

Abstract
Pediatric embryonal brain tumors can be difficult to classify. Atypical teratoid rhabdoid tumors (ATRT) contain rhabdoid cells, while primitive neuroectodermal tumors (PNETs) are composed of “small round blue cells.” Loss of INI1 is a common event in ATRT; therefore, we investigated if the loss of INI1 protein expression was also observed in central nervous system (CNS) PNET and pineoblastoma. A histological review of 42 CNS PNETs and six pineoblastomas was performed. INI1 expression was assessed by immunohistochemistry. Sequencing was performed on the mutational hotspots of INI1. INI1‐immunonegative tumors were further investigated using fluorescence in situ hybridization. Epithelial membrane antigen (EMA) protein expression was assessed in six CNS PNETs to further define the phenotype. Five CNS PNETs without rhabdoid cell morphology were immuno‐negative for both INI1 and EMA. Of these primary CNS PNET patients, three died <11 months postdiagnosis, which was dissimilar to the INI1‐immunopositive primary CNS PNETs where 18/24 (75%) patients were alive 1 year postdiagnosis. We have identified a small subgroup of CNS PNETs which lack INI1 protein expression, but have no evidence of rhabdoid cell morphology. INI1 protein loss may occur through mechanisms other than gene deletion. INI1 immunohistochemistry should be performed for all CNS PNET cases.
Keywords: ATRT, CNS PNET, EMA, INI1, pineoblastoma, rhabdoid
Introduction
Histopathological classification of pediatric malignant brain tumors, especially those with primitive cells, can be difficult. Imprecise histogenetic classification due to overlapping histological appearances and a lack of a lineage‐specific markers hamper a correct diagnosis. Definitions for histological and genetic subgroups for supratentorial World Health Organization (WHO) grade IV embryonal tumors are at present in their infancy and a proportion of tumors are extremely hard to categorize and assign a specific diagnosis to, with features overlapping two or more classifications. In the past decade, significant progress has been made in the histological subclassification of the infratentorial primitive neuroectodermal tumor (PNET), medulloblastoma 13, 29. Importantly, these histological subtypes are associated with prognostic implications 12, 13, 16, 31, 32. Lessons learned from the histopathological subtyping of medulloblastoma are now being applied to the less well‐characterized WHO grade IV tumors of the brain. In recent years, histological subtypes for central nervous system (CNS) PNET have emerged. These include the “classic” CNS PNET (NOS) containing “small round blue cells” of undifferentiated or poorly differentiated neuroepithelial cells with scant cytoplasm; the CNS neuroblastoma, where neuronal differentiation is present; and CNS ganglioneuroblastoma with the additional presence of ganglion cells. Two rarer entities are also described, the medulloepithelioma characterized by papillary, tubular arrangements of neoplastic neuroepithelium which mimics the embryonic neural tube; and the ependymoblastomas which are characterized by multilayered rosettes 29.
Current research has suggested further refinements and identified new entities within the CNS PNET group. The term embryonal tumor with abundant neuropil and true rosettes (ETANTR) was first described by Eberhart in 2000. This subgroup of tumors contains rounded rosettes (resembling Flexner–Wintersteiner rosettes) which appear distinct from the rosettes of ependymoblastoma which are more elongated in structure 9, 11, 14, 15. Another distinguishing feature separating ETANTR from ependymoblastoma is the lack of abundant neuropil in ependymoblastoma. Although potentially separate entities by histological examination, a recent study identified focal amplification at 19q13.42 in 37/40 (93%) ETANTR and ependymoblastomas, suggesting that these two tumor classifications are in fact a single biological entity 26. Finally, Hasselblatt et al have described two intracranial non‐rhabdoid neuroectodermal tumors with loss of INI1 protein expression and positive epithelial membrane antigen (EMA) immuno‐staining. A new term “cribrifrom neuroepithelial tumor (CRINET)” was suggested for these tumors which are characterized by cribriform strands and trabeculae, lack of rhabdoid cells, and favorable prognosis. These two patients were in complete remission 5 years postoperatively, which is dissimilar to the extremely poor prognosis observed for atypical teratoid rhabdoid tumor (ATRT) patients 18.
In 2011, Raffel and Rutka questioned whether CNS PNET was still a useful term for diagnostic classification 37. Increasingly sophisticated technologies give the prospect of defining genetic subgroups of CNS PNET. Our previous study identified a subgroup of CNS PNETs in older children with loss of CDKN2A/B (9p21.3), while a study by Li et al identified a subgroup of 11/45 (24%) CNS PNETs with amplification of the C19MC polycistron, a large proportion of which had “variant” histology (defined as ependymal or ependymoblastic differentiation and distinct rosette structures) and gain of chromosome 2 28, 33. 19q13.41 amplification and variant histology have also been found in previous studies featuring fewer tumor samples 15, 26, 30, 36, 46. Patients with tumors harboring the C19MC amplicon had a significantly poorer prognosis 28. Interestingly, in 2010, Behdad and Perry found that 18/33 CNS PNETs had large cell components and half of the tumors studied had amplifications of MYCC or MYCN. Polysomies of chromosomes 2 and 8 were also a common feature 1. All three elements linked with a poorer patient prognosis. The high frequency of MYCC and MYCN amplifications and presence of large cells have not previously been noted in other studies of CNS PNET. Taken together, these studies show the genetic heterogeneity of CNS PNETs.
Of all the embryonal CNS tumors, ATRT has one of the worst prognoses and is usually fatal within 2 years 8, 11. Misclassification of these tumors as CNS PNET/Medulloblastoma can occur due to their complex histologic patterns. The classic morphological features displayed in ATRT include eccentric vesicular nuclei, prominent nucleoli and eosinophilic cytoplasmic inclusions, defining rhabdoid cells. While both CNS PNETs and ATRTs can contain primitive cells, they can also differentiate along many different lineages. For example CNS PNETs have the capacity for neuronal, astrocytic, muscular and melanocytic differentiation, while ATRT displays differentiation along epithelial, mesenchymal, neuronal or glial lines 29. In 2002, a workshop on childhood ATRT was held, where leading experts in the field discussed the criteria needed for the diagnosis of ATRT 4. It was established that not all ATRTs have mutation of INI1 and if histological features in addition to immunostaining are consistent with ATRT, absence of the mutation is not enough to change the diagnosis from ATRT. Also discussed was the issue of INI1 mutation in tumors of PNET histology, and it was agreed that if INI1 mutation occurred even without the presence of a rhabdoid cell component, this would be sufficient to categorize as an ATRT.
