

Abstract
White matter abnormalities on magnetic resonance imaging (MRI) are associated with dementia and include white matter hyperintensities (WMH; also termed leukoaraiosis) and visible perivascular spaces (PVS). We review the potential role of impaired drainage of interstitial fluid in the pathogenesis of WMH and PVS. Whereas the volume of extracellular space in the grey matter is tightly controlled, fluid accumulates and expands the extracellular spaces of the white matter in acute hydrocephalus, vasogenic edema and WMH. Although there are no conventional lymphatic vessels in the brain, there is very effective lymphatic drainage for fluid and solutes along restricted pathways in the basement membranes of cerebral capillaries and arteries in young individuals. Lymphatic drainage of the brain is impaired with age and in association with apolipoprotein E ε4, risk factors for Alzheimer's disease and cerebral amyloid angiopathy (CAA). Deposition of proteins in the lymphatic drainage pathways in the walls of cerebral arteries with age is recognized as protein elimination failure angiopathy (PEFA), as in CAA and cerebral autosomal dominant arteriopathy and leukoencephalopathy (CADASIL). Facilitating perivascular lymphatic drainage from the aging brain may play a significant role in the prevention of CAA, WMH and Alzheimer's disease and may enhance the efficacy of immunotherapy for Alzheimer's disease.
Keywords: Alzheimer's disease, amyloid‐β, CADASIL, cerebral amyloid angiopathy, perivascular drainage, perivascular spaces, protein elimination failure angiopathy (PEFA), white matter hyperintensities
Introduction
Leukoaraiosis, seen as cerebral white matter hyperintensities (WMH) on magnetic resonance imaging (MRI) or low attenuation on computed tomographic scanning (CT), is frequent in patients with dementia. However, the causes are unclear and appear to be multiple. It has long been assumed that WMH are due to arteriosclerotic small vessel disease and infarction but another strong candidate is emerging as a result of MRI, clinical and pathological studies. This strong candidate is the failure of elimination of interstitial fluid (ISF) from white matter, especially associated with cerebral amyloid angiopathy (CAA) 92, but also related to other disorders of small cerebral blood vessels such as cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL) 16. Pathological studies and detection of dilated perivascular spaces (PVS) by MRI in the white matter in association with evidence of CAA‐related intracerebral hemorrhage support a relationship between CAA and impaired drainage of ISF from the white matter. In the wider context, failure of elimination of ISF from the aging brain indicates a failure in disposal of soluble metabolites from the brain with a consequent failure of homoeostasis of the neuronal environment with important implications for the pathogenesis of dementia 16.
In this review, we first set the scene with a description of the clinical and radiological aspects of leukoaraiosis and dilated PVS in relation to intracerebral hemorrhage and lobar microbleeds putatively because of CAA. This is followed by a brief account of the production and elimination of cerebrospinal fluid (CSF) and ISF from the brain and the interrelationships between CSF and ISF. A more detailed account of how ISF and solutes drain from the brain along basement membranes in the walls of cerebral capillaries and arteries follows; such perivascular pathways represent the lymphatic drainage pathways of the brain. Major pathologies of ISF drainage include its failure with age and the deposition of amyloid‐β (Aβ) and other proteins, such as CADASIL‐derived material, within basement membranes of the perivascular drainage pathways. Evidence will be presented that perivascular drainage of ISF and solutes is impaired with age, CAA and Alzheimer's disease (AD) and in individuals possessing the ε4 allele of apolipoprotein E (APOE4).
The failure of trials of anti‐Aβ immunotherapy to show significant clinical improvement in AD has been extensively discussed. Despite evidence that insoluble plaques of Aβ are removed from the brain in immunized patients 79 and in experimental animals 97, there is an increase in severity of CAA and WMH in association with immunotherapy 79. We therefore review the possible role of immune complexes in the impairment of perivascular elimination of Aβ from the brain. Finally, we examine the generic concept of protein elimination failure angiopathy (PEFA) that includes Aβ and other amyloid angiopathies together with prion angiopathies and CADASIL. In this way, we seek to emphasize the wide range of pathologies that are associated with failure of perivascular drainage of ISF and solutes from the brain, particularly in the aging population.
Clinical Significance of WMH and Dilated PVS
The term leukoaraiosis [from the Greek λευκος (leukos): white; and αραιός (araios): rarefied] was coined by Hachinski et al in 1986 48 to describe changes in the hemispheric cerebral white matter seen on neuroimaging: initially as areas of low attenuation on CT and subsequently as hyperintensities on T2‐weighted or Fluid Attenuated Inversion Recovery (FLAIR) MRI. The term was developed to avoid ascribing a particular pathological cause to these increasingly detected imaging findings, which may reflect a range of pathological processes in different populations (eg, in younger patients may be due to leukodystrophy or inflammatory demyelination). Leukoaraiosis is common in patients with cognitive impairment including dementia as well as those with symptomatic cerebrovascular disease 29. In this review, we focus on leukoaraiosis in older individuals, in whom the changes have been presumed to be due to cerebrovascular disease, principally affecting small vessels. Here, we assess to what extent it is possible to correlate fluid levels in white matter on MRI with dilated PVS (Figure 1) and cerebral amyloid angiopathy.
Figure 1.
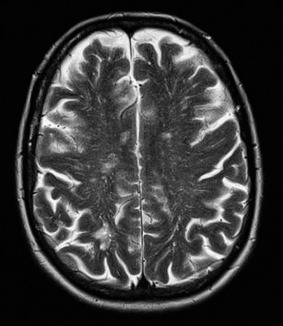
T2‐weighted axial image showing MRI‐visible PVS in the hemisphere white matter in a patient with CAA‐related ICH.
Leukoaraiosis can be defined as white matter abnormality (low density on CT, hyperintensity on T2‐weighted or FLAIR MRI), which may be patchy or confluent. These white matter appearances on MRI have variously been termed white matter abnormalities, white matter changes and WMH; a recent consensus group recommended the term “white matter hyperintensities of presumed vascular origin” 111. The term leukoaraiosis nevertheless has the advantage of being applicable to both CT and MRI. Here, we will mainly focus on imaging changes seen on MRI, which will be referred to as “white matter hyperintensities” (WMH).
White Matter Hyperintensities
WMH are commonly detected on MRI in various populations: in the general population, the prevalence is between about 10% and 20% in the seventh decade of life and nearly 100% in the ninth decade. WMH are more common and extensive in patients with cardiovascular risk factors and symptomatic cerebrovascular disease 69. WMH appear to be of clinical relevance because of a strong relationship with vascular risk factors and cognitive impairment in cross‐sectional studies. WMH also contribute to gait disturbance and falls 9. There is now also a strong evidence that WMH are important for clinical prognosis: a systematic review of 958 published prospective studies found strong evidence of an increased risk of stroke, cognitive decline (especially in the executive function and processing speed domains), dementia and death 29. WMH were significantly associated with an increased risk of stroke, both in the general population and in high‐risk populations with a history of stroke or vascular disease, even after adjusting for the potential confounding effect of vascular risk factors known to be associated with WMH (eg, hypertension) 29. WMH were associated with an increased risk of dementia in the general population but not in high‐risk populations 29. The mechanisms underlying this association with future cognitive risk may include direct damage to white matter subcortical networks or aggravation of neurodegenerative processes including AD. CT‐defined leukoaraiosis has been associated with an increased risk of intracerebral bleeding in patients with stroke treated with high intensity oral anticoagulation with warfarin 43.
