

Abstract
Antibodies to aquaporin‐4 (called NMO‐IgG or AQP4‐Ab) constitute a sensitive and highly specific serum marker of neuromyelitis optica (NMO) that can facilitate the differential diagnosis of NMO and classic multiple sclerosis. NMO‐IgG/AQP4‐Ab seropositive status has also important prognostic and therapeutic implications in patients with isolated longitudinally extensive myelitis (LETM) or optic neuritis (ON). In this article, we comprehensively review and critically appraise the existing literature on NMO‐IgG/AQP4‐Ab testing. All available immunoassays—including tissue‐based (IHC), cell‐based (ICC, FACS) and protein‐based (RIPA, FIPA, ELISA, Western blotting) assays—and their differential advantages and disadvantages are discussed. Estimates for sensitivity, specificity, and positive and negative likelihood ratios are calculated for all published studies and accuracies of the various immunoassay techniques compared. Subgroup analyses are provided for NMO, LETM and ON, for relapsing vs. monophasic disease, and for various control groups (eg, MS vs. other controls). Numerous aspects of NMO‐IgG/AQP4‐Ab testing relevant for clinicians (eg, impact of antibody titers and longitudinal testing, indications for repeat testing, relevance of CSF testing and subclass analysis, NMO‐IgG/AQP4‐Ab in patients with rheumatic diseases) as well as technical aspects (eg, AQP4‐M1 vs. AQP4‐M23‐based assays, intact AQP4 vs. peptide substrates, effect of storage conditions and freeze/thaw cycles) and pitfalls are discussed. Finally, recommendations for the clinical application of NMO‐IgG/AQP4‐Ab serology are given.
Keywords: aquaporin‐4 antibodies, brainstem encephalitis, Devic disease, Devic syndrome, diagnosis, diagnostic assays, immunoassays, inter‐assay comparison, longitudinally extensive transverse myelitis, neuromyelitis optica, NMO‐IgG, NMO spectrum disorders, optic neuritis, serology, Sjögren syndrome, systemic lupus erythematosus
Introduction
Neuromyelitis optica (NMO) is a severely disabling inflammatory disorder of the central nervous system (CNS) of putative autoimmune etiology which predominantly affects the optic nerves and the spinal cord 14, 29, 42, 69, 147, 155, 156. NMO usually follows a relapsing course without marked remission between relapses, and accumulation of irreversible deficits and rapid progression of disability are thus frequent 69, 156. NMO was first described in the 19th century and for many decades was considered a clinical subtype of multiple sclerosis (MS) 44, 46, 47, 48, 49, 50. However, in 2004, Lennon et al described a novel serum IgG autoantibody in a subset of patients with NMO binding to astrocytic endfeet adjacent to the microvasculature, the Virchow‐Robin spaces and the pia mater 90. Subsequently, aquaporin‐4 (AQP4), the most abundant water channel in the CNS, was identified as the target antigen 91. This antibody (termed NMO‐IgG or AQP4‐Ab) was found almost exclusively in patients with NMO and its formes frustes but not in patients with classical MS 90, 91. This finding together with evidence from histopathological and immunological studies (including passive transfer experiments in animal models) supports the concept of NMO as a humorally mediated autoimmune disease in NMO‐IgG/AQP4‐Ab‐positive patients that is pathogenetically distinct from MS 43, 57. In the present review, we give an overview of the diagnostic tests currently available for the detection of NMO‐IgG/AQP4‐Ab and critically appraise their limitations.
Disorders Associated with NMO‐IgG/AQP4‐Ab
Apart from classical NMO, NMO‐IgG/AQP4‐Ab have been found in patients with Asian opticospinal MS (OSMS) 90, 106, 107, 116, 141, 142, 153, in patients with isolated longitudinally extensive transverse myelitis (LETM) 154, in patients with isolated optic neuritis (ON) 61, 105, 124 and in rare patients with isolated brainstem encephalitis (mainly affecting the medulla oblongata) 69, 71, 143, diencephalitis (mainly affecting the hypothalamus) 69, 127 or posterior reversible encephalopathy 39, 101. NMO‐IgG/AQP4‐Ab have been demonstrated also in patients with NMO and supratentorial brain lesions, some of whom even met the magnetic resonance imaging (MRI) criteria for MS 69, 79, 108, 126; such lesions had previously been considered an exclusion criterion for NMO 156. The discovery of NMO‐IgG/AQP4‐Ab and the demonstration of brain lesions in patients with NMO resulted in a revision of the diagnostic criteria for NMO in 2006 52, 157.
The spectrum of clinico‐radiological findings associated with NMO‐IgG/AQP4‐Ab may be even broader in children. Whereas most brain lesions in adults with NMO‐IgG/AQP4‐Ab remain clinically silent 126, the largest pediatric study thus far performed in children found episodic cerebral symptoms in 45% of NMO‐IgG/AQP4‐Ab‐positive patients, including encephalopathy, seizures, ataxia, ophthalmoparesis, intractable vomiting and hiccups 111. Another study reported brain or brainstem symptoms in five out of seven NMO‐IgG/AQP4‐Ab‐positive children 97. No major difference in the seroprevalence of the antibody was found between adults and children 6, 97.
Spinal cord lesions usually extend over three or more segments in patients with NMO 69, 157. However, several studies have shown that short lesions occasionally occur in NMO‐IgG/AQP4‐Ab‐positive NMO, in particular if MRI is performed very early during lesion evolution or as a residual sign denoting lesion resolution 69, 137, 157. However, the overall frequency of NMO‐IgG/AQP4‐Ab among patients with non‐longitudinally extensive myelitis (NETM) is very low (Supporting Information Table S1).
As the presence of NMO‐IgG/AQP4‐Ab in all of these conditions and the high rate of conversion of NMO‐IgG/AQP4‐Ab‐positive patients with LETM 154, ON 105 or brainstem encephalitis 69 to clinically definite NMO suggests a shared pathogenesis, it has been proposed to subsume these disorders under the title of “AQP4 autoimmune channelopathies” or “AQP4 encephalomyelitis” 49. Others proposed to refer to these disorders as “limited or inaugural forms of NMO”, “high risk syndromes for NMO” (HRS) or “NMO spectrum disorders” (NMOSD) 49, 158. However, the etiopathogenesis of NMO‐IgG/AQP4‐Ab‐negative LETM, ON and brainstem encephalitis is heterogeneous and not all NMO‐IgG/AQP4‐Ab‐negative patients convert to NMO 49.
Importantly, NMO‐IgG/AQP4‐Ab have also been found in patients with NMOSD in the setting of connective tissue disorders (CTD) such as systemic lupus erythematosus or Sjögren syndrome with roughly the same frequency as in patients with uncomplicated NMOSD, but not in patients with CTD without NMOSD 64, 123, 128 or with CTD and neurological symptoms other than NMOSD 64, 161. Although a contribution of pathomechanisms associated with CTD such as vasculitis cannot be fully ruled out, the latter findings suggest that NMOSD might be independently caused by NMO‐IgG/AQP4‐Ab in these patients. The strong association of NMO‐IgG/AQP4‐Ab‐positive NMO with CTD suggests that the two conditions might arise from the same general autoimmune predisposition. Similarly, NMO‐IgG/AQP4‐Ab have been occasionally described in association with a number of further autoimmune disorders associated with NMOSD such as, among others, myasthenia gravis (MG) 53, 67, 77, 112 or celiac disease 40, 56, 69.
Laboratory Tests for NMO‐IgG/AQP4‐Ab
To date, almost 60 studies reporting on more than 40 different immunoassays for the detection of NMO‐IgG/AQP4‐Ab in patients with NMO have been described in the English literature. While some are in‐house assays, others are commercially available. The sensitivity of these assays, which employ different immunological techniques, varies broadly, but virtually all confirm a high specificity of the antibody for NMO. A comparison of results obtained in various studies evaluating these tests can be found in Tables 1, 2, 3 and Supporting Information Table S1. Depending on the diagnostic substrates used, those tests can be divided into tissue‐based assays, cell‐based assays and protein‐based assays.
Table 1.
Tissue‐based assays. Sensitivities, specificities and positive and negative likelihood ratios; results from 34 studies. Abbreviations: NMO = neuromyelitis optica; MS = multiple sclerosis; OD = neurological and non‐neurological disease controls other than MS; HC = healthy controls; pLR = positive likelihood ratio; nLR = negative likelihood ratio; CI = 95% confidence interval
| Assay subtype | Sensitivity (%, CI, N) NMO | Specificity (%, CI, N) NMO vs. all controls | Specificity (%, CI, N) NMO vs. MS | Specificity (%, CI, N) NMO vs. OD + HC | PLR (CI) NMO vs. MS | PLR (CI) NMO vs. OD + HC | NLR (CI) NMO vs. MS | NLR (CI) NMO vs. OD + HC |
|---|---|---|---|---|---|---|---|---|
| Fluoroimmunohistochemistry (IHC‐F) | ||||||||
| 1. Original assay | ||||||||
| Lennon et al 90 | 73.3 (57.8–84.9), 45 | 97.94 (92.03–99.64), 97 | 95.12 (82.19–99.15), 41 | 100 (92–100), 56 | 15 (3.8–58.6) | ∞ (n.a.‐n.a.) | 0.28 (0.17–0.46) | 0.27 (0.17–0.44) |
| Wingerchuk et al 157 | 76.1 (65.7–84.3), 88 | 93.75 (77.78–98.91), 32 | 93.75 (77.78–98.91), 32 | n.d. | 12.2 (3.2–46.9) | n.d. | 0.25 (0.17–0.37) | n.d. |
| Weinshenker et al 154 | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. |
| Matiello et al 105 | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. |
| Scott et al 137 | 75 (21.9–98.7), 4 | 96.43 (79.76–99.81), 28 | 100 (51.68–98.45), 6 | n.d. | ∞ (n.a.‐n.a.) | n.d. | 0.25 (0.05–1.36) | n.d. |
| Adoni et al 1 | 64.3 (44.1–80.7), 28 | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. |
| Smith et al 140 | n.d. | 100 (96.42–100), 130 | 100 (96.42–99.93), 130 | n.d. | n.d. | n.d. | n.d. | n.d. |
| McKeon et al 110 | 57.5 (41–72.6), 40 | 99.71 (98.85–99.95), 695 | 100 (98.11–99.96), 249 | 99.48 (97.91–99.91), 382 | ∞ (n.a.‐n.a.) | 110 (26.9–449) | 0.43 (0.3–0.62) | 0.43 (0.3–0.62) |
| Kalluri et al 73 | 63.6 (31.6–87.6), 11 | 66.67 (12.53–98.23), 3 | 66.67 (12.53–98.23), 3 | n.d. | 1.9 (0.4–10) | n.d. | 0.55 (0.18–1.68) | n.d. |
| Petzold et al 124 | 55.6 (22.7–84.7), 9 | 100 (84.98–100), 28 | 100 (84.98–99.67), 28 | n.d. | ∞ (n.a.‐n.a.) | n.d. | 0.44 (0.21–0.91) | n.d. |
| Waters et al 152 | 48.6 (31.7–65.7), 35 | 100 (94.61–100), 85 | 100 (88.83–99.77), 39 | 100 (90–100), 44 | ∞ (n.a.‐n.a.) | ∞ (n.a.‐n.a.) | 0.51 (0.37–0.7) | 0.51 (0.37–0.7) |
| Sum | 66.5 (60.4–72.2), 260 | 99.27 (98.51–99.66), 1098 | 99.05 (97.67–99.65), 528 | 99.59 (98.34–99.93), 482 | 70.3 (29.3–169) | 160 (40.1–641) | 0.34 (0.29–0.4) | 0.34 (0.29–0.4) |
| Asian patients | ||||||||
| Lennon et al 90 | n.d. | 71.43 (47.69–87.81), 21 | 62.5 (35.87–83.72), 16 | 100 (46.29–100), 5 | n.d. | n.d. | n.d. | n.d. |
| Nakashima et al 116 | 63.2 (38.6–82.8), 19 | 87.5 (60.41–97.8), 16 | 84.62 (53.66–97.29), 13 | n.d. | 4.1 (1.1–15.4) | n.d. | 0.44 (0.23–0.83) | n.d. |
| Matsuoka et al 106 | n.d. | 90.85 (84.84–94.72), 153 | 84.62 (75.19–91.03), 91 | 100 (92.62–100), 61 | n.d. | n.d. | n.d. | n.d. |
| Hayakawa et al 32 | 61.9 (38.7–81.1), 21 | 95.65 (83.96–99.24), 46 | 95.65 (83.96–99.24), 46 | n.d. | 14.2 (3.5–57.4) | n.d. | 0.4 (0.23–0.69) | n.d. |
| Matsushita et al 107 | 37.5 (19.6–59.2), 24 | 87.84 (77.67–93.95), 74 | 87.84 (77.67–93.95), 74 | n.d. | 3.1 (1.4–6.9) | n.d. | 0.71 (0.51–0.98) | n.d. |
| Apiwattanakul et al 5 | 40 (13.7–72.6), 10 | 100 (51.68–100), 6 | 100 (46.29–98.13), 5 | n.d. | ∞ (n.a.‐n.a.) | n.d. | 0.6 (0.36–1) | n.d. |
| Sum | 51.4 (39.5–63), 74 | 89.56 (85.52–92.6), 316 | 86.53 (81.46–90.42), 245 | 100 (93.15–100), 66 | 3.8 (2.6–5.6) | ∞ (n.a.‐n.a.) | 0.56 (0.44–0.71) | 0.49 (0.39–0.62) |
| Children | ||||||||
| Banwell et al 6 | 47.1 (23.9–71.5), 17 | 100 (90–100), 44 | 100 (89.33–99.78), 41 | n.d. | ∞ (n.a.‐n.a.) | n.d. | 0.53 (0.34–0.83) | n.d. |
| Lotze et al 97 | 77.8 (40.2–96.1), 9 | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. |
| Sum | 57.7 (37.2–76), 26 | 100 (90–100), 44 | 100 (89.33–99.78), 41 | n.d. | ∞ (n.a.‐n.a.) | n.d. | 0.42 (0.27–0.66) | n.d. |
| Total, original assay | 62.8 (57.5–67.8), 360 | 97.19 (96.17–97.95), 1458 | 95.33 (93.59–96.63), 814 | 99.64 (98.54–99.94), 548 | 13.4 (9.7–18.5) | 172 (43–688) | 0.39 (0.34–0.45) | 0.37 (0.32–0.42) |
| 2. Independent assays | ||||||||
| Jarius et al 51 | 61.1 (43.5–76.4), 36 | 99.27 (95.4–99.96), 137 | 98.75 (92.27–99.93), 80 | 100 (90.4–100), 46 | 48.9 (6.9–348.9) | ∞ (n.a.‐n.a.) | 0.39 (0.26–0.59) | 0.39 (0.26–0.59) |
| Waters et al 151 | 58.3 (36.9–77.2), 24 | 98.72 (92.09–99.93), 78 | 100 (88.57–99.76), 38 | 97.5 (85.27–99.87), 40 | ∞ (n.a.‐n.a.) | 23.3 (3.3–166) | 0.42 (0.26–0.67) | 0.43 (0.27–0.69) |
| Marignier et al 102 | 53.8 (33.8–72.9), 26 | 95.15 (88.5–98.2), 103 | 90.38 (78.2–96.41), 52 | 100 (89.79–100), 43 | 5.6 (2.3–13.9) | ∞ (n.a.‐n.a.) | 0.51 (0.33–0.78) | 0.46 (0.3–0.7) |
| Bizzoco et al 10 | 57.1 (20.2–88.2), 7 | 99.94 (99.61–100), 1672 | 99.82 (98.84–99.99), 556 | 100 (99.51–100), 979 | 318 (40.5–2494) | ∞ (n.a.‐n.a.) | 0.43 (0.18–1.01) | 0.43 (0.18–1.01) |
| Fazio et al 26 | 39.4 (23.4–57.8), 33 | 96.77 (90.19–99.16), 93 | 100 (79.95–99.54), 20 | 95.52 (86.63–98.84), 67 | ∞ (n.a.‐n.a.) | 8.8 (2.7–28.8) | 0.61 (0.46–0.8) | 0.63 (0.48–0.83) |
| Fazio et al 226 | 46.7 (28.8–65.4), 30 | 95.7 (88.74–98.61), 93 | 95 (73.06–99.74), 20 | 95.52 (86.63–98.84), 67 | 9.3 (1.3–65.3) | 10.4 (3.2–33.5) | 0.56 (0.39–0.79) | 0.56 (0.4–0.79) |
| Jarius et al 62 | 65.6 (46.8–80.8), 32 | 99 (93.76–99.95), 100 | 98.48 (90.73–99.92), 66 | 100 (87.36–100), 34 | 43.3 (6.1–308) | ∞ (n.a.‐n.a.) | 0.35 (0.22–0.57) | 0.34 (0.21–0.55) |
| De Vidi et al 19 | 37.5 (24.3–52.7), 48 | 100 (87.99–100), 36 | 100 (84.98–99.67), 28 | 100 (59.77–100), 8 | ∞ (n.a.‐n.a.) | ∞ (n.a.‐n.a.) | 0.63 (0.51–0.78) | 0.63 (0.51–0.78) |
| Jarius et al 66 | 65.5 (51.8–77.2), 58 | 98.31 (93.4–99.71), 118 | 97.7 (91.16–99.6), 87 | 100 (86.27–100), 31 | 28.5 (7.2–114) | ∞ (n.a.‐n.a.) | 0.35 (0.25–0.5) | 0.34 (0.24–0.48) |
| Granieri et al 31 | 95 (73.1–99.7), 20 | 95.77 (87.33–98.9), 71 | 97.56 (85.59–99.87), 41 | 93.33 (76.49–98.84), 30 | 39 (5.6–271) | 14.3 (3.7–54.8) | 0.05 (0.01–0.34) | 0.05 (0.01–0.34) |
| Delavance et al 21 | 85.1 (71.1–93.3), 47 | 99.83 (98.93–99.99), 604 | 92.31 (62.09–99.6), 13 | 100 (99.19–100), 591 | 11.1 (1.7–73.2) | ∞ (n.a.‐n.a.) | 0.16 (0.08–0.32) | 0.15 (0.08–0.3) |
| Alvarenga et al 2 | n.d. | 100 (62.88–100), 9 | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. |
| Sum | 60.1 (54.8–65.2), 361 | 99.29 (98.91–99.55), 3114 | 98.7 (97.73–99.28), 1001 | 99.54 (99.09–99.77), 1936 | 46.3 (26.8–80) | 129 (67–250) | 0.4 (0.35–0.45) | 0.4 (0.35–0.45) |
| Asian patients | ||||||||
| Chan et al 12 | 61.1 (36.1–81.7), 18 | 100 (96.07–100), 118 | 100 (89.09–99.77), 40 | 100 (91.43–100), 52 | ∞ (n.a.‐n.a.) | ∞ (n.a.‐n.a.) | 0.39 (0.22–0.7) | 0.39 (0.22–0.7) |
| Kim et al 83 | 44.4 (15.3–77.4), 9 | 94.29 (85.27–98.15), 70 | 70 (35.37–91.91), 10 | 98.33 (89.86–99.91), 60 | 1.5 (0.5–5) | 26.7 (3.3–213) | 0.79 (0.39–1.61) | 0.56 (0.31–1.01) |
| Long et al 92 | 70 (55.2–81.7), 50 | 92.22 (84.11–96.55), 90 | 87.72 (75.71–94.51), 57 | 100 (85.87–100), 30 | 5.7 (2.8–11.7) | ∞ (n.a.‐n.a.) | 0.34 (0.22–0.52) | 0.3 (0.2–0.46) |
| Long et al 92 | 62 (47.2–75), 50 | 93.33 (85.5–97.26), 90 | 89.47 (77.81–95.65), 57 | 100 (85.87–100), 30 | 5.9 (2.7–13) | ∞ (n.a.‐n.a.) | 0.42 (0.29–0.61) | 0.38 (0.27–0.54) |
| Sum | 63.8 (54.7–72), 127 | 95.38 (92.56–97.2), 368 | 90.24 (84.38–94.14), 164 | 99.42 (96.31–99.97), 172 | 6.5 (4–10.5) | 110 (15.5–778) | 0.4 (0.32–0.51) | 0.36 (0.29–0.45) |
| Total, independent assays | 61.1 (56.6–65.4), 488 | 98.88 (98.46–99.19), 3482 | 97.51 (96.4–98.3), 1165 | 99.53 (99.1–99.76), 2108 | 24.5 (17–35.3) | 129 (69.1–240) | 0.4 (0.36–0.45) | 0.39 (0.35–0.44) |
| Conventional immunohistochemistry (IHC‐C) | ||||||||
| Saiz et al 135 | 62.5 (35.9–83.7), 16 | 100 (96.42–100), 130 | 100 (96.34–99.93), 127 | n.d. | ∞ (n.a.‐n.a.) | n.d. | 0.38 (0.2–0.72) | n.d. |
Table 2.
