||
| Rat collagenase injection 27 | |||
| Rat and mouse endovascular perforation 15, 20, 62, 88 | |||
| Smaller infarcts, edema, BBB breakdown, and neurological deficits in NLRP3−/− mice | Microglial and endothelial cells | Mouse tMCAo 95 | |
| Infiltrating leukocytes express high levels of NLRP3 and inhibition of the inflammasome lessens brain injury | Infiltrating leukocytes | Mouse tMCAo 44 | |
| NLRP1 | Increased mRNA and/or protein expression in stroke brains | Neurons | Rat and mouse tMCAo 23, 58 |
| The NLRP1 inflammasome complex was shown to form in neurons, whilst a neutralizing antibody reduces infarct size | Neurons, Astrocytes, Microglia | Mouse thromboembolic stroke 1 | |
| AIM2 | mRNA upregulation in the peri‐infarct region | NA | Rat tMCAo 58 |
| Smaller infarcts, less neurological deficits and dampened neuroinflammation in AIM2−/− mice | NA | Mouse tMCAo 19 | |
| NLRC4 | mRNA upregulation in the peri‐infarct region | NA | Rat tMCAo 58 |
| Smaller infarcts, less neurological deficits and dampened neuroinflammation in NLRC4−/− mice | NA | Mouse tMCAo 19 |
NOD‐ LRR‐ PYD‐containing (NLRP), Absent in melanoma 2 (AIM2), NOD‐ LRR‐ CARD‐containing 4 (NLRC4), transient (t), embolic (e), permanent (p) middle cerebral artery occlusion (MCAo), blood brain barrier (BBB).
The NLRP1 molecule was the first inflammasome sensor to be imaged within murine ischemic brain tissue, where it was found to be ubiquitously expressed in neurons and astrocytes, but specifically upregulated in microglia within 6 hours of thromboembolic stroke 1. In mice, NLRP3 has been localized to microglia and the endothelium of ischemic and hemorrhagic brain tissue 70, 95, whilst in rat tMCAo models, it was found to be upregulated in neurons 58. The most recent study concerning inflammasomes in stroke used flow cytometry to identify cells expressing NLRP3 44. In this study the authors used fluorescence activated cell sorting and found that NLRP3 mRNA was specifically expressed within infiltrating myeloid cells. It is well established that inflammasomes are functional in many cell types relevant to stroke—see review 60—though functional readouts for inflammasome activation in the brain are currently lacking.
Using size exclusion chromatography of ischemic brain lysates, an NLRP1 inflammasome complex was found in fractions above 600 kDa, along with the interacting molecules ASC, XIAP and caspase 1 1. NLRP1 was the first inflammasome to be characterized and is unique in its double death domain structure. The most established activator of NLRP1 is the Bacillus anthracis virulence factor and metalloproteinase, anthrax lethal toxin, which acts by cleaving the N‐terminal domain, causing oligomerization and caspase‐1 activation. It is suggested NLRP1 is present in neurons in a pre‐assembled state that is sensitized to neuronal stress, leading to activation under metabolic disruption 90. Interestingly, glucose starvation and hypoxia (both conditions of paramount importance to ischemic stroke pathology) have been shown to activate NLRP1 through intracellular ATP depletion 64. In a thromboembolic stroke model, the application of an anti‐NLRP1 antibody inhibited the formation of the NLRP1 inflammasome and reduced brain lesion volume by 18% 1. Since this finding in 2009 no further studies have addressed NLRP1 activation in stroke and focus has turned toward characterizing stroke pathology in transgenic animals.
NLRP3−/− mice subjected to tMCAo are protected from ischemic damage, with better functional outcomes and reduced inflammatory pathology 95. Lesion volumes and neurological deficit scores were reduced by around 40% in NLRP3 deficient mice, correlating with less endothelial damage and blood brain barrier (BBB) breakdown. This is contrary to results from Denes et al 19 which suggest NLRP3 does not contribute to MCAo injury in mice. The discrepancy between these results could be explained by differences in occlusion time causing modifications of the inflammatory response. Bruton's tyrosine kinase (BTK) physically associates with NLRP3 and ASC, and these interactions are essential for inflammasome activation in response to multiple stimuli 44. Both BTK and NLRP3 are expressed within infiltrating myeloid cells present in the ischemic penumbra 1 day post‐stroke. Moreover, pharmacological inhibition of BTK activity reduces lesion volumes in an IL‐1β specific manner. This finding suggests that infiltrating leukocytes express NLRP3 and that this directly contributes to ischemic brain injury. Many of the NLRP3 activating DAMPs are present within the ischemic brain. Oxygen depletion leads to a metabolic switch toward glycolysis, causing lactic acid and proton build up and subsequent extracellular acidosis. The NLRP3 inflammasome is able to sense acidic conditions through an uncharacterized mechanism leading to K+ efflux and formation of the inflammasome complex 79. Similarly, ATP is released by dying cells and is a well‐established activator of the NLRP3 inflammasome through P2X7R activation. The latter mechanism has been shown to be an important facet of hemorrhagic stroke. The P2X7R antagonist brilliant blue G is protective in both SAH and ICH models, resulting in smaller lesions, reduced neuronal death, and fewer infiltrating neutrophils 15, 27. One of the most injurious aspects of hemorrhagic stroke is the toxicity of parenchymal blood components 3. Interestingly, the red blood cell breakdown product haem activates the NLRP3 inflammasome through the production of mitochondrial ROS 21. In ICH, genetic deletion of the haem scavenging protein hemopexin accentuates brain injury, whereas, mitochondrial ROS scavengers reduce brain injury through NLRP3 downregulation 69, 70. A major predictor of mortality in ischemic and hemorrhagic stroke patients is the development of cerebral edema. Cytotoxic edema occurs foremost through accumulation of intracellular ions causing influx of water molecules. This cell swelling may also be a mechanism by which NLRP3 becomes active within the brain, as hypotonic environments are well established activators of the inflammasome 16. Common amongst studies investigating NLRP3 in preclinical stroke models is the lack of pre‐existing stroke co‐morbidities. The co‐morbidities commonly linked with stroke, such as obesity and atherosclerosis, have significant NLRP3‐dependent inflammatory changes 12. Whether these inflammatory changes systemically prime the NLRP3 inflammasome and affect stroke outcomes is unknown. However, it is known that hypertensive rats have a higher number of cells expressing NLRP3 in the brain compared with normotensive counterparts 66.