Initially described in 1987 by Rorke, ATRT was the first pediatric brain tumor to have a candidate tumor suppressor gene identified. The breakthrough discovery of the loss of INI1 in the majority of ATRT cases has made an important contribution to the diagnosis of childhood brain tumors 2, 23, 40. INI1 (also known as BAF47, SMARCB1 or SNF5 at chromosome position 22q11.2) is related to the SWI/SNF5 family, with the encoded protein engaging in a complex relieving chromatin structures and limiting aberrant cell division. Hence, the loss of INI1 inhibits the normal transcriptional machineries access to its target. Two recent studies have identified alternative tumorigenic events in ATRT. A second member of the SMARC gene family (SMARCA4, 19p13.2) was found to be inactivated by mutation in INI1/SMARCB1‐positive ATRTs 19, 43. The morphology of ATRT may overlap with CNS PNET and the status of INI1 expression can aid in the correct diagnosis of embryonal brain tumor. When a diagnosis between ATRT and PNET cannot easily be distinguished by morphology and standard immunohistochemistry, the loss of INI1 expression supports the diagnosis of ATRT. Deletions or mutations of INI1 are common in ATRT and previous mutational screens of the nine exons have identified mutational hotspots in sporadic ATRTs involving exons 5 and 9 2, 10, 21. Classification of ATRTs as PNETs by pathologists due to the existence of a primitive neuroepithelial component within tumors has been noted in a number of previous studies 2, 3, 7, 41; however, ATRT can usually be distinguished from MB/CNS PNET using a immunohistochemical stain for EMA which is positive in ATRT 29. Recently, a study defined that INI1‐deficient tumors and rhabdoid tumors were convergent, but not fully overlapping entities 6. Interestingly, two separate studies have identified other brain tumors (more specifically primitive neuroectodermal tumors and choroid plexus carcinomas) which were INI1‐immunonegative but lacked rhabdoid morphology 6, 17. Furthermore, outside the category of “brain tumors,” several other tumor types have been found which lack INI1 protein expression and rhabdoid morphology. These include chordomas 34, chondrosarcomas 25 and epitheloid sarcomas 20, 24, 27, 35, 38.
These studies highlight the existence of INI1‐immunonegative tumors lacking both INI1 protein expression and rhabdoid features. The aggressive biological behavior of brain tumors lacking INI1 protein expression and the poor patient outcome if treated by conventional therapy regimens potentially means that PNET patients with tumors lacking INI1 expression could therefore benefit from the intensified therapies administered to rhabdoid tumor patients.
In this study, immunohistochemistry was used to identify whether loss of INI1 protein expression occurred in the CNS PNETs and pineoblastomas of the cohort. A mutational screen of exons 5 and 9 of the INI1 gene was performed to identify mutations. This study highlights the importance of screening the expression status of INI1 in pediatric patients with newly diagnosed embryonal tumors.
Materials and methods
Tissue microarray construction
Samples and clinical information were collected from nine Children's Cancer and Leukaemia Group (CCLG) registered centers in the UK. The study met with Multiple Centre Research Committee approval (04/MRE 04/72) and samples were consented for in accordance with national banking procedures and the UK human tissue act (2004). Tumors were histopathologically reviewed at each center according to the WHO criteria of the time for an original diagnosis and were subsequently reviewed centrally by three neuropathologists upon collection (JL, KR, M‐AB). Forty‐eight formalin‐fixed paraffin‐embedded (FFPE) tumor tissue blocks were available for inclusion in a tissue microarray (TMA). Cores (0.6 mm width and 4 μm depth) were taken in triplicate from representative areas of viable tumor tissue.
Re‐evaluation of the tumor cohort for the presence of rhabdoid cell morphology
Whole histological sections (stained with hematoxylin and eosin) from the original diagnostic material were re‐reviewed (JL) for the 48 tumors of the study to rule out the existence of a rhabdoid cell component within the tumors that may have been missed during the routine diagnosis and review process.
Immunohistochemistry for INI1
Forty‐eight tumors had scorable results for INI1. Forty‐two CNS PNETs (34 primaries, 6 recurrences and 2 without clinical information) and six pineoblastomas (5 primaries and 1 lacking clinical information) were included in this analysis. CNS PNET patient age at diagnosis ranged from 6 to 190 months, with a mean age of 74 months, while pineoblastoma patient age at diagnosis ranged from 24 to 183 months, with a mean age of 109 months. TMA slides were de‐paraffinized in xylene and hydrated using decreasing concentrations of ethanol. Antigen retrieval was performed in a pressure cooker for 1 min at full pressure in sodium citrate buffer (pH 6.0), followed by an endogenous peroxidise block for 5 min (Dako Cambridgeshire, UK). INI1 (mouse mAb for BAF47, 612110, BD Biosciences Pharmingen, Oxford, UK) (1:200) was incubated overnight at 4°C. Dako Chemate Envision Detection Kit with DAB chromogen was used for detection. Sections were then counterstained with Harris Haematoxylin (Surgipath, Cambridgeshire, UK), dehydrated and a coverslip was mounted using DPX. Tonsil was used as a positive control, while a negative control consisted of primary antibody substituted for antibody diluent. Results were visualized using an Olympus BX14 light microscope (Essex, UK). Scoring for INI1 consisted of positive or negative tumor cell nuclei.
Immunohistochemistry for EMA
EMA protein expression was assessed using immunohistochemistry for five tumors originally diagnosed as CNS PNET, which lacked rhabdoid features (post histological review) and which were found to be INI1‐immunonegative. Patient age at diagnosis ranged from 6 to 148 months, with a mean age of 68.6 months. We also assessed EMA protein expression 1 tumor reclassified as an ATRT which was histologically biphasic with areas of both rhabdoid and PNET cells. Slides were de‐paraffinized in xylene and hydrated using decreasing concentrations of ethanol. Antigen retrieval was performed using a trypsin enzymatic antigen retrieval kit (Abcam, ab970, Cambridge, UK) with slides incubated at room temperature with 100 μL of a 1:1 dilution trypsin solution for 20 minutes. Following a normal goat serum block (1:5 NGS : PBS) for 20 minutes and an endogenous peroxidise block for 5 minutes (Dako), EMA primary antibody (clone E29, IS629, 1:1200) was incubated for 30 minutes at room temperature. Dako Chemate Envision Detection Kit (Dako) with DAB chromogen was used for detection. Sections were then counterstained with Harris Haematoxylin (Surgipath), dehydrated and a coverslip was mounted using DPX. Tonsil was used as a positive control, while a negative control consisted of primary antibody substituted for antibody diluent. Scoring for EMA consisted of either positive or negative cytoplasmic or membranous staining. All five tumors were scorable.