Cell‐based assays. Sensitivities, specificities and positive and negative likelihood ratios; results from 33 studies and 39 test series. Abbreviations: NMO = neuromyelitis optica; MS = multiple sclerosis; OD = neurological and non‐neurological disease controls other than MS; HC = healthy controls; pLR = positive likelihood ratio; nLR = negative likelihood ratio; CI = 95% confidence interval
| Assay subtype | Sensitivity (%, CI, N) NMO | Specificity (%, CI, N) NMO vs. all controls | Specificity (%, CI, N) NMO vs. MS | Specificity (%, CI, N) NMO vs. OD + HC | PLR (CI) NMO vs. MS | PLR (CI) NMO vs. OD + HC | NLR (CI) NMO vs. MS | NLR (CI) NMO vs. OD + HC |
|---|---|---|---|---|---|---|---|---|
| Fluoroimmunocytochemistry (ICC‐F) | ||||||||
| a. In‐house | ||||||||
| Lennon et al 91 | 100 (31–100), 3 | 100 (31–100), 3 | n.d. | 100 (31–100), 3 | n.a. (‐) | ∞ (n.a.‐n.a.) | n.a. (n.a.‐n.a.) | 0 (n.a.‐n.a.) |
| Waters et al 151 | 80 (58.7–92.4), 25 | 100 (94.15–100), 78 | 100 (88.57–99.76), 38 | 100 (89.09–100), 40 | ∞ (n.a.‐n.a.) | ∞ (n.a.‐n.a.) | 0.2 (0.09–0.44) | 0.2 (0.09–0.44) |
| Mader et al 99 | 96.7 (81–99.8), 30 | 99.15 (96.61–99.85), 234 | 99.22 (95.08–99.96), 128 | 99.06 (94.1–99.95), 106 | 124 (17.5–872) | 103 (14.6–722) | 0.03 (0–0.21) | 0.03 (0–0.21) |
| Mader et al 99 | 70 (50.4–84.6), 30 | 99.57 (97.27–99.98), 234 | 99.22 (95.08–99.96), 128 | 100 (95.64–100), 106 | 89.6 (12.5–640) | ∞ (n.a.‐n.a.) | 0.3 (0.17–0.52) | 0.3 (0.17–0.52) |
| Waters et al 152 | 68.6 (50.6–82.6), 35 | 100 (94.61–100), 85 | 100 (88.83–99.77), 39 | 100 (90–100), 44 | ∞ (n.a.‐n.a.) | ∞ (n.a.‐n.a.) | 0.31 (0.19–0.51) | 0.31 (0.19–0.51) |
| Sum | 78.9 (70.4–85.5), 123 | 99.53 (98.5–99.88), 634 | 99.4 (97.61–99.9), 333 | 99.67 (97.86–99.98), 299 | 131 (32.9–524) | 236 (33.3–1672) | 0.21 (0.15–0.3) | 0.21 (0.15–0.3) |
| Asian patients | n.d. | n.d. | ||||||
| Takahashi et al 142 | 90.9 (69.4–98.4), 22 | 100 (95.9–100), 113 | 100 (92.84–99.86), 63 | 100 (91.11–100), 50 | ∞ (n.a.‐n.a.) | ∞ (n.a.‐n.a.) | 0.09 (0.02–0.34) | 0.09 (0.02–0.34) |
| Matsuoka et al 106 | n.d. | 90.85 (84.84–94.72), 153 | 84.62 (75.19–91.03), 91 | 100 (92.62–100), 61 | n.d. | n.d. | n.d. | n.d. |
| Tanaka et al 144 | n.d. | 82.42 (72.72–89.31), 91 | 69.81 (55.49–81.26), 53 | 100 (88.57–100), 38 | n.d. | n.d. | n.d. | n.d. |
| Tanaka et al 145 | 54.3 (36.9–70.8), 35 | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. |
| Matsushita et al 107 | 41.4 (24.1–60.9), 29 | 88.51 (83.56–92.16), 235 | 81.76 (74.39–87.43), 148 | 100 (94.73–100), 87 | 2.3 (1.3–4) | ∞ (n.a.‐n.a.) | 0.72 (0.53–0.99) | 0.59 (0.43–0.8) |
| Kim et al 81 | 92.9 (64.2–99.6), 14 | 100 (81.5–100), 22 | 100 (81.5–99.58), 22 | n.d. | ∞ (n.a.‐n.a.) | n.d. | 0.07 (0.01–0.46) | n.d. |
| Isobe et al 38 | 41.4 (24.1–60.9), 29 | 100 (95.94–100), 114 | n.d. | 100 (95.94–100), 114 | n.d. | ∞ (n.a.‐n.a.) | n.d. | 0.59 (0.43–0.8) |
| Yoshimura et al 160 | 84.4 (74–91.4), 77 | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. |
| Katsumata et al 76 | 33.3 (1.8–87.5), 3 | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. |
| Yang et al 159 | 73.6 (59.4–84.3), 53 | 96.52 (90.81–98.88), 115 | 95.35 (87.87–98.5), 86 | 100 (85.44–100), 29 | 15.8 (6–41.7) | ∞ (n.a.‐n.a.) | 0.28 (0.18–0.44) | 0.26 (0.17–0.41) |
| Chan et al 212 | 77.8 (51.9–92.6), 18 | 100 (96.07–100), 118 | 100 (89.09–99.77), 40 | 100 (91.43–100), 52 | ∞ (n.a.‐n.a.) | ∞ (n.a.‐n.a.) | 0.22 (0.09–0.52) | 0.22 (0.09–0.52) |
| Sum | 69.6 (63.8–74.9), 280 | 93.65 (91.87–95.07), 961 | 87.87 (84.62–90.53), 503 | 100 (98.9–100), 431 | 5.7 (4.4–7.3) | ∞ (n.a.‐n.a.) | 0.35 (0.29–0.42) | 0.3 (0.25–0.36) |
| Children | ||||||||
| Rostasy et al 132 | 25 (4.5–64.4), 8 | 100 (95.16–100), 95 | 100 (89.79–99.79), 43 | 100 (91.43–100), 52 | ∞ (n.a.‐n.a.) | ∞ (n.a.‐n.a.) | 0.75 (0.5–1.12) | 0.75 (0.5–1.12) |
| Total, in‐house assays | 71.5 (66.9–75.8), 411 | 96.21 (95.16–97.05), 1690 | 92.83 (90.87–94.41), 879 | 99.87 (99.17–99.99), 782 | 10 (7.8–12.8) | 559 (78.8–3970) | 0.31 (0.27–0.36) | 0.29 (0.25–0.34) |
| b. Commercial (Euroimmun) | ||||||||
| Jarius et al 62 | 78.1 (59.6–90.1), 32 | 100 (95.39–100), 100 | 100 (93.15–99.86), 66 | 100 (87.36–100), 34 | ∞ (n.a.‐n.a.) | ∞ (n.a.‐n.a.) | 0.22 (0.11–0.42) | 0.22 (0.11–0.42) |
| Marnetto et al 104 | 100 (75.9–100), 16 | 100 (93.15–100), 66 | 100 (87.99–99.75), 36 | 100 (85.87–100), 30 | ∞ (n.a.‐n.a.) | ∞ (n.a.‐n.a.) | 0 (n.a.‐n.a.) | 0 (n.a.‐n.a.) |
| Granieri et al 31 | 95 (73.1–99.7), 20 | 100 (93.6–100), 71 | 100 (89.33–99.78), 41 | 100 (85.87–100), 30 | ∞ (n.a.‐n.a.) | ∞ (n.a.‐n.a.) | 0.05 (0.01–0.34) | 0.05 (0.01–0.34) |
| Závada et al 161 | n.d. | 98.68 (91.89–99.93), 76 | n.d. | 100 (93.93–100), 75 | n.d. | n.d. | n.d. | n.d. |
| Waters et al 152 | 60 (42.2–75.7), 35 | 100 (94.61–100), 85 | 100 (88.83–99.77), 39 | 100 (90–100), 44 | ∞ (n.a.‐n.a.) | ∞ (n.a.‐n.a.) | 0.4 (0.27–0.6) | 0.4 (0.27–0.6) |
| Sum | 78.6 (69.2–85.9), 103 | 99.75 (98.38–99.99), 398 | 100 (97.42–99.95), 182 | 100 (97.79–100), 213 | ∞ (n.a.‐n.a.) | ∞ (n.a.‐n.a.) | 0.21 (0.14–0.3) | 0.21 (0.14–0.3) |
| Asian patients | ||||||||
| Kim et al 82 | 78.1 (65.7–87.1), 64 | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. |
| Kim et al 83 | 55.6 (22.7–84.7), 9 | 90 (54.12–99.48), 10 | 90 (54.12–99.48), 10 | n.d. | n.d. | n.d. | n.d. | n.d. |
| Zhong et al 163 | 80.8 (67–89.9), 52 | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. |
| Kang et al 75 | 100 (51.7–100), 6 | 95 (85.18–98.7), 60 | 94.74 (80.93–99.08), 38 | 95.45 (75.12–99.76), 22 | 19 (4.9–73.2) | 22 (3.2–149) | 0 (n.a.‐n.a.) | 0 (n.a.‐n.a.) |
| Long et al 95 | 76.9 (60.3–88.3), 39 | 100 (92.84–100), 63 | 100 (89.56–99.78), 42 | 100 (69.87–100), 12 | ∞ (n.a.‐n.a.) | ∞ (n.a.‐n.a.) | 0.23 (0.13–0.41) | 0.23 (0.13–0.41) |
| Long et al 94 | 88.6 (74.7–95.7), 44 | 98.25 (93.18–99.7), 114 | 95.65 (83.96–99.24), 46 | 100 (91.58–100), 53 | 20.4 (5.2–79.4) | ∞ (n.a.‐n.a.) | 0.12 (0.05–0.27) | 0.11 (0.05–0.25) |
| Apiwattanakul et al 5 | 60 (27.4–86.3), 10 | 83.33 (36.48–99.12), 6 | 100 (46.29–98.13), 5 | n.d. | ∞ (n.a.‐n.a.) | n.d. | 0.4 (0.19–0.85) | n.d. |
| Long et al 93 | 76.9 (60.3–88.3), 39 | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. |
| Etemadifar et al 24 | 100 (19.8–100), 2 | 100 (51.68–100), 6 | 100 (51.68–98.45), 6 | n.d. | ∞ (n.a.‐n.a.) | n.d. | 0 (n.a.‐n.a.) | n.d. |
| Etemadifar et al 25 | 50 (32.2–67.8), 32 | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. |
| Sum | 76.1 (70.8–80.8), 297 | 97.3 (94.27–98.81), 259 | 96.6 (91.83–98.74), 147 | 98.85 (92.87–99.94), 87 | 22.4 (9.4–53.1) | 66.2 (9.4–465) | 0.25 (0.2–0.31) | 0.24 (0.2–0.29) |
| Total, commercial assay | 76.8 (72.2–80.7), 400 | 98.78 (97.52–99.43), 657 | 98.48 (96.28–99.44), 329 | 99.67 (97.86–99.98), 300 | 50.5 (21.1–121) | 230 (32.5–1631) | 0.24 (0.2–0.29) | 0.23 (0.19–0.27) |
| Flow cytometry (FACS) | ||||||||
| a. HEK293 cells | ||||||||
| Fazio et al 26 | 30.3 (16.2–48.9), 33 | 96.77 (90.19–99.16), 93 | 95 (73.06–99.74), 20 | 97.01 (88.68–99.48), 67 | 6.1 (0.8–44.1) | 10.2 (2.4–43.9) | 0.73 (0.57–0.93) | 0.72 (0.57–0.91) |
| De Vidi et al 19 | 37.5 (24.3–52.7), 48 | 100 (87.99–100), 36 | 100 (84.98–99.67), 28 | 100 (59.77–100), 8 | ∞ (n.a.‐n.a.) | ∞ (n.a.‐n.a.) | 0.63 (0.51–0.78) | 0.63 (0.51–0.78) |
| Ketelslegers et al 78 | 55.6 (38.3–71.7), 36 | 99.09 (96.39–99.84), 219 | 98.73 (95.03–99.78), 158 | 100 (92.62–100), 61 | 43.9 (10.7–179) | ∞ (n.a.‐n.a.) | 0.45 (0.31–0.65) | 0.44 (0.31–0.63) |
| Waters et al 152 | 71.4 (53.5–84.8), 35 | 100 (94.61–100), 85 | 100 (88.83–99.77), 39 | 100 (90–100), 44 | ∞ (n.a.‐n.a.) | ∞ (n.a.‐n.a.) | 0.29 (0.17–0.49) | 0.29 (0.17–0.49) |
| Asian patients | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. |
| Isobe et al 38 | 51.7 (32.9–70.1), 29 | 100 (95.94–100), 114 | n.d. | 100 (95.94–100), 114 | n.d. | ∞ (n.a.‐n.a.) | n.d. | 0.48 (0.33–0.7) |
| Sum, HEK293 | 48.6 (41.2–56.1), 181 | 99.09 (97.75–99.66), 547 | 98.78 (96.17–99.68), 245 | 99.32 (97.3–99.88), 294 | 39.7 (12.8–124) | 71.5 (17.8–287) | 0.52 (0.45–0.6) | 0.52 (0.45–0.6) |
| b. LN18 cells | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. |
| Kalluri et al 73 | 81.8 (47.8–96.8), 11 | 66.67 (12.53–98.23), 3 | 66.67 (12.53–98.23), 3 | n.d. | 2.5 (0.5–12.7) | n.d. | 0.27 (0.06–1.19) | n.d. |
| Kalluri et al 73 | 61.1 (36.1–81.7), 18 | 100 (88.57–100), 38 | 100 (88.57–99.76), 38 | n.d. | ∞ (n.a.‐n.a.) | n.d. | 0.39 (0.22–0.7) | n.d. |
Table 3.
Protein‐based assays. Sensitivities, specificities and positive and negative likelihood ratios; results from 14 studies and 18 test series. Abbreviations: NMO = neuromyelitis optica; MS = multiple sclerosis; OD = neurological and non‐neurological disease controls other than MS; HC = healthy controls; pLR = positive likelihood ratio; nLR = negative likelihood ratio; CI = 95% confidence interval
| Assay subtype | Sensitivity (%, CI, N) NMO | Specificity (%, CI, N) NMO vs. all controls | Specificity (%, CI, N) NMO vs. MS | Specificity (%, CI, N) NMO vs. OD + HC | PLR (CI) NMO vs. MS | PLR (CI) NMO vs. OD + HC | NLR (CI) NMO vs. MS | NLR (CI) NMO vs. OD + HC |
|---|---|---|---|---|---|---|---|---|
| Radioimmunoprecipitation assays (RIPA) | ||||||||
| Paul et al 123 | 56.8 (39.6–72.5), 37 | 98.21 (95.65–99.34), 280 | 97.22 (92.6–99.11), 144 | 99.17 (94.81–99.96), 121 | 20.4 (7.5–55.8) | 68.7 (9.6–494) | 0.44 (0.3–0.64) | 0.44 (0.3–0.64) |
| Fazio et al 26 | 33.3 (18.6–51.9), 33 | 96.77 (90.19–99.16), 93 | 100 (79.95–99.54), 20 | 97.01 (88.68–99.48), 67 | ∞ (n.a.‐n.a.) | 11.2 (2.6–47.6) | 0.67 (0.53–0.85) | 0.69 (0.54–0.88) |
| Fluoroimmunoprecipitation assays (FIPA) | ||||||||
| a. M1/M23‐EGFP, protein A | ||||||||
| Waters et al 151 | 76 (54.5–89.8), 25 | 100 (94.15–100), 78 | 100 (88.57–99.76), 38 | 100 (89.09–100), 40 | ∞ (n.a.‐n.a.) | ∞ (n.a.‐n.a.) | 0.24 (0.12–0.48) | 0.24 (0.12–0.48) |
| Waters et al 152 | 45.7 (29.2–63.1), 35 | 100 (94.61–100), 85 | 100 (88.83–99.77), 39 | 100 (90–100), 44 | ∞ (n.a.‐n.a.) | ∞ (n.a.‐n.a.) | 0.54 (0.4–0.73) | 0.54 (0.4–0.73) |
| Jarius et al 61 | 58.8 (33.5–80.6), 17 | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. |
| Sum | 58.4 (46.7–69.4), 77 | 100 (97.13–100), 163 | 100 (94.08–99.88), 77 | 100 (94.55–100), 84 | ∞ (n.a.‐n.a.) | ∞ (n.a.‐n.a.) | 0.42 (0.32–0.55) | 0.42 (0.32–0.55) |
| b. M1‐EGFP, protein G | ||||||||
| McKeon et al 110 | 32.5 (19.1–49.2), 40 | 99.28 (98.23–99.73), 695 | 99.6 (97.43–99.98), 249 | 98.95 (97.15–99.66), 382 | 80.9 (10.9–602) | 31 (10.6–90.6) | 0.68 (0.55–0.84) | 0.68 (0.55–0.84) |
| Kalluri et al 73 | 72.7 (39.3–92.7), 11 | 66.67 (12.53–98.23), 3 | 66.67 (12.53–98.23), 3 | n.d. | 2.2 (0.4–11.3) | n.d. | 0.41 (0.12–1.44) | n.d. |
| Waters et al 152 | 45.7 (29.2–63.1), 35 | 97.65 (90.96–99.59), 85 | 97.44 (84.92–99.87), 39 | 97.73 (86.49–99.88), 44 | 17.8 (2.5–127.4) | 20.1 (2.8–144) | 0.56 (0.41–0.76) | 0.56 (0.41–0.76) |
| Sum | 43 (32.5–54.1), 86 | 98.98 (97.91–99.52), 783 | 98.97 (96.76–99.73), 291 | 98.83 (97.12–99.57), 426 | 41.7 (13.2–132) | 36.7 (14.9–90.7) | 0.58 (0.48–0.7) | 0.58 (0.48–0.7) |
| Western blot assay | ||||||||
| Marnetto et al 104 | 81.3 (53.7–95), 16 | 96.97 (88.52–99.47), 66 | 94.44 (79.99–99.03), 36 | 100 (85.87–100), 30 | 14.6 (3.7–57.3) | ∞ (n.a.‐n.a.) | 0.2 (0.07–0.56) | 0.19 (0.07–0.53) |
| Marnetto et al 104 | 12.5 (2.2–39.6), 16 | 90.91 (80.61–96.25), 66 | 97.22 (83.8–99.85), 36 | 83.33 (64.55–93.7), 30 | 4.5 (0.4–46.1) | 0.8 (0.2–3.7) | 0.9 (0.74–1.09) | 1.05 (0.82–1.34) |
| Enzyme linked immunosorbent assay (ELISA) | ||||||||
| a. In‐house assays | ||||||||
| Asian patients | ||||||||
| Hayakawa et al 32 | 71.4 (47.7–87.8), 21 | 97.64 (94.28–99.13), 212 | 95.65 (83.96–99.24), 46 | 98.19 (94.39–99.53), 166 | 16.4 (4.1–65.3) | 39.5 (12.5–125) | 0.3 (0.15–0.59) | 0.29 (0.15–0.57) |
| Kim et al 82 | 71.9 (59.1–82.1), 64 | 98.09 (94.85–99.39), 209 | 96.19 (89.97–98.77), 105 | 100 (95.56–100), 104 | 18.9 (7.1–50) | ∞ (n.a.‐n.a.) | 0.29 (0.2–0.43) | 0.28 (0.19–0.41) |
| b. Commercial, RSR | n.d. | n.d. | ||||||
| Jarius et al 66 | 75.8 (63.4–85.1), 66 | 98.69 (94.87–99.77), 153 | 99.08 (94.26–99.95), 109 | 97.73 (86.49–99.88), 44 | 82.6 (11.7–584) | 33.3 (4.8–232) | 0.24 (0.16–0.37) | 0.25 (0.16–0.38) |
| Waters et al 152 | 51.4 (34.3–68.3), 35 | 100 (94.61–100), 85 | 100 (88.83–99.77), 39 | 100 (90–100), 44 | ∞ (n.a.‐n.a.) | ∞ (n.a.‐n.a.) | 0.49 (0.35–0.69) | 0.49 (0.35–0.69) |
| Asian patients | n.d. | n.d. | ||||||
| Isobe et al 38 | 48.3 (29.9–67.1), 29 | 100 (95.94–100), 114 | n.d. | 100 (95.94–100), 114 | n.d. | ∞ (n.a.‐n.a.) | n.d. | 0.52 (0.37–0.74) |
| Isobe et al 38 | n.d. | 100 (97.37–100), 178 | n.d. | 100 (97.37–100), 178 | n.d. | n.d. | n.d. | n.d. |
| Apiwattanakul et al 5 | 50 (20.1–79.9), 10 | 83.33 (36.48–99.12), 6 | 100 (46.29–98.13), 5 | n.d. | ∞ (n.a.‐n.a.) | n.d. | 0.5 (0.27–0.93) | n.d. |
| Kim et al 83 | 55.6 (22.7–84.7), 9 | 95.71 (87.16–98.89), 70 | 70 (35.37–91.91), 10 | 100 (92.5–100), 60 | 1.9 (0.6–5.8) | ∞ (n.a.‐n.a.) | 0.63 (0.27–1.45) | 0.44 (0.21–0.91) |
| Total, commercial ELISA | 61.7 (53.4–69.5), 149 | 99.01 (97.75–99.6), 606 | 97.55 (93.44–99.21), 163 | 99.77 (98.54–99.99), 440 | 25.2 (9.5–66.9) | 271.7 (38.2–1932.4) | 0.39 (0.32–0.48) | 0.38 (0.31–0.47) |
Tissue‐based assays
Immunohistochemical (IHC) assays utilize micrometer‐thick microtome or cryostat sections as a substrate, taken from tissues or a composite of tissues known to express the target antigen of interest at high levels. The tissue sections are mounted on microscopy slides, often chemically pretreated, blocked to avoid unspecific reactions and subsequently incubated with dilutions of the patient's serum. Finally, a secondary antibody to human IgG labeled with a fluorescent (fluoroimmunohistochemistry, IHC‐F) or non‐fluorescent (conventional IHC, IHC‐C) dye is applied to visualize bound patient antibodies. The diagnosis is then made upon the recognition of antibody‐ and tissue‐specific binding patterns.