Our group have shown that both absent in melanoma 2 (AIM2) and NLRC4 contribute to damage after ischemic stroke, although the mechanisms leading to their involvement remain elusive 19. When subjected to MCAo, infarct volumes were significantly smaller in NLRC4−/− and AIM2−/− mice compared with wild type (WT) C57BL/6 mice. NLRC4−/− and AIM2−/− mice showed a dampened immune response to stroke, with fewer active microglia and leukocytes observed within the ipsilateral hemisphere. AIM2 is a cytosolic double‐stranded DNA (dsDNA) sensor that can react to both pathogenic and endogenous dsDNA. The AIM2 sensor molecule is composed of a pyrin domain and a DNA‐binding HIN domain, and thus recruitment of caspase‐1 to the inflammasome complex requires ASC 29, 40. How AIM2 becomes active during stroke is unclear. However, AIM2 contributes to autoinflammatory disease through recognition of endogenous dsDNA. Moreover, similar to NLRP3, the AIM2 inflammasome has recently been described as a sensor of glycolysis and could contribute to stroke damage through this pathway 93. NLRC4, on the other hand, possesses a CARD domain and can directly interact with procaspase‐1 to cause cell death, although the presence of ASC is required for cytokine processing 72. Bacterial PAMPs such as flagellin, and rod and needle units of bacterial type III secretion systems are the most cited activators of the NLRC4 inflammasome 30. NLRC4 responds to these exogenous molecules indirectly through interacting proteins termed NLR‐family, apoptosis inhibitory proteins (NAIPs). It is currently unclear whether NAIPs can respond to endogenous molecules, although NLRC4 is known to respond to hyperosmotic conditions 42, which may be present in the post‐stroke brain. Interestingly, NLRC4 does not respond to necrotic cell death 45 but its role in sterile inflammation is becoming increasingly apparent, with reported links to acute coronary syndrome 47, breast cancer progression and diabetic nephropathy 53, 99.
Caspase‐1 activation after stroke
Studies evaluating caspase 1 activity in the stroke affected brain also give some insight into inflammasome activity in the disease. Following a permanent model of stroke, the proteolytic activity of caspase‐1 is increased 30 minutes after occlusion with a second wave of activation 12 hours later 7. The increase of caspase‐1 expression has been described in neurons and astrocytes after thromboembolic stroke and observed later in microglia (24 h post stroke) 1. Other studies have demonstrated a role of caspase‐1 after stroke by using transgenic mice. Caspase‐1 knockout mice and mice expressing a dominant‐negative caspase‐1 exhibit a reduction of brain damage compared with wild type after experimental stroke 32, 86. Moreover, intra‐cerebroventricular administration of Ac‐YVAD‐cmk or VRT‐018858, both caspase‐1 inhibitors, provides protective effects in experimental stroke models 38, 78, 83. Beneficial effects of caspase‐1 inhibition are also observed in a model of oxygen and glucose deprivation in rat organotypic hippocampal slices 81. Altogether, these studies demonstrate a key role of caspase‐1 during stroke.
Caspase 11 and non‐canonical inflammasome after stroke
In addition to the canonical mode of inflammasome activation, caspase‐4 and −5 in humans and caspase‐11 in mice can participate in IL‐1/IL‐18 processing and cell death, through the “non‐canonical inflammasome” 36, 80. Initial studies showed that activation of caspase‐11 promoted subsequent activation of caspase‐1 and −3 and that caspase‐11 knockout mice exhibited markedly reduced apoptosis following a permanent model of MCAO 49. In more recent studies, caspase‐11 has been described to detect intracellular LPS and promote cell death and the release of IL‐1α 37, 52. Caspase‐11 can also lead to caspase‐1 dependent IL‐1β release and caspase‐1 independent pyroptosis via direct cleavage of gasdermin D (GSDMD—see below) by caspase‐11 51. However, the precise role of caspase‐11 in the context of sterile inflammation is not well understood and further studies are required in order to fully delineate the role of the non‐canonical inflammasome during stroke.
IL‐1 Independent Roles of Inflammasomes in Stroke
Pyroptosis
As shown in Figure 1, one of the defining features of inflammasome activation is the induction of a pro‐inflammatory programmed cell death termed pyro‐ (fire/fever) ptosis (falling). The two hallmark features of pyroptosis are DNA damage and membrane rupture, the former akin to apoptosis whilst the latter in complete contrast 8. Another distinction from apoptosis is the initiation of pyroptosis through the inflammatory caspases, namely caspase 1 or caspase 4/5/11, and the subsequent release of pro‐inflammatory cytosolic components. The process of pyroptosis is essential for the clearance of pathogens in vivo and has recently been shown to retain bacteria within intracellular traps during the lytic cell death process 48. With pyroptosis strongly linked with IL‐1 signaling, it may well have emerged as a vehicle to disseminate the now termed canonical DAMPs 73. We know these canonical DAMPs play implicit roles during sterile inflammation, yet it is still unclear how pyroptosis is involved in stroke.