Fluorescence in situ hybridization (FISH)
FISH was performed for seven INI1 immuno‐negative tumors with available tissue. Whole sections (3 μm) were used. Probes were derived from BAC clones RP11‐431G10 (1p36) and RP11‐71G19 (22q11.2) (Invitrogen, Carlsbad, CA, USA). BAC DNA (100 ng) was labeled with either Biotin (1p36 probe) or Digoxigenin using the Bioprime DNA Kit (Invitrogen). Briefly, slides were de‐paraffinized in xylene and washed in 100% ethanol. Slides were incubated in formalin for 1 h and washed in warm running water. Slides were placed in a steamer with citrate buffer for 1 h at >90°C. Next, tissue was digested in 8 mg/mL Pepsin (Dako, Ely, UK) in 0.1 M HCl for 30 minutes. Tissue was fixed using Carnoy's mild fixative for 30 minutes then air dried. Probe mixtures were diluted 1:3.3 in DenHyb buffer (Insitus Biotechnologies, Albuquerque, NM, USA), applied to tissue and a cover slip was added. Slides were incubated in a hybridizer at 90°C for 12 minutes, then 37°C overnight. Following a wash in 2× SSC for 30 minutes and cover slip removal, slides were then washed in 4 M urea for 2 minutes and 2× SSC for 1 minute. Samples were blocked in 2.5% bovine serum albumin (BSA) for 30 minutes at room temperature. Two antibody solutions were prepared: (i) Solution 1: 0.6 μL anti‐DIG (Roche, Penzberg, Germany) with 0.75 μL streptavidin‐Cy3 (Roche) in 150 μL 2.5% BSA; and (ii) Solution 2: 0.3 μL of Alexa Fluor 488 goat anti mouse IgG (Roche) in 150 μL 2.5% BSA. Solution 1 (145 μL) was added to slides under a cover slip and incubated for 35 minutes at 37°C. Coverslips were removed and slides were washed in 4× SSCT for 10 minutes. Solution 2 (145 μL) was added to slides under a coverslip and incubated for 35 minutes at 37°C. Coverslips were removed in 4× SSCT and slides were washed in PBS for 6 minutes. Following a wash in distilled water for 1 minute, slides were air dried. DAPI (20 μL) (750 mg/mL, Vector Labs, Burlingame, CA, USA) was applied to each slide and a cover slip was applied and sealed. A minimum of 100 cells were scored by two independent investigators and a majority result was identified from the nuclei examined.
INI1 sequencing
DNA was extracted from 26 snap‐frozen CNS PNETs, one pineoblastomas and seven constitutional samples (patients’ paired blood). DNA extraction was carried out as described earlier 39. DNA samples were assessed for mutations within hotspots of the INI1 gene (exons 5 and 9). PCR primers were designed, exon 5 (Forward 5'‐TGTGCAGAGAGAGAGGCTGA‐3’, Reverse 5'‐CAAAACTATGCCCCGATGTC‐3’) and exon 9 (Forward 5'‐CTCTGTTCCCACCCCTACAC‐3’, Reverse 5’ TGTGCCAACCTTGTTCACAT‐3’). PCR products were amplified using BioMix Red (Bioline, London, UK) with an annealing temperature of 62○C for 35 cycles. PCR products were purified with 0.3 U of Shrimp Alkaline Phosphatase (Promega, Southampton, UK) and 1.5 U Exonuclease I (NEB, Hertfordshire, UK) by incubating for 8 minutes at 37°C and 15 minutes at 72°C. Sequencing was conducted with 1 μL of PCR product with Big Dye V1.1 under the manufacturer's protocol. Mutational analysis was performed using Chromaslite v2.01 (Technelysium Pty Ltd., Tewantin, Australia) where a comparison of the normal sequence and the test sequence was made. Sixteen samples screened for mutations were also included in the INI1 immunohistochemical study. Clinical information has not been included for the additional samples in the mutational screen.
Statistics
Statistical analysis was performed using SPSSv16 (IBM, Armonk, NY, USA). Clinical information for primary CNS PNET patients was entered into SSPS including age, metastatic status at diagnosis, length of patient survival, and whether the patient was presently alive or deceased. With age a continuous variable, an independent samples t‐test was performed to identify if loss of INI1 protein expression was associated with age. The Fisher's exact test was performed to investigate if loss of INI1 protein expression was associated with metastasis at diagnosis. Lastly, to test if INI1 protein loss was associated with patient survival, a univariate survival curve was constructed in SPSS using the Kaplan–Meier method and a comparison made by the Log Rank test (Mantel–Cox).
Results
INI1 immunohistochemistry, FISH and mutational screen
Of 48 scorable tumors, 42/48 (87.5%) did not have rhabdoid features (Figure 1A and Table 1). There were 5/48 (10.4%) that contained rhabdoid cells (Figure 1B) and 1/48 (2.1%) primary CNS PNET was biphasic with components including areas of both rhabdoid and PNET cells (Figure 1C); these were subsequently reclassified as ATRT. Of the 42 tumors with no rhabdoid features, 37/42 (88.1%) were INI1 positive (Figure 1D) and 5/42 (11.9%) were INI1 negative (Figure 1E). Two INI1‐negative CNS PNETs without rhabdoid features had copy‐number results for INI1 [from a previous SNP array study 33, validated by FISH] (Table 1). While one tumor had two copies of the INI1 gene present, the other showed heterozygous loss. All six tumors with rhabdoid features were INI1‐immunonegative (Figure 1F) (including both rhabdoid and PNET cell regions of the biphasic tumor, Figure 1G). There were 5/6 INI1‐immunonegative tumors with rhabdoid cells that had gene copy‐number data (Table 1). There were 3/5 (60%) that had two copies of INI1 (Figure 2B) while 2/5 (40%) had homozygous loss. A mutational screen was performed for exons 5 and 9 of INI1 to identify whether mutation was an event in the pathogenesis of CNS PNET and pineoblastoma. Twenty‐six CNS PNETs, one pineoblastoma and seven paired constitutional DNAs were assessed; however, no mutations were identified (Table 1).