IHC‐F
NMO‐IgG/AQP4‐Ab were first discovered by means of a standard indirect immunofluorescence (IIF) assay, which used a composite of adult mouse tissues as substrate and which was already well established for the detection of a broad range of other CNS autoantibodies 90. In this assay, NMO‐IgG/AQP4‐Ab were identified by their distinctive binding to structures adjacent to the microvasculature, the Virchow‐Robin spaces and the pia mater on cerebellum tissue cryosections (Figure 1). Later, several independent studies confirmed IHC‐F as a useful tool for the detection of NMO‐IgG/AQP4‐Ab (Table 1) 1, 51, 102. One of the major advantages of this type of assay is its broad availability, for it can be performed by all laboratories familiar with IIF, a technique widely used in clinical immunology. Moreover, IHC is the only method that permits the detection of coexisting paraneoplastic or CTD‐associated antibodies, which might be of differential diagnostic relevance—in particular in NMO‐IgG/AQP4‐Ab‐negative patients—and must therefore not be overlooked 70. However, some serious limitations apply. First, results are observer‐dependent and thus subjective, ie, they require interpretation by a human rater. This is problematic, as rare sera from non‐NMO patients and even healthy controls may show binding patterns that mimic NMO‐IgG/AQP4‐Ab, eg, anti‐endothelial antibodies. In our experience, pre‐adsorption of sera with guinea pig liver powder, which results in elimination of most non‐CNS‐specific antibodies but not of NMO‐IgG, can therefore be important to avoid false‐positive results 51, although this procedure might cause some loss of sensitivity. Alternatively, counterstaining of samples with suspected AQP4‐Ab positivity with AQP4‐specific monoclonal antibodies can be helpful in unclear cases. Secondly, the sensitivity of the IIF assay has been found in independent studies to be much lower than that of some of the recombinant assays described below (Tables 1 and 5) 32, 62, 72, 107, 142, 151, 152. Third, only semiquantitative results can be obtained (by means of serum titration), and end‐titers are again observer‐dependent. Finally, testing by IHC‐F can be labor‐intensive and time‐consuming, in particular if including pre‐absorption and titration of sera. It may therefore not represent the method of choice if high‐throughput analysis is demanded. While binding of circulating NMO‐IgG to other tissues in the CNS and in peripheral organs such as kidney, stomach or muscle has been described 68, 90, studies that formally demonstrate a significant increase in sensitivity following from the use of composite tissue substrates are lacking; however, use of composite substrates might potentially increase the specificity of this type of assay, in particular if applied in laboratories not familiar with NMO‐IgG testing. From our own experience, mouse cerebellum (as used in most studies) might be preferable to monkey cerebellum; however, two studies that directly compared these two substrates produced conflicting results (Table 1) 26, 92. Recently, it has been proposed that the fine filamentous white matter staining observed with a majority of NMO‐IgG positive sera, in particular on primate tissue, may possibly be useful if applied as an additional positivity criterion 31, 104. To date, 19 studies have evaluated the originally described IHC‐F assay 90 and 14 studies have reported on independent yet similar IHC‐F assays (Table 1). The authors found the assay to be 37.5%–95% (median 61.11%) sensitive for NMO samples and to be 93.33%–100% (median 100%) specific for that diagnosis based on controls with diseases other than NMO or MS and on healthy controls (Table 1). The specificity for NMO vs. MS (not CIS or OSMS) was lower (87%–100%; median 97.67%) (Table 1). However, as discussed above, MS and NMO share common clinico‐radiological features and recent studies found that 30%–40% of patients with NMO were initially wrongly diagnosed as having MS in the past 69, 113; therefore, assessment of assay specificity should not be primarily based on MS controls. Overall, 848 IHC‐F results from NMO patients and 2656 from controls other than MS or NMOSD have been reported in the literature (Table 1), 524 (61.8%) and 12 (0.5%) of which were positive, respectively. As a potential confounder, however, it must be kept in mind that some patients may have been tested in more than one study.
Figure 1.
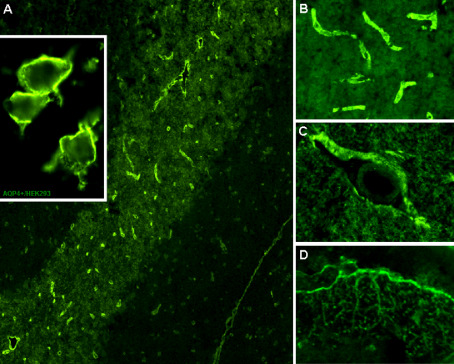
Binding of serum NMO‐IgG/AQP4‐Ab to adult mouse cerebellum as demonstrated by immunohistochemistry (A) and to the surface of cultured human embryonic kidney cells (HEK293) transfected with AQP4 as demonstrated by immunocytochemistry (A, inset). Magnified images show staining of the microvasculature (B), the Virchow‐Robin spaces (C) and the pia mater (D). Bound IgG was visualized using a goat anti‐human IgG secondary antibody labeled with fluorescein isothiocyanate. Reprinted by permission from Macmillan Publishers Ltd: Nature Reviews Neurology ©2010 43.
Table 5.
Direct comparisons of immunohistochemical and recombinant assays for NMO‐IgG/AQP4‐Ab serology. Abbreviations: ELISA = enzyme‐linked immunosorbent assay; FIPA = fluoroimmunoprecipitation assay; ICC = immunocytochemistry; IHC = immunohistochemistry; NMO = neuromyelitis optica; RIPA = radioimmunoprecipitation assay; CI = 95% confidence interval
| Immunohistochemistry | Recombinant assays | ||||
|---|---|---|---|---|---|
| Sensitivity (%, CI, N) | Specificity (%, CI, N) | Sensitivity (%, CI, N) | Specificity (%, CI, N) | ||
| NMO | All controls | NMO | All controls | ||
| Waters et al 151, a,b | IHC vs. ICC | 58.3 (36.9–77.2), 24 | 98.72 (92.09–99.93), 78 | 80 (58.7–92.4), 25 | 100 (94.15–100), 78 |
| Waters et al 151, b | IHC vs. Protein A‐FIPA (EGFP) | 76 (54.5–89.8), 25 | 100 (94.15–100), 78 | ||
| Jarius et al 62, a | IHC vs. ICC | 65.6 (46.8–80.8), 32 | 99 (93.76–99.95), 100 | 78.1 (59.6–90.1), 32 | 100 (95.39–100), 100 |
| Matsushita et al 107, a | IHC vs. ICC | 37.5 (19.6–59.2), 24 | 87.84 (77.67–93.95), 74 | 41.4 (24.1–60.9), 29 | 88.51 (83.56–92.16), 235 |
| Chan et al 12, a | IHC vs. ICC | 61.1 (36.1–81.7), 18 | 100 (96.07–100), 118 | 77.8 (51.9–92.6), 18 | 100 (96.07–100), 118 |
| Apiwatanakul et al 5, a | IHC vs. ICC | 40 (13.7–72.6), 10 | 100 (51.68–100), 6 | 60 (27.4–86.3), 10 | 83.33 (36.48–99.12), 6 |
| Kim et al 83 c,d | IHC vs. ICC | 44.4 (15.3–77.4), 9 | 94.29 (85.27–98.15), 70 | 55.6 (22.7–84.7), 9 | 90 (54.12–99.48), 10 |
| Takahashi et al 142 | IHC vs. ICC | Out of 21 (87%) AQP4‐Ab‐positive samples (ICC), 15 were positive for NMO‐IgG (IHC‐F). | |||
| Granieri et al 31 | IHC vs. ICC | 95 (73.1–99.7), 20 | 95.77 (87.33–98.9), 71 | 95 (73.1–99.7), 20 | 100 (93.6–100), 71 |
| Waters et al 152, c | IHC vs. ICC | 48.6 (31.7–65.7), 35 | 100 (94.61–100), 85 | 68.6 (50.6–82.6), 35 | 100 (94.61–100), 85 |
| Waters et al 152, c | IHC vs. ICC | 60 (42.2–75.7), 35 | 100 (94.61–100), 85 | ||
| Waters et al 152, c | IHC vs. Protein A‐FIPA (EGFP) | 45.7 (29.2–63.1), 35 | 100 (94.61–100), 85 | ||
| Waters et al 152, c | IHC vs. Protein G‐FIPA (GFP) | 45.7 (29.2–63.1), 35 | 97.65 (90.96–99.59), 85 | ||
| McKeon et al 110, c,f | IHC vs. Protein G‐FIPA (GFP) | 57.5 (41–72.6), 40 | 99.71 (98.85–99.95), 695 | 32.5 (19.1–49.2), 40 | 99.28 (98.23–99.73), 695 |
| Kalluri et al 73, a | IHC vs. Protein G‐FIPA (GFP) | 63.6 (31.6–87.6), 11 | 66.67 (12.53–98.23), 3 | 72.7 (39.3–92.7), 11 | 66.67 (12.53–98.23), 3 |
| Kalluri et al 73, a | IHC vs. FACS | 81.8 (47.8–96.8), 11 | 66.67 (12.53–98.23), 3 | ||
| De Vidi et al 19, a | IHC vs. FACS | 37.5 (24.3–52.7), 48 | 100 (87.99–100), 36 | 37.5 (24.3–52.7), 48 | 100 (87.99–100), 36 |
| Fazio et al 26, c | IHC mouse vs. FACS | 39.4 (23.4–57.8), 33 | 96.77 (90.19–99.16), 93 | 30.3 (16.2–48.9), 33 | 96.77 (90.19–99.16), 93 |
| Fazio et al 26, c | IHC mouse vs. RIPA | 33.3 (18.6–51.9), 33 | 96.77 (90.19–99.16), 93 | ||
| Fazio et al 26, c | IHC primate vs. FACS | 46.7 (28.8–65.4), 30 | 95.7 (88.74–98.61), 93 | See above | See above |
| Fazio et al 26, c | IHC primate vs. RIPA | See above | See above | ||
| Hayakawa et al 32, c | IHC vs. ELISA | 61.9 (38.7–81.1), 21 | 95.65 (83.96–99.24), 46 | 71.4 (47.7–87.8), 21 | 97.64 (94.28–99.13), 212 |
| Jarius et al 66, c,e | IHC vs. ELISA | 65.6 (46.8–80.8), 32 | 99 (93.76–99.95), 100 | 75.8 (63.4–85.1), 66 | 98.69 (94.87–99.77), 153 |
| Median | 57.5 | 98.7 | 68.6 | 99.2 | |
All IHC‐positive patients were also positive in the corresponding recombinant assay.
All FIPA‐positive patients were also positive in the ICC assay; one additional patient was positive in the ICC assay but not in the FIPA.
Some IHC‐positive patients were not positive in one or more of the recombinant assays and vice versa.
Only four NMO samples were positive in all three assays; 2 × ON and 1 × OND only IHC‐positive; 2 OND only (weakly) CBA‐positive; 4 × ON and 2 × OSMS only ELISA‐positive; 1 × NNO only CBA‐ and ELISA‐positive.
10 × IHC‐negative but ELISA‐ and CBA‐positive; 3 × ELISA‐negative but IHC‐ and CBA‐positive; two controls (RRMS) positive only in the ELISA; two additional controls positive only in the IHC assay.
30% of samples were positive only in the IHC assay and 5% only in the FIPA; in a second cohort tested using the same assays, 76 out of 331 FIPA positive samples (23%) were negative in the IHC assay and at least 167 out of 498 IHC positives were negative when measured by FIPA (as clinical data were not available for the negative patients, this cohort was not included in Tables 1–4 in the present study). Such strong discrepancies were not found in a later IHC/FIPA comparison published by the same authors (152).
Conventional immunohistochemistry
Conventional immunohistochemistry is still used by some laboratories for the detection of paraneoplastic antibodies. To date, only one group has employed this method to detect NMO‐IgG/AQP4‐Ab 135. The authors reported a sensitivity of avidin‐biotin IHC similar to that observed in most IHC‐F studies (Table 1).
Cell‐based assays (CBA)
CBA utilize cell lines such as human embryonic kidney (HEK) cells or Chinese hamster ovary (CHO) cells that have been transfected with the antigen of interest. Mock‐transfected cells (transfected with the vector alone) or (theoretically less suitably) non‐transfected cells from the same cell line are used as control substrate. As these cells do not naturally express the antigen, binding of patient serum to the antigen‐transfected but not to the mock‐ or non‐transfected cells indicates the presence of antibodies specific for the respective target antigen in the patient serum.
Fluoroimmunocytochemistry
A first recombinant immunocytochemical assay (ICC) for the detection of NMO‐IgG/AQP4‐Ab was presented in 2005 by Lennon et al, who stably transfected an HEK293 cell line with a transgene encoding green fluorescent protein (GFP) fused to full‐length human M1‐AQP4 91. This early assay was used to confirm that NMO‐IgG binding colocalizes with the sites of AQP4‐Ab expression, but not yet as a large‐scale diagnostic test. Other groups later demonstrated that this method (applied with small modifications) can be utilized as a highly sensitive serological test for NMO‐IgG/AQP4‐Ab 12, 62, 141, 142, 151. Whereas Waters et al used non‐stably transfected HEK cells expressing both human AQP4‐Ab isoforms (M1 and M23) fused to enhanced GFP (EGFP). Takahashi et al used the same cell line stably transfected with unmodified full‐length human AQP4 (M1) 141, 142, 151. However, both assays yielded very high sensitivity and specificity, exceeding those obtained in the IHC assay in a direct comparison (Tables 2 and 5) 141, 142, 151. The exact reason why the performance of the CBAs was higher than that of IHC is unknown. The primary sequences of murine AQP4 (used as substrate in the IHC assay) and human AQP4 (used in the CBA) differ slightly. In a recent study, cells transfected with mouse AQP4 indeed showed lower AQP4‐Ab binding capacity than cells transfected with human AQP4 146. In particular, human NMO‐IgG/AQP4‐Ab might not bind well to the M21 isoform of mouse AQP4 104. Similarly, differences in the relative ratio of the AQP4 isoforms M1 and M23, which determines the rate of orthogonal AQP4 arrays in the plasma membrane, could play a role. Moreover, a higher expression rate of AQP4 in transfected cells than in normal tissue might apply. Finally, tissue AQP4 is anchored in the basal lamina via the dystrophin‐associated protein complex, a large membrane assembly that connects the cytoskeleton of astroctyes, the main AQP4‐Ab‐expressing cells in the brain, to the extracellular matrix 3; AQP4 might thus not be as easily accessible in tissue sections as under cell culture conditions.
Some limitations apply to this type of assay. First, the use of non‐stably transfected cells requires the cell line to be maintained over time and freshly transfected prior to testing. This challenges the reproducibility of the assay and restricts its availability to a few specialized laboratories. Even in “stably” transfected cells the expression rate may decline over time. Secondly, fusion of AQP4 to GFP or EGFP, as used in many CBAs, might not only alter the structure of the protein itself but might also hamper the formation of orthogonal arrays (in particular, if attached to the N‐terminus) 99. This could directly influence antigen recognition and, in consequence, assay performance, and seems dispensable 62, 142. Thirdly, similar to IHC, ICC is a semiquantitative and observer‐dependent method. This may pose a problem if weakly positive samples are tested, although the chance of false‐positive results is lower than in IHC, because mock‐ or non‐transfected cells are used as control substrates. Finally, rheumatic, paraneoplastic or new autoantibody reactivities, which may play a role in NMO‐IgG/AQP4‐Ab‐negative patients, cannot be detected in this type of assay.
To overcome some of these problems, a new assay was recently developed that includes large‐scale production of AQP4‐transfected cells on millimetre‐sized cover glasses, which are stored in liquid nitrogen until used (Figure 1) 69. This procedure theoretically guarantees that the same lot can be used over many years, which is important for long‐term monitoring. Because cells are provided as ready‐made microscopy slides with up to 10 wells, this improved CBA may prove suitable for high throughput analysis. Moreover, optional combination with IHC‐F within the same well (as a cell‐tissue composite mosaic) allows two independent methods to be applied in parallel 62, 103; this can provide qualitative confirmation of AQP4‐Ab positivity and makes it possible to look for non‐AQP4‐specific antibodies in the same session. So far, 15 studies from 11 independent groups have evaluated this commercial CBA and reported a median sensitivity of 78.13% for NMO (range 50%–100%) and a median specificity of 100% (range 95.45%–100%) based on non‐MS/non‐NMOSD disease controls and healthy controls (Tables 2 and Supporting Information Table S1). Overall, 400 CBA results from NMO patients and 300 from non‐MS/NMOSD controls have been reported in the literature, respectively, 307 (76.8%) and one (0.5%) of which, respectively, were rated positive for NMO‐IgG/AQP4‐Ab (Tables 2 and Supporting Information Table S1); again, some patients may have been tested in more than one study.
Flow cytometry (FACS)
Kalluri et al stably transfected the human astrocytoma cell line LN18 using a lentiviral vector to overexpress human AQP4 73. The transfected cells were then incubated with patient sera at 1/100 dilution and analyzed for binding of NMO‐IgG/AQP4‐Ab by FACS. The authors reported a sensitivity of 69% in a cohort of 29 patients with NMO (Table 3) 73. Others used AQP4‐transfected HEK293 cells as utilized in the CBA described above, but obtained lower sensitivity rates (Table 3) 19, 26, 78. This type of assay is potentially suitable for large‐scale analysis and allows for quantification of results, which is useful for long‐term studies and for monitoring NMO‐IgG/AQP4‐Ab titers under therapy, but the techniques applied might preclude its broad use. Moreover, results have to be corrected for background binding to mock‐transfected control cells. This could result in underestimating NMO‐IgG/AQP4‐Ab titers if non‐AQP4‐specific, high‐titer antibodies binding to both transfected and control cells are present. Like all other recombinant assays, FACS assays are not able to detect autoantibodies other than AQP4‐Ab. Very recently, a new FACS assay based on HEK293 cells transfected with the short M23 isoform of AQP4 coupled to EGFP has been reported with apparently preferential sensitivity as found in a direct comparison with IHC, two CBAs, and two FIPAs 152; however, as in many other studies, the total number of controls was too small to allow the specificity (and, in consequence, the sensitivity) of this new type of assay to be definitely appraised. Given the overall very promising results, independent confirmation in unselected, larger cohorts is now recommended 27.
Other cell‐based assays
The potential applications of cell‐based enzyme‐linked immunosorbent assays (cell‐ELISA) go far beyond hybridoma screening 98. Only recently, a neuroblastoma cell line‐based cell‐ELISA employed for screening for anti‐neuronal antibodies has been reported 162. Like FACS assays, cell‐ELISAs generate quantitative results and allow high‐throughput analysis. However, controlling for non‐specific binding is more difficult in these types of assays than in conventional ICC qualitatively analyzed by a human rater. To date, no cell‐ELISAs for the detection of NMO‐IgG/AQP4‐Ab have been published 98.
Protein‐based assays
Recombinant AQP4 protein can be used for radioactive or fluorescence‐based immunoprecipitation assays (RIPA/FIPA), Western blotting (WB) and ELISA. Although protein‐based assays yielded a higher sensitivity than IHC‐F in several studies, some investigators reported a lower sensitivity than found with CBAs (Tables 2 and 3).
Radioimmunoprecipitation assays
A first RIPA for the detection of NMO‐IgG/AQP4‐Ab was published by Paul et al in 2007 123. This assay used full‐length human AQP4 (M1) labeled with radioactive 35S‐methionine. Following incubation of patient serum with the recombinant AQP4 protein, protein A beads were added to bind immune complexes formed by patient IgG and AQP4, which were then transferred to filter plates. A scintillation counter was used to measure the amount of bound radioactive AQP4, which was taken as an indirect indicator of the amount of AQP4‐Ab contained in the patient serum. This was the first large‐scale study to prove that AQP4 is the main target of NMO‐IgG in the majority of patients with NMOSD. However, it yielded lower sensitivity (63%) and specificity (98.3%) than some of the recombinant assays that were developed later and should thus no longer be used. A more recent study by Fazio et al found even lower sensitivity in an Italian population using an independent but similar RIPA 26. The lower sensitivity may be partly explained by the use of a reticulocyte lysate‐based cell‐free in vitro transcription/translation system to express AQP4, which may well have affected protein conformation.