Figure 1.
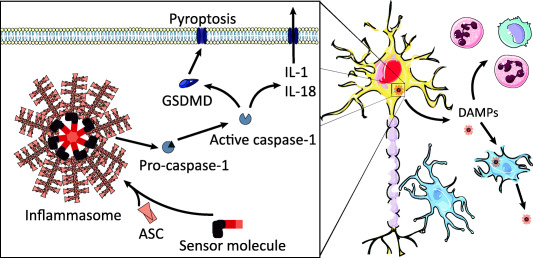
The detection of pro‐inflammatory stimuli leads to the formation of macromolecular inflammasome complexes. Sensor molecules react to an array of damage associated molecular patterns (DAMPs) by recruiting a scaffold protein termed apoptosis‐associated speck forming protein containing a CARD (ASC). ASC recruits the zymogen caspase‐1 causing auto‐cleavage and activation. Active caspase‐1 imparts a pro‐inflammatory lytic cell death termed pyroptosis through cleavage of gasdermin D (GSDMD). The N‐terminus of GSDMD forms pores in the plasma membrane which allows passage of proinflammatory cytokines, such as interleukin (IL‐) 1 and 18 that are also processed by inflammasomes. During stroke, inflammasomes contribute to brain injury through the spread of inflammation, leading to microglial activation and ingress of circulating immune cells. Inflammasomes themselves can be released during pyroptosis and propagate an inflammasome activation cascade within tissue.
The caspase 1/11 dependent cell death process may contribute enormously to damage post‐stroke independently of IL‐1 signaling. Caspase 1/11−/− mice have reduced infarcts after MCAo, yet IL‐1β deficient mice are not protected 10, 86. Pyroptosis may be the principal cell death mechanism of neurons within the ischemic core during stroke. Neurons are the earliest source of caspase 1 activation, where it has been identified to precede pro‐apoptotic caspase 3 activity 7. Moreover, the expression of a neuronal dominant negative caspase 1 is known to lessen stroke brain injury 32. The pyroptosome forming ASC protein has also been shown to substantially contribute to acute brain injury, with knockout mice exhibiting reduced infarct volumes and improved neurological outcome independently of IL‐1β levels 19. However, pyroptosis can occur irrespective of ASC pyroptosome assembly 13, thus the true extent of pyroptotic cell death cannot be inferred solely through the use of ASC‐deficient mice. Despite strong evidence implicating multiple inflammasomes in CNS disease 90, there is no conclusive evidence describing pyroptosis within the brain. More studies are required to evaluate the activity of caspase 11 and GSDMD during acute brain injury.
In 2015 multiple articles were released which described the previously uncharacterized GSDMD protein as an effector of pyroptosis 39, 50, 89. Cells lacking GSDMD were unable to undergo pyroptotic cell death or secrete canonically processed IL‐1β. Kayagaki et al (2015) found that, in the absence of GSDMD, IL‐1β failed to be processed when LPS was delivered intracellularly, suggesting GSDMD may also initiate non‐canonical inflammasome formation 50. Cleavage of GSDMD releases an N‐terminal fragment which has recently been shown to associate with the plasma membrane and form 13 nm pores 2, 67, 85. Interestingly, Shi et al (2015) showed GSDMD to be the effector of pyroptosis for all of the previously mentioned stroke‐related inflammasomes bar NLRP1 89, suggesting that inflammasomes formed within the brain induce pyroptosis through GSDMD. However, it is currently unclear if pyroptosis occurs within brain cells at all and whether it would follow the same route as described for myeloid cells.
Inflammasome dependent protein release
On top of the processing of IL‐1β, IL‐18 and GSDMD, caspase 1 activation also facilitates the release of other DAMPs. In fact, the principal output of caspase 1 activation may indeed be the induction of pyroptosis and the subsequent release of DAMPs. Genetic knockout of IL‐1β in mice still renders the species susceptible to endotoxemia 24. Whereas caspase 1/11−/− mice given an 800 µg dose of LPS do not enter septic shock and have a far greater survival rate than their WT counterparts 63. More recent evidence suggests caspase 11 is crucial for LPS induced sepsis and this may occur through HMGB1 signalling 51, 57, 92. HMGB1 released during pyroptosis is hyperacetylated at its nuclear localization sequence 68, which has previously been shown to cause cytoplasmic redistribution and facilitation of its secretion 9. Interestingly, HMGB1 levels are elevated in stroke patient serum and neutralizing antibodies are protective in different experimental models of stroke 65, 75.
Unlike IL‐1β, IL‐1α is biologically active in its pro‐form and its release is heavily dependent of cell death 22, 35. Despite lacking a caspase‐1 cleavage site, IL‐1α secretion can be triggered through inflammasome activation 35. In contrast to IL‐1β, inflammasome dependent IL‐1α release does not require membrane permeabilization 35, 74, suggesting that GSDMD cleavage is dispensable for its secretion. Despite this, IL‐1α secretion is proposed to heavily rely on caspase‐1 dependent processes when; (i) the stimulus does not cause intracellular calcium levels to rise, (ii) IL‐1α is sequestered by the cytosolic IL‐1RII 35, 77. Studies have shown there to be a substantial reduction in the amount of IL‐1α released from caspase1/11−/− macrophages and monocytes stimulated with LPS 56, 63, and this reduction in IL‐1α release is likely to be caspase 11 dependent 51. IL‐1α expression precedes IL‐1β in ischemic brain tissue and is known to be a critical mediator of acute brain inflammation, which is extensively reviewed elsewhere 11, 76. HMGB1 and IL‐1a posses the potential to cause a positive NLRP3 inflammasome signalling loop within the parenchyma post‐stroke, with the former associated with inflammasome activation and the latter in priming 94.