Figure 1.
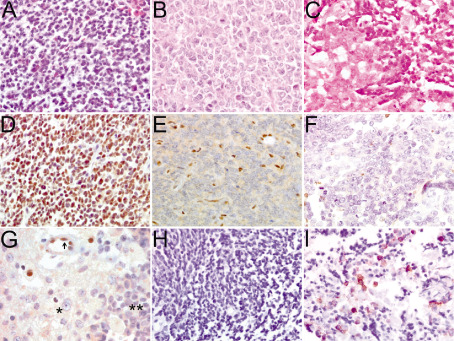
Histopathology and immunohistochemisty of tumors originally diagnosed as CNS PNET. Small round blue cells of CNS PNET 13 stained with hematoxylin and eosin (A). Rhabdoid cells of an ATRT with distinctive prominent nucleoli, vesicular chromatin and eosinophilic cytoplasmic inclusions, CNS PNET 11 (B). Biphasic tumor with rhabdoid features and characteristic large nuclei cells on the left and small primitive cells consistent with a PNET classification on the right, CNS PNET 6 (C). INI1‐immunopositive CNS PNET 13 (D). INI1 immunonegative tumor cells with positive stromal cells, CNS PNET 3 (E). INI1‐immunonegative ATRT (CNS PNET 11) (F). Biphasic tumor with INI1‐immunonegative rhabdoid cells (*), INI‐immunonegative PNET cells (**) and INI1‐immunopositive endothelial stromal cells (arrow), CNS PNET 6 (G). EMA‐immunonegative CNS PNET 4 (H). Biphasic tumor with EMA‐immunopositive rhabdoid cells, CNS PNET 6 (I). Magnification × 40. ATRT = atypical teratoid rhabdoid tumor; CNS = central nervous system; EMA = epithelial membrane antigen; PNET = primitive neuroectodermal tumor.
Table 1.
CNS PNET and pineoblastoma immunohistochemistry cohort. Abbreviations: CNS PNET = central nervous system primitive neuroectodermal tumor; PB = pineoblastoma; Neg = negative; Pos = positive; CN = copy number; FISH = fluorescence in situ hybridization; m = months; F = female; M = male; D = deceased; A = alive; n/a = no tissue; Wt = wild type.
| ID | INI1 IHC | Rhabdoid features? | INI1 CN by SNP array’ | INI1 FISH result | Hotspot sequencing | Primary or recurrence | Age (m) | Gender | Location | Metastasis | Follow‐up (m) | Censor |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CNS PNET1 | Neg | No | 2 | Normal | — | P | 6 | F | Left temporal and parietal lobes | M3 | 10 | D |
| CNS PNET2 | Neg | No | — | n/a | — | — | 17 | M | Supratentorial | — | — | — |
| CNS PNET3 | Neg | No | — | n/a | — | P | 46 | F | Supratentorial | M0 | 11 | D |
| CNS PNET4 | Neg | No | — | n/a | — | P | 126 | F | Cerebral | M0 | 0.5 | D |
| CNS PNET5 | Neg | No | 1 | Heterozygous Loss | Wt | R | 148 | F | Frontal and parietal lobes | — | 36 | D |
| CNS PNET6 | Neg | Yes—Biphasic | — | Normal | — | P | 30 | M | Parietal lobe | M0 | 6 | D |
| CNS PNET7 | Neg | Yes | — | n/a | — | P | 12 | M | Temporal and parietal Lobes | M3 | 8 | D |
| CNS PNET8 | Neg | Yes | 2 | Normal | Wt | P | 37 | M | Right temporal lobe | M4 | 6 | D |
| CNS PNET9 | Neg | Yes | — | Normal | — | P | 38 | F | Left lateral ventricle | M0 | 16 | D |
| CNS PNET10 | Neg | Yes | — | Homozygous Loss | — | P | 87 | F | Parietal lobe | M0 | 10 | D |
| CNS PNET11 | Neg | Yes | — | Homozygous Loss | — | R | 101 | F | Parietal lobe | M0 | 153 | A |
| CNS PNET12 | Pos | No | — | — | — | P | 6 | M | Cerebral | M0 | — | — |
| CNS PNET13 | Pos | No | 3 | — | Wta | P | 10 | F | Right parietal and frontal lobe | M2 | 41 | D |
| CNS PNET14 | Pos | No | 2 | — | Wt | R | 10 | F | Right parietal and frontal lobe | M2 | 41 | D |
| CNS PNET15 | Pos | No | — | — | — | P | 16 | F | Parietal lobe | M3 | 6 | D |
| CNS PNET16 | Pos | No | 2 | — | Wt | P | 20 | F | Right frontal lobe | M0 | 0 | D |
| CNS PNET17 | Pos | No | — | — | — | P | 32 | F | Leptomeningeal | M3 | 8 | D |
| CNS PNET18 | Pos | No | — | — | — | P | 39 | F | Right parietal lobe | M0 | 165 | A |
| CNS PNET19 | Pos | No | — | — | — | P | 43 | F | Parietal lobe | M0 | 264 | A |
| CNS PNET20 | Pos | No | — | — | — | P | 45 | F | Suprasellar | — | 12 | D |
| CNS PNET21 | Pos | No | 4 | — | Wta | P | 53 | F | Right frontal, parietal and temporal lobes | M0 | 62 | A |
| CNS PNET22 | Pos | No | 2 | — | Wt | P | 59 | M | Frontal lobe | M2 | 9 | D |
| CNS PNET23 | Pos | No | 2 | — | Wta | P | 61 | M | Left frontal lobe | M0 | 21 | D |
| CNS PNET24 | Pos | No | — | — | — | P | 62 | M | Left frontal lobe | M0 | 38 | D |
| CNS PNET25 | Pos | No | — | — | — | — | 64 | M | Supratentorial | — | — | — |
| CNS PNET26 | Pos | No | — | — | — | P | 68 | F | Right frontal lobe | — | — | — |
| CNS PNET27 | Pos | No | 3 | — | Wt | P | 71 | F | Right frontal and parietal lobes | M0 | 7 | D |
| CNS PNET28 | Pos | No | 3 | — | — | P | 85 | F | Frontal lobe | M0 | 112 | A |
| CNS PNET29 | Pos | No | 1 | — | — | P | 91 | F | Supratentorial | M1 | 31 | A |
| CNS PNET30 | Pos | No | — | — | — | P | 99 | F | Parietal lobe | M0 | 8 | D |
| CNS PNET31 | Pos | No | — | — | — | P | 105 | F | Left occipital lobe | — | 21 | D |
| CNS PNET32 | Pos | No | 2 | — | Wt | P | 107 | M | Left hemisphere | M2 | 71 | D |