Fluoroimmunoprecipitation assays
Waters et al established a highly sensitive and specific immunoprecipitation assay that employed EGFP‐coupled M1‐ and M23‐AQP4, which was extracted from transfected HEK293 cells (Table 3) 151. The cell lysate was incubated with patient serum, and antibody‐antigen complexes were captured using protein A beads. After washing, the amount of bound EGFP‐AQP was determined using a fluorescence plate reader and used as an indirect measure for bound AQP4‐IgG. This type of assay makes it possible to screen large numbers of samples and provides the quantitative data required for long‐term monitoring of AQP4 levels 54. The employment of EGFP‐AQP4 fusion proteins as used in the FIPA and in some of the CBAs allows convenient identification of transfected cells as well as quantification of AQP4‐Ab by reference to EGFP standards, but bears the risk of false‐positive results due to rare patient antibodies binding to those fluorophores. Whereas the robust specificity argues against such coexisting antibodies being a major confounder in this FIPA (Table 3 and Supporting Information Table S1) 151, Apiwattanakul et al found a false‐positive rate of 5% owing to anti‐GFP antibodies in a similar FIPA employing protein G instead of protein A 4. To control for false‐positive results, the authors recommend reassessing positive samples using the respective fluorophore alone as target antigen instead of the fusion protein. It is unclear why this assay yielded positive AQP4‐Ab results in only 331 out of 557 cases previously tested positive for NMO‐IgG according to an IIF test but detected AQP4‐Ab in 76 out of 4943 samples previously tested negative for NMO‐IgG in the same IIF assay 4. This is different from other recombinant assays, which were more sensitive than IIF and demonstrated a good correspondence of AQP4‐Ab and NMO‐IgG results (Table 5). Moreover, no such discrepancy was reported in a follow‐up paper by the same authors based on an identical cut‐off 152. Another FIPA study employing EGFP‐tagged M1‐AQP4 did not find binding to EGFP alone or to EGFP‐tagged antigens other than NMO 151. Assay sensitivity may also be limited in this type of assay by the large size of the EGFP fluorophore, which could prevent the formation of AQP4 molecules into orthogonal array particles (OAPs) believed by some to contain major NMO‐IgG/AQP4‐Ab epitopes 118. When directly compared, FIPA testing using EGFP‐coupled AQP4 yielded a lower sensitivity in two studies than a CBA using untagged AQP4 151, 152. As a major limitation, FIPA is labor‐intense and time‐consuming and the cell culture facilities required restrict its use to a few specialized laboratories. So far five studies have used FIPA; sensitivities for NMO ranged between 52% and 76% (median 52.27%) and specificities between 97.73% and 100% (median 99.48%) (Tables 3 and Supporting Information Table S1).
Western blotting
A combined immunoprecipitation and Western blotting assay (WBA) was published by Lennon et al 91. Pooled patient and control sera were incubated with the clarified lysate of HEK293 cells transfected with human full‐length AQP4‐GFP. Protein G‐agarose beads were then added to bind IgG/AQP‐GFP complexes from AQP4‐IgG positive samples. After washing and resuspension, the immune complexes were released, electrophoresed and transferred to nitrocellulose paper. An antibody to GFP and a horseradish peroxidase‐labeled secondary antibody were used to detect GFP. Binding to GFP was then visualized autoradiographically by enhanced chemiluminescence and used as an indirect indicator of bound AQP4‐Ab. Employing mouse tissue homogenate as substrate, Marnetto et al recently found AQP4‐Ab in 13 out of 16 NMO samples in a WBA 104. All positive samples bound to mouse M1‐AQP4 but only two recognized the M21 isoform of mouse AQP4 104. In general, ready‐made WBAs require no sophisticated technical resources and are already widely used for the detection of a number of paraneoplastic antibodies in neurology. A potential limitation is the use of denatured AQP4, which might cause non‐specific binding. Iorio et al recently confirmed that NMO‐IgG/AQP4‐Ab from some patients with NMO recognize both denatured AQP4 M1 and denatured M23 monomers, but found a significantly lower sensitivity (68%; in a series of selected patients with extremely high titers of NMO‐IgG/AQP4‐Ab) than with “native” tetramers (90%) and cell‐membrane bound AQP4 (100%) 37. In that study, none out of 85 controls bound to denatured monomeric AQP4.
Enzyme‐linked immunosorbent assays
Whereas all recombinant assays described above either used cells transfected with AQP4 or cell extracts derived from lysis of such cells, Hayakawa et al employed for the first time purified AQP4 protein (His‐tagged at both the C‐ and the N‐terminus) for use in an ELISA 32. In general, this type of assay is easy to use, allows large‐scale analysis and has the potential to be automated. For the Hayakawa study, the protein was expressed in a Sf9/baculovirus system, which is preferential to Escherichia coli‐based expression systems when it comes to producing low background antigens for immunoassays. The authors discussed that the use of rat AQP4 instead of human AQP4, the primary sequences of which differ, might have resulted in false‐negatives. A human AQP4 ELISA developed shortly thereafter yielded similar sensitivity but better specificity in an independent cohort 82.
The first commercial ELISA (RSR Ltd, Cardiff, UK) for AQP4‐Ab recently became available. Human M1‐AQP4 coated onto ELISA plate wells is incubated with patient sera and biotinylated AQP4. Because of the divalent nature of IgG, AQP4‐Ab ideally interact both with coated AQP4 and with AQP4‐biotin (so‐called “bridge‐ELISA”). Assay sensitivity for NMO ranged between 48.3% and 75.8% (median 51.4%) in five independent studies, in three of which all patients were Asian; specificity rates varied between 97.73% and 100% (median 100%; NMO vs. non‐MS/non‐NMOSD controls) (Tables 3 and Supporting Information Table S1). Overall, results from 149 NMO patients tested in this commercial ELISA have been published, 92 of whom came up positive (61.7%), and 440 non‐MS/non‐NMOSD controls, only one of whom was positive (0.23%). This assay was more sensitive than IHC but equally specific in direct comparison in two independent studies 66, 152, but missed several NMOSD samples that were positive for AQP4‐Ab in at least two independent CBAs in both of those studies 66, 152. Whether biotinylation hampered AQP4‐Ab binding in the false‐negative cases, or whether other factors played a role, is unknown. Interestingly, a few of the false negatives yielded higher values than most of the controls, but did not exceed the cut‐off recommended by the manufacturer 66, 152; however, lowering the cut‐off resulted in loss of specificity 152. In our hands, this assay yielded very good intra‐run yet only moderate inter‐run variability 66. While ELISAs, by providing quantitative results, are potentially suitable for long‐term measurement of AQP4‐Ab serum concentrations (the possible indications for which may include monitoring of disease activity or treatment response), this advantage was partly challenged by the fact that some sera harbored AQP4‐Ab at concentrations that exceeded the upper reference range of the standard curve 66. Predilution of sera might enable users to circumvent this problem; according to the manufacturer's instructions, however, not all sera will dilute in the same way. A potential problem may be the occasional presence of antibodies to biotin or biotinylated proteins in normal human sera 13, 18, which theoretically could act as anti‐reagent antibodies hampering both antibody‐antigen interaction and streptavidin‐biotin complex formation.
To improve assay sensitivity, the development of ELISAs with membrane‐expressed AQP4 as substrate has been proposed 27.
Assay Accuracy
NMO‐IgG/AQP4‐Ab in patients with NMO
To date, around 60 studies (many of which reported on more than one assay type; Tables 1, 2, 3) have been published that report on the frequency of NMO‐IgG/AQP4‐Ab in NMO and/or the antibodies' specificity for this condition. Overall, ∼15 000 test results have been reported, most of them from control patients with diseases other than NMOSD. Patient and/or control numbers were low in some studies; therefore, confidence intervals (as provided below and in Tables 1, 2, 3) are generally more meaningful than absolute data on sensitivities and specificities (Supporting Information Table S1) when it comes to rating assay accuracy.
Sensitivity
Fifty‐three of the 59 studies analyzed for this review included patients classified as NMO (the remaining studies included patients with OSMS, LETM, ON, NETM and/or rheumatic disorders). These studies reported results from 83 test series and around 40 independent assays using eight different methods (IHC, ICC, FACS, RIPA, FIPA, WB, ELISA). Sensitivities for NMO varied between 12.5% and 100% with a median of 62.25% (Tables 1, 2, 3, 4 and Supporting Information Table 1S). This wide inter‐study variance in sensitivities may reflect not only technical differences among the various immunoassays and among study populations, but very likely also an unintended selection bias due to low NMO sample size in some of the studies. Accordingly, 95% confidence interval width was more than 40% in 29 test series (median 34.4%). When only those 15 series that included more than 40 NMO patients were taken into account, the median sensitivity was 73.58% and the median 95% confidence interval (CI) width was 22%.
Table 4.
Comparison of mean sensitivities, specificities and likelihood ratios of NMO‐IgG/AQP4‐Ab serology found using six different immunoassay types as reported in the literature (based on 12 703 individual test results; see Tables 1–3 and Supporting Information Table S1 for details). Abbreviations: NMO = neuromyelitis optica; MS = multiple sclerosis; OD = neurological and non‐neurological disease controls other than MS; HC = healthy controls; pLR = positive likelihood ratio; nLR = negative likelihood ratio; CI = 95% confidence interval
| Assay type | Sensitivity (%, CI, N) | Specificity (%, CI, N) | Specificity (%, CI, N) | Specificity (%, CI, N) | PLR (CI) | PLR (CI) | NLR (CI) | NLR (CI) |
|---|---|---|---|---|---|---|---|---|
| NMO | NMO vs. all controls | NMO vs. MS | NMO vs. OD + HC | NMO vs. MS | NMO vs. OD + HC | NMO vs. MS | NMO vs. OD + HC | |
| IHC | 61.8 (58.5–65), 864 | 98.42 (98.03–98.74), 5070 | 96.82 (95.95–97.51), 2106 | 99.55 (99.19–99.76), 2656 | 19.4 (15.2–24.7) | 137 (77.6–241) | 0.39 (0.36–0.42) | 0.38 (0.35–0.41) |
| ICC | 74.1 (70.9–77.1), 811 | 96.93 (96.13–97.58), 2347 | 94.37 (92.88–95.57), 1208 | 99.82 (99.26–99.97), 1082 | 13.2 (10.4–16.7) | 401 (100–1602) | 0.27 (0.24–0.3) | 0.26 (0.23–0.29) |
| FACS | 51.4 (44.5–58.3), 210 | 98.98 (97.68–99.58), 588 | 98.6 (96.21–99.55), 286 | 99.32 (97.3–99.88), 294 | 36.8 (13.8–98) | 75.6 (18.9–303) | 0.49 (0.43–0.56) | 0.49 (0.43–0.56) |
| RIPA | 45.7 (33.9–58), 70 | 97.86 (95.65–99), 373 | 97.56 (93.48–99.22), 164 | 98.4 (95.03–99.59), 188 | 18.7 (6.9–50.9) | 28.6 (9–90.4) | 0.56 (0.45–0.7) | 0.55 (0.44–0.68) |
| FIPA | 50.3 (42.4–58.2), 163 | 99.15 (98.27–99.61), 946 | 99.18 (97.43–99.79), 368 | 99.02 (97.59–99.64), 510 | 61.7 (19.8–192) | 51.3 (21.2–124) | 0.5 (0.43–0.58) | 0.5 (0.43–0.58) |
| ELISA | 65.4 (58.9–71.4), 234 | 98.54 (97.54–99.15), 1027 | 96.82 (94.04–98.37), 314 | 99.44 (98.46–99.82), 710 | 20.5 (11.1–38) | 116 (43.5–310) | 0.36 (0.3–0.43) | 0.35 (0.29–0.42) |
Based on all 2384 reported test results from NMO patients, 1525 of which were positive, an estimated prevalence of NMO‐IgG/AQP4‐Ab in NMO of ∼64% (95% CI 62–65.1) can be calculated (matching the mean of the sensitivities of all test series), with lower values for tissue‐ and protein‐based assays (61.8% and 56.5%) than for CBAs (69.4%); this is in line with results from direct comparative studies (see Comparative studies and Table 5). It should be noted as a caveat that some patients may have been included in more than one series.
Several independent studies found a higher frequency of NMO‐IgG/AQP4‐Ab in patients with relapsing NMO than in patients with monophasic NMO, both in adults 69, 78 and in children 6. However, this does not imply that NMO‐IgG/AQP4‐Ab turn positive only some time after disease onset, but rather that monophasic NMO is pathogenetically different from relapsing NMO [eg, caused by acute disseminated encephalomyelitis (ADEM)].
Specificity
Samples from disease or healthy controls were tested in 49 out of the 59 studies analyzed for this review (Tables 1, 2, 3, 4 and Supporting Information Table S1). Specificity ranged between 62.50% (such low values were mainly driven by inclusion of patients with OSMS, which is now considered to be identical with NMO in many cases) and 100% for NMO vs. MS (median of 99.08%), between 83.33% and 100% for NMO vs. non‐MS/non‐NMOSD controls (median 100%), and between 66.67% and 100% for NMO vs. all controls with a median of 98.86%; median 95% CI width was 14.7 for NMO vs. MS, 10 for NMO vs. non‐MS/non‐NMOSD controls, and 8.09 for NMO vs. all controls. Based on the total number of 10483 test results reported in the 49 studies, specificity of 96.48% for NMO vs. MS, 99.4% for NMO vs. non‐MS/non‐NMOSD and 98.22% for all controls can be calculated.
As an important drawback, the number of control samples was too low in many studies to allow proper specificity assessment (median 85; range 3–1672; <100 in 55% of all test series), including a recent two‐center multi‐assay comparison trial (n = 85) 152. Only three out of the 59 (5%) studies included more than 500 controls 10, 21, 110, and only one of these 10 included more than 1000. In particular, the number of samples from patients with MS, the most important differential diagnosis of NMO, was very low in most studies (median 39). However, exact data on assay specificity are essential, since most patients tested for NMO‐IgG/AQP4‐Ab are probably MS patients as suggested by the extremely high number of tests for NMO‐IgG/AQP4‐Ab currently performed per year [eg, more than 23 000 at only two centers 152]. Given the low prevalence of NMO (∼1.5/100 000) compared to classical MS (∼120/100 000), specificity rates well above 99% are required to avoid an unfavorably high ratio of false‐positive to true‐positive results, which could render NMO‐IgG/AQP4‐Ab testing useless and even harmful if not used in well‐selected populations. Future studies evaluating NMO‐IgG/AQP4‐Ab assay accuracy (including currently planned multicenter comparison trials) should therefore include a sufficient number of MS controls (eg, >1000), and patient numbers should ideally (although not essentially) reflect the true NMO/MS prevalence ratio to permit calculation of predictive values (see Predictive values). When only test series that included more than 100 controls were considered (n = 28), median specificity was 99.28% (range 88.51%–100%) for NMO vs. all controls (median CI width 5.56%).
Specificity of NMO vs. all controls was higher in CBAs (median specificity of all reported test series 100%) than in tissue‐based (median 98.12%) and protein‐based assays (median 98.09%).
As a possible major confounder, misclassification of NMOSD as MS may have been an issue in some studies (see Test performance: influence of clinical misclassification).
Predictive values
As the ratio of NMO samples to MS control samples was in accordance with the prevalence ratio of the two diseases (∼1/80) in virtually none of the published test series (median 1/1.6), calculation of predictive values (PV) would not be appropriate. This drawback can be partly compensated by calculation of likelihood ratios (see the following section). The single study that included a sufficient number of MS controls (1/79.4) found a positive PV for NMO vs. MS of 0.8 and a negative PV of 0.9946 in an IHC‐F assay employing rat cerebellum tissue sections; as a major drawback, however, NMO sample numbers were low (n = 7) in that study and CIs broad (0.299–0.99 and 0.983–0.999) 10.
Likelihood ratios
Based on assay sensitivities and specificities as given in Tables 1, 2, 3 (columns 1–4), we calculated positive and negative likelihood ratios (pLR, nLR) for each assay (see Tables 1, 2, 3, columns 5–8). By convention, tests with pLRs over 10 and/or nLR <0.1 are considered clinically useful 20. The pLRs for NMO vs. MS ranged between 1.5 and ∞ among studies, with an extremely high median of 106.5, and were >10 in 54 out of the 68 test series (79.4%); the nLRs for NMO vs. MS ranged between 0 and 0.9 (median 0.39) but were below 0.1 only in eight out of the 68 test series (11.8%). When only non‐MS/non‐NMOSD controls were considered (in order to control for misclassification of NMOSD as MS; see Test performance: influence of clinical misclassification), the median pLR was ∞ and pLRs were >10 in 53 out of the 55 test series (96.3%); nLR ranged between 0 and 1.05 (median 0.385) and was below 0.1 only in seven out of the 55 test series (12.7%).
Test performance: influence of clinical misclassification
As mentioned above, NMO patients were frequently misdiagnosed as having MS in the past. In a European cohort of 175 NMOSD patients, around 40% had initially been diagnosed with MS, mainly before NMO‐IgG/AQP4‐Ab testing became available 69. A North American study reported a rate of false diagnosis of around 30% 113. This was caused both by a lack of awareness regarding NMO, which is a very rare condition compared to MS, in the past and by partial overlap of the clinico‐radiological features of NMO and MS, in particular in the early stages. Moreover, there is a significant overlap between the diagnostic criteria. In a recent Japanese study 17 out of the 26 seropositive patients initially diagnosed with MS met both McDonald criteria for MS and Wingerchuk's 2006 criteria for NMO 38. These factors may explain why assay specificity for NMO was significantly (P < 0.0001; Fisher's exact test) higher if calculated against the non‐MS/non‐NMOSD disease controls and healthy controls than if calculated against the MS group (see Specificity). In line with this hypothesis, specificity was higher if patients with a clinically isolated syndrome suggestive of MS (CIS) but no definite MS and (mainly Asian) patients classified as having “OSMS,” which is now considered to be identical to NMO in many cases (in particular, if associated with LETM), were excluded from the analysis (total MS group vs. NMO: median specificity of all studies 99.08%, mean specificity based on 4518 reported test results 96.48%; MS without CIS and OSMS vs. NMO: median specificity of all studies 100%, specificity based on 766 reported test results 98.27%). Among 226 test results from patients classified as OSMS, 76 (33.63%) were positive for NMO‐IgG/AQP4 (the lower positivity rate than in the NMO group is partly be explained by the inclusion of patients with classical MS presenting with NETM and ON in the OSMS group in some studies). When only European or North‐American cohorts were considered, the median specificity of all studies was 100% (NMO vs. non‐MS/NMOSD); based on the total number of 4008 tests results from non‐MS/NMOSD patients reported in those studies, specificity was 99.3%. In several studies, “false‐positive” MS patients had ON, LETM and/or brainstem encephalitis, which are compatible with a diagnosis of AQP4 autoimmune encephalomyelitis. Considering that differentiating NMO and MS based on clinico‐radiological findings can be difficult, future studies investigating assay specificity should therefore should also include a large number of non‐MS controls.
NMO‐IgG/AQP4‐Ab in patients with isolated LETM
The frequency of NMO‐IgG/AQP4‐Ab ranged between 0% and 100% with a median of 53.3% (Supporting Information Table S1). In total, 731 test results from patients with LETM were reported, of which 333 were positive (45.1%). While not all studies differentiated between monophasic and recurrent LETM, some reported a higher frequency of NMO‐IgG/AQP4‐Ab in patients with relapsing LETM 6, 12, 81, 128, 154, similar to what has been found in patients with definite NMO (Table 6).
Table 6.
Frequency of NMO‐IgG/AQP4‐Ab in monophasic vs. relapsing NMOSD (percentages in parentheses). Abbreviations: NMO = neuromyelitis optica; LETM = longitudinally extensive transverse myelitis; ON = optic neuritis
| NMO | Monophasic/first attack | LETM | Monophasic/first attack | ON | Monophasic/first attack | |
|---|---|---|---|---|---|---|
| Relapsing | Relapsing | Relapsing | ||||
| Jarius et al 69 | 92/114 (81) | 0/5 (0) | 30/35 (86) | 10/14 (71) | — | — |
| Banwell et al 6 | 7/9 (78) | 1/7 (14) | 1/1 (100) | 0/9 (0) | 1/5 (20) | 0/8 (0) |
| Ketelslegers et al 78 | 20/27 (74) | 0/9 (0) | — | — | — | — |
| Kim et al 81 | — | — | 7/15 (47) | 2/35 (6) | — | — |
| Long et al, CNN 94 | — | — | — | — | 3/8 (38) | 1/5 (20) |
| Chan et al 12 | — | — | 6/12 (50) | 0/2 (0) | 2/9 (22) | 1/14 (7) |
| Pittock et al 128 | — | — | 31/44 (71) | 10/31 (32) | — | — |
| Jarius et al 61 | — | — | — | — | 5/50 (10) | 3/89 (3.4) |
| Petzold et al 124 | — | — | — | — | 2/36 (6) | 2/41 (5) |
| Sum | 119/150 (79) | 1/21 (5) | 75/107 (70) | 22/91 (24) | 13/108 (12) | 7/157 (4) |
| p (Fisher's exact test, two‐tailed) | <0.0001 | <0.0001 | 0.021 | |||
“—”: Study did not include patients with the respective diagnosis or did not distinguish between relapsing and monophasic disease.