Self‐propagating inflammasomes
ASC speck formation may have pleiotropic roles in the propagation of inflammation, the best characterized being the aforementioned processing of zymogens and the induction of pyroptosis. There is some evidence to suggest the supramolecular ASC assembly is able to stably interact with hydrophobic molecules and may act to sequester bacterial PAMPs or endogenous DAMPs and deliver these pro‐inflammatory signals to neighboring phagocytes, which in the brain would be microglial cells 84.
During pyroptosis the inflammasome complex is released into the extracellular milieu where the platform can potentiate tissue inflammation 5, 31. Using in vitro assembled ASC specks, two groups have shown that specks are extremely stable and can activate extracellular caspase 1. Within the necrotic regions of the damaged stroke brain this would enable activated inflammasomes to capture any unprocessed zymogens released during necrosis, maximizing the inflammatory response in the process. ASC specks were described as having “prionoid” activity by Franklin et al 31 as they were shown to nucleate inflammasome formation when ingested by phagocytic cells. As previously discussed, there is evidence for inflammasome activation in stroke and ASC specks have been quantified within the ischemic penumbra 55. This data begs the question; what role do extracellular inflammasome complexes play in the stroked brain? Interestingly, there is evidence that inflammasome targeting antibodies lessen neuronal damage after stroke and traumatic brain injury in mice 1, 82. As antibodies cannot enter the cytosol of live cells, these neutralizing antibodies may be limiting the acute spread of inflammation via effects on extracellular inflammasomes. Both inflammasome complexes and anti‐ASC antibodies have been identified in the serum of patients suffering with inflammatory illnesses 5, 31, suggesting ASC specks can have long‐lasting physiological effects. As ASC is an implicit mediator of stroke damage, it may also propagate an acute injury into chronic inflammation, possibly contributing toward inflammatory‐driven post‐stroke sequelae such as depression and cognitive decline.
Conclusion
Sterile inflammation contributes enormously to brain damage during stroke. Inflammasomes, which are central effectors of sterile inflammation, are expressed and activated after stroke leading to IL‐1 processing and cell death through pyroptosis. The role of IL‐1 in the pathogenesis of stroke is well described, however inflammasome‐induced cell death in acute brain injury is less understood. Moreover, release of inflammasome complexes could represent an inflammasome dependent inflammatory drive after stroke. Further understanding of the molecular mechanisms of inflammasome activation within the brain after stroke is required in order to elucidate therapeutic targets. The development of therapeutic strategies to target inflammasomes will, one day, provide new opportunities in the management of stroke.
References
- 1. Abulafia DP, Vaccari JP, de R, Lozano JD, Lotocki G, Keane RW Dietrich WD (2009) Inhibition of the inflammasome complex reduces the inflammatory response after thromboembolic stroke in mice. J Cereb Blood Flow Metab 29:534–544. [DOI] [PubMed] [Google Scholar]
- 2. Aglietti RA, Estevez A, Gupta A, Gonzalez M, Liu PS, Kayagaki N et al (2016) GsdmD p30 elicited by caspase‐11 during pyroptosis forms pores in membranes. Proc Natl Acad Sci U S A 113:7858–7863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Aronowski J, Zhao X (2011) Molecular pathophysiology of cerebral hemorrhage: secondary brain injury. Stroke 42:1781–1786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Van Asch CJJ, Luitse MJA, Rinkel GE, van der Tweel I, Algra A, Klijn CJM (2010) Incidence, case fatality, and functional outcome of intracerebral haemorrhage overtime, according to age, sex, and ethnic origin: a systematic review and meta‐analysis. Lancet Neurol 9:167–176. [DOI] [PubMed] [Google Scholar]
- 5. Baroja‐Mazo A, Martin‐Sanchez F, Gomez AI, Martinez CM, Amores‐Iniesta J, Compan V et al (2014) The NLRP3 inflammasome is released as a particulate danger signal that amplifies the inflammatory response. Nat Immunol 15:738. [DOI] [PubMed] [Google Scholar]
- 6. Bauernfeind FG, Horvath G, Stutz A, Alnemri ES, MacDonald K, Speert D et al (2009) Cutting edge: NF‐kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J Immunol 183:787–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Benchoua a, Guégan C, Couriaud C, Hosseini H, Sampaïo N, Morin D et al (2001) Specific caspase pathways are activated in the two stages of cerebral infarction. J Neurosci 21:7127–7134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bergsbaken T, Fink SL, Cookson BT (2009) Pyroptosis: host cell death and inflammation. Nat Rev Microbiol 7:99–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bonaldi T, Talamo F, Scaffidi P, Ferrera D, Porto A, Bachi A et al (2003) Monocytic cells hyperacetylate chromatin protein HMGB1 to redirect it towards secretion. embo J 22:5551–5560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Boutin H, LeFeuvre RA, Horai R, Asano M, Iwakura Y, Rothwell NJ (2001) Role of IL‐1 alpha and IL‐1 beta in ischemic brain damage. J Neurosci 21:5528–5534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Brough D, Denes A (2015) Interleukin‐1alpha and brain inflammation. IUBMB Life 67:323–330. [DOI] [PubMed] [Google Scholar]