| CNS PNET33 | Pos | No | 3 | — | Wt | R | 107 | M | Supratentorial | M2 | 71 | D |
| CNS PNET34 | Pos | No | — | — | — | P | 116 | F | Supratentorial | M0 | 25 | D |
| CNS PNET35 | Pos | No | 2 | — | Wta | P | 122 | M | Right parietal lobe | M0 | 58 | A |
| CNS PNET36 | Pos | No | 2 | — | Wt | R | 122 | M | Right parietal lobe | M0 | 58 | A |
| CNS PNET37 | Pos | No | — | — | — | P | 123 | M | Left hemisphere | M0 | 16 | D |
| CNS PNET38 | Pos | No | — | — | — | P | 141 | F | Parietal lobe | M4 | 58 | D |
| CNS PNET39 | Pos | No | 2 | — | Wta | P | 141 | M | Right frontal lobe | M3 | 30 | D |
| CNS PNET40 | Pos | No | 2 | — | Wt | R | 141 | M | Right frontal lobe | M3 | 30 | D |
| CNS PNET41 | Pos | No | — | — | — | P | 186 | M | Right parietal lobe | M0 | 256 | A |
| CNS PNET42 | Pos | No | 2 | — | — | P | 190 | F | Cerebral | M0 | 35 | A |
| PB1 | Pos | No | 3 | — | Wt | P | 24 | M | Pineal gland | M0 | 180 | A |
| PB2 | Pos | No | — | — | — | P | 45 | F | Pineal gland | M3 | 35 | D |
| PB3 | Pos | No | — | — | — | P | 98 | M | Pineal gland | M3 | 59 | D |
| PB4 | Pos | No | — | — | — | P | 140 | F | Pineal gland | M0 | 12 | A |
| PB5 | Pos | No | — | — | — | — | 163 | M | Pineal gland | — | — | — |
| PB6 | Pos | No | — | — | — | P | 183 | M | Pineal gland | M3 | 37 | D |
Constitutional blood sample also tested.
From previous SNP array paper ref.
—, no information.
Figure 2.
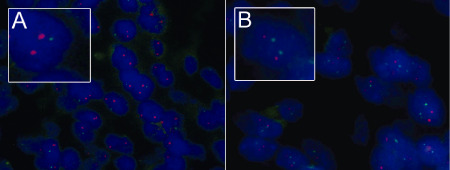
INI1 FISH on a CNS PNET and a reclassified ATRT. CNS PNET 5 with heterozygous loss of INI1 (A) and biphasic CNS PNET 6 with normal copy number for INI1 (B). 22q11.2 (INI1) green probe and 1p36 (EPHB2) red probe. ATRT = atypical teratoid rhabdoid tumor; CNS = central nervous system; FISH = fluorescence in situ hybridization; PNET = primitive neuroectodermal tumor.
EMA Immunohistochemistry
To further investigate the correct classification of five INI1 immunonegative CNS PNETs with no evidence of rhabdoid cell morphology, immunohistochemistry was used to investigate the protein expression of EMA. EMA may help to distinguish between ATRT and CNS PNET. ATRT may be positive for EMA protein expression while CNS PNET is negative. There were 5/5 (100%) INI1‐immunonegative CNS PNETs that were negative for EMA (Figure 1H). Additionally, EMA protein expression was assessed in a biphasic tumor which was reclassified as ATRT postreview. The tumor was positive for EMA immunostaining in the rhabdoid cell component while the PNET component was immunonegative (Figure 1I).
Statistical analysis linking INI1 protein expression status to clinical factors
Following the exclusion of six tumors originally diagnosed as CNS PNET which were subsequently reclassified as ATRT (due to INI1‐immunonegativity and the presence of rhabdoid cells, which included the biphasic tumor), statistical analysis of the remaining primary CNS PNET cohort (excluding pineoblastomas) was performed to test whether the lack of INI1 protein expression was linked to clinical factors. INI1‐immunonegativity was not associated with patient age, metastatic status or patient prognosis. Interestingly, of the INI1‐immunonegative primary CNS PNETs with clinical information, all three had died <11 months postdiagnosis, which was dissimilar to the INI1‐immunopositive primary CNS PNETs where 18/24 (75%) patients were alive 1 year postdiagnosis. The INI1‐immunopositive CNS PNET patients had a median survival of 27.5 months compared with the INI1‐immunonegative CNS PNETs with a median of 10.5 months. The 3 and 5‐year survival of the INI1‐immunopositive CNS PNETs were 9/24 (37.5%) and 5/24 (20.8%), respectively. Of note were 5/23 INI1‐immunopositive CNS PNET patients still presently alive, 58, 112, 165, 256 and 264 months postdiagnosis.
Discussion
The recent discovery of INI1‐negative pediatric brain tumors lacking rhabdoid morphology led us to investigate the status of INI1 in our relatively large cohort of CNS PNETs and pineoblastomas 6, 17. We evaluated the tumor cohort for INI1 loss and mutation, INI1 protein expression and reassessed whether rhabdoid morphology was histologically present within the tumors. Following the exclusion of 6 INI1‐immunonegative tumors originally diagnosed as CNS PNET and later reclassified as ATRT, we identified 5/36 (13.8%) INI1‐immunonegative CNS PNETs with no evidence of the rhabdoid morphology, suggesting reclassification to ATRT. Our study is therefore the first to explore INI1 loss in CNS PNETs and pineoblastomas, with the results suggesting that INI1 could be a potential tumor suppressor gene in a small subset of CNS PNET with extremely poor prognosis.