NMO‐IgG/AQP4‐Ab in patients with isolated ON
In the 46 test series that included patients with ON, the frequency of NMO‐IgG/AQP4‐Ab ranged between 0% and 75% with a median of 20% (Supporting Information Table S1). In total, 891 test results from patients with ON were reported, of which 127 were positive (14.3%). Results from several studies indicate that NMO‐IgG/AQP4‐Ab are much less frequent in patients with isolated monophasic ON 6, 12, 61, 105, 124. In a large cohort, we found the antibody in three out of 89 patients (3.4%) with a single attack of ON but in five out of 50 (10%) with relapsing ON using a CBA. Results from other studies are summarized in Table 6.
NMO‐IgG/AQP4‐Ab in patients with HRS other than LETM or ON
It is of importance that NMO‐IgG/AQP4‐Ab were also found in some patients presenting with NETM (Table S1). While NMO‐IgG/AQP4‐Ab were very rare in those with isolated NETM (465 results reported, five [1.1%] positive), they were rather frequent among patients with NETM and a history of ON (103 results reported, 17 [16.5%] positive). This is well in line with findings from a recent study in which 7.3% of patients with a history of NMO or LETM presented with NETM on MRI at least once over the course of disease (probably depending on MRI timing, since short lesions in NMO might represent lesions in either evolution or resolution or, alternatively, residual atrophy) 69. Therefore, NETM patients should preferably not be used as disease controls when it comes to assessing the specificity of diagnostic assays; the same applies to patients with isolated brainstem encephalitis, especially if involving regions with high AQP4 expression such as the medulla oblongata or the diencephalon. For OSMS, see Test performance: influence of clinical misclassification.
NMO‐IgG/AQP4‐Ab in the total HRS group
When LETM, ON and NETM + ON, which are considered to confer a risk of conversion to NMO, were analyzed together, the median frequency of NMO‐IgG/AQP4‐Ab was 38.5% (range 0%–100%; n = 64 test series). Overall, 1829 results from patients with HRS were reported, 509 of which were positive (27.8%).
Comparative Assay Accuracy
Inter‐study comparison
A consistent finding across almost all studies is the higher sensitivity of NMO‐IgG/AQP4‐Ab testing in patients with definite NMO than in patients with HRS. Moreover, a higher frequency of NMO‐IgG/AQP4‐Ab in NMO was found in CBAs (median of all reported test series 73.58%) than in the original IHC‐F assay (median 61.51%) (Tables 4 and 5).
However, striking differences still exist among studies with regard to assay accuracy (see Tables 1, 2, 3 and Supporting Information Table S1). These differences may reflect variations in sex, age and ethnic background (Caucasian vs. Asian) of both disease and control subjects; diagnostic criteria [Wingerchuk 1999 vs. 2006 156, 157; Paty 1988/1991 vs. McDonald 2001, 2005, or 2010 vs. Kira 2003 84, 109, 122, 129, 130; Wingerchuk 2006 without the need for AQP4‐Ab positivity but no brain lesions at onset and LETM in all patients vs. Wingerchuk 2006 including the serological criterion and thus possibly including patients with NETM or brain lesions at onset; see Supporting Information Table S1 for a detailed summary]; disease status and treatment status at the time of blood sampling; the proportion of monophasic NMO and HRS patients (with monophasic cases being less frequently associated with AQP4‐Ab 54, 61, 78; see Table 6); and, in qualitative assays (IHC, ICC), rater experience. Moreover, as mentioned above, some patients with NMO may have been wrongly classified as MS in the past 69, 113; in fact, signs and symptoms of optic nerve and spinal cord involvement were reported in some of the NMO‐IgG/AQP4‐Ab‐positive MS patients (see legend to Supporting Information Table S1 for details). Finally, sample numbers are crucial when it comes to fine differences in assay accuracy.
Furthermore, similar studies may differ with regard to small methodological details including pre‐absorption procedures, fixation, blocking procedures, choice of cell lines, tissue type and preparation, animal species and age, cell culture conditions, stable vs. non‐stable transfection, transfection methods, transfection rates (and their variations over time), cell lysis buffers, starting serum dilutions, incubation times, secondary antibody conjugates and other detection substrates, cut‐off values, or the use of either protein A or G in IP assays. Also factors that can potentially affect epitope confirmation including the ability of AQP4 to form OAPs have to be considered, such as the use of either native or denatured AQP4 (as in WB) protein, the use of M1 or M23 AQP4, untagged AQP4 or AQP4 coupled to fluorophores or biotin, the absence or presence of HIS tags, the coupling or tagging site (N‐terminal vs. C‐terminal) and the type (and thus size) of fluorophores (GPF, EGFP, EmGFP, etc) (see Supporting Information Table S1 for an overview of substrates and fluorophores used). Such methodological issues may explain some of the marked differences concerning sensitivity and specificity found among studies that did not differ with regard to the genetic background of the patients included or the diagnostic criteria for NMO applied 123, 142, 145, 151. The fact that so many studies directly comparing IHC‐F to newly developed recombinant assays consistently found a higher sensitivity of the recombinant assays strongly suggests issues inherent to that method in general (Tables 1, 2, 3, 4, 5) 32, 62, 72, 107, 142, 151.
It is worth mentioning that the methods applied in some studies deviated in possibly important details from those used in previous studies from the same groups employing similar assays (eg, EGFP‐M1 + M23‐CBA vs. untagged M1‐CBA); this may possibly explain some of the slight variations in test accuracy observed between studies. However, other factors such as differences in study populations may have played a role as well.
Comparative studies
To exclude the possibility that differences in accuracy between the various methods reflect differences in study populations or other confounders rather than differences in assay performance, direct comparisons are highly desirable. So far, more than 15 studies have been performed that directly compared at least two different methods in the same study population; as mentioned above, almost all demonstrated a higher sensitivity of recombinant assays than of tissue‐based assays (see Table 5 for details). However, no studies included all assays currently available; accordingly, no assay can currently considered best.
Moreover, interpretation of those comparative studies is hampered by several confounders. First, the number of relevant control samples was not sufficient in some studies. In consequence, it is difficult to appraise whether samples detected by only one assay are true positives or possibly false positives. Second, sample selection criteria were not mentioned in some studies. This is relevant since an unintended bias toward high‐ or medium‐titer samples could mask differences in assay sensitivity, which may become apparent only if low‐titer samples are tested. Third, some studies compared quantitative and qualitative assays, with the latter being observer‐dependent; in consequence, such comparisons are necessarily non‐objective to some extent. Finally, not all comparative studies were performed independent of the manufacturer or patent holder.
AQP4‐Ab levels in NMO‐IgG‐negative (according to IHC‐F) but AQP4‐Ab‐positive (as detected in recombinant assays) samples were investigated in two studies. Interestingly (although the reasons are not well understood), AQP4‐Ab levels were found to be in the mid‐ or even high range as assessed by FACS or ELISA in many of the NMO‐IgG‐negative patients in both of these studies 66, 73.
AQP4‐M1‐ vs. AQP4‐M23‐based assays
Two isoforms of AQP4 exist in humans, termed M1‐AQP4 and M23‐AQP4. In common with other aquaporins, AQP4 forms tetramers. While M23‐AQP4 tetramers organize into large so‐called OAPs, M1‐AQP4 is thought to inhibit and limit the formation of M23 tetramers to such arrays. In CHO cells transfected with M1‐AQP4, most tetramers are present as singlets, only very few (<5%) are organized to small OAPs (2–12 tetramers). In M23 cells, large rafts are detectable in the plasma membrane, most of them containing >100 individual AQP4 tetramers. In cells transfected with both M1 and M23, AQP4 forms arrays of intermediate size, similar to those found in astrocytic endfeet. Based on preliminary evidence suggesting that larger OAPs could enhance NMO‐IgG/AQP4‐Ab binding or that AQP4‐Ab might partly bind to confirmational epitopes linked to OAP formation, it has been suggested that the use of M23 in diagnostic assays might be preferential 16, 99, 118.
In 2008, Hayakawa et al presented an assay that for the first time employed solely M23‐AQP4, although rat instead of human AQP4 was used 32. The availability of purified AQP4 enabled the authors to confirm by direct competition that their patient's sera indeed recognized M23‐AQP4; serum from an ELISA‐positive NMO patient turned negative after pre‐incubation with the recombinant M23‐AQP4 protein. However, identical or even higher sensitivities and specificities were later reported in ELISA studies employing M1‐AQP4 (Table 3) 66, 82. M23‐transfected cells were also found to be slightly less sensitive than M1‐transfected cells in a FACS study 73. However, differences in study populations might also account for this effect; studies directly comparing assays in the same cohort are therefore crucial. In a commercial HEK293 CBA (Euroimmun, Luebeck, Germany), we detected no differences in assay sensitivity between M1‐ and M23‐transfected cells employed in parallel in the same cohort of patients, although binding to M23‐transfected cells was associated with stronger signal intensity (Sven Jarius, unpublished data). In line with this finding, a recent ELISA study reported similar positivity rates between denatured M23‐AQP4 and denatured M1‐AQP4, although the average optical density (OD) value was ∼20% higher with AQP4‐M23, resulting in an improved signal‐to‐noise ratio 82. Similarly, no difference in sensitivity was found on direct comparison between an M1‐AQP4‐based FIPA and an M1 + M23‐AQP4‐FIPA 152. Similarly, Crane et al recently performed affinity studies using AQP4‐transfected human astrocyte‐derived U87MG cells and found binding to both isoforms, although consistently stronger binding to M23 16.
It should be noted as a limitation that some of the M23‐based assays used to compare the sensitivity of M23 vs. M1 employed fluorophore‐coupled M23‐AQP4 as substrate (Supporting Information Table S1). Binding of EGFP to the N‐terminus of AQP4 might yet hamper OAP formation 99. A single study that used C‐terminal EmGFP in a HEK293‐CBA indeed reported higher sensitivity of M23 than of M1 99. However, as mentioned above, we did not find a difference between the two isoforms when using untagged AQP4 in a CBA.
Testing purified Fab fragments of NMO‐IgG/AQP4‐Ab, Crane et al found patterns of M1‐ vs. M23‐specific binding similar to those of intact NMO‐IgG/AQP4‐Ab; this could suggest that structural differences (eg, changes in the AQP4 epitope upon array assembly) and not bivalent cross‐linking of whole IgG result in the greater binding affinity to OAPs 16, which would be in line with the very small size of AQP4 compared to IgG. However, Verkman et al also showed a wide variation in NMO‐IgG/AQP4‐Ab binding intensity to M1‐ vs. M23‐AQP4 between patients and even between recombinant monoclonal AQP4‐Abs generated from different plasma cell clones of a single patient 16. Whether differences in the proportion of affinity of antibodies binding to M1 vs. M23 correlate with clinical parameters such as disease activity or severity, treatment response and prognosis is currently being studied.
Given that many studies employing M1‐AQP‐transfected cells and lysates derived from those cells yielded high sensitivities and specificities (Supporting Information Table S1), there is no doubt that M1‐AQP‐transfected cell are suitable substrates in diagnostic NMO‐IgG/AQP4‐Ab assays. However, it has been controversial whether NMO‐IgG/AQP4‐Ab in fact recognizes M1‐AQP4 or rather M23‐AQP4, which could be produced to some degree in M1‐transfected cells by leaky scanning 131. Minor OAP formation has indeed been demonstrated by freeze‐fracture electron microscopy in M1‐transfected CHO cells, and M23‐AQP4 has been found in M1‐transfected HeLa cells 28, 131. However, a recent study did not find evidence for M23 expression in M1‐transfected HEK293 cells as used in diagnostic assays 37. Moreover, several studies demonstrated binding of NMO‐IgG/AQP4‐Ab‐positive samples to both M1 and M23 tetramers as well as to M1 in the absence of high‐order arrays 15, 37. These findings challenge an earlier paper that concluded from an analysis of seven samples in a HeLa cell assay that M23 high‐order arrays are the exclusive target of NMO‐IgG 118.
AQP4 peptides vs. intact AQP4
Qualitative evidence from CBA experiments strongly suggests that NMO‐IgG/AQP4‐Ab binds mainly to extracellular epitopes of both M1 and M23 AQP4 34, 37, 142, 146, 151. Data from peptide‐based ELISA studies are contradictory. While one study claimed preferential binding to intracellular epitopes based on an analysis of 11 synthetic peptides, spanning the entire intracellular and extracellular domains of the AQP4 molecule 74, a more recent study found that binding to intracellular loop B, the N‐terminus or the C‐terminus of AQP4 is not disease‐specific but is present also in almost 50% of control samples 37. By contrast, binding to both peptides and GST fusion proteins corresponding to extracellular loop C was 100% disease‐specific (n = 85) in the same study. Slightly lower specificity was observed with the extracellular loops E and A. Both loop C, which is highly flexible 35, 36, and loop E had been previously suggested as target epitopes in NMO 125, 146. While the idea of improving assay specificity by using peptide‐based assays seems tantalizing, alterations of the tertiary structure and the limited flexibility of immobilized peptides are likely to influence assay accuracy. While being disease‐specific, the above‐mentioned loop‐C‐specific ELISA yielded a sensitivity of only 31% in a series of selected, high‐titer samples 37.
Special Issues
NMO‐IgG/AQP4‐Ab in the CSF
Assays for the detection of cerebrospinal fluid (CSF) antibodies have to take into consideration possible matrix effects, such as a higher concentration of ions and a lower amount of total protein compared to serum, which can affect antigen/antibody interactions and even cause false‐negative results unless diluted samples are used. To the best of our knowledge, none of the assays published thus far has been formally optimized for the detection of NMO‐IgG/AQP4‐Ab in the CSF. However, qualitative assays such as IHC are thought to be rather robust against such effects.
Interestingly, Klawiter et al recently reported on three patients in whom NMO‐IgG/AQP4‐Ab was detected only in the CSF, not in the serum 86. This would indicate that in patients with negative NMO‐IgG/AQP4‐Ab status, lumbar puncture is advisable. However, this is controversial. First, in that IHC‐F study serum samples were tested at a dilution of 1:128, although lower serum titers are not unusual in NMO and a dilution of 1:60 was used in previous studies using the same IHC‐F 51, 90; some serum samples may even come up positive for NMO‐IgG only when re‐tested at 1:10 or even undiluted. Second, the author discussed the possibility that coexisting serum autoantibodies could have masked the typical NMO‐IgG binding pattern. Recombinant assays are more robust against interfering autoantibodies and, in addition, are more sensitive than IHC‐F. However, no recombinant assays were used to confirm the seronegative antibody status in those three patients. In line with the findings of Klawitter et al, a more recent Chinese study reported AQP4‐Ab‐CSF‐positivity in 15 out of the 24 (54%) AQP4‐serum‐negative NMO patients using a CBA; however, the authors reported specificity of only 88%, which raises severe methodological concerns 95.
By contrast, a large study from our laboratory, which analyzed 87 paired serum and CSF samples from 37 patients with NMOSD and 42 controls with other neurological diseases, found AQP4‐Ab in ∼70% of CSF samples from AQP4‐Ab‐positive patients with NMOSD using the same CBA but neither in any of the serum‐negative patients studied nor in any of the controls, suggesting that testing of CSF samples might not be needed in the majority of cases 60. Acute disease relapse in the 30 days before lumbar puncture, AQP4‐Ab serum titers >1:250, and blood‐CSF barrier dysfunction, but not treatment status, predicted CSF AQP4‐Ab positivity in this study.
Calculation of antibody indices (AI) allows determination of the intrathecal production (IP) of specific antibodies 55, 60. Based on AI calculation, IP of AQP4‐Ab was detectable in only one of 23 samples (4.3%) in a recent study 60. This sample was obtained during an acute relapse of ON. However, 20 out of the 23 AQP4‐IgG CSF‐positive samples with normal AQP4‐AI values were also taken during disease attacks; AQP4‐AI elevation thus seems not to be a reliable disease activity marker. The infrequency of intrathecal AQP4‐IgG production suggests that in patients with NMOSD, AQP4‐Ab‐producing B cell clones usually reside in the systemic compartment. CSF AQP4‐Ab may thus reflect passive diffusion of serum AQP4‐Ab into the CSF 60. In line with this hypothesis, Takahashi et al, in a study on 12 Japanese patients, found that titers of CSF AQP4‐IgG were almost proportional to serum AQP4‐IgG in NMO, although, as a limitation, that study had not taken into account possible blood‐CSF barrier disruption. While Bennett et al reported an additional single case of low level intrathecal NMO‐IgG/AQP4 synthesis 7, Kalluri et al found normal AI in seven out of seven patients using the same FACS assay 73.
Given the contrasting results and the methodological concerns described above, more studies are required before CSF testing in seronegative patients can be generally recommended, at least from an economic point of view and, in particular, if testing for CSF NMO‐IgG/AQP4‐Ab would require repeat lumbar puncture, an invasive and potentially harmful procedure.
Testing for AQP4‐IgM and AQP4‐IgA
Determination of AQP4‐IgM antibodies is challenging. IgG antibodies can hamper the detection of IgM in immunoassays. IgG, if present in excess, can supersede IgM binding to the same antigen due to usually higher affinity, causing false‐negative results 30, 41, 136. Moreover, rheumatoid factors—antibodies of the IgM class directed against the Fc portion of IgG, which are mainly found in patients with autoimmune connective tissue disorders but also in up to 5% of healthy individuals—can react with IgG specifically bound to its antigen, causing false‐positive IgM results 114. It is therefore recommendable to remove antibodies of the IgG class from serum specimens prior to determining antibodies of the IgM class by ultracentrifugation, chromatography, immunoadsorption to protein A or G, or preferably, by immunoprecipitation. The only study performed after depletion of IgG found a frequency of AQP4‐IgM in patients with NMOSD of ∼10% in a CBA, but in none of 66 controls 59. In three patients, titers were higher after depletion of total IgG from the samples, and one sample was positive only after precipitation of total IgG. Importantly, all AQP4‐IgM‐positive patients were also positive for AQP4‐IgG in that study and none of the AQP4‐IgG‐negative samples were positive for AQP4‐IgM. Routine testing for AQP4‐IgM may thus not be justified; at least unless new data suggesting a definite diagnostic or prognostic impact of AQP4‐IgM determinations becomes available. AQP4‐IgM antibodies were also found in several other studies, which used other CBAs including FACS 73, 99, 151; however, the methodological concerns delineated above apply and may possibly explain the limited specificity of AQP4‐IgM found in one of these studies 99. There is no established indication for AQP4‐IgA or ‐IgE testing.
Testing for AQP4‐IgG subclasses
NMO‐IgG/AQP4‐IgG belong mainly to the complement‐activating IgG1 subclass, although IgG2, Ig3 and IgG4 antibodies have been occasionally detected, mostly with low frequency and at low titer 38, 73, 151. To date, no study has shown a significant correlation of NMO‐IgG/AQP4‐IgG subclasses with disease activity, treatment response or prognosis. Accordingly, there is currently no indication for routine assessment of NMO‐IgG/AQP4‐IgG subclasses.
AQP4‐Ab titers and disease activity
There is growing evidence that anti‐AQP4 titers may reflect disease activity. A retrospective longitudinal assessment of AQP4‐Ab in 96 samples obtained over a median of 5 years in eight patients with NMOSDs (6 × NMO; 2 × relapsing LETM) demonstrated significantly higher autoantibody serum levels during relapse than during remission as measured in a FIPA 54. Clinical attacks were preceded by a continuous rise in levels of AQP4‐Ab (but not of other autoantibodies), and acute disease activity was followed by an intraindividual decline of serum levels 54. Importantly, however, absolute AQP4‐Ab levels at relapse varied widely both intra‐ and interindividually and, accordingly, no general threshold value for triggering clinical relapse was established. Kim et al recently reported on the serial measurement of serum AQP4‐Ab levels by ELISA in individual patients during the long‐term course of the disease and also showed a strong correlation between antibody levels and disease activity 82. Most relapses were associated with high or rising AQP4‐Ab levels. Again, absolute AQP4‐Ab levels varied both between individuals and intraindividually over time 82. In a cross‐sectional Japanese study, including 35 individuals with NMOSD, high anti‐AQP4 titers, as determined in a CBA, coincided with complete blindness and extensive spinal cord and brain involvement 142. Three other studies found significantly higher serum titers during relapse using a CBA, a commercial ELISA and a FIPA, respectively 60, 66. AQP4‐Ab were also shown to be more frequently present in the CSF during relapse 60. Moreover, a decrease in AQP‐Ab serum concentrations in response to various immunosuppressive therapies was found in several studies 54, 82, 142. In patients treated with rituximab, the reappearance of even low B‐cell numbers was associated with an increase in AQP4‐Ab values and a high relapse risk. CD19+ cell counting might thus be an alternative to AQP4‐Ab testing in those patients 54. Possibly as a result of the drug not affecting plasma cells (including so‐called long‐lived ones), NMO‐IgG/AQP4‐Ab may remain detectable in rituximab‐treated patients even if CD19 cell counts are below the detection limit. A recent study confirmed that rituximab significantly lowers NMO‐IgG/AQP4‐IgG antibody levels, but found a transient increase in several patients 2 weeks after the first injection, raising concerns over the risk for an early BAFF‐mediated clinical worsening in patients with NMO receiving that drug 117. A particularly strong decline in Ab levels occurs following plasma exchange (∼85% ± 15% in a recent ELISA study) 80.