- 12. Brough D, Tyrrell PJ, Allan SM (2011) Regulation of interleukin‐1 in acute brain injury. Trends Pharmacol Sci 32:617–622. [DOI] [PubMed] [Google Scholar]
- 13. Broz P, von Moltke J, Jones JW, Vance RE, Monack DM (2010) Differential Requirement for Caspase‐1 Autoproteolysis in Pathogen‐Induced Cell Death and Cytokine Processing. Cell Host Microbe 8:471–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chen GY, Nuñez G (2010) Sterile inflammation: sensing and reacting to damage. Nat Rev Immunol 10:826–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chen S, Ma Q, Krafft PR, Hu Q, Rolland W, Sherchan P et al (2013) P2X7R/cryopyrin inflammasome axis inhibition reduces neuroinflammation after SAH. Neurobiol Dis 58:296–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Compan V, Baroja‐Mazo A, López‐Castejón G, Gomez AI, Martínez CM, Angosto D et al (2012) Cell volume regulation modulates nlrp3 inflammasome activation. Immunity 37:487–500. [DOI] [PubMed] [Google Scholar]
- 17. Corbyn Z (2014) A growing global burden. Nature 510:S2–S3. [DOI] [PubMed] [Google Scholar]
- 18. Denes A, Lopez‐Castejon G, Brough D (2012) Caspase‐1: is IL‐1 just the tip of the ICEberg?. Cell Death Dis 3:e338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Denes A, Coutts G, Lénárt N, Cruickshank SM, Pelegrin P, Skinner J et al (2015) AIM2 and NLRC4 inflammasomes contribute with ASC to acute brain injury independently of NLRP3. Proc Natl Acad Sci U S A 112:4050–4055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Dong Y, Fan C, Hu W, Jiang S, Ma Z, Yan X et al (2016) Melatonin attenuated early brain injury induced by subarachnoid hemorrhage via regulating NLRP3 inflammasome and apoptosis signaling. J Pineal Res 60:253–262. [DOI] [PubMed] [Google Scholar]
- 21. Dutra FF, Alves LS, Rodrigues D, Fernandez PL, de Oliveira RB, Golenbock DT et al (2014) Hemolysis‐induced lethality involves inflammasome activation by heme. Proc Natl Acad Sci U S A 111:E4110–E4118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. England H, Summersgill HR, Edye ME, Rothwell NJ, Brough D (2014) Release of interleukin‐1 alpha or interleukin‐1 beta depends on mechanism of cell death. J Biol Chem 289:15942–15950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Fann DY‐W, Santro T, Manzanero S, Widiapradja A, Cheng Y‐L, Lee S‐Y et al (2014) Intermittent fasting attenuates inflammasome activity in ischemic stroke. Exp Neurol 257:17. [DOI] [PubMed] [Google Scholar]
- 24. Fantuzzi G, Zheng H, Faggioni R, Benigni F, Ghezzi P, Sipe JD et al (1996) Effect of endotoxin in IL‐1 beta‐deficient mice. J Immunol 157:291–296. [PubMed] [Google Scholar]
- 25. Feigin VL, Lawes CMM, Bennett DA, Anderson CS (2003) Stroke epidemiology: a review of population‐based studies of incidence, prevalence, and case‐fatality in the late 20th century. Lancet Neurol 2:43–53. [DOI] [PubMed] [Google Scholar]
- 26. Feigin VL, Forouzanfar MH, Krishnamurthi R, Mensah G. a, Connor M, Bennett D et al (2014) Global and regional burden of stroke during 1990‐2010: findings from the global burden of disease study 2010. Lancet 383:245–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Feng L, Chen Y, Ding R, Fu Z, Yang S, Deng X et al (2015) P2X7R blockade prevents NLRP3 inflammasome activation and brain injury in a rat model of intracerebral hemorrhage: involvement of peroxynitrite. J Neuroinflamm 12:190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Fernandes‐Alnemri T, Wu J, Yu J‐W, Datta P, Miller B, Jankowski W et al (2007) The pyroptosome: a supramolecular assembly of ASC dimers mediating inflammatory cell death via caspase‐1 activation. Cell Death Differ 14:1590–1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Fernandes‐Alnemri T, Yu J‐W, Datta P, Wu J, Alnemri ES (2009) AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature 458:509–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Franchi L, Kamada N, Nakamura Y, Burberry A, Kuffa P, Suzuki S et al (2012) NLRC4‐driven production of IL‐1β discriminates between pathogenic and commensal bacteria and promotes host intestinal defense. Nat Immunol 13:449–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Franklin BS, Bossaller L, De Nardo D, Ratter JM, Stutz A, Engels G et al (2014) The adaptor ASC has extracellular and “prionoid” activities that propagate inflammation. Nat Immunol 15:727–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Friedlander RM, Gagliardini V, Hara H, Fink KB, Li W, MacDonald G et al (1997) Expression of a dominant negative mutant of interleukin‐1 beta converting enzyme in transgenic mice prevents neuronal cell death induced by trophic factor withdrawal and ischemic brain injury. J Exp Med 185:933–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Gauberti M, Montagne A, Marcos‐Contreras OA, Le Béhot A, Maubert E, Vivien D (2013) Ultra‐sensitive molecular MRI of vascular cell adhesion molecule‐1 reveals a dynamic inflammatory penumbra after strokes. Stroke 44:1988–1996. [DOI] [PubMed] [Google Scholar]