Finally, WMH may be a useful biomarker or “intermediate phenotype,” which could help to identify new genetic or environmental risk factors and potentially function as a surrogate end point in clinical trials 111.
The pathological substrates of WMH include tissue rarefaction associated with loss of myelin and axons, enlarged (dilated) PVS (see: Pathology of Dilated PVS) and mild gliosis. These lesions are located in deep hemispheric white matter, typically sparing the subcortical U‐fibers, and are often seen in association with vessels affected by small vessel disease including arteriolosclerosis. However, the pathological basis of WMH is heterogeneous 44 as the basis of increased signal on T2‐weighted and FLAIR MRI is simply an increase in free tissue water content, which is not specific for any particular pathological process. Furthermore, the degree of pathological heterogeneity appears to reduce as the severity of MRI‐defined lesions increases from small punctate lesions to confluent WMH; the latter almost always have an ischemic appearance with myelin loss, gliosis and small areas of infarction 36.
Pathophysiological mechanisms underlying WMH
Despite their high prevalence and clear clinical relevance, the pathophysiological mechanisms underlying WMH remain unclear. One hypothesis is that damage to small medullary perforating vessels, which supply the white matter, causes chronic hypoperfusion, with associated disruption of the blood–brain barrier, leading to both ischemic injury and chronic leakage of potentially toxic plasma proteins into the white matter. However, which comes first (ischemia or blood–brain barrier dysfunction) remains controversial. Evidence supporting an ischemic mechanism of WMH comes from the observation that some forms of hypoxic‐ischemic injury cause selective white matter injury (Figure 2). The anatomy of the blood supply to cerebral white matter is also consistent with a vulnerability of deep white matter regions to reduced perfusion and ischemic injury. Moreover, cerebral blood flow is reduced in areas of WMH in patients with small vessel disease 83.
Figure 2.
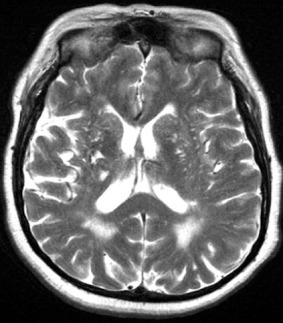
T2‐weighted MRI axial image showing confluent WMH in association with extensive deep (basal ganglia) MRI‐visible perivascular spaces and lacunar infarcts.
An alternative hypothesis is that WMH (known to be associated with increased tissue water content) are due to alterations in CSF flow. Normal pressure hydrocephalus is known to be associated with WMH and experimental hydrocephalus causes changes in the white matter that can be reversed by shunting 78. However, it is also possible that WMH causes ex vacuo dilatation of the ventricular system suggesting a possible reverse causation. It is suggested that the increased accumulation of CSF in the ventricles raises interstitial pressure in the periventricular parenchyma and causes ischemia of the white matter 93. The potential contribution of impaired ISF drainage to WMH outside the context of hydrocephalus remains even more uncertain.
Dilated Perivascular Spaces
Dilated PVS seen on MRI (Figure 1) have recently emerged as a new MRI marker of small vessel disease, as they are associated with other MRI features of small vessel disease, including lacunes and WMH 30. Dilated PVS have particular interest in the study of ISF dynamics as they are closely linked to the established clearance pathways for ISF and solutes along basement membranes 15, 16 and are enlarged in patients with conditions known to impair ISF drainage, including AD with associated CAA 92. There is no detectable PVS around the arteries in the cerebral cortex 127 whereas PVS may dilate in the white matter and are thought to indicate failure of drainage of fluid and solutes from the white matter 92 (see Pathology of dilated PVS section).
Recent data in cohorts with intracerebral hemorrhage (ICH) (another end result of small vessel disease, but in this case causing fragility) indicate that the anatomical distribution of MRI‐visible dilated PVS is linked to the underlying small vessel pathological process: in patients with lobar ICH attributed to probable CAA, severe dilatation of PVS was noted in the cerebral hemispheric white matter, while in other ICH cases (including deep ICH attributed to arteriolosclerosis‐related rupture of deep basal ganglia perforating small vessels) severe dilatation of PVS was found in the basal ganglia (Figure 2) 21. A similar topographical association of hemispheric white matter PVS with radiologically defined CAA was also noted in a memory clinic cohort, supporting this hypothesis 72. The clinical relevance of dilated PVS is less well established than that of WMH, with no clear association shown with cognitive function 54 and no available prospective data on the risk of future stroke or cognitive impairment.
If dilated PVS in the hemispheric white matter are due to impaired drainage of ISF then they should logically be associated with markers of severe leptomeningeal and cortical arteriolar amyloid deposition in CAA. Indeed, a recent study showed that dilated PVS in hemispheric white matter were strongly linked to one such marker of severe superficial CAA, namely cortical superficial siderosis (because of recurrent subarachnoid bleeding and hemosiderin deposition from fragile, amyloid‐laden superficial vessels) 22. Another study used an amyloid‐binding Positron Emission Tomography (PET) ligand and showed that increased amyloid binding was associated with the severity of WMH in patients with CAA (but not AD), supporting the hypothesis that vascular amyloid may aggravate or cause white matter injury in CAA 47. However, ischemic injury to white matter associated with CAA in small vessels perforating the cerebral cortex is also possible 45 as an alternative mechanism to impaired ISF drainage. An effect of amyloid on the pathogenesis of WMH is also supported by the finding that in AD, CAA, mild cognitive impairment and white matter changes correlate with serum levels of Aβ1–40 peptide (the main constituent of vascular amyloid deposits in CAA) 46.
Physiology and Pathology of CSF and ISF of the Brain
CSF and ISF are the major extracellular fluids associated with the central nervous system (CNS). The total volume of CSF in humans is 140 mL with 30 mL in the ventricles and 110 mL within the cerebral and spinal subarachnoid spaces; ISF has an estimated total volume of 280 mL in the CNS 6.
Production and drainage of CSF
CSF is produced by the choroid plexuses with a probable contribution from brain ISF 59. Following circulation through the ventricular system, CSF enters the subarachnoid space and passes directly into the blood through arachnoid granulations and villi. An estimated 50% of CSF drains to cervical lymph nodes 60 in smaller mammals along channels that pass from the subarachnoid space through the cribriform plate of the ethmoid bone to join nasal lymphatics 66. This pathway appears to be a major pathway in small mammals and is also present in humans 60, 66, 116.
Pathology of CSF
The major pathologies of the CSF are leptomeningitis, infiltration by neoplastic cells (carcinomatous/neoplastic meningitis), and more important in the context of this paper, hydrocephalus because of impaired drainage of CSF. In the acute stages of hydrocephalus, interstitial edema of the periventricular white matter may be observed 112 which suggests that there is restricted capacity for ISF drainage from the white matter that cannot cope with the excess of CSF in hydrocephalus. This may be a further indication of the limited capacity of ISF drainage from the white matter that is present in children with hydrocephalus and may be a factor in the pathogenesis of leukoaraiosis in aging adults.
Lymphatic drainage of Interstitial Fluid
ISF is generated from the blood and from metabolic activity within the brain tissue at an estimated rate of 0.1–0.3 μL/minute/g in the rat 1. In the 1980s, Helen Cserr and her group showed that radiolabeled human serum albumin, injected into ISF of the rat brain, drained rapidly to cervical lymph nodes on the same side of the neck 103. The drainage pathway was associated with the walls of the arteries within the brain, leptomeningeal arteries and the upper part of the internal carotid artery in the neck. Absence of radiolabeled human serum albumin in the lower part of the internal carotid suggested that most of the tracer had drained to the cervical lymph nodes. The speed of perivascular drainage was comparable with lymphatic drainage of other organs in the body and only 15% of tracer leaked from the ISF drainage pathways in artery walls into the CSF 103.