In conclusion, serial NMO‐IgG/AQP4‐Ab measurement could possibly facilitate the monitoring of NMOSD patients, for relapses are often preceded by a marked increase in antibody serum levels. However, no general threshold value for triggering clinical relapse exists, and, rising serum AQP4‐Ab levels are not accompanied by clinical relapses in all cases, suggesting that apart from AQP4‐Ab other factors such as blood–brain barrier damage, cytokine profiles, or T‐cell activation may play a role as well. Moreover, the need to test at very close intervals challenges the practical feasibility of such an approach. Furthermore, some assays provide only semiquantitative results and/or data on inter‐run reproducibility are lacking.
AQP4‐Ab titers and clinical phenotype
AQP4‐Ab seropositivity has been found to be more frequent in patients with relapsing than in patients with monophasic NMO 6, 69, 78 (Table 6), in patients presenting with either myelitis or ON at onset than in those presenting with simultaneous myelitis and ON at onset 69; in those with unilateral ON at onset than in those with bilateral ON 69; and in patients with coexisting autoimmunity 69. However, none of these criteria distinguishes sharply between seropositive and seronegative patients. Moreover, the diagnosis of monophasic NMO largely depends on the follow‐up period; in a recent multicenter study, the latency interval between first and second attack in patients meeting Wingerchuk's 2006 criteria ranged between 1 and 216 months. Apart from a more pronounced spinal lesion load in seropositive patients, no significant differences with regard to spinal or brain MRI at disease onset or later were found between seropositive and seronegative patients in the same study 69. Accordingly, the decision to test for AQP4‐Ab or not should not be based on any of such clinical or MRI features if the patient otherwise presents with a condition compatible with a diagnosis of NMO.
However, different recommendations apply in conditions in which NMO‐IgG/AQP4‐Ab are much less frequent than in clinically definite NMO and, in consequence, the risk of an unfavorably high ratio of false‐positive to true‐positive results is especially high, for example isolated, monophasic ON or brainstem encephalitis. In such patients, testing in at least two assays (and in the case of discrepant results, a third one), which is generally recommended, seems mandatory.
Median serum levels as measured by ELISA did not differ significantly between patients with NMO‐IgG/AQP4‐Ab‐positive NMO and patients with isolated LETM or isolated ON in four independent studies based on CBA, FIPA and ELISA data, respectively 61, 66, 82, 142. A CBA study suggested correlation between NMO/AQP4‐Ab serum levels and spinal cord lesion length, the presence of lesions extending over >3 segments and permanent visual loss in Japanese patients 142; however, contrasting results emerged from another Japanese study employing IHC, ELISA and FACS analysis 38.
Specificity of AQP4‐Ab in rheumatic patients
Sera from patients with rheumatic disorders (RDs) are commonly used as controls in studies evaluating diagnostic antibody assays, for RDs are often associated with wide range, polyclonal B‐cell activation, which may result in non‐specific reactions (“sticky samples”). It is therefore a potential drawback that not all studies included RD controls when evaluating assay specificity. However, those which included RD controls found almost no false‐positive results in this special control population.
Moreover, NMO has been shown to be frequently associated with other autoimmune disorders, including RD such as systemic lupus erythematosus (SLE) and Sjögren syndrome (SS) 64, 128, 149. By contrast, NMO‐IgG/AQP4‐Ab seems to be rare among patients with RD 76, 161. A recent study which analyzed serum samples from 109 neurological patients with established CTD, possible CTD, or vasculitis by means of a CBA found NMO‐IgG/AQP4‐Ab exclusively in patients with CTD and NMOSD but in none of 69 samples from patients with CTD or vasculitis and neurological disorders other than NMO, LETM or recurrent ON 64. Moreover, the frequency of NMO‐IgG/AQP4‐Ab in patients with NMO and RD did not differ from that found in patients with NMO but no RD (31 out of the 40, 78%) 64. Table 7 summarizes the results from six studies that investigated the frequency and specificity of NMO‐IgG/AQP4‐Ab in patients with RD.
Table 7.
NMO‐IgG/AQP4‐Ab in patients with rheumatic diseases (percentages in parentheses). CTD = connective tissue disorders; RD = rheumatic diseases; SLE = systemic lupus erythematosus; SS = Sjögren syndrome; NMOSD = neuromyelitis optica spectrum disorders; ICC‐F = fluoroimmunocytochemistry; RIPA = radioimmunoprecipitation assay; IHC‐F = fluoroimmunohistochemistry; Hu = human; M1 = M1 isoform of human AQP4; M23 = M23 isoform of human AQP4
| NMO‐IgG/AQP4 in patients with RD and NMOSD | NMO‐IgG/AQP4 in patients with RD and neurological disorders other than NMOSD | NMO‐IgG/AQP4 in patients with RD but no neurological disorders | Assay type and substrate | |
|---|---|---|---|---|
| Jarius et al 64, definite CTD | 16/54a (30) | 0/33 (0) | — | ICC‐F (Hu, M1) |
| Jarius et al 64, possible CTD | 15/42b (36) | 0/23 (0) | — | ICC‐F (Hu, M1) |
| Jarius et al 64, vasculitis | — | 0/13c (0) | — | ICC‐F (Hu, M1) |
| Katsumata et al 76, SLE/SS | 2/6d (33) | — | — | ICC‐F (Hu, M23) |
| Paul et al 123, CTD | — | — | 0/45e (0) | RIPA (HuM1‐35S‐methionine) |
| Paul et al 123, vasculitis | — | — | 0/6f (0) | RIPA (HuM1‐35S‐methionine) |
| Pittock et al, SLE/SS 128, cohort 1 | 5/5 (100) | 0/8 (0) | 0/25 (0) | IHC‐F (mouse) |
| Pittock et al, SLE/SS 128, cohort 2 | 5/14 (36) | 0/6 (0) | 0/10 (0) | IHC‐F (mouse) |
| Wandinger et al 149, definite SLE/SS | 8/11g (73) | 0/39 | 0/42 (0) | IIC‐F (Hu, M1) |
| Wandinger et al 149, definite SLE/SS | 7/11h (64) | 0/39 | 0/42 (0) | IHC‐F (mouse) |
| Závada, definite SLE | — | 1/50i | j | ICC‐F (Hu, M1) |
| Sum, test results | 58/143 (41) | 1/211 (0.4) | 0/170 (0) | |
| Sum, patients | 58/132 (44) | 1/172 (0.6) | 0/128 (0) |
Fifty‐four patients with SLE (n = 41), primary SS (6), SLE with secondary SS (2), systemic sclerosis (1), systemic sclerosis with SS (1), scleroderma en coup‐de‐sabre (1), CREST syndrome (1), and Sharp syndrome with biopsy‐proven polymyositis (1).
Forty‐two patients with various neurological syndromes (Table 1), who were positive for auto‐antibodies usually associated with CTD but who did not meet the formal criteria for any CTD based on the data available for analysis (anti‐nuclear antibodies in 40/42, SS‐A and/or SS‐B in 13, cardiolipin and anti‐β2‐glycoprotein antibodies in 16, double‐stranded DNA antibodies in nine, ribonucleoprotein antibodies in nine, lupus anticoagulants in three, Scl‐70 antibodies in two, histone antibodies in three, and centromer antibodies in five; three patients were, in addition, positive for rheumatoid factor and one for single‐strand DNA antibodies and circulating immune complexes; further features of CTD such as Raynaud's phenomenon, sicca symptoms, polyserositis, nephritis, or arthralgia were present in 16 patients; in addition, Coombs‐positive anemia was reported in one, and other hematological disturbances in seven).
Thirteen patients with vasculitis (7 × primary arteritis of the CNS, 4 × giant cell arteritis, 1 × leukocytoclastic vasculitis, and 1 × post‐infectious systemic vasculitis involving the CNS).
3 × active SLE, 3 × active SS.
11 × rheumatoid arthritis; 16 × SS, 8 × systemic lupus erythematosus, 9 × Wegener disease, 1 × Bechterew disease.
5 × “vasculitis”, 1 × Wegener disease.
7 × SLE, 4 × SS.
7 × SLE, 4 × SS.
The only positive patient had non‐longitudinally extensive transverse myelitis (NETM); NETM occurs in up to 7% of NMO patients at least once over the course of disease (see ref 69 and NMO‐IgG/AQP4‐Ab in patients with HRS other than LETM or ON).
Samples from patients with RD but no neurological disorders were reportedly negative, but data were not shown.
Seronegative NMO
With differences in assay sensitivity of up to 20% between the various diagnostic assays currently available 62, 83, 151, 152, methodological issues are certainly still among the most common causes of seronegativity in patients with NMO. Furthermore, NMO could be etiologically heterogeneous, representing a common phenotype of various autoimmune or infectious diseases, as indicated by a number of epidemiological and clinical differences between seropositive and seronegative patients 49, 69 [an overview of the differential diagnosis of NMO is provided elsewhere 17]. Evidence for a role of autoantibodies also in “seronegative” NMO comes from the finding that complement‐dependent astrocyte cell death induced by serum from NMO‐IgG/AQP4‐Ab‐seronegative patients with NMO was more pronounced that that induced by serum from patients with MS or healthy donors 134. Moreover, an effect of plasma exchange was reported in some AQP4‐Ab‐negative patients 11. However, it remains unknown whether these findings signify the presence of as yet unidentified, novel antibodies in AQP4‐Ab‐negative NMO or of low‐titer or low‐affinity NMO‐IgG/AQP4‐Ab not detectable with current techniques. In myasthenia gravis, another autoimmune disorder with proven humoral pathogenesis, it took almost 25 years until a more sensitive class of immunoassays was developed; subsequently, two‐thirds of patients previously classified as “seronegative” were found to harbor low‐affinity acetylcholine receptor (AChR) serum antibodies 88. Attempts to improve the sensitivity of AQP4‐Ab testing as well as to identify novel autoantigens in NMO are in progress. Recently, antibodies to MOG‐IgG as well as paraneoplastic antibodies such as CV2/CMRP5 have been detected in a small subset of seronegative patients with NMOSD or NMO‐like disease 23, 63, 70, 85, 100, 132, 133. Importantly, a lack of NMO‐IgG/AQP4‐Ab seropositivity does not rule out a diagnosis of NMO 157. Besides clinical and MRI (longitudinal extensive myelitis, no brain lesions meeting Paty criteria at disease onset) features, CSF analysis [negative oligoclonal bands (OCB) or OCB conversion 8, 9, 65, measles, rubella, zoster (MRZ) reaction 58, 69] can facilitate the differential diagnosis of MS and seronegative NMO.
Repeat testing in seronegative patients
Using the standard IHC assay, Lotze et al 97 found NMO‐IgG/AQP4‐Ab in only three out of five, one out of three, one out of three, four out of five and three out of three repetitive samples (median follow‐up since first testing 1.8 years), respectively, taken from five children with NMOSD. Treatment with immunosuppressants resulted in NMO‐IgG seronegativity in that study. Matsuoka et al 106 observed seroconversion from positive to negative or vice versa during follow‐up in 15% of their patients using a CBA, and Kim et al 82 in 55% using an ELISA. Given the therapeutic and diagnostic impact of NMO‐IgG/AQP4‐Ab seropositivity, repeat testing in patients with an initially negative test result therefore seems recommendable. However, the decision on how often to test for NMO‐IgG/AQP4‐Ab certainly has to take into account the differential performance of the various assays currently available: In sharp contrast to the findings discussed above, Takahashi et al found AQP4‐Ab in all of 25 samples from three patients taken over a period of 27, 39 and 69 months, respectively, using an ICC assay 142. Treatments during the observation period included intravenous methylprednisolone (IVMP), prednisolone and azathioprine. In another study, a FIPA was used to analyze longitudinal samples from eight patients with NMOSD obtained during relapse or remission over a median period of 62 months. NMO‐IgG/AQP4‐Ab was detectable in 95 out of the 96 serum samples in this assay, both in untreated patients and in all but one sample taken during immunosuppressive or immunomodulatory therapy. Treatment regimes included high dose IVMP, prednisolone, dexamethasone, rituximab, azathioprine, mitoxantrone, cyclosphosphamide, interferon beta and glatiramer acetate 54. While one study reported seronegativity after plasma exchange 33, AQP4‐Ab remained detectable in two others 54, 142.
NMO‐IgG/AQP4‐Ab before the onset of NMO
In most patients with NMO and MG, MG preceded NMO 53, 67, 89. In seven patients with this rare combination of autoimmune disorders, serum samples taken for AChR testing prior to the onset of NMO were available for retrospective NMO‐IgG/AQP4‐Ab serology 53, 89, 115, 119. Interestingly, NMO‐IgG/AQP4‐Ab was present in six out of seven of these patients, at 3–14 years before the first clinically apparent attack of NMO. Pathogenetically, this indicates that NMO‐IgG/AQP4‐Ab alone may not be sufficient to cause CNS damage; additional factors (such as blood–brain barrier damage and/or T‐cell activation) may be required. This is in line with the fact that NMO‐IgG/AQP4‐Ab remains detectable during remission in the majority of patients with established NMO, sometimes at a relatively high level 54, 66, 82, 142. Diagnostically and with regard to studies investigating assay accuracy, this means that NMO‐IgG/AQP4‐Ab seropositivity in patients without NMOSD does not always denote insufficient assay specificity; in such cases, repeat testing and confirmatory testing with methodologically independent assays with high accuracy is recommended.
High‐dose hook effect
The so‐called high‐dose hook effect (HDE) is a well‐known phenomenon in laboratory medicine causing false‐negative or falsely low results in immunoassays. Whether the simultaneous rather than sequential incubation of the coated antigen with both patient serum and AQP4‐biotin in the bridge‐ELISA could cause an HDE (this would require the patient's IgG to be capable of binding two AQP4‐biotin molecules) has not been addressed in the studies published so far 38, 66. HDE only rarely occurs in solid‐phase assays such as IHC or ICC assays. While HDE is caused by a saturating excess in antigen concentration in ELISA, which prevents sandwich or bridge formation, the cause of HDE in IHC and ICC is less well understood. Among other explanations, it has been speculated that HDE in IIF might be caused by anti‐immunoglobulin conjugates being unable to reach their antigenic determinants on tightly clustered immunoglobulin molecules 22. Long et al did not find evidence for a prozone effect with the commercial CBA (Euroimmun) when re‐testing sera that were negative at 1:4 and the standard 1:10 dilution at 1:32 and 1:120 dilutions 95, 96.
Effect of storage conditions and freeze/thaw cycles
The number of patients tested for NMO‐IgG/AQP4‐Ab has increased considerably in recent years. As tests are still only available at relatively few centers worldwide, however, samples often need to be sent elsewhere for testing. Moreover, samples may be tested repeatedly and thus undergo several freeze/thaw cycles. Recent data from a small preliminary study suggest that AQP4‐IgG are relatively stable over a period of at least 8 days at room temperature or 4°C and that AQP4‐IgG levels are not affected to any significant degree by repeat freeze/thaw cycles 45. Shipment at room temperature might thus be justified if other shipment options are not available, provided that temperatures do not exceed 18°C, serum is separated prior to shipment, and tests are performed within a few days after blood sampling. As a limitation, it should be noted that borderline samples or hemolytic samples, which were not investigated in that study, might be more sensitive to storage conditions. Moreover, if proteins other than AQP4‐IgG—such as cytokines—need to be analyzed in addition to AQP4‐Ab, it is highly recommended that the samples be stored at −80°C or shipped on dry ice and that freeze/thaw cycles are avoided 45.
Summary and Outlook
Misclassification of NMO‐IgG/AQP4‐Ab‐mediated autoimmunity as classical MS may result in treatment with interferon beta or natalizumab, which are thought to be ineffective or even harmful in patients with NMO 54, 87, 120, 121, 138, 139, 145, 148, 150. Conversely, misclassification of MS as NMO may result in treatment with potentially harmful immunosuppressive drugs not approved for the treatment of MS. Therefore, high assay accuracy is crucial. Moreover, treatment decisions should never be based solely on NMO‐IgG/AQP4‐Ab treatment status. Currently, CBAs (ICC, FACS) seem to be most sensitive and specific. Assays with low sensitivity such as immunohistochemistry on brain tissue sections (IHC‐F) should not be used as screening assays but might be useful as second‐line confirmatory assays. The diagnostic impact of testing CSF for NMO‐IgG/AQP4‐Ab is controversial; serum samples are currently the specimen of choice. There is no established indication of AQP‐IgM or ‐IgA serology. Longitudinal measurement of NMO‐IgG/AQP4‐Ab is potentially useful (especially in patients treated with rituximab), but might not be feasible in practice due to the close test intervals needed, missing threshold values, and insufficient inter‐run reproducibility of some assays. Given the low prevalence of AQP4‐Ab‐positive NMO compared to that of MS, most studies did not include sufficiently large control cohorts to assess assay specificity in any definitive way. Confirmation in a second (and, in the case of discrepant results, a third), methodologically independent assay with high test accuracy is generally recommended and is particularly important in patients presenting with conditions only rarely associated with NMO‐IgG/AQP4‐Ab such as isolated ON or brainstem encephalitis. The present analysis is helpful by summarizing results from studies using the same type of assay. However, large‐scale, multicenter studies in unselected cohorts are warranted. Assay improvement remains an important goal, as well as the development of standardized and easy‐to‐use assays, which would make NMO‐IgG/AQP4‐Ab testing more widely available.
Conflict of Interest Statement
The authors declare that they have no conflict of interest.
AUTHOR CONTRIBUTIONS
S.J. designed the study, analysed the data and wrote the manuscript. S.J. and B.W. were involved in collecting the data and in revising the manuscript for important intellectual content.
Supporting information
Table S1. Sample numbers, positivity rates, and differences in methodology and diagnostic criteria; data from 59 studies including 93 test series.
Acknowledgments
The work of S.J. was supported by a research fellowship from the European Committee for Treatment and Research in Multiple Sclerosis (ECTRIMS). The work of B.W. was supported by research grants from Merck Serono.