- 34. Gelderblom M, Leypoldt F, Steinbach K, Behrens D, Choe C‐U, Siler DA et al (2009) Temporal and spatial dynamics of cerebral immune cell accumulation in stroke. Stroke 40:1849–1857. [DOI] [PubMed] [Google Scholar]
- 35. Groß O, Yazdi AS, Thomas CJ, Masin M, Heinz LX, Guarda G et al, (2012) Inflammasome activators induce interleukin‐1α secretion via distinct pathways with differential requirement for the protease function of caspase‐1. Immunity 36:388–400. [DOI] [PubMed] [Google Scholar]
- 36. Guo H, Callaway JB, Ting JP (2015) Inflammasomes: mechanism of action, role in disease, and therapeutics. Nat Med 21:677–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hagar JA, Powell DA, Aachoui Y, Ernst RK, Miao EA (2013) Cytoplasmic LPS activates caspase‐11: implications in TLR4‐independent endotoxic shock. Science 341:1250–1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hara H, Friedlander RM, Gagliardini V, Ayata C, Fink K, Huang Z et al (1997) Inhibition of interleukin 1beta converting enzyme family proteases reduces ischemic and excitotoxic neuronal damage. Proc Natl Acad Sci U S A 94:2007–2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. He W‐T, Wan H, Hu L, Chen P, Wang X, Huang Z et al (2015) Gasdermin D is an executor of pyroptosis and required for interleukin‐1β secretion. Cell Res 25:1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hornung V, Ablasser A, Charrel‐Dennis M, Bauernfeind F, Horvath G, Caffrey DR et al (2009) AIM2 recognizes cytosolic dsDNA and forms a caspase‐1‐activating inflammasome with ASC. Nature 458:514–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Iadecola C, Anrather J (2011) The immunology of stroke: from mechanisms to translation. Nat Med 17:796–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ip WKE, Medzhitov R (2015) Macrophages monitor tissue osmolarity and induce inflammatory response through NLRP3 and NLRC4 inflammasome activation. Nat Commun 6:6931–6942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ishrat T, Mohamed IN, Pillai B, Soliman S, Fouda AY, Ergul A et al (2015) Thioredoxin‐interacting protein: a novel target for neuroprotection in experimental thromboembolic stroke in mice. Mol Neurobiol 51:766–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ito M, Shichita T, Okada M, Komine R, Noguchi Y, Yoshimura A et al (2015) Bruton's tyrosine kinase is essential for NLRP3 inflammasome activation and contributes to ischaemic brain injury. Nat Commun 6:8360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Iyer SS, Pulskens WP, Sadler JJ, Butter LM, Teske GJ, Ulland TK et al (2009) Necrotic cells trigger a sterile inflammatory response through the Nlrp3 inflammasome. Proc Natl Acad Sci U S A 106:20388–20393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Jin R, Yang G, Li G (2010) Inflammatory mechanisms in ischemic stroke: role of inflammatory cells. J Leukoc Biol 87:779–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Johansson Å, Eriksson N, Becker RC, Storey RF, Himmelmann A, Hagström E et al (2015) NLRC4 inflammasome is an important regulator of interleukin‐18 levels in patients with acute coronary syndromes: genome‐wide association study in the platelet inhibition and patient outcomes trial (PLATO). Circ Cardiovasc Genet 8:498–506. [DOI] [PubMed] [Google Scholar]
- 48. Jorgensen I, Zhang Y, Krantz BA, Miao EA (2016) Pyroptosis triggers pore‐induced intracellular traps (PITs) that capture bacteria and lead to their clearance by efferocytosis. J Exp Med 213:2113–2128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kang SJ, Wang S, Hara H, Peterson EP, Namura S, Amin‐Hanjani S et al (2000) Dual role of caspase‐11 in mediating activation of caspase‐1 and caspase‐3 under pathological conditions. J Cell Biol 149:613–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kayagaki N, Stowe IB, Lee BL, O'Rourke K, Anderson K, Warming S et al (2015) Caspase‐11 cleaves gasdermin D for non‐canonical inflammasome signalling. Nature 526:666–671. [DOI] [PubMed] [Google Scholar]
- 51. Kayagaki N, Warming S, Lamkanfi M, Walle L, Vande Louie S, Dong J et al (2011) Non‐canonical inflammasome activation targets caspase‐11. Nature 479:117–121. [DOI] [PubMed] [Google Scholar]
- 52. Kayagaki N, Wong MT, Stowe IB, Ramani SR, Gonzalez LC, Akashi‐Takamura S et al (2013) Noncanonical inflammasome activation by intracellular LPS independent of TLR4. Science 341:1246–1249. [DOI] [PubMed] [Google Scholar]
- 53. Kolb R, Phan L, Borcherding N, Liu Y, Yuan F, Janowski AM et al (2016) Obesity‐associated NLRC4 inflammasome activation drives breast cancer progression. Nat Commun 7:13007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kono S, Kurata T, Sato K, Omote Y, Hishikawa N, Yamashita T et al (2015) Neurovascular protection by telmisartan via reducing neuroinflammation in stroke‐resistant spontaneously hypertensive rat brain after ischemic stroke. J Stroke Cerebrovasc Dis 24:537–547. [DOI] [PubMed] [Google Scholar]
- 55. Krey L, Lühder F, Kusch K, Czech‐Zechmeister B, Könnecke B, Fleming Outeiro T et al (2015) Knockout of silent information regulator 2 (SIRT2) preserves neurological function after experimental stroke in mice. J Cereb Blood Flow Metab 35:2080–2088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Kuida Lippke JA, Ku G, Harding MW, Livingston DJ, Su MSS, Flavell RAK (1995) Altered cytokine export and apoptosis in mice deficient in interleukin‐1β converting enzyme. Science 267:2000–2003. [DOI] [PubMed] [Google Scholar]
- 57. Lamkanfi M, Sarkar A, Vande Walle L, Vitari AC, Amer AO, Wewers MD et al (2010) Inflammasome‐dependent release of the alarmin HMGB1 in endotoxemia. J Immunol 185:4385–4392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Lammerding L, Slowik A, Johann S, Beyer C, Zendedel A (2016) Poststroke Inflammasome expression and regulation in the peri‐infarct area by gonadal steroids after transient focal ischemia in the rat brain. Neuroendocrinology 103:460–475. [DOI] [PubMed] [Google Scholar]
- 59. Latz E, Xiao TS, Stutz A (2013) Activation and regulation of the inflammasomes. Nat Rev Immunol 13:397–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Lénárt N, Brough D, Dénes Á (2016) Inflammasomes link vascular disease with neuroinflammation and brain disorders. J Cereb Blood Flow Metab 36:1668–1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Li C, Wang J, Fang Y, Liu Y, Chen T, Sun H et al (2016) Nafamostat mesilate improves function recovery after stroke by inhibiting neuroinflammation in rats. Brain Behav Immun 56:230–245. [DOI] [PubMed] [Google Scholar]
- 62. Li J, Chen J, Mo H, Chen J, Qian C, Yan F et al (2016) Minocycline protects against nlrp3 inflammasome‐induced inflammation and p53‐associated apoptosis in early brain injury after subarachnoid hemorrhage. Mol Neurobiol 53:2668–2678. [DOI] [PubMed] [Google Scholar]
- 63. Li P, Allen H, Banerjee S, Franklin S, Herzog L, Johnston C et al (1995) Mice deficient in IL‐1??‐converting enzyme are defective in production of mature IL‐1?? and resistant to endotoxic shock. Cell 80:401–411. [DOI] [PubMed] [Google Scholar]
- 64. Liao KC, Mogridge J (2013) Activation of the Nlrp1b inflammasome by reduction of cytosolic ATP. Infect Immun 81:570–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Liu K, Mori S, Takahashi HK, Tomono Y, Wake H, Kanke T et al (2007) Anti‐high mobility group box 1 monoclonal antibody ameliorates brain infarction induced by transient ischemia in rats. faseb J 21:3904–3916. [DOI] [PubMed] [Google Scholar]
- 66. Liu W, Yamashita T, Kurata T, Kono S, Hishikawa N, Deguchi K et al (2015) Protective effect of telmisartan on neurovascular unit and inflammasome in stroke‐resistant spontaneously hypertensive rats. Neurol Res 37:491–501. [DOI] [PubMed] [Google Scholar]
- 67. Liu X, Zhang Z, Ruan J, Pan Y, Magupalli VG, Wu H et al (2016) Inflammasome‐activated gasdermin D causes pyroptosis by forming membrane pores. Nature 535:. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Lu B, Nakamura T, Inouye K, Li J, Tang Y, Lundback P et al (2012) Novel role of PKR in inflammasome activation and HMGB1 release. Nature 488:670–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Ma B, Day JP, Phillips H, Slootsky B, Tolosano E, Doré S (2016) Deletion of the hemopexin or heme oxygenase‐2 gene aggravates brain injury following stroma‐free hemoglobin‐induced intracerebral hemorrhage. J Neuroinflamm 13:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Ma Q, Chen S, Hu Q, Feng H, Zhang JH, Tang J (2014) NLRP3 inflammasome contributes to inflammation after intracerebral hemorrhage. Ann Neurol 75:209–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Macrez R, Ali C, Toutirais O, Le Mauff B, Defer G, Dirnagl U et al (2011) Stroke and the immune system: from pathophysiology to new therapeutic strategies. Lancet Neurol 10:471–480. [DOI] [PubMed] [Google Scholar]
- 72. Mariathasan S, Newton K, Monack DM, Vucic D, French DM, Lee WP et al (2004) Differential activation of the inflammasome by caspase‐1 adaptors ASC and Ipaf. Nature 430:213–218. [DOI] [PubMed] [Google Scholar]
- 73. Martin SJ (2016) Cell death and inflammation: the case for IL‐1 family cytokines as the canonical DAMPs of the immune system. Febs J 283:2599–615. [DOI] [PubMed] [Google Scholar]
- 74. Martín‐sánchez F, Diamond C, Zeitler M, Gomez‐sanchez A, Peñalver M, Paszek P et al (2016) Inflammasome‐dependent IL‐1 β release depends upon membrane permeabilisation. Cell Death Differ 1:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Muhammad S, Barakat W, Stoyanov S, Murikinati S, Yang H, Tracey KJ et al (2008) The HMGB1 receptor RAGE mediates ischemic brain damage. J Neurosci 28:12023–12031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Murray KN, Parry‐Jones AR, Allan SM (2015) Interleukin‐1 and acute brain injury. Front Cell Neurosci 9:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Di Paolo NC, Shayakhmetov DM (2016) Interleukin 1α and the inflammatory process. Nat Immunol 17:906–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Rabuffetti M, Sciorati C, Tarozzo G, Clementi E, Manfredi aa, Beltramo M (2000) Inhibition of caspase‐1‐like activity by Ac‐Tyr‐Val‐Ala‐Asp‐chloromethyl ketone induces long‐lasting neuroprotection in cerebral ischemia through apoptosis reduction and decrease of proinflammatory cytokines. J Neurosci 20:4398–4404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Rajamäki K, Nordström T, Nurmi K, Åkerman KEO, Kovanen PT, Öörni K et al (2013) Extracellular acidosis is a novel danger signal alerting innate immunity via the NLRP3 inflammasome. J Biol Chem 288:13410–13419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Rathinam VAK, Fitzgerald KA (2016) Inflammasome complexes: emerging mechanisms and effector functions. Cell 165:792–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Ray AM, Owen DE, Evans ML, Davis JB, Benham CD (2000) Caspase inhibitors are functionally neuroprotective against oxygen glucose deprivation induced CA1 death in rat organotypic hippocampal slices. Brain Res 867:62–69. [DOI] [PubMed] [Google Scholar]
- 82. De Rivero Vaccari JP, Lotocki G, Alonso OF, Bramlett HM, Dietrich WD, Keane RW (2009) Therapeutic neutralization of the NLRP1 inflammasome reduces the innate immune response and improves histopathology after traumatic brain injury. J Cereb Blood Flow Metab 29:1251–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Ross J, Brough D, Gibson RM, Loddick SA, Rothwell NJ (2007) A selective, non‐peptide caspase‐1 inhibitor, VRT‐018858, markedly reduces brain damage induced by transient ischemia in the rat. Neuropharmacology 53:638–642. [DOI] [PubMed] [Google Scholar]
- 84. Sahillioʇlu AC, Özören N (2015) Artificial loading of ASC specks with cytosolic antigens. PLoS One 10:e0134912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Sborgi L, Rühl S, Mulvihill E, Pipercevic J, Heilig R (2016) GSDMD pore formation in the plasma membrane constitutes the mechanism of pyroptotic cell death. embo J 1:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Schielke GP, Yang GY, Shivers BD, Betz AL (1998) Reduced ischemic brain injury in interleukin‐1 beta converting enzyme‐deficient mice. J Cereb Blood Flow Metab 18:180–185. [DOI] [PubMed] [Google Scholar]
- 87. Schroder K, Tschopp J (2010) The inflammasomes. Cell 140:821–832. [DOI] [PubMed] [Google Scholar]
- 88. Shao A, Wu H, Hong Y, Tu S, Sun X, Wu Q et al (2016) Hydrogen‐rich saline attenuated subarachnoid hemorrhage‐induced early brain injury in rats by suppressing inflammatory response: possible involvement of nf‐κb pathway and nlrp3 inflammasome. Mol Neurobiol 53:3462–3476. [DOI] [PubMed] [Google Scholar]
- 89. Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H et al (2015) Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 526:660–665. [DOI] [PubMed] [Google Scholar]
- 90. Walsh JG, Muruve DA, Power C (2014) Inflammasomes in the CNS. Nat Rev Neurosci 15:84–97. [DOI] [PubMed] [Google Scholar]
- 91. Wang X, Li R, Wang X, Fu Q, Ma S (2015) Umbelliferone ameliorates cerebral ischemia–reperfusion injury via upregulating the PPAR gamma expression and suppressing TXNIP/NLRP3 inflammasome. Neurosci Lett 600:16. [DOI] [PubMed] [Google Scholar]
- 92. Willingham SB, Bergstralh DT, O'Connor W, Morrison AC, Taxman DJ, Duncan JA et al (2007) Microbial pathogen‐induced necrotic cell death mediated by the inflammasome components CIAS1/cryopyrin/NLRP3 and ASC. Cell Host Microbe 2:147–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Xie M, Yu Y, Kang R, Zhu S, Yang L, Zeng L et al (2016) PKM2‐dependent glycolysis promotes NLRP3 and AIM2 inflammasome activation. Nat Commun 7:3280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Xu J, Jiang Y, Wang J, Shi X, Liu Q, Liu Z et al (2014) Macrophage endocytosis of high‐mobility group box 1 triggers pyroptosis. Cell Death Differ 21:1229–1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Yang F, Wang Z, Wei X, Han H, Meng X, Zhang Y et al (2014) NLRP3 deficiency ameliorates neurovascular damage in experimental ischemic stroke. J Cereb Blood Flow Metab 34:660–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Yang Z, Zhong L, Xian R, Yuan B (2015) MicroRNA‐223 regulates inflammation and brain injury via feedback to NLRP3 inflammasome after intracerebral hemorrhage. Mol Immunol 65:267–276. [DOI] [PubMed] [Google Scholar]
- 97. Yang‐Wei Fann D, Lee S‐Y, Manzanero S, Tang S‐C, Gelderblom M, Chunduri P et al (2013) Intravenous immunoglobulin suppresses NLRP1 and NLRP3 inflammasome‐mediated neuronal death in ischemic stroke. Cell Death Dis 4:e790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Yuan B, Shen H, Lin L, Su T, Zhong S, Yang Z (2015) Recombinant adenovirus encoding NLRP3 RNAi attenuate inflammation and brain injury after intracerebral hemorrhage. J Neuroimmunol 287:71–75. [DOI] [PubMed] [Google Scholar]
- 99. Yuan F, Kolb R, Pandey G, Li W, Sun L, Liu F et al (2016) Involvement of the NLRC4‐inflammasome in diabetic nephropathy. PLoS One 11:e0164135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Zhang N, Zhang X, Liu X, Wang H, Xue J, Yu J et al (2014) Chrysophanol inhibits NALP3 inflammasome activation and ameliorates cerebral ischemia/reperfusion in mice. Mediat Inflamm 2014:370530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Zhao J, Mou Y, Bernstock JD, Klimanis D, Wang S, Spatz M et al (2015) Synthetic oligodeoxynucleotides containing multiple telemeric ttaggg motifs suppress inflammasome activity in macrophages subjected to oxygen and glucose deprivation and reduce ischemic brain injury in stroke‐prone spontaneously hypertensive rats. PLoS One 10:e0140772. [DOI] [PMC free article] [PubMed] [Google Scholar]