In more recent investigations of the ISF drainage pathways, soluble lysine‐fixable fluorescein‐labeled 3 kDa dextran and Fluorescein Isothiocyanate (FITC)‐labeled ovalbumin (40 kDa) were injected separately into the grey matter of the corpus striatum in mouse brains. The distribution of the tracers was defined at 5 minutes, 30 minutes, 3 h and 24 h after injections. By 5 minutes, dextran had spread diffusely in the brain parenchyma and was within the basement membranes of capillary and artery walls (Figure 3) 15. At 30 minutes, dextran had largely disappeared from brain parenchyma and from the walls of capillaries and arteries. At 3 h, dextran was present in a punctate form within perivascular macrophages associated with capillaries and arteries 15. Further analysis by confocal microscopy following intracerebral injection of fluorescent dextrans and fluorescent soluble Aβ revealed that tracers in the extracellular spaces of the brain parenchyma rapidly enter bulk flow pathways in basement membranes in the walls of cerebral capillaries. From there, tracers drain into basement membranes of smooth muscle cells within the tunica media of cerebral arteries (Figure 3) 15, 114. These experiments defined the 100–150 nm thick basement membranes as the route for lymphatic drainage of ISF and soluble metabolites from the brain. The findings are highly relevant to the pathology of human neurodegenerative disease as Aβ is deposited in this pathway in CAA (Figure 4) 16 and relevant to neuroimmunology as antigens may drain from the brain to regional lymph nodes in the neck as part of the induction of neuroimmunological reactions 68, 118.
Figure 3.
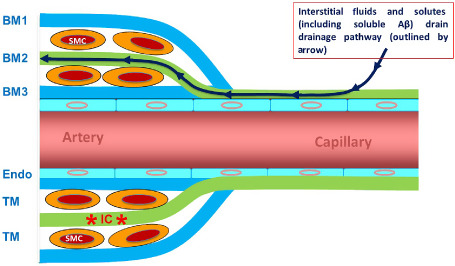
Perivascular drainage. ISF and solutes, including Aβ, diffuse through the extracellular spaces of the brain and then drain out of the brain along basement membranes (BM) in the walls of capillaries and arteries. The drainage pathway is along the BM colored green: that is capillary endothelial BM and the BM of smooth muscle cells (SMC) in the mid‐zone of the tunica media (TM) of the artery (BM2). In the artery wall, BM on the outer aspect (BM1) and on the endothelial aspect (BM3) is not part of the drainage pathway. With reference to Figure 8, immune complexes (*IC*) are trapped within the BM2 pathway and obstruct perivascular drainage. ENDO, endothelium.
Figure 4.
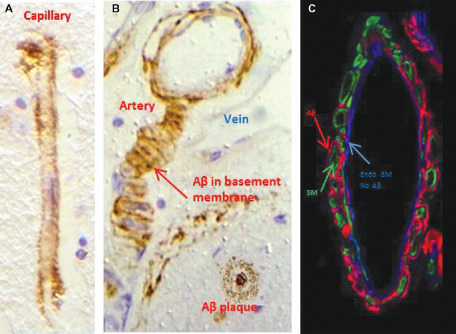
Cerebral amyloid angiopathy. Aβ (brown) is deposited in basement membranes in the walls of capillaries (A) and arteries (B,C). Although the basement membranes of smooth muscle cells (SM) in the tunica media contain Aβ (see figures B and C), the endothelial basement membrane (Endo BM) is almost devoid of Aβ. Immunocytochemistry for Aβ (brown or red) and smooth muscle actin (green). Collagen IV in basement membrane in (C) is blue.
Relationship between CSF and ISF
The brain is directly related to the CSF on its ventricular and pial surfaces. CSF appears to play a major role as a buoyancy fluid for the brain. Although there is a well‐characterized barrier between the blood and the brain, tracers injected into the CSF diffuse into the surface of the brain and spinal cord as shown by Goldman in 1913 42. Injection studies by Rennels et al in the 1980s 89 showed that horseradish peroxidase injected into the cisterna magna diffused from the subarachnoid space into the brain alongside arteries in the cerebral hemispheres to reach capillary level. Fluid entry appeared to be driven by vascular pulsations and was termed convective tracer influx 89. Entry of tracers from the CSF into ISF was confirmed by the injection of fluorescent tracers that revealed an entry pathway from the CSF into the brain along extravascular compartments between cerebral arteries and brain parenchyma 57, 58. Entry of low molecular weight tracers from the paravascular pathway into the brain parenchyma is mediated by aquaporin‐4 associated with astrocytes in the brain 57. Because of its association with astrocytes and the eventual drainage of tracers to cervical lymph nodes, the name of the system has been changed from the original terms of convective tracer influx to the “glymphatic system.” The relationship between the glymphatic system and the periarterial basement membrane drainage pathway described in Figure 3 15 for elimination of solutes from the extracellular space of the brain is unclear. However, it seems that the glymphatic system is a slow equilibration between CSF and ISF. On the other hand, drainage of ISF and solutes along basement membranes in the walls of the arteries is a much more rapid response system that may play a signaling role between solutes in the ISF and smooth muscle cells in the walls of the arteries.
Pathology of Perivascular Drainage of ISF and Solutes from the Brain
The pathology of perivascular drainage of ISF and solutes from the brain relates to two major areas: first, neuroimmunology 68 and second, the failure of ISF drainage in hydrocephalus and perhaps more significantly in the aging brain, CAA and AD 116.
Antigens drain from the brain to regional lymph nodes in the neck with ISF along basement membranes in the walls of capillaries and arteries 15, 103. However, this pathway is too narrow to allow the migration of antigen‐presenting cells from the brain to cervical lymph nodes 15, 16. The absence of such effective migration of antigen‐presenting cells may be a major factor in the relative immunological privilege of the brain 18, 118. Evidence for a significant role for cervical lymph nodes in neuroimmunological reactions in the brain comes from experiments whereby the severity of experimental autoimmune encephalomyelitis (EAE) is reduced by cervical lymphadenectomy 87, 108. Antigen‐presenting cells do appear to migrate from the CSF to cervical lymph nodes via nasal lymphatics and appear to play a role in the induction of immunological tolerance 68.
Deposition of amyloid in perivascular drainage pathways in CAA
CAA is a major pathological manifestation of impaired ISF drainage 16. The presence of CAA in transgenic mice that produce human Aβ solely in the brain is taken as firm evidence that CAA is due to the failure of drainage of Aβ from the brain 53. A number of amyloidogenic proteins are deposited in the basement membranes in the walls of cerebral arteries, the most common of which is Aβ associated with AD 90. Other amyloids such as cystatin, transthyretin and certain forms of prion protein and the Notch‐3 associated protein in CADASIL are also deposited in cerebral vessel walls 16. These disorders have been grouped under the collective term PEFA 16. One of the major pathological effects of impairment of perivascular drainage of ISF and solutes from the brain with age and CAA appears to be loss of homoeostasis of the extracellular environment in the brain, so that metabolites such as soluble Aβ and probably other metabolites accumulate within brain tissue in AD 71, 76. Such a change in the extracellular environment of neurons may result in oxidative stress and ultimately in dementia.
The distribution of Aβ in the walls of cerebral capillaries and arteries in the early stages of CAA mirrors exactly the pathways by which ISF and solutes drain from the brain 16 (Figures 3 and 4). Electron microscope studies indicate that Aβ is initially deposited as fibrils in the central portion (lamina densa) of basement membranes surrounding smooth muscle cells 120, although at the time the authors suggested that the Aβ was derived from vascular myocytes rather than from the brain. One of the major questions about CAA is exactly why amyloid is deposited in basement membranes in the walls of cerebral capillaries and arteries. What interferes with the perivascular drainage of soluble Aβ that causes it to precipitate as fibrillar amyloid in vascular basement membranes?
Age and possession of the ε4 allele of APOE4 are major risk factors for CAA and for AD and the next section of this review shows how both these factors impede the drainage of ISF and solutes along perivascular pathways.
Impairment of perivascular drainage of ISF and solutes with age and in the presence of apoE4
Age and possession of APOE4 allele are the strongest risk factors for the development of sporadic AD and CAA. Experiments described here show that age, CAA and possession of APOE4 are associated with impaired perivascular drainage of ISF and solutes, including Aβ.
Perivascular drainage of Aβ was assessed by confocal microscopy following intracerebral injection of 0.5 μL fluorescent dextran or soluble human Aβ40 over a period of 2 minutes into the hippocampus in three groups of animals. First: To test the effects of age on perivascular drainage, intracerebral injections of fluorescent dextran were performed on young and aged wild‐type C57B1/6 mice, at 3, 7 and 22 months of age 49. Second: To assess the effect of CAA on perivascular drainage, fluorescent dextran was injected into the brains of Tg2576 transgenic mice with CAA 49. Third: To test the effects of APOE4 on perivascular drainage, intracerebral injection of soluble Aβ40 was performed in targeted replacement (TRE) mice carrying the human APOE ε3 or APOE ε4 allele 50.
The results of these studies showed that perivascular drainage was significantly (P < 0.001–P < 0.05) impaired in capillaries and arteries:
-
(i)
in 22‐month‐old wild‐type mice compared with 3‐ and 7‐month‐old animals [furthermore, age‐related changes in the levels of laminin, fibronectin and perlecan in vascular basement membranes were observed in these animals 49 ] and
-
(ii)
in 7‐ and 22‐month‐old Tg2576 mice with CAA 49.
-
(iii)
In addition, Aβ40 aggregated in periarterial drainage pathways in APOE ε4 mice, but not in APOE ε3 or wild‐type mice (Figure 5) 50. The number of Aβ deposits was significantly higher in the hippocampi of APOE ε4 mice than in the APOE ε3 mice, at both 3 and 16 months of age, indicating that clearance of Aβ from the brain was disrupted in APOE ε4 mice 50.
Figure 5.
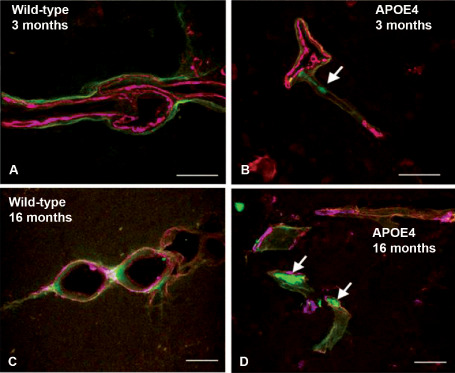
Leptomeningeal arteries from wild‐type and APOE ε4 mice following intracerebral injection of fluorescent Aβ (green). Within 5 minutes of the injection, Aβ was observed along basement membranes, colocalizing (pink) with collagen IV (blue) in young, 3‐month‐old wild‐type mice in (A). Aggregation of Aβ is seen as early as 3 months in the APOE4 mice (arrow in B). Aβ continues to drain along basement membranes of aged wild‐type mice (C), but appears as large deposits in 16 months in the APOE ε4 mice (D, arrows). Scale bars = 10 μm in A; 25 μm in B, C and D. Reproduced from reference 50.
In summary, the results of these studies suggest that age, possession of APOE ε4 and the presence of CAA all result in impaired perivascular drainage of solutes, including Aβ, from the brain. This raises the question as to exactly why these factors interfere with perivascular drainage and whether these data apply directly to humans.
Age changes in arteries and distribution of Aβ in plaques and CAA
Aged changes in human cerebral arteries are well recognized, with generalized intimal thickening and fibrosis of the tunica media in arteriosclerosis 115. Theoretical and experimental studies suggest that the motive force for perivascular drainage of ISF and solutes is derived from vascular pulsations 2, 98. As arteries age, they become stiffer 115 which would imply that the amplitude of vascular pulsations is reduced. Such a reduction in pulsations may reduce the motive force for perivascular drainage and the consequent slowing of drainage may allow Aβ fibril formation to occur within the basement membranes of the perivascular pathways, particularly as the nature of the basement membrane proteins change with age. Deposition of Aβ in basement membranes in CAA appears to further impede perivascular drainage 49.
ApoE colocalizes with Aβ within plaques in the brain parenchyma and in CAA 106. It is thought that ApoE is a transport molecule for Aβ. Our experiments suggest that Aβ is not transported along perivascular pathways as efficiently in the presence of apoE4 compared to apoE3 50. The exact reasons for this are not clear but may be related to a difference in association between the isoforms of APOE and basement membrane proteins or an effect of APOE genotype on composition of basement membranes.
There are regional differences in the deposition of Aβ as plaques and CAA in the brains of aging and AD patients 105. CAA is observed predominantly in the parietal and occipital cerebral cortex, less so in the basal ganglia and thalamus and rarely in vessels of the brain stem. Experiments in which Aβ was injected into different areas of aged mouse brains showed that perivascular drainage from the hippocampus became less efficient with age, whereas there was little change in perivascular drainage from the thalamus with age 51. These results suggest that there is a differential effect of age on perivascular drainage in different regions of the brain and this may have some bearing on the distribution of CAA and the deposition of amyloid as plaques in the brain in AD.
Biochemical and morphological changes in blood vessels of the grey matter that contribute to impaired drainage of ISF
Although the mechanisms underlying disrupted perivascular drainage in the aged and ApoE4 brain have not been fully elucidated, multiple lines of evidence support a role for morphological and biochemical changes in blood vessel walls in the pathogenesis of CAA. In vitro, aggregation of Aβ is inhibited and preformed fibrils are destabilized in the presence of the basement membrane proteins laminin, nidogens and collagen IV. By contrast, incubation of Aβ with heparan sulphate proteoglycans, such as agrin and perlecan, accelerate its aggregation. In mice, brain regions that are vulnerable to CAA show age‐related decreases in laminin, collagen IV and nidogen 2 and increased levels of fibronectin and perlecan 51. Decreased amounts of collagen IV have been reported in small diameter vessels (<50 μm) in the grey matter in AD, compared with age‐matched controls 23. Conversely, the levels of heparan sulphate proteoglycans were increased in AD brains 99. These data suggest that age‐related changes in the expression of individual basement membrane proteins may contribute to a pro‐amyloidogenic environment within the vessel wall.
Less is known about the effect of ApoE genotype on the biochemistry of cerebral blood vessels, but a significant reduction in the surface area of agrin immunoreactivity has been reported in the brains of AD patients homozygous for the APOE ε4 allele 94. Changes in the levels of collagen IV between 3 and 16 months are greater in TRE4 than in TRE3 mice 50, suggesting that APOE genotype may also influence the degree to which expression of basement membrane proteins changes over the lifespan.
Age‐related changes in the morphology of grey matter vessels include splitting, duplication and thickening of the basement membrane, particularly in areas of the brain that are susceptible to AD pathology and CAA 51, 126. The contribution of basement membrane abnormalities to the pathogenesis of CAA is suggested by the observation that an increase in thickness of basement membranes precedes endogenous CAA development in transforming growth factor‐β (TGF‐β) transgenic mice 121. Increased rigidity in the artery walls, elongation and tortuosity, as well as a reduction in smooth muscle cells are also observed in the aging brain 63. This reduction in vascular elasticity and contractile force may diminish the amplitude of the arterial pulse and slow the driving force for perivascular clearance of ISF and Aβ, leading to its accumulation as CAA.
Pathology of dilated PVS
Dilatation of PVS occurs in the white matter of the cerebral hemispheres (Figure 1), in the midbrain and in the grey matter of the basal ganglia. In some cases, large giant lacunae form in these areas of the brain 95; in most cases, however, the dilatation is more moderate.
PVS, which is the potential space between the outer aspect of an artery wall and the brain parenchyma, can be expanded experimentally by the intracerebral injection of Indian ink particles 128 or fluorescent polystyrene beads (fluorospheres) 15. However, the PVS does not appear to function as a fluid drainage pathway in the same way as the basement membrane pathways within the artery walls themselves. Injection of pharmacological preparations into the brain to expand PVS (intrastriatal convection‐enhanced delivery) has been developed to create a reservoir for slow release of drugs for the treatment of neurological disorders 5.
There is evidence that dilatation of PVS in the white matter is associated with CAA 92. Figure 6 shows how branches of the leptomeningeal arteries on the surface of the brain pass through the cerebral cortex, often without branching, to supply the underlying white matter 33. Perivascular drainage of fluid and solutes from the white matter seems to be impaired by CAA in the parent leptomeningeal or cortical arteries with consequent dilatation of PVS in the white matter 92 (Figure 6). Dilatation of PVS in the cortical grey matter is rarely seen.
Figure 6.
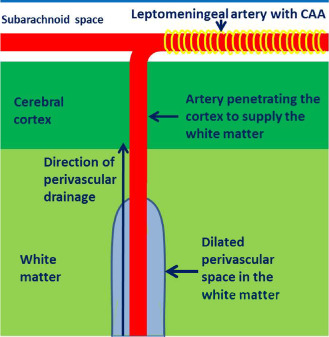
Proposed mechanism for CAA‐induced dilatation of perivascular spaces in the white matter. Branches of the leptomeningeal arteries penetrate the cortex (often without branching) to supply the subcortical white matter. CAA in the walls of the leptomeningeal arteries (yellow banding) obstructs the drainage of interstitial fluid and solutes resulting in retention of fluid in dilated perivascular spaces around arteries in the white matter.
The other major site for dilated PVS is the grey matter of the basal ganglia. This is evident in several diseases involving small cerebral arteries. The structure of arteries in the basal ganglia differs from arteries in the cerebral cortex 88, 127. As arteries enter the surface of the cerebral cortex, the pia mater is reflected from the surface of the brain on to the vessels in the subarachnoid space, thus separating the subarachnoid space from the cerebral cortex 55, 113, 127. There is no PVS around cortical arteries in the normal brain as layers of smooth muscle, basement membrane and one layer of leptomeninges are all compressed together 127. In the basal ganglia, however, there are two layers of leptomeninges surrounding the artery with a PVS between the layers 88. This anatomical arrangement may explain why there is no expansion of PVS in the cortex, whereas perivascular lacunae are frequently seen in the basal ganglia 95, 113.
WMH and CAA in Aβ Immunotherapy Trials for AD
Aβ immunotherapy impairs the elimination of solutes from the brain
Trials of immunotherapy for AD in humans were introduced after successful studies in human amyloid precursor protein (hAPP) transgenic mice showed that insoluble Aβ plaques were removed from the brain following immunization with Aβ42 97. However, despite the clearance of plaques in patients with AD 39, 73, 79, active immunization seemed to increase, rather than decrease, the amount of arterial CAA both in transgenic animals and in humans 79, 119. It appears that Aβ can be solubilized from the plaques but becomes trapped in the perivascular drainage pathway 85, 117, manifesting as an increase in the severity of CAA. The dynamics of the events resulting in increased CAA following immunotherapy are not clear but it could reflect either increasing flow of Aβ out of the brain in the perivascular drainage pathway or impaired drainage and elimination of soluble Aβ as it becomes impeded by aggregated Aβ and possibly by immune complexes. In addition, human Aβ immunotherapy trials, both active 8, 41 and passive 64, 65, encountered side effects comprising focal abnormalities in the cerebral white matter on imaging, interpreted as amyloid‐related imaging abnormalities, a lymphocyte response in the CSF and variable deterioration in neurological function 102. The pathophysiology underlying these side effects is as yet unclear; they may be due partly to the impaired drainage of ISF associated with the increase severity of CAA but may also be due to the effects of immune complexes in relation to the cerebral vasculature.
In CAA, Aβ is seen initially in the basement membranes (Figure 4) and then occupies the whole thickness of the walls of arteries and capillaries, suggesting that insoluble Aβ is deposited in the ISF drainage pathway 16, 18, 24, 40.
Active immunization of humans and mice with Aβ results in decreased plaque load that correlates with the anti‐Aβ titre in serum, but the exact mechanism by which antibodies remove Aβ is not clear 10. It seems likely that anti‐Aβ IgG enters the brain and may form Aβ immune complexes, interfering with vascular function and impairing the perivascular drainage of Aβ. Immune complexes formed in the brain parenchyma generate a robust and long‐lasting inflammatory response, characterized by increased expression of the microglia markers CD11b, CD68 and FcRII/III, but this feature is absent in the immunized Fcγ receptor‐null mice 104.
Immune complexes in the walls of arteries disrupt perivascular drainage of ISF and solutes
We tested the hypothesis that immune complexes disrupt the perivascular drainage of solutes along the walls of arteries in normal brains but not in brains of mice lacking Fcγ receptor. The schedule for the experiments is summarized in Figure 7. Using the presence of complement C3, IgG and ovalbumin as markers for immune complex formation 101, we demonstrated that by 24 h, immune complexes are localized to the basement membranes of the cerebral vasculature 17. This response is slower than the one seen systemically, where immune complexes form within 10 minutes 70.
Figure 7.
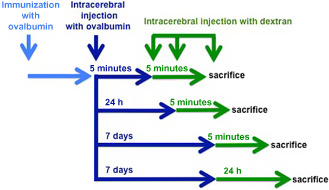
Schematic diagram of the temporal sequence of immunization and injection experiments. Wild‐type BALB/c mice were immunized with ovalbumin (OVA) and then injected with OVA in the striatum at different time points. A soluble fixable fluorescent dextran was then injected intracerebrally, 5 minutes or 24 h or 7 days post‐immunization. Reproduced from reference 17 Original Publisher: BioMed Central.
Normally, when solutes similar in molecular weight to Aβ are injected in the grey matter, they diffuse through the extracellular spaces of the brain and drain along the basement membranes of capillaries and arteries within 5 minutes 15. In the animals in which immune complexes had been allowed only 5 minutes to form, there was no change in the pattern of distribution of soluble dextran of 3 kDa at 5 minutes after its injection. At this time, immune complexes were not detected in the cerebrovascular basement membranes, permitting drainage of solutes in a physiological manner.
At 24 h after the formation of immune complexes, confocal microscopy revealed immune complexes in arterial basement membranes (Figures 3 and 8). Quantitative assessment of perivascular drainage in artery walls demonstrated that the drainage of solutes from the brain was significantly impaired by the presence of immune complexes in perivascular drainage pathways 17.
Figure 8.
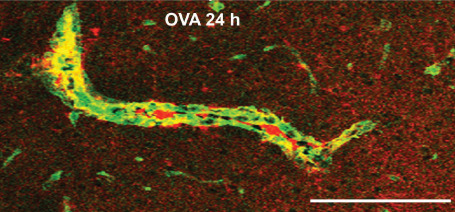
Immune complexes in basement membranes in the walls of cerebral capillaries and arteries. Active immunization with ovalbumin (OVA) was followed by intracerebral challenge with OVA and left in situ for 24 h. The figure shows a longitudinal section of an artery in the striatum at 24 h after injection of antigen. There is colocalization (yellow) of complement (green) with laminin (red) in the basement membranes surrounding smooth muscle cells in the tunica media. Scale bar = 60 μm. Reproduced from reference 17 Original Publisher: BioMed Central.
Normally, by 24 h after its injection, dextran has been cleared from the extracellular spaces and is present only in perivascular macrophages around arteries in the ipsilateral hemisphere 15. When left in situ for 24 h in mice with previously formed immune complexes, dextran is taken up by macrophages in the ipsilateral parenchyma, around veins, and by meningeal macrophages. This suggests that, in the presence of inflammation induced by immune complexes, solutes may become sequestered by inflammatory cells and not cleared from the brain parenchyma.
At 7 days following the formation of immune complexes, the pattern of drainage of soluble dextran was remarkably different. A striking feature was the presence of dextran in the cellular infiltrates around veins. This pattern of distribution suggests that the dextran tracer is diverted from the extracellular space toward the larger perivascular spaces around veins where it is taken up by activated macrophages. We did not observe cuffs of dextran around capillaries or arteries. There was a trend for the number of capillaries and arteries with dextran associated with their basement membranes to decrease in the animals in which immune complexes had been left for 7 days, but this did not reach statistical significance. These results suggest that even in the presence of inflammatory cells associated with the process of immune complex formation and the taking up of dextran by macrophages, perivascular drainage had returned to normal by 7 days.
In animals without the Fcγ receptor, perivascular drainage was normal, with dextran tracer draining along the basement membranes of capillaries and arteries, with very few veins involved. This is consistent with the absence of inflammatory cells recruited around veins in immunized Fcγ‐deficient mice 104.
There are no reports of immune complexes forming in the brains of humans immunized against Aβ. However, it is possible that Aβ solubilized from plaques may encounter Aβ‐specific antibodies, generated in the process of immunotherapy, in the walls of cerebral blood vessels. We know that there are high levels of complement in transgenic mice that harbor Aβ mutations and that complement colocalizes with the microvascular Aβ 35. Increased levels of C3 were also found to be associated with increased hemorrhages following experimental immunotherapy 26, 52, 86, 109, suggesting that immune complex formation may explain these side effects. It has been reported that, following immunotherapy with Aβ in mouse models, small amounts of anti‐Aβ antibodies enter the brain and form immune complexes that are cleared from the brain into the blood via the neonatal Fcγ receptors present ubiquitously in the endothelia 28. The risk of intracerebral hemorrhage may increase if immune complexes are formed in the basement membranes of capillaries and arteries affected by CAA. The vessel wall is already structurally weak from the deposition of Aβ and is predisposed to aneurysm formation and rupture 19, 74, 75. Recently, it has been shown that active immunotherapy using Aβ derivatives and alum in adult but not in aged Tg2576 mice results in a reduction of Aβ burden, without worsening of CAA or inducing microhemorrhages 4. If a number of blood vessels are still unaffected by the morphological changes associated with aging and deposition of Aβ, it may be sufficient to cope with the drainage of the Aβ solubilized from plaques, without rupturing the vascular wall.
It remains unclear whether immune complexes are formed in the extracellular spaces of the brain and then follow the perivascular drainage route, or whether they form in situ within the arterial basement membranes, or indeed whether immune complexes are formed at all after immunization with Aβ. Our results suggest that the absence of Fcγ receptors restores the ability of normal perivascular clearance of solutes, supporting recent studies that demonstrate that deletion of Fcγ receptors prevents the cognitive impairment associated with AD 37.
Future prophylactic and therapeutic strategies in aging and CAA will need to take into account the possibility that immune complexes may interfere with perivascular drainage in patients treated with Aβ immunotherapy.
PEFA in CAA and CADASIL
As discussed earlier, we have previously proposed reasons why both CAA and CADASIL are categorized as PEFA disorders 16. Although different types of PEFA involve failure of removal of specific proteins, they share several risk factors, pathological processes and a final common pathway, which also typify them as angiopathies. CAAs are characterized by the vascular deposition of different proteins such as Aβ, cystatin C and transthyretin 91. In prion disease, insoluble prion protein accumulates mainly in the brain parenchyma but the perivascular clearance of anchorless prion protein induces PEFA‐CAA 16. The most common type of PEFA‐CAA is associated with the deposition of Aβ, which increases in old age and is observed in over 90% of subjects with AD. All CAAs including the Aβ type, however, foremost entail the parenchymal production or impaired metabolism of specific protein(s), which aggregate to accumulate in predominantly cortical areas within the brain. Several processes including uptake by macrophages or microglia are engaged in the clearance of insoluble and degraded proteins from the cerebrum. However, the perivascular lymphatic drainage pathways are an important route for the bulk removal of degraded proteins and solutes along with the ISF. With increasing age and likely greater volumes of solutes and degraded proteins, this unique perivascular system gradually loses its efficiency 49, 116. Walls of cerebral arteries exhibiting arteriosclerosis also stiffen with age and lose their elasticity over time. As a consequence of such weakening, arteries fail to revert to their original form following the pulse wave, leaving them dilated and with a stretched internal elastic lamina 116. Age‐related change within vascular basement membranes, such as increased collagen in arteries that form the perivascular pathways 14, is another factor that contributes to the inflexibility of the vessel wall and favors deposition of insoluble proteins and aggregates such as Aβ 7, 107.
CADASIL is characterized by widespread arteriolosclerosis, multiple lacunar infarcts and microinfarcts within subcortical grey and the deep white matter 123. There is an exclusive and widespread distribution of the granular osmiophilic material (GOM) in the walls of leptomeningeal arteries and cerebral arterioles. In moderately advanced cases, an abundance of GOM is readily detected as electron‐dense masses sited at intervals around vascular smooth muscle cells and along capillaries. The particulate rather amorphous, 0.2–2 μm‐sized Periodic acid‐Schiff (PAS) positive granular deposits accumulate in arteries (Figure 9) but may also be found in capillary basement membranes, frequently associated with pericytes 34. Analogous to PEFA‐CAA, the loss of arterial smooth muscle cells in CADASIL reduces vascular tone and amplitude of pulsations for the propagation of perivascular lymphatic drainage and results in further accumulation of other insoluble proteins along with GOM. These deposits predominantly contain NOTCH3 fragments but may accrue specific extracellular matrix proteins produced by vascular smooth muscle cells and capillaries 123. The significance of the localization of GOM in the vasculature in terms of the pathogenesis of CADASIL is still unclear. It is likely that Extracellular Domain of the NOTCH3 Receptor (N3ECD)‐containing GOM is secreted by the vascular smooth muscle and endothelial cells, possibly to remove potentially cytotoxic, aggregation‐prone mutant N3ECD, but its deposition and failure of subsequent removal from the vessel walls incorporates CADASIL within the spectrum of PEFA.
Figure 9.
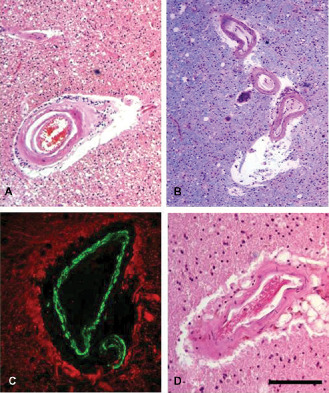
White matter changes and dilated PVS in non‐amyloid microangiopathies (eg, CADASIL) and CAA. Lobar or intracerebral hemorrhages occur in CAA (unlike in CADASIL) whereas white matter disease and microbleeds (cortical and subcortical) are common to both disorders. Examination of the white matter reveals vascular degenerative processes progressing from loss of smooth muscle cells, wall thickening, fibrinoid necrosis to hyalinosis. Dilated PVS occur because of arterial vascular degeneration and impaired perivascular drainage of fluid and solutes in both CADASIL (A and C) and CAA (B and D). Dilated PVS in the white matter is consistently observed to a greater extent in CADASIL compared with CAA. Sections were stained with Hematoxylin and Eosin (H&E) (A, B, D) and double immunofluorescence (C). In C, the wall of an arteriole is labeled with smooth muscle α‐actin (green) with glial fibrillary acidic protein (GFAP)‐labeled perivascular astrocytic processes (red). Scale bars = 70 μm in A and B; 150 μm in C and D.
In CADASIL, vascular smooth muscle cells carrying copies of the mutated NOTCH3 receptor appear affected by age‐related physiological changes and there is lack of normal replacement of cells such that continued cell‐to‐cell signaling is impaired causing ultimate breakdown in the integrity of the vascular wall. These changes impact on function of vascular smooth muscle cells leading to abnormal arterial tone or contractibility 31, 61. The lack of muscular tone in turn would reduce the motive force for perivascular drainage of solutes in the walls of long‐perforating arteries downstream to the white matter. Although such aberrations occur in key targeted arterial segments defined by the cerebrovascular architecture 77 they presumably spread along the length of the vessel and extend to other domains within the microvascular network 81. Focal vascular insufficiency in end arteries likely makes the poorly perfused regions such as the white matter susceptible to infarcts.
The small deposits of GOM may also adhere to microvascular walls, even inertly, but their accumulation in perivascular drainage routes suggests that perivascular drainage of fluid and solutes is impaired in CADASIL. One of the consequences of this could be the characteristic enlargement of PVS 20. The dilated PVS result from an even greater burden and insufficiency in the drainage of ISF and degraded protein products into the lymphatic system 116. However, such enlargement could also be attributed to the loss of white matter in tandem with the increased tortuosity and kinking of normally perforating vessels with focal loss of cellular elements within artery walls.
White matter changes are particularly prominent in CADASIL. This is because GOM is widespread even along white matter end arteries and capillaries whereas in Aβ CAA, Aβ deposition is largely restricted to cortical regions and the white matter changes may result from effects of deposits within the cortex that affect perfusion of the deep white matter as a consequence.
White matter damage and PEFA
Diffuse and focal white matter changes are a surrogate of small vessel diseases of the brain and common to both CADASIL and CAA (Figures 1 and 10). They manifest as cerebral WMH on T2‐weighted MRI or leukoaraiosis as a decreased signal on CT. There is some controversy as to whether deep or periventricular lesions are of more importance; this may depend on the definition of boundaries between the periventricular and deep white matter 67. The U‐fibers within the white matter are frequently spared but the pathological features comprise several patterns of alterations including pallor or swelling of myelin, loss of oligodendrocytes, axons and myelin fibers, foci of cavitation with or without macrophages and areas of reactive astrogliosis 100. However, white matter changes primarily reflected by increased fluid mobility within the tissue may incorporate a range of pathological entities including white matter rarefaction, incomplete infarction, lacunar infarcts, microinfarcts, dilated PVS and loss of myelin as well as axonal (or wallerian) degeneration. Several imaging studies have demonstrated independent associations between cerebral white matter changes and disability, comorbidity and cognitive dysfunction in older age 29. Previous neuroimaging studies indicated that dilated PVS increase with age and their size in the white matter of the frontal lobe is related to cognitive impairment 27, 84. Diffusion tensor imaging studies 82 suggest that tissue microstructural changes in white matter tracts and subcortical regions, for example putamen and thalamus, are related to worsening clinical outcomes in sporadic small vessel diseases and CADASIL. Extensive WMH in CADASIL also appear to be associated with increased brain volume 124. The progression of the microangiopathy with increasing age may also promote the dilation of PVS throughout the whole brain but with variable extent according to cerebral location. The severity of dilated PVS in the temporal lobes and subinsular areas was, however, found to be specifically related to the extent of WMH 125. The signal changes relate not only to loss of white matter tissue but also to increased fluidity and global increase in water content of the cerebral tissue.
Figure 10.
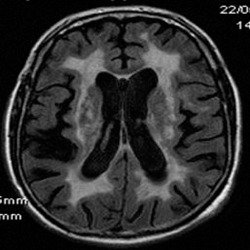
WMH in CADASIL. FLAIR MRI scan shows the extent of confluent white matter changes in a 56‐year‐old CADASIL subject carrying the p.Arg182Cys mutation.
While some studies have yielded contradictory results on the exact association between WMH and CAA 96, it is clear that the frequency of WMH is increased in CAA compared with normally aging subjects without evidence of CAA 3. However, in both CAA and CADASIL, age‐related arteriosclerotic changes coupled with the characteristic deposition of insoluble proteins along long‐perforating arteries may affect perfusion of the deeper structures, particularly the white matter 56. The lack of perfusion as attested in moderate to severe CAA leads to a chronic hypoxic state in the deep white matter with induction of essential control factors such as the hypoxia‐inducible factor 1α 38.
White matter infarcts in CADASIL
Recent imaging studies in CADASIL examined the spatial relationship between the development of incident lacunes or small infarcts and WMH in relation to the vascular anatomy to reveal the mechanistic links between the two lesion types. Remarkably, the majority (>90%) of lacunes develop at the edge of WMH rather within or outside and they also develop proximal to a WMH along the course of perforating vessels supplying the respective brain region 32. These findings suggest that the mechanisms of lacunes and WMH are intimately connected but they identify the edge of WMH as a predilection site for lacunes. As discussed earlier, it is possible that white matter changes in CAA may arise because of several mechanisms; it is plausible that microinfarcts and border zone infarcts associated with CAA 80 are progenitors of changes in the deep white matter.
Impaired blood flow and cellular mechanisms in CADASIL
In CADASIL, the widespread white matter axonal changes, particularly in the frontal lobe, may arise from differential stenosis and sclerosis of arterioles 25, possibly affecting certain axon bundles connecting to targets in the subcortical structures, specifically degeneration of thalamocortical pathways causing cortical atrophy 62. Lesions within the white matter also include spongiosis, that is vacuolation 122. Consistent with this finding, increased volumes of PVS were related to white matter myelin protein changes 122, indicating that reduction in white matter volume is possibly one factor that causes dilated PVS.
The cellular mechanisms involved in loss of myelin and abnormalities in axons in CADASIL are yet to be determined. Lack of integrity of artery walls and endothelia is likely to reduce both blood flow and volume in affected frontotemporal white matter (centrum semiovale) and subcortical grey matter structures and affect the hemodynamic reserve by decreasing the vasodilatory response. As supported by previous studies 20, this indicates that blood flow and volume are reduced in demented CADASIL subjects compared with those with no cognitive impairment. Endothelial cell abnormalities and angiopathic changes may also contribute to white matter damage and blood–brain barrier disruption can cause osmotic demyelination. Disruption of the blood–brain barrier would also result in increased permeability of the vessel wall and leakage of adverse factors, such as macrophages, lymphocytes and complement 123. Neuronal apoptosis has been described predominantly in neocortical layers III and V as a crucial mechanism for cell death in CADASIL 110. It is unclear whether this occurs secondary to the cerebral hypoperfusion, particularly in the white matter, but it is likely to contribute to cortical atrophy and to affect frontal lobe cognitive functions 20.
Another factor that could possibly impact the white matter changes in both CADASIL and CAA is the occlusion of veins and venules by collagenous thickening of the vessel walls. Venous collagenosis increases with age and it has been demonstrated that perivenous collagenosis increases in concert with leukoaraiosis 11. The presence of apoptotic cells in white matter adjacent to areas of leukoaraiosis suggests that such lesions are dynamic, with progressive cell loss and expansion 11. In turn, vascular stenosis caused by collagenosis may induce chronic ischemia or edema in the deep white matter leading to capillary loss and more widespread effects on the brain 12, 13.
Conclusions
In this review, we have examined the clinical and pathological evidence for the failure of elimination of ISF and solutes from the brain as a significant cause of pathology, particularly in the aging brain. This failure is reflected in the PEFA that extends from the common amyloid angiopathies to the less common CADASIL. The failure of ISF drainage, particularly from the aging brain and compounded by amyloid angiopathy, is a significant factor in the pathogenesis of dementia. Development of therapeutic strategies that facilitate the drainage of ISF and soluble metabolites from the aging brain would be a fruitful path to follow for the prevention and treatment of dementia.
Acknowledgments
CAH and ROC thank Alzheimer's Research UK for supporting their research and Dr Jessica Telling for her contribution to obtaining data for Figure 8. RNK's work is supported by grants from the UK Medical Research Council (MRC), G0500247, Newcastle Centre for Brain Ageing and Vitality (BBSRC, EPSRC, ESRC and MRC, LLHW) and Alzheimer's Research UK. Tissue for this study was collected by the Newcastle Brain Tissue Resource, which is funded in part by a grant from the UK MRC (G0400074), by the Newcastle NIHR Biomedical Research Centre in Ageing and Age Related Diseases award to the Newcastle upon Tyne Hospitals NHS Foundation Trust and by a grant from the Alzheimer's Society and ART as part of the Brains for Dementia Research Project. We thank Prof Kalaria and the University of Newcastle, UK for funding open access for this paper.
References
- 1. Abbott NJ (2004) Evidence for bulk flow of brain interstitial fluid: significance for physiology and pathology. Neurochem Int 45:545–552. [DOI] [PubMed] [Google Scholar]
- 2. Arbel‐Ornath M, Hudry E, Eikermann‐Haerter K, Hou S, Gregory JL, Zhao L et al (2013) Interstitial fluid drainage is impaired in ischemic stroke and Alzheimer's disease mouse models. Acta Neuropathol 126:353–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Arvanitakis Z, Leurgans SE, Wang Z, Wilson RS, Bennett DA, Schneider JA (2011) Cerebral amyloid angiopathy pathology and cognitive domains in older persons. Ann Neurol 69:320–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Asuni AA, Boutajangout A, Scholtzova H, Knudsen E, Li YS, Quartermain D et al (2006) Vaccination of Alzheimer's model mice with Abeta derivative in alum adjuvant reduces Abeta burden without microhemorrhages. Eur J Neurosci 24:2530–2542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Barua NU, Bienemann AS, Hesketh S, Wyatt MJ, Castrique E, Love S, Gill SS (2012) Intrastriatal convection‐enhanced delivery results in widespread perivascular distribution in a pre‐clinical model. Fluids Barriers CNS 9:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bergsneider M (2001) Evolving concepts of cerebrospinal fluid. Neurosurg Clin N Am 36:631–638. [PubMed] [Google Scholar]
- 7. Berzin TM, Zipser BD, Rafii MS, Kuo‐Leblanc V, Yancopoulos GD, Glass DJ et al (2000) Agrin and microvascular damage in Alzheimer's disease. Neurobiol Aging 21:349–355. [DOI] [PubMed] [Google Scholar]
- 8. Boche D, Nicoll JA (2008) The role of the immune system in clearance of Abeta from the brain. Brain Pathol 18:267–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Briley DP, Wasay M, Sergent S, Thomas S (1997) Cerebral white matter changes (leukoaraiosis), stroke, and gait disturbance. J Am Ger Soc 45:1434–1438. [DOI] [PubMed] [Google Scholar]
- 10. Brody DL, Holtzman DM (2008) Active and passive immunotherapy for neurodegenerative disorders. Ann Rev Neurosci 31:175–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Brown WR, Moody DM, Challa VR, Thore CR, Anstrom JA (2002) Apoptosis in leukoaraiosis lesions. J Neurol Sci 203–204:169–171. [DOI] [PubMed] [Google Scholar]
- 12. Brown WR, Moody DM, Thore CR, Challa VR, Anstrom JA (2007) Vascular dementia in leukoaraiosis may be a consequence of capillary loss not only in the lesions, but in normal‐appearing white matter and cortex as well. J Neurol Sci 257:62–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Brown WR, Moody DM, Thore CR, Anstrom JA, Challa VR (2009) Microvascular changes in the white mater in dementia. J Neurol Sci 283:28–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Candiello J, Cole GJ, Halfter W (2010) Age‐dependent changes in the structure, composition and biophysical properties of a human basement membrane. Matrix Biol 29:402–410. [DOI] [PubMed] [Google Scholar]
- 15. Carare RO, Bernardes‐Silva M, Newman TA, Page AM, Nicoll JAR, Perry VH, Weller RO (2008) Solutes, but not cells, drain from the brain parenchyma along basement membranes of capillaries and arteries. Significance for cerebral amyloid angiopathy and neuroimmunology. Neuropathol Appl Neurobiol 34:131–144. [DOI] [PubMed] [Google Scholar]
- 16. Carare RO, Hawkes CA, Jeffrey M, Kalaria RN, Weller RO (2013) Review: cerebral amyloid angiopathy, prion angiopathy, CADASIL and the spectrum of protein elimination failure angiopathies (PEFA) in neurodegenerative disease with a focus on therapy. Neuropathol Appl Neurobiol 39:593–611. [DOI] [PubMed] [Google Scholar]
- 17. Carare RO, Teeling JL, Hawkes CA, Puntener U, Weller RO, Nicoll JA, Perry VH (2013) Immune complex formation impairs the elimination of solutes from the brain: implications for immunotherapy in Alzheimer's disease. Acta Neuropathol Commun 1:48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Carare RO, Hawkes CA, Weller RO (2014) Afferent and efferent immunological pathways of the brain. Anatomy, function and failure. Brain