References
- 1. Adoni T, Lino AM, Marchiori PE, Kok F, Callegaro D (2008) Seroprevalence of NMO‐IgG antibody in Brazilian patients with neuromyelitis optica. Arq Neuropsiquiatr 66(2b):295–297. [DOI] [PubMed] [Google Scholar]
- 2. Alvarenga MP, Alvarenga RM, Santos AM, Thuler LC (2012) Anti‐AQP(4) antibody in idiopathic acute transverse myelitis with recurrent clinical course: frequency of positivity and influence in prognosis. J Spinal Cord Med 35:251–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Amiry‐Moghaddam M, Frydenlund DS, Ottersen OP (2004) Anchoring of aquaporin‐4 in brain: molecular mechanisms and implications for the physiology and pathophysiology of water transport. Neuroscience 129:999–1010. [DOI] [PubMed] [Google Scholar]
- 4. Apiwattanakul M, McKeon A, Fryer JP, Hinson SR, Pittock SJ, Lennon VA (2009) AQP4‐IgG immunoprecipitation assay optimization. Clin Chem 55:592–594. [DOI] [PubMed] [Google Scholar]
- 5. Apiwattanakul M, Asawavichienjinda T, Pulkes T, Tantirittisak T, Hemachudha T, Horta ES et al (2012) Diagnostic utility of NMO/AQP4‐IgG in evaluating CNS inflammatory disease in Thai patients. J Neurol Sci 320:118–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Banwell B, Tenembaum S, Lennon VA, Ursell E, Kennedy J, Bar‐Or A et al (2008) Neuromyelitis optica‐IgG in childhood inflammatory demyelinating CNS disorders. Neurology 70:344–352. [DOI] [PubMed] [Google Scholar]
- 7. Bennett JL, Lam C, Kalluri SR, Saikali P, Bautista K, Dupree C et al (2009) Intrathecal pathogenic anti‐aquaporin‐4 antibodies in early neuromyelitis optica. Ann Neurol 66:617–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bergamaschi R (2003) Importance of cerebrospinal fluid examination in differential diagnosis of Devic's neuromyelitis optica by multiple sclerosis. Neurol Sci 24:95–96. [DOI] [PubMed] [Google Scholar]
- 9. Bergamaschi R, Tonietti S, Franciotta D, Candeloro E, Tavazzi E, Piccolo G et al (2004) Oligoclonal bands in Devic's neuromyelitis optica and multiple sclerosis: differences in repeated cerebrospinal fluid examinations. Mult Scler 10:2–4. [DOI] [PubMed] [Google Scholar]
- 10. Bizzoco E, Lolli F, Repice AM, Hakiki B, Falcini M, Barilaro A et al (2009) Prevalence of neuromyelitis optica spectrum disorder and phenotype distribution. J Neurol 256:1891–1898. [DOI] [PubMed] [Google Scholar]
- 11. Bonnan M, Brasme H, Diaby MM, Vlaicu M, Le Guern V, Zuber M (2009) [Severe bouts of neuromyelitis optica: dramatic improvement after plasma exchanges.]. Rev Neurol (Paris) 165:479–481. [DOI] [PubMed] [Google Scholar]
- 12. Chan KH, Kwan JS, Ho PW, Ho JW, Chu AC, Ramsden DB (2010) Aquaporin‐4 autoantibodies in neuromyelitis optica spectrum disorders: comparison between tissue‐based and cell‐based indirect immunofluorescence assays. J Neuroinflammation 7:50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chen T, Hedman L, Mattila PS, Jartti L, Jartti T, Ruuskanen O et al (2012) Biotin IgM antibodies in human blood: a previously unknown factor eliciting false results in biotinylation‐based immunoassays. PLoS ONE 7:e42376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Collongues N, Marignier R, Zephir H, Papeix C, Blanc F, Ritleng C et al (2010) Neuromyelitis optica in France: a multicenter study of 125 patients. Neurology 74:736–742. [DOI] [PubMed] [Google Scholar]
- 15. Crane JM, Bennett JL, Verkman AS (2009) Live cell analysis of aquaporin‐4 M1/M23 interactions and regulated orthogonal array assembly in glial cells. J Biol Chem 284:35850–35860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Crane JM, Lam C, Rossi A, Gupta T, Bennett JL, Verkman AS (2011) Binding affinity and specificity of neuromyelitis optica autoantibodies to aquaporin‐4 M1/M23 isoforms and orthogonal arrays. J Biol Chem 286:16516–16524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cree BA, Goodin DS, Hauser SL (2002) Neuromyelitis optica. Semin Neurol 22:105–122. [DOI] [PubMed] [Google Scholar]
- 18. Dale GL, Gaddy P, Pikul FJ (1994) Antibodies against biotinylated proteins are present in normal human serum. J Lab Clin Med 123:365–371. [PubMed] [Google Scholar]
- 19. De Vidi I, Boursier G, Delouche N, Portales P, Cadars E, Bouthier M et al (2011) Strategy for anti‐aquaporin‐4 auto‐antibody identification and quantification using a new cell‐based assay. Clin Immunol 138:239–246. [DOI] [PubMed] [Google Scholar]
- 20. Deeks JJ, Altman DG (2004) Diagnostic tests 4: likelihood ratios. BMJ 329:168–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Dellavance A, Alvarenga RR, Rodrigues SH, Kok F, de Souza AW, Andrade LE (2012) Anti‐aquaporin‐4 antibodies in the context of assorted immune‐mediated diseases. Eur J Neurol 19:248–252. [DOI] [PubMed] [Google Scholar]
- 22. Dubois‐Galopin F, Beauvillain C, Dubois D, Pillet A, Renier G, Jeannin P et al (2007) New markers and an old phenomenon: prozone effect disturbing detection of filaggrin (keratin) autoantibodies. Ann Rheum Dis 66:1121–1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ducray F, Roos‐Weil R, Garcia PY, Slesari J, Heinzlef O, Chatelain D et al (2007) Devic's syndrome‐like phenotype associated with thymoma and anti‐CV2/CRMP5 antibodies. J Neurol Neurosurg Psychiatry 78:325–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Etemadifar M, Abtahi MA, Razmjoo H, Abtahi SH, Dehghani AR, Abtahi ZA et al (2012) Anti‐aquaporin‐4 IgG in patients presenting with unilateral optic neuritis: a cohort study. Int J Prev Med 3:612–615. [PMC free article] [PubMed] [Google Scholar]
- 25. Etemadifar M, Mollabashi M, Chitsaz A, Behnamfar O, Bahrami E, Minagar A, Maghzi AH (2012) Seroprevalence of NMO‐IgG among patients with neuromyelitis optica and opticospinal multiple sclerosis. Clin Neurol Neurosurg 114:17–20. [DOI] [PubMed] [Google Scholar]
- 26. Fazio R, Malosio ML, Lampasona V, De Feo D, Privitera D, Marnetto F et al (2009) Antiacquaporin 4 antibodies detection by different techniques in neuromyelitis optica patients. Mult Scler 15:1153–1163. [DOI] [PubMed] [Google Scholar]
- 27. Fujihara K (2012) Comment: sensitivity and clinical relevance of available anti‐aquaporin‐4 antibody assays. Neurology 78:669. [Google Scholar]
- 28. Furman CS, Gorelick‐Feldman DA, Davidson KG, Yasumura T, Neely JD, Agre P, Rash JE (2003) Aquaporin‐4 square array assembly: opposing actions of M1 and M23 isoforms. Proc Natl Acad Sci U S A 100:13609–13614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ghezzi A, Bergamaschi R, Martinelli V, Trojano M, Tola MR, Merelli E et al (2004) Clinical characteristics, course and prognosis of relapsing Devic's Neuromyelitis Optica. J Neurol 251:47–52. [DOI] [PubMed] [Google Scholar]
- 30. Graham DA, Foster JC, Mawhinney KA, Elvander M, Adair BM, Merza M (1999) Detection of IgM responses to bovine respiratory syncytial virus by indirect ELISA following experimental infection and reinfection of calves: abolition of false positive and false negative results by pre‐treatment of sera with protein‐G agarose. Vet Immunol Immunopathol 71:41–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Granieri L, Marnetto F, Valentino P, Frau J, Patanella AK, Nytrova P et al (2012) Evaluation of a multiparametric immunofluorescence assay for standardization of neuromyelitis optica serology. PLoS ONE 7:e38896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hayakawa S, Mori M, Okuta A, Kamegawa A, Fujiyoshi Y, Yoshiyama Y et al (2008) Neuromyelitis optica and anti‐aquaporin‐4 antibodies measured by an enzyme‐linked immunosorbent assay. J Neuroimmunol 196:181–187. [DOI] [PubMed] [Google Scholar]
- 33. Heerlein K, Jarius S, Jacobi C, Rohde S, Storch‐Hagenlocher B, Wildemann B (2009) Aquaporin‐4 antibody positive longitudinally extensive transverse myelitis following varicella zoster infection. J Neurol Sci 276:184–186. [DOI] [PubMed] [Google Scholar]
- 34. Hinson SR, Pittock SJ, Lucchinetti CF, Roemer SF, Fryer JP, Kryzer TJ, Lennon VA (2007) Pathogenic potential of IgG binding to water channel extracellular domain in neuromyelitis optica. Neurology 69:2221–2231. [DOI] [PubMed] [Google Scholar]
- 35. Hiroaki Y, Tani K, Kamegawa A, Gyobu N, Nishikawa K, Suzuki H et al (2006) Implications of the aquaporin‐4 structure on array formation and cell adhesion. J Mol Biol 355:628–639. [DOI] [PubMed] [Google Scholar]
- 36. Ho JD, Yeh R, Sandstrom A, Chorny I, Harries WE, Robbins RA et al (2009) Crystal structure of human aquaporin 4 at 1.8 A and its mechanism of conductance. Proc Natl Acad Sci U S A 106:7437–7442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Iorio R, Fryer JP, Hinson SR, Fallier‐Becker P, Wolburg H, Pittock SJ, Lennon VA (2013) Astrocytic autoantibody of neuromyelitis optica (NMO‐IgG) binds to aquaporin‐4 extracellular loops, monomers, tetramers and high order arrays. J Autoimmun 40:21–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Isobe N, Yonekawa T, Matsushita T, Kawano Y, Masaki K, Yoshimura S et al (2012) Quantitative assays for anti‐aquaporin‐4 antibody with subclass analysis in neuromyelitis optica. Mult Scler 18:1541–1551. [DOI] [PubMed] [Google Scholar]
- 39. Ito S (2009) Posterior reversible encephalopathy syndrome in neuromyelitis optica spectrum disorders. Neurology 73:1604. author reply ‐5. [DOI] [PubMed] [Google Scholar]
- 40. Jacob S, Zarei M, Kenton A, Allroggen H (2005) Gluten sensitivity and neuromyelitis optica: two case reports. J Neurol Neurosurg Psychiatry 76:1028–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Jankowski M, Gut W, Imbs D, Switalski L (1979) Study of interaction between IgG and IgM antibodies against rubella virus by the immunofluorescence method. Acta Microbiol Pol 28:63–69. [PubMed] [Google Scholar]
- 42. Jarius S, Wildemann B (2007) [Neuromyelitis optica.]. Nervenarzt 78:1365–1377. [DOI] [PubMed] [Google Scholar]
- 43. Jarius S, Wildemann B (2010) AQP4 antibodies in neuromyelitis optica: diagnostic and pathogenetic relevance. Nat Rev Neurol 6:383–392. [DOI] [PubMed] [Google Scholar]
- 44. Jarius S, Wildemann B (2011) An early case of neuromyelitis optica: on a forgotten report by Jacob Lockhart Clarke, FRS. Mult Scler 17:1384–1386. [DOI] [PubMed] [Google Scholar]
- 45. Jarius S, Wildemann B (2011) Effect of storage conditions and freeze/thaw cycles on aquaporin‐4 antibody (NMO‐IgG) serum levels. Clin Chem Lab Med 49:2121–2122. [DOI] [PubMed] [Google Scholar]
- 46. Jarius S, Wildemann B (2012) The case of the Marquis de Causan (1804): an early account of visual loss associated with spinal cord inflammation. J Neurol 259:1354–1357. [DOI] [PubMed] [Google Scholar]
- 47. Jarius S, Wildemann B (2012) An early British case of neuromyelitis optica (1850). BMJ 345:e6430. [DOI] [PubMed] [Google Scholar]
- 48. Jarius S, Wildemann B (2012) “Noteomielite” accompanied by acute amaurosis (1844). An early case of neuromyelitis optica. J Neurol Sci 313:182–184. [DOI] [PubMed] [Google Scholar]
- 49. Jarius S, Wildemann B (2013) The history of neuromyelitis optica. J Neuroinflammation 10:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Jarius S, Wildemann B (2013) On the contribution of Thomas Clifford Allbutt, F.R.S., to the early history of neuromyelitis optica. J Neurol 260:100–104. [DOI] [PubMed] [Google Scholar]
- 51. Jarius S, Franciotta D, Bergamaschi R, Wright H, Littleton E, Palace J et al (2007) NMO‐IgG in the diagnosis of neuromyelitis optica. Neurology 68:1076–1077. [DOI] [PubMed] [Google Scholar]
- 52. Jarius S, Paul F, Franciotta D, Aktas O, Hohlfeld R, Zipp F, Vincent A (2007) Revised diagnostic criteria for neuromyelitis optica—incorporation of NMO‐IgG status. Nat Clin Pract Neurol 3:E1. [DOI] [PubMed] [Google Scholar]
- 53. Jarius S, Paul F, Martins da Silva A, Leite MI, Waters P, Littleton E et al (2007) Neuromyelitis optica and longitudinally extensive transverse myelitis following thymectomy for myasthenia gravis. Mult Scler 13(Suppl 2):P534. [Google Scholar]
- 54. Jarius S, Aboul‐Enein F, Waters P, Kuenz B, Hauser A, Berger T et al (2008) Antibody to aquaporin‐4 in the long‐term course of neuromyelitis optica. Brain 131(Pt 11):3072–3080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Jarius S, Franciotta D, Bergamaschi R, Rauer S, Wandinger KP, Petereit HF et al (2008) Polyspecific, antiviral immune response distinguishes multiple sclerosis and neuromyelitis optica. J Neurol Neurosurg Psychiatry 79:1134–1136. [DOI] [PubMed] [Google Scholar]
- 56. Jarius S, Jacob S, Waters P, Jacob A, Littleton E, Vincent A (2008) Neuromyelitis optica in patients with gluten sensitivity associated with antibodies to aquaporin‐4. J Neurol Neurosurg Psychiatry 79:1084. [DOI] [PubMed] [Google Scholar]
- 57. Jarius S, Paul F, Franciotta D, Waters P, Zipp F, Hohlfeld R et al (2008) Mechanisms of disease: aquaporin‐4 antibodies in neuromyelitis optica. Nat Clin Pract Neurol 4:202–214. [DOI] [PubMed] [Google Scholar]
- 58. Jarius S, Eichhorn P, Jacobi C, Wildemann B, Wick M, Voltz R (2009) The intrathecal, polyspecific antiviral immune response: specific for MS or a general marker of CNS autoimmunity? J Neurol Sci 280:98–100. [DOI] [PubMed] [Google Scholar]
- 59. Jarius S, Franciotta D, Bergamaschi R, Wildemann B, Wandinger KP (2010) Immunoglobulin M antibodies to aquaporin‐4 in neuromyelitis optica and related disorders. Clin Chem Lab Med 48:659–663. [DOI] [PubMed] [Google Scholar]
- 60. Jarius S, Franciotta D, Paul F, Ruprecht K, Bergamaschi R, Rommer PS et al (2010) Cerebrospinal fluid antibodies to aquaporin‐4 in neuromyelitis optica and related disorders: frequency, origin, and diagnostic relevance. J Neuroinflammation 7:52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Jarius S, Frederikson J, Waters P, Paul F, Akman‐Demir G, Marignier R et al (2010) Frequency and prognostic impact of antibodies to aquaporin‐4 in patients with optic neuritis. J Neurol Sci 298:158–162. [DOI] [PubMed] [Google Scholar]
- 62. Jarius S, Probst C, Borowski K, Franciotta D, Wildemann B, Stoecker W, Wandinger KP (2010) Standardized method for the detection of antibodies to aquaporin‐4 based on a highly sensitive immunofluorescence assay employing recombinant target antigen. J Neurol Sci 291:52–56. [DOI] [PubMed] [Google Scholar]
- 63. Jarius S, Warth A, Wandinger KP, Schnabel PA, Hoffmann H, Wildemann B, Muley T (2010) Antibodies to aquaporin‐4 in non‐small cell lung cancer: a study on 50 patients. Neurol Sci 31:871–872. [DOI] [PubMed] [Google Scholar]
- 64. Jarius S, Jacobi C, de Seze J, Zephir H, Paul F, Franciotta D et al (2011) Frequency and syndrome specificity of antibodies to aquaporin‐4 in neurological patients with rheumatic disorders. Mult Scler 17:1067–1073. [DOI] [PubMed] [Google Scholar]
- 65. Jarius S, Paul F, Franciotta D, Ruprecht K, Ringelstein M, Bergamaschi R et al (2011) Cerebrospinal fluid findings in aquaporin‐4 antibody positive neuromyelitis optica: results from 211 lumbar punctures. J Neurol Sci 306:82–90. [DOI] [PubMed] [Google Scholar]
- 66. Jarius S, Franciotta D, Paul F, Bergamaschi R, Rommer PS, Ruprecht K et al (2012) Testing for antibodies to human aquaporin‐4 by ELISA: sensitivity, specificity, and direct comparison with immunohistochemistry. J Neurol Sci 320:32–37. [DOI] [PubMed] [Google Scholar]
- 67. Jarius S, Paul F, Franciotta D, de Seze J, Munch C, Salvetti M et al (2012) Neuromyelitis optica spectrum disorders in patients with myasthenia gravis: ten new aquaporin‐4 antibody positive cases and a review of the literature. Mult Scler 18:1135–1143. [DOI] [PubMed] [Google Scholar]
- 68. Jarius S, Paul F, Ruprecht K, Wildemann B (2012) Low vitamin B12 levels and gastric parietal cell antibodies in patients with aquaporin‐4 antibody‐positive neuromyelitis optica spectrum disorders. J Neurol 259:2743–2745. [DOI] [PubMed] [Google Scholar]
- 69. Jarius S, Ruprecht K, Wildemann B, Kuempfel T, Ringelstein M, Geis C et al (2012) Contrasting disease patterns in seropositive and seronegative neuromyelitis optica: a multicentre study of 175 patients. J Neuroinflammation 9:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Jarius S, Wandinger KP, Borowski K, Stoecker W, Wildemann B (2012) Antibodies to CV2/CRMP5 in neuromyelitis optica‐like disease: case report and review of the literature. Clin Neurol Neurosurg 114:331–335. [DOI] [PubMed] [Google Scholar]
- 71. Jarius S, Lauda F, Wildemann B, Tumani H (2013) Steroid‐responsive hearing impairment in NMO‐IgG/aquaporin‐4‐antibody‐positive neuromyelitis optica. J Neurol 260:663–664. [DOI] [PubMed] [Google Scholar]
- 72. Kalluri SR, Srivastava R, Cepok S, Menge T, Cree B, Berthele A, Hemmer B (2009) A novel bio‐assay to determine and characterize the antibodies to aquaporin‐4. Neurology 72(Suppl 3):A189. [Google Scholar]
- 73. Kalluri SR, Illes Z, Srivastava R, Cree B, Menge T, Bennett JL et al (2010) Quantification and functional characterization of antibodies to native aquaporin 4 in neuromyelitis optica. Arch Neurol 67:1201–1208. [DOI] [PubMed] [Google Scholar]
- 74. Kampylafka EI, Routsias JG, Alexopoulos H, Dalakas MC, Moutsopoulos HM, Tzioufas AG (2011) Fine specificity of antibodies against AQP4: epitope mapping reveals intracellular epitopes. J Autoimmun 36:221–227. [DOI] [PubMed] [Google Scholar]
- 75. Kang ES, Min JH, Lee KH, Kim BJ (2012) Clinical usefulness of cell‐based indirect immunofluorescence assay for the detection of aquaporin‐4 antibodies in neuromyelitis optica spectrum disorder. Ann Lab Med 32:331–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Katsumata Y, Kawachi I, Kawaguchi Y, Gono T, Ichida H, Hara M, Yamanaka H (2012) Semiquantitative measurement of aquaporin‐4 antibodies as a possible surrogate marker of neuromyelitis optica spectrum disorders with systemic autoimmune diseases. Mod Rheumatol 22:676–684. [DOI] [PubMed] [Google Scholar]
- 77. Kay CS, Scola RH, Lorenzoni PJ, Jarius S, Arruda WO, Werneck LC (2008) NMO‐IgG positive neuromyelitis optica in a patient with myasthenia gravis but no thymectomy. J Neurol Sci 275:148–150. [DOI] [PubMed] [Google Scholar]
- 78. Ketelslegers IA, Modderman PW, Vennegoor A, Killestein J, Hamann D, Hintzen RQ (2011) Antibodies against aquaporin‐4 in neuromyelitis optica: distinction between recurrent and monophasic patients. Mult Scler 17:1527–1530. [DOI] [PubMed] [Google Scholar]
- 79. Kim JE, Kim SM, Ahn SW, Lim BC, Chae JH, Hong YH et al (2011) Brain abnormalities in neuromyelitis optica. J Neurol Sci 302:43–48. [DOI] [PubMed] [Google Scholar]
- 80. Kim SH, Kim W, Huh SY, Lee KY, Jung IJ, Kim HJ (2013) Clinical efficacy of plasmapheresis in patients with neuromyelitis optica spectrum disorder and effects on circulating anti‐aquaporin‐4 antibody levels. J Clin Neurol 9:36–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Kim SM, Waters P, Woodhall M, Kim JY, Kim JE, Yang JW et al (2013) Utility of aquaporin‐4 antibody assay in patients with neuromyelitis optica spectrum disorders. Mult Scler 19:1060–1067. [DOI] [PubMed] [Google Scholar]
- 82. Kim W, Lee JE, Li XF, Kim SH, Han BG, Lee BI et al (2012) Quantitative measurement of anti‐aquaporin‐4 antibodies by enzyme‐linked immunosorbent assay using purified recombinant human aquaporin‐4. Mult Scler 18:578–586. [DOI] [PubMed] [Google Scholar]