Our data add to previous evidence that loss of INI1 expression is not exclusive to tumors with rhabdoid cell morphology 6, 17, 20, 24, 25, 27, 34, 35, 38. Recently, one study identified a small proportion of brain tumors, 4/39 (10.2%), with loss of INI1 protein expression lacking rhabdoid features 6. This has previously been found in INI1‐immunonegative brain tumors with indeterminate histology, where eight cases were hard to distinguish histologically between CNS PNET and ATRT, and 7/8 (87.5%) did not have loss or mutation of the INI1 gene 23. A separate study also identified nine INI1‐immunonegative brain tumors lacking the morphological characteristics of ATRT and these were found to be aggressive in nature and have a poor response to therapy 17. Genetic analysis was not undertaken to test for INI1 gene deletion or mutation. Mutational hotspots on the INI1 gene include exons 5 and 9, and have been identified previously in sporadic pediatric brain tumors, especially ATRT 5, 6, 17, 22, 42, 44, 45. In the present study, a mutational screen of exons 5 and 9 of the INI1 gene was performed in the CNS PNET/pineoblastoma cohort, demonstrating that the loss of INI1 protein expression was not due to mutations residing in the mutational hotspots of the INI1 gene. Screening the seven remaining exons would further confirm whether INI1 gene mutation was a component of the lost protein expression within this subset of CNS PNETs; however, mutations in sporadic brain tumors within the other exons of INI1 is rare 2, 47. For two of the five (40%) INI1‐immunonegative CNS PNETs, copy‐number results were available from our previous genetic study and current FISH analyses 33. One tumor was diploid at the INI1 locus while a second tumor had heterozygous loss of INI1, suggesting that although some CNS PNETs have copy number‐driven changes in the expression of INI1, this does not completely account for the lack of protein expression. Other mechanisms, possibly methylation or silencing/degradation due to the involvement of miRNAs, may play roles in the loss of INI1 protein expression within this subgroup of tumors. The loss of INI1 in CNS PNET 5 which lacked rhabdoid features also provides further evidence that INI1 loss is not exclusive to cells with rhabdoid morphology. On the other hand, CNS PNETs 8 and 9, which were subsequently reclassified as ATRT due to this investigation, had two copies of INI1, which show that not all rhabdoid cells have INI1 loss. Of note, the six pineoblastomas included in the immunohistochemical study were all INI1‐immunopositive and did not have rhabdoid cells present. Of particular interest was CNS PNET 6, originally diagnosed as a CNS PNET and later reclassified as an ATRT. The tumor was biphasic with distinct areas of rhabdoid cells adjacent to areas of PNET cells. Cells in both regions were INI1‐immunonegative, and we hypothesize that the genetic switches that determine a rhabdoid morphology in this case are downstream from the loss of INI1 protein expression.
Negative staining for EMA would have been more characteristic of CNS PNET rather than ATRT or CRINET in the INI1‐immunonegative tumors of the present study. Interestingly, none of the INI1‐immunonegative primary CNS PNET patients survived longer than 11 months, whereas all patients presently alive (n = 9) had primary tumors with positive INI1 staining (n = 9, followed up between 21–264 months with median follow up of 112 months). Therefore, with the evidence from this study and others, we propose that INI1 immunohistochemistry is performed in all newly diagnosed embryonal brain tumors; with the loss of INI1 protein expression being regarded as a marker of poor prognosis, which is independent of tumor diagnosis/classification 6, 17. These results highlight the importance of histological reviews and immunohistochemical screening for prognostication in high‐grade brain tumors, and suggest that basing tumor classification merely on conventional histomorphology is not sufficient.
In summary, our data corroborate that loss of INI1 protein expression is not exclusive to intrinsic brain tumors with rhabdoid cell morphology. We have identified a fraction of INI1‐negative CNS PNETs lacking the rhabdoid features consistent with a diagnosis of ATRT. The poor outcome of this fraction of patients further demonstrates the importance of a molecular classification for embryonal brain tumors rather than a purely morphological classification. This paper adds evidence to the concept that high‐grade embryonal tumors are a spectrum of diseases rather than being clearly defined entities. INI1 is a potential tumor suppressor gene in CNS PNET, in addition to its loss being a marker of poor prognosis. These findings warrant further investigation in a larger cohort of samples. Future studies need to focus on the discovery of mechanisms of INI1 inactivation in embryonal brain tumors, other than copy‐number loss or mutation. The characterization and definition of clearly separate tumor entities should contribute to patients receiving an optimal treatment.
Acknowledgments
We thank Dr Keith Robson (KR) and Dr Marie‐Anne Brundler (M‐AB) for histopathological review of the tumor cohort. We also thank The Samantha Dickson Brain Tumour Trust and Gentlemen's Night Out for funding. Furthermore, we thank the CCLG centres and the CCLG Biological Studies Committee for patient samples and clinical data.
References
- 1. Behdad A, Perry A (2010) Central nervous system primitive neuroectodermal tumors: a clinicopathologic and genetic study of 33 cases. Brain Pathol 20:441–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Biegel JA, Zhou JY, Rorke LB, Stenstrom C, Wainwright LM, Fogelgren B (1999) Germ‐line and acquired mutations of INI1 in atypical teratoid and rhabdoid tumors. Cancer Res 59:74–79. [PubMed] [Google Scholar]
- 3. Biegel JA, Fogelgren B, Zhou JY, James CD, Janss AJ, Allen JC et al (2000) Mutations of the INI1 rhabdoid tumor suppressor gene in medulloblastomas and primitive neuroectodermal tumors of the central nervous system. Clin Cancer Res 6:2759–2763. [PubMed] [Google Scholar]
- 4. Biegel JA, Kalpana G, Knudsen ES, Packer RJ, Roberts CW, Thiele CJ et al (2002) The role of INI1 and the SWI/SNF complex in the development of rhabdoid tumors: meeting summary from the workshop on childhood atypical teratoid/rhabdoid tumors. Cancer Res 62:323–328. [PubMed] [Google Scholar]
- 5. Biegel JA, Tan L, Zhang F, Wainwright L, Russo P, Rorke LB (2002) Alterations of the hSNF5/INI1 gene in central nervous system atypical teratoid/rhabdoid tumors and renal and extrarenal rhabdoid tumors. Clin Cancer Res 8:3461–3467. [PubMed] [Google Scholar]
- 6. Bourdeaut F, Freneaux P, Thuille B, Lellouch‐Tubiana A, Nicolas A, Couturier J et al (2007) hSNF5/INI1‐deficient tumours and rhabdoid tumours are convergent but not fully overlapping entities. J Pathol 211:323–330. [DOI] [PubMed] [Google Scholar]
- 7. Burger PC, Yu IT, Tihan T, Friedman HS, Strother DR, Kepner JL et al (1998) Atypical teratoid/rhabdoid tumor of the central nervous system: a highly malignant tumor of infancy and childhood frequently mistaken for medulloblastoma: a Pediatric Oncology Group study. Am J Surg Pathol 22:1083–1092. [DOI] [PubMed] [Google Scholar]
- 8. Chen ML, McComb JG, Krieger MD (2005) Atypical teratoid/rhabdoid tumors of the central nervous system: management and outcomes. Neurosurg Focus 18(6A):E8. [PubMed] [Google Scholar]
- 9. Dunham C, Sugo E, Tobias V, Wills E, Perry A (2007) Embryonal tumor with abundant neuropil and true rosettes (ETANTR): report of a case with prominent neurocytic differentiation. J Neurooncol 84:91–98. [DOI] [PubMed] [Google Scholar]
- 10. Eaton KW, Tooke LS, Wainwright LM, Judkins AR, Biegel JA (2011) Spectrum of SMARCB1/INI1 mutations in familial and sporadic rhabdoid tumors. Pediatr Blood Cancer 56:7–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Eberhart CG, Brat DJ, Cohen KJ, Burger PC (2000) Pediatric neuroblastic brain tumors containing abundant neuropil and true rosettes. Pediatr Dev Pathol 3:346–352. [DOI] [PubMed] [Google Scholar]
- 12. Eberhart CG, Kepner JL, Goldthwaite PT, Kun LE, Duffner PK, Friedman HS et al (2002) Histopathologic grading of medulloblastomas: a Pediatric Oncology Group study. Cancer 94:552–560. [DOI] [PubMed] [Google Scholar]
- 13. Ellison D (2002) Classifying the medulloblastoma: insights from morphology and molecular genetics. Neuropathol Appl Neurobiol 28:257–282. [DOI] [PubMed] [Google Scholar]
- 14. Fuller C, Fouladi M, Gajjar A, Dalton J, Sanford RA, Helton KJ (2006) Chromosome 17 abnormalities in pediatric neuroblastic tumor with abundant neuropil and true rosettes. Am J Clin Pathol 126:277–283. [DOI] [PubMed] [Google Scholar]
- 15. Gessi M, Giangaspero F, Lauriola L, Gardiman M, Scheithauer BW, Halliday W et al (2009) Embryonal tumors with abundant neuropil and true rosettes: a distinctive CNS primitive neuroectodermal tumor. Am J Surg Pathol 33:211–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Giangaspero F, Perilongo G, Fondelli MP, Brisigotti M, Carollo C, Burnelli R et al (1999) Medulloblastoma with extensive nodularity: a variant with favorable prognosis. J Neurosurg 91:971–977. [DOI] [PubMed] [Google Scholar]
- 17. Haberler C, Laggner U, Slavc I, Czech T, Ambros IM, Ambros PF et al (2006) Immunohistochemical analysis of INI1 protein in malignant pediatric CNS tumors: lack of INI1 in atypical teratoid/rhabdoid tumors and in a fraction of primitive neuroectodermal tumors without rhabdoid phenotype. Am J Surg Pathol 30:1462–1468. [DOI] [PubMed] [Google Scholar]
- 18. Hasselblatt M, Oyen F, Gesk S, Kordes U, Wrede B, Bergmann M et al (2009) Cribriform neuroepithelial tumor (CRINET): a nonrhabdoid ventricular tumor with INI1 loss and relatively favorable prognosis. J Neuropathol Exp Neurol 68:1249–1255. [DOI] [PubMed] [Google Scholar]
- 19. Hasselblatt M, Gesk S, Oyen F, Rossi S, Viscardi E, Giangaspero F et al (2011) Nonsense mutation and inactivation of SMARCA4 (BRG1) in an atypical teratoid/rhabdoid tumor showing retained SMARCB1 (INI1) expression. Am J Surg Pathol 35:933–935. [DOI] [PubMed] [Google Scholar]
- 20. Hornick JL, Dal Cin P, Fletcher CD (2009) Loss of INI1 expression is characteristic of both conventional and proximal‐type epithelioid sarcoma. Am J Surg Pathol 33:542–550. [DOI] [PubMed] [Google Scholar]
- 21. Jackson EM, Sievert AJ, Gai X, Hakonarson H, Judkins AR, Tooke L et al (2009) Genomic analysis using high‐density single nucleotide polymorphism‐based oligonucleotide arrays and multiplex ligation‐dependent probe amplification provides a comprehensive analysis of INI1/SMARCB1 in malignant rhabdoid tumors. Clin Cancer Res 15:1923–1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Janson K, Nedzi LA, David O, Schorin M, Walsh JW, Bhattacharjee M et al (2006) Predisposition to atypical teratoid/rhabdoid tumor due to an inherited INI1 mutation. Pediatr Blood Cancer 47:279–284. [DOI] [PubMed] [Google Scholar]
- 23. Judkins AR, Mauger J, Ht A, Rorke LB, Biegel JA (2004) Immunohistochemical analysis of hSNF5/INI1 in pediatric CNS neoplasms. Am J Surg Pathol 28:644–650. [DOI] [PubMed] [Google Scholar]
- 24. Kim HJ, Kim MH, Kwon J, Kim JY, Park K, Ro JY (2011) Proximal‐type epithelioid sarcoma of the vulva with INI1 diagnostic utility. Ann Diagn Pathol [Epub ahead of print] PMID:21724432. [DOI] [PubMed] [Google Scholar]
- 25. Kohashi K, Oda Y, Yamamoto H, Tamiya S, Oshiro Y, Izumi T et al (2008) SMARCB1/INI1 protein expression in round cell soft tissue sarcomas associated with chromosomal translocations involving EWS: a special reference to SMARCB1/INI1 negative variant extraskeletal myxoid chondrosarcoma. Am J Surg Pathol 32:1168–1174. [DOI] [PubMed] [Google Scholar]
- 26. Korshunov A, Remke M, Gessi M, Ryzhova M, Hielscher T, Witt H et al (2010) Focal genomic amplification at 19q13.42 comprises a powerful diagnostic marker for embryonal tumors with ependymoblastic rosettes. Acta Neuropathol 120:253–260. [DOI] [PubMed] [Google Scholar]
- 27. Kosemehmetoglu K, Kaygusuz G, Bahrami A, Raimondi SC, Kilicarslan K, Yildiz Y, Folpe AL (2011) Intra‐articular epithelioid sarcoma showing mixed classic and proximal‐type features: report of 2 cases, with immunohistochemical and molecular cytogenetic INI‐1 study. Am J Surg Pathol 35:891–897. [DOI] [PubMed] [Google Scholar]
- 28. Li M, Lee KF, Lu Y, Clarke I, Shih D, Eberhart C et al (2009) Frequent amplification of a chr19q13.41 microRNA polycistron in aggressive primitive neuroectodermal brain tumors. Cancer Cell 16:533–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Louis D, Ohgaki H, Wiestler OD, Cavenee WK (2007) WHO Classification of Tumours of the Central Nervous System, 4th edn. International Agency for Research on Cancer: Lyon. [Google Scholar]
- 30. Manjila S, Ray A, Hu Y, Cai DX, Cohen ML, Cohen AR (2011) Embryonal tumors with abundant neuropil and true rosettes: 2 illustrative cases and a review of the literature. Neurosurg Focus 30:E2. [DOI] [PubMed] [Google Scholar]
- 31. McManamy CS, Lamont JM, Taylor RE, Cole M, Pearson AD, Clifford SC, Ellison DW (2003) Morphophenotypic variation predicts clinical behavior in childhood non‐desmoplastic medulloblastomas. J Neuropathol Exp Neurol 62:627–632. [DOI] [PubMed] [Google Scholar]
- 32. McManamy CS, Pears J, Weston CL, Hanzely Z, Ironside JW, Taylor RE et al (2007) Nodule formation and desmoplasia in medulloblastomas‐defining the nodular/desmoplastic variant and its biological behavior. Brain Pathol 17:151–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Miller S, Rogers HA, Lyon P, Rand V, Adamowicz‐Brice M, Clifford SC et al (2011) Genome‐wide molecular characterization of central nervous system primitive neuroectodermal tumor and pineoblastoma. Neuro Oncol 13:866–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mobley BC, McKenney JK, Bangs CD, Callahan K, Yeom KW, Schneppenheim R et al (2010) Loss of SMARCB1/INI1 expression in poorly differentiated chordomas. Acta Neuropathol 120:745–753. [DOI] [PubMed] [Google Scholar]
- 35. Modena P, Lualdi E, Facchinetti F, Galli L, Teixeira MR, Pilotti S, Sozzi G (2005) SMARCB1/INI1 tumor suppressor gene is frequently inactivated in epithelioid sarcomas. Cancer Res 65:4012–4019. [DOI] [PubMed] [Google Scholar]
- 36. Pfister S, Remke M, Castoldi M, Bai AH, Muckenthaler MU, Kulozik A et al (2009) Novel genomic amplification targeting the microRNA cluster at 19q13.42 in a pediatric embryonal tumor with abundant neuropil and true rosettes. Acta Neuropathol 117:457–464. [DOI] [PubMed] [Google Scholar]
- 37. Raffel C, Rutka JT (2011) Central nervous system primitive neuroectodermal tumors: still a useful classification? Neurosurg Focus 30:Introduction. [DOI] [PubMed] [Google Scholar]
- 38. Raoux D, Peoc'h M, Pedeutour F, Vaunois B, Decouvelaere AV, Folpe AL (2009) Primary epithelioid sarcoma of bone: report of a unique case, with immunohistochemical and fluorescent in situ hybridization confirmation of INI1 deletion. Am J Surg Pathol 33:954–958. [DOI] [PubMed] [Google Scholar]
- 39. Rogers HA, Miller S, Lowe J, Brundler MA, Coyle B, Grundy RG (2009) An investigation of WNT pathway activation and association with survival in central nervous system primitive neuroectodermal tumours (CNS PNET). Br J Cancer 100:1292–1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Rorke LB (1987) Classification of central nervous system tumors in children. Prog Exp Tumor Res 30:57–60. [DOI] [PubMed] [Google Scholar]
- 41. Rorke LB, Packer RJ, Biegel JA (1996) Central nervous system atypical teratoid/rhabdoid tumors of infancy and childhood: definition of an entity. J Neurosurg 85:56–65. [DOI] [PubMed] [Google Scholar]
- 42. Schmitz U, Mueller W, Weber M, Sevenet N, von Delattre O, Deimling A (2001) INI1 mutations in meningiomas at a potential hotspot in exon 9. Br J Cancer 84:199–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Schneppenheim R, Fruhwald MC, Gesk S, Hasselblatt M, Jeibmann A, Kordes U et al (2010) Germline nonsense mutation and somatic inactivation of SMARCA4/BRG1 in a family with rhabdoid tumor predisposition syndrome. Am J Hum Genet 86:279–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Sevenet N, Lellouch‐Tubiana A, Schofield D, Hoang‐Xuan K, Gessler M, Birnbaum D et al (1999) Spectrum of hSNF5/INI1 somatic mutations in human cancer and genotype‐phenotype correlations. Hum Mol Genet 8:2359–2368. [DOI] [PubMed] [Google Scholar]
- 45. Versteege I, Sevenet N, Lange J, Rousseau‐Merck MF, Ambros P, Handgretinger R et al (1998) Truncating mutations of hSNF5/INI1 in aggressive paediatric cancer. Nature 394:203–206. [DOI] [PubMed] [Google Scholar]
- 46. Wang Y, Chu SG, Xiong J, Cheng HX, Chen H, Yao XH (2011) Embryonal tumor with abundant neuropil and true rosettes (ETANTR) with a focal amplification at chromosome 19q13.42 locus: further evidence of two new instances in China. Neuropathology 31:639–647. [DOI] [PubMed] [Google Scholar]
- 47. Weber M, Stockhammer F, von Schmitz U, Deimling A (2001) Mutational analysis of INI1 in sporadic human brain tumors. Acta Neuropathol 101:479–482. [DOI] [PubMed] [Google Scholar]