- 83. Kim YJ, Jung SW, Kim Y, Park YJ, Han K, Oh EJ (2012) Detection of anti‐aquaporin‐4 antibodies in neuromyelitis optica: comparison of tissue‐based and cell‐based indirect immunofluorescence assays and ELISA. J Clin Lab Anal 26:184–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Kira J, Kanai T, Nishimura Y, Yamasaki K, Matsushita S, Kawano Y et al (1996) Western versus Asian types of multiple sclerosis: immunogenetically and clinically distinct disorders. Ann Neurol 40:569–574. [DOI] [PubMed] [Google Scholar]
- 85. Kitley J, Woodhall M, Waters P, Leite MI, Devenney E, Craig J et al (2012) Myelin‐oligodendrocyte glycoprotein antibodies in adults with a neuromyelitis optica phenotype. Neurology 79:1273–1277. [DOI] [PubMed] [Google Scholar]
- 86. Klawiter EC, Alvarez E 3rd, Xu J, Paciorkowski AR, Zhu L, Parks BJ et al (2009) NMO‐IgG detected in CSF in seronegative neuromyelitis optica. Neurology 72:1101–1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Kleiter I, Hellwig K, Berthele A, Kumpfel T, Linker RA, Hartung HP et al (2012) Failure of natalizumab to prevent relapses in neuromyelitis optica. Arch Neurol 69:239–245. [DOI] [PubMed] [Google Scholar]
- 88. Leite MI, Jacob S, Viegas S, Cossins J, Clover L, Morgan BP et al (2008) IgG1 antibodies to acetylcholine receptors in “seronegative” myasthenia gravis. Brain 131(Pt 7):1940–1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Leite MI, Coutinho E, Lana‐Peixoto M, Apostolos S, Waters P, Sato D et al (2012) Myasthenia gravis and neuromyelitis optica spectrum disorder: a multicenter study of 16 patients. Neurology 78:1601–1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Lennon VA, Wingerchuk DM, Kryzer TJ, Pittock SJ, Lucchinetti CF, Fujihara K et al (2004) A serum autoantibody marker of neuromyelitis optica: distinction from multiple sclerosis. Lancet 364:2106–2112. [DOI] [PubMed] [Google Scholar]
- 91. Lennon VA, Kryzer TJ, Pittock SJ, Verkman AS, Hinson SR (2005) IgG marker of optic‐spinal multiple sclerosis binds to the aquaporin‐4 water channel. J Exp Med 202:473–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Long Y, Hu X, Peng F, Lu Z, Wang Y, Yang Y, Qiu W (2012) Neuromyelitis optica immunoglobulin G in Chinese patients detected by immunofluorescence assay on a monkey brain substrate. Neuroimmunomodulation 19:20–24. [DOI] [PubMed] [Google Scholar]
- 93. Long Y, Lu Z, Hu X (2012) Serum‐positive and ‐negative AQP4 antibody NMO in Chinese patients. Can J Neurol Sci 39:232–235. [DOI] [PubMed] [Google Scholar]
- 94. Long Y, Qiu W, Hu X, Peng F, Lu Z, Wang Y, Yang Y (2012) Anti‐aquaporin‐4 antibody in Chinese patients with central nervous system inflammatory demyelinating disorders. Clin Neurol Neurosurg 114:1131–1134. [DOI] [PubMed] [Google Scholar]
- 95. Long Y, Qiu W, Lu Z, Bao J, Wu A, Wang Y et al (2012) Aquaporin 4 antibodies in the cerebrospinal fluid are helpful in diagnosing chinese patients with neuromyelitis optica. Neuroimmunomodulation 19:96–102. [DOI] [PubMed] [Google Scholar]
- 96. Long Y, Qiu W, Lu Z, Peng F, Hu X (2013) Clinical features of Chinese patients with multiple sclerosis with aquaporin‐4 antibodies in cerebrospinal fluid but not serum. J Clin Neurosci 20:233–237. [DOI] [PubMed] [Google Scholar]
- 97. Lotze TE, Northrop JL, Hutton GJ, Ross B, Schiffman JS, Hunter JV (2008) Spectrum of pediatric neuromyelitis optica. Pediatrics 122:e1039–e1047. [DOI] [PubMed] [Google Scholar]
- 98. Lourenco EV, Roque‐Barreira MC (2010) Immunoenzymatic quantitative analysis of antigens expressed on the cell surface (cell‐ELISA). Methods Mol Biol 588:301–309. [DOI] [PubMed] [Google Scholar]
- 99. Mader S, Lutterotti A, Di Pauli F, Kuenz B, Schanda K, Aboul‐Enein F et al (2010) Patterns of antibody binding to aquaporin‐4 isoforms in neuromyelitis optica. PLoS ONE 5:e10455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Mader S, Gredler V, Schanda K, Rostasy K, Dujmovic I, Pfaller K et al (2011) Complement activating antibodies to myelin oligodendrocyte glycoprotein in neuromyelitis optica and related disorders. J Neuroinflammation 8:184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Magana SM, Matiello M, Pittock SJ, McKeon A, Lennon VA, Rabinstein AA et al (2009) Posterior reversible encephalopathy syndrome in neuromyelitis optica spectrum disorders. Neurology 72:712–717. [DOI] [PubMed] [Google Scholar]
- 102. Marignier R, De Seze J, Vukusic S, Durand‐Dubief F, Zephir H, Vermersch P et al (2008) NMO‐IgG and Devic's neuromyelitis optica: a French experience. Mult Scler 14:440–445. [DOI] [PubMed] [Google Scholar]
- 103. Marnetto F, Granieri L, Sala A, Frau J, Patanella AK, Gilli F et al (2009) Validation of a multi‐parametric immunofluorescence assay for the detection of anti‐AQP4 antibodies in the diagnosis of neuromyelitis optica. Mult Scler 15:P154. [Google Scholar]
- 104. Marnetto F, Hellias B, Granieri L, Frau J, Patanella AK, Nytrova P et al (2009) Western blot analysis for the detection of serum antibodies recognizing linear Aquaporin‐4 epitopes in patients with Neuromyelitis Optica. J Neuroimmunol 217:74–79. [DOI] [PubMed] [Google Scholar]
- 105. Matiello M, Lennon VA, Jacob A, Pittock SJ, Lucchinetti CF, Wingerchuk DM, Weinshenker BG (2008) NMO‐IgG predicts the outcome of recurrent optic neuritis. Neurology 70:2197–2200. [DOI] [PubMed] [Google Scholar]
- 106. Matsuoka T, Matsushita T, Kawano Y, Osoegawa M, Ochi H, Ishizu T et al (2007) Heterogeneity of aquaporin‐4 autoimmunity and spinal cord lesions in multiple sclerosis in Japanese. Brain 130(Pt 5):1206–1223. [DOI] [PubMed] [Google Scholar]
- 107. Matsushita T, Isobe N, Matsuoka T, Shi N, Kawano Y, Wu XM et al (2009) Aquaporin‐4 autoimmune syndrome and anti‐aquaporin‐4 antibody‐negative opticospinal multiple sclerosis in Japanese. Mult Scler 15:834–847. [DOI] [PubMed] [Google Scholar]
- 108. Matsushita T, Isobe N, Piao H, Matsuoka T, Ishizu T, Doi H et al (2010) Reappraisal of brain MRI features in patients with multiple sclerosis and neuromyelitis optica according to anti‐aquaporin‐4 antibody status. J Neurol Sci 291:37–43. [DOI] [PubMed] [Google Scholar]
- 109. McDonald WI, Compston A, Edan G, Goodkin D, Hartung HP, Lublin FD et al (2001) Recommended diagnostic criteria for multiple sclerosis: guidelines from the International Panel on the diagnosis of multiple sclerosis. Ann Neurol 50:121–127. [DOI] [PubMed] [Google Scholar]
- 110. McKeon A, Fryer JP, Apiwattanakul M, Lennon VA, Hinson SR, Kryzer TJ et al (2009) Diagnosis of neuromyelitis spectrum disorders: comparative sensitivities and specificities of immunohistochemical and immunoprecipitation assays. Arch Neurol 66:1134–1138. [DOI] [PubMed] [Google Scholar]
- 111. McKeon A, Lennon VA, Lotze T, Tenenbaum S, Ness JM, Rensel M et al (2008) CNS aquaporin‐4 autoimmunity in children. Neurology 71:93–100. [DOI] [PubMed] [Google Scholar]
- 112. McKeon A, Lennon VA, Jacob A, Matiello M, Lucchinetti CF, Kale N et al (2009) Coexistence of myasthenia gravis and serological markers of neurological autoimmunity in neuromyelitis optica. Muscle Nerve 39:87–90. [DOI] [PubMed] [Google Scholar]
- 113. Mealy MA, Wingerchuk DM, Greenberg BM, Levy M (2012) Epidemiology of neuromyelitis optica in the United States: a multicenter analysis. Arch Neurol 69:1176–1180. [DOI] [PubMed] [Google Scholar]
- 114. Meurman OH, Ziola BR (1978) IgM‐class rheumatoid factor interference in the solid‐phase radioimmunoassay of rubella‐specific IgM antibodies. J Clin Pathol 31:483–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Mori M, Kawaguchi N, Uzawa A, Nemoto Y, Masuda S, Kuwabara S (2012) Seroconversion of anti‐aquaporin‐4 antibody in NMO spectrum disorder: a case report. J Neurol 259:980–981. [DOI] [PubMed] [Google Scholar]
- 116. Nakashima I, Fujihara K, Miyazawa I, Misu T, Narikawa K, Nakamura M et al (2006) Clinical and MRI features of Japanese patients with multiple sclerosis positive for NMO‐IgG. J Neurol Neurosurg Psychiatry 77:1073–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Nakashima I, Takahashi T, Cree BA, Kim HJ, Suzuki C, Genain CP et al (2011) Transient increases in anti‐aquaporin‐4 antibody titers following rituximab treatment in neuromyelitis optica, in association with elevated serum BAFF levels. J Clin Neurosci 18:997–998. [DOI] [PubMed] [Google Scholar]
- 118. Nicchia GP, Mastrototaro M, Rossi A, Pisani F, Tortorella C, Ruggieri M et al (2009) Aquaporin‐4 orthogonal arrays of particles are the target for neuromyelitis optica autoantibodies. Glia 57:1363–1373. [DOI] [PubMed] [Google Scholar]
- 119. Nishiyama S, Ito T, Misu T, Takahashi T, Kikuchi A, Suzuki N et al (2009) A case of NMO seropositive for aquaporin‐4 antibody more than 10 years before onset. Neurology 72:1960–1961. [DOI] [PubMed] [Google Scholar]
- 120. Palace J, Leite MI, Nairne A, Vincent A (2010) Interferon Beta treatment in neuromyelitis optica: increase in relapses and aquaporin 4 antibody titers. Arch Neurol 67:1016–1017. [DOI] [PubMed] [Google Scholar]
- 121. Papeix C, Vidal JS, de Seze J, Pierrot‐Deseilligny C, Tourbah A, Stankoff B et al (2007) Immunosuppressive therapy is more effective than interferon in neuromyelitis optica. Mult Scler 13:256–259. [DOI] [PubMed] [Google Scholar]
- 122. Paty DW, Oger JJ, Kastrukoff LF, Hashimoto SA, Hooge JP, Eisen AA et al (1988) MRI in the diagnosis of MS: a prospective study with comparison of clinical evaluation, evoked potentials, oligoclonal banding, and CT. Neurology 38:180–185. [DOI] [PubMed] [Google Scholar]
- 123. Paul F, Jarius S, Aktas O, Bluthner M, Bauer O, Appelhans H et al (2007) Antibody to aquaporin 4 in the diagnosis of neuromyelitis optica. Plos Med 4:e133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Petzold A, Pittock S, Lennon V, Maggiore C, Weinshenker BG, Plant GT (2010) Neuromyelitis optica‐IgG (aquaporin‐4) autoantibodies in immune mediated optic neuritis. J Neurol Neurosurg Psychiatry 81:109–111. [DOI] [PubMed] [Google Scholar]
- 125. Pisani F, Mastrototaro M, Rossi A, Nicchia GP, Tortorella C, Ruggieri M et al (2011) Identification of two major conformational aquaporin‐4 epitopes for neuromyelitis optica autoantibody binding. J Biol Chem 286:9216–9224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Pittock SJ, Lennon VA, Krecke K, Wingerchuk DM, Lucchinetti CF, Weinshenker BG (2006) Brain abnormalities in neuromyelitis optica. Arch Neurol 63:390–396. [DOI] [PubMed] [Google Scholar]
- 127. Pittock SJ, Weinshenker BG, Lucchinetti CF, Wingerchuk DM, Corboy JR, Lennon VA (2006) Neuromyelitis optica brain lesions localized at sites of high aquaporin 4 expression. Arch Neurol 63:964–968. [DOI] [PubMed] [Google Scholar]
- 128. Pittock SJ, Lennon VA, de Seze J, Vermersch P, Homburger HA, Wingerchuk DM et al (2008) Neuromyelitis optica and non organ‐specific autoimmunity. Arch Neurol 65:78–83. [DOI] [PubMed] [Google Scholar]
- 129. Polman CH, Reingold SC, Edan G, Filippi M, Hartung HP, Kappos L et al (2005) Diagnostic criteria for multiple sclerosis: 2005 revisions to the “McDonald Criteria”. Ann Neurol 58:840–846. [DOI] [PubMed] [Google Scholar]
- 130. Polman CH, Reingold SC, Banwell B, Clanet M, Cohen JA, Filippi M et al (2011) Diagnostic criteria for multiple sclerosis: 2010 revisions to the McDonald criteria. Ann Neurol 69:292–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Rossi A, Pisani F, Nicchia GP, Svelto M, Frigeri A (2010) Evidences for a leaky scanning mechanism for the synthesis of the shorter M23 protein isoform of aquaporin‐4: implication in orthogonal array formation and neuromyelitis optica antibody interaction. J Biol Chem 285:4562–4569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Rostasy K, Mader S, Hennes E, Schanda K, Gredler V, Guenther A et al (2013) Persisting myelin oligodendrocyte glycoprotein antibodies in aquaporin‐4 antibody negative pediatric neuromyelitis optica. Mult Scler 19:1052–1059. [DOI] [PubMed] [Google Scholar]
- 133. Rostasy K, Mader S, Schanda K, Huppke P, Gartner J, Kraus V et al (2012) Anti‐myelin oligodendrocyte glycoprotein antibodies in pediatric patients with optic neuritis. Arch Neurol 69:752–756. [DOI] [PubMed] [Google Scholar]
- 134. Sabater L, Giralt A, Boronat A, Hankiewicz K, Blanco Y, Llufriu S et al (2009) Cytotoxic effect of neuromyelitis optica antibody (NMO‐IgG) to astrocytes: an in vitro study. J Neuroimmunol 215:31–35. [DOI] [PubMed] [Google Scholar]
- 135. Saiz A, Zuliani L, Blanco Y, Tavolato B, Giometto B, Graus F (2007) Revised diagnostic criteria for neuromyelitis optica (NMO): application in a series of suspected patients. J Neurol 254:1233–1237. [DOI] [PubMed] [Google Scholar]
- 136. Salonen EM, Vaheri A, Suni J, Wager O (1980) Rheumatoid factor in acute viral infections: interference with determination of IgM, IgG, and IgA antibodies in an enzyme immunoassay. J Infect Dis 142:250–255. [DOI] [PubMed] [Google Scholar]
- 137. Scott TF, Kassab SL, Pittock SJ (2006) Neuromyelitis optica IgG status in acute partial transverse myelitis. Arch Neurol 63:1398–1400. [DOI] [PubMed] [Google Scholar]
- 138. Shimizu J, Hatanaka Y, Hasegawa M, Iwata A, Sugimoto I, Date H et al (2010) IFNbeta‐1b may severely exacerbate Japanese optic‐spinal MS in neuromyelitis optica spectrum. Neurology 75:1423–1427. [DOI] [PubMed] [Google Scholar]
- 139. Shimizu Y, Yokoyama K, Misu T, Takahashi T, Fujihara K, Kikuchi S et al (2008) Development of extensive brain lesions following interferon beta therapy in relapsing neuromyelitis optica and longitudinally extensive myelitis. J Neurol 255:305–307. [DOI] [PubMed] [Google Scholar]
- 140. Smith CH, Waubant E, Langer‐Gould A (2009) Absence of neuromyelitis optica IgG antibody in an active relapsing‐remitting multiple sclerosis population. J Neuroophthalmol 29:104–106. [DOI] [PubMed] [Google Scholar]
- 141. Takahashi T, Fujihara K, Nakashima I, Misu T, Miyazawa I, Nakamura M et al (2006) Establishment of a new sensitive assay for anti‐human aquaporin‐4 antibody in neuromyelitis optica. Tohoku J Exp Med 210:307–313. [DOI] [PubMed] [Google Scholar]
- 142. Takahashi T, Fujihara K, Nakashima I, Misu T, Miyazawa I, Nakamura M et al (2007) Anti‐aquaporin‐4 antibody is involved in the pathogenesis of NMO: a study on antibody titre. Brain 130(Pt 5):1235–1243. [DOI] [PubMed] [Google Scholar]
- 143. Takahashi T, Miyazawa I, Misu T, Takano R, Nakashima I, Fujihara K et al (2008) Intractable hiccup and nausea in neuromyelitis optica with anti‐aquaporin‐4 antibody: a herald of acute exacerbations. J Neurol Neurosurg Psychiatry 79:1075–1078. [DOI] [PubMed] [Google Scholar]
- 144. Tanaka K, Tani T, Tanaka M, Saida T, Idezuka J, Yamazaki M et al (2007) Anti‐aquaporin 4 antibody in selected Japanese multiple sclerosis patients with long spinal cord lesions. Mult Scler 13:850–855. [DOI] [PubMed] [Google Scholar]
- 145. Tanaka M, Tanaka K, Komori M (2009) Interferon‐beta(1b) treatment in neuromyelitis optica. Eur Neurol 62:167–170. [DOI] [PubMed] [Google Scholar]
- 146. Tani T, Sakimura K, Tsujita M, Nakada T, Tanaka M, Nishizawa M, Tanaka K (2009) Identification of binding sites for anti‐aquaporin 4 antibodies in patients with neuromyelitis optica. J Neuroimmunol 211:110–113. [DOI] [PubMed] [Google Scholar]
- 147. Trebst C, Berthele A, Jarius S, Kumpfel T, Schippling S, Wildemann B, Wilke C (2011) [Diagnosis and treatment of neuromyelitis optica. Consensus recommendations of the Neuromyelitis Optica Study Group]. Nervenarzt 82:768–777. [DOI] [PubMed] [Google Scholar]
- 148. Uzawa A, Mori M, Hayakawa S, Masuda S, Kuwabara S (2010) Different responses to interferon beta‐1b treatment in patients with neuromyelitis optica and multiple sclerosis. Eur J Neurol 17:672–676. [DOI] [PubMed] [Google Scholar]
- 149. Wandinger KP, Stangel M, Witte T, Venables P, Charles P, Jarius S et al (2010) Autoantibodies against aquaporin‐4 in patients with neuropsychiatric systemic lupus erythematosus and primary Sjogren's syndrome. Arthritis Rheum 62:1198–1200. [DOI] [PubMed] [Google Scholar]
- 150. Warabi Y, Matsumoto Y, Hayashi H (2007) Interferon beta‐1b exacerbates multiple sclerosis with severe optic nerve and spinal cord demyelination. J Neurol Sci 252:57–61. [DOI] [PubMed] [Google Scholar]
- 151. Waters P, Jarius S, Littleton E, Leite MI, Jacob S, Gray B et al (2008) Aquaporin‐4 antibodies in neuromyelitis optica and longitudinally extensive transverse myelitis. Arch Neurol 65:913–919. [DOI] [PubMed] [Google Scholar]
- 152. Waters PJ, McKeon A, Leite MI, Rajasekharan S, Lennon VA, Villalobos A et al (2012) Serologic diagnosis of NMO: a multicenter comparison of aquaporin‐4‐IgG assays. Neurology 78:665–671. discussion 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Weinshenker BG, Wingerchuk DM, Nakashima I, Fujihara K, Lennon VA (2006) OSMS is NMO, but not MS: proven clinically and pathologically. Lancet Neurol 5:110–111. [DOI] [PubMed] [Google Scholar]
- 154. Weinshenker BG, Wingerchuk DM, Vukusic S, Linbo L, Pittock SJ, Lucchinetti CF, Lennon VA (2006) Neuromyelitis optica IgG predicts relapse after longitudinally extensive transverse myelitis. Ann Neurol 59:566–569. [DOI] [PubMed] [Google Scholar]
- 155. Wildemann B, Jarius S, Paul F (2013) [Neuromyelitis optica.]. Nervenarzt 84:436–441. [DOI] [PubMed] [Google Scholar]
- 156. Wingerchuk DM, Hogancamp WF, O'Brien PC, Weinshenker BG (1999) The clinical course of neuromyelitis optica (Devic's syndrome). Neurology 53:1107–1114. [DOI] [PubMed] [Google Scholar]
- 157. Wingerchuk DM, Lennon VA, Pittock SJ, Lucchinetti CF, Weinshenker BG (2006) Revised diagnostic criteria for neuromyelitis optica. Neurology 66:1485–1489. [DOI] [PubMed] [Google Scholar]
- 158. Wingerchuk DM, Lennon VA, Lucchinetti CF, Pittock SJ, Weinshenker BG (2007) The spectrum of neuromyelitis optica. Lancet Neurol 6:805–815. [DOI] [PubMed] [Google Scholar]
- 159. Yang Y, Huang DH, Wu WP, Wu L, Chen LF, Wu Q (2013) The role of aquaporin‐4 antibodies in Chinese patients with neuromyelitis optica. J Clin Neurosci 20:94–98. [DOI] [PubMed] [Google Scholar]
- 160. Yoshimura S, Isobe N, Matsushita T, Yonekawa T, Masaki K, Sato S et al (2013) Distinct genetic and infectious profiles in Japanese neuromyelitis optica patients according to anti‐aquaporin 4 antibody status. J Neurol Neurosurg Psychiatry 84:29–34. [DOI] [PubMed] [Google Scholar]
- 161. Zavada J, Nytrova P, Wandinger KP, Jarius S, Svobodova R, Probst C et al (2013) Seroprevalence and specificity of NMO‐IgG (anti‐aquaporin 4 antibodies) in patients with neuropsychiatric systemic lupus erythematosus. Rheumatol Int 33:259–263. [DOI] [PubMed] [Google Scholar]
- 162. Zhang X, Shu H, Zhang F, Tian X, Dong Y (2007) Cell‐ELISA detection of antineuronal antibodies in central nervous system involvement in systemic lupus erythematosus. Ann Rheum Dis 66:530–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Zhong XN, Wang HH, Bao J, Li R, Long YM, Lu ZQ et al (2012) Relationship between neuromyelitis optica‐IgG status and spinal cord magnetic resonance imaging in patients with neuromyelitis optica. Chin Med J (Engl) 125:270–274. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Sample numbers, positivity rates, and differences in methodology and diagnostic criteria; data from 59 studies including 93 test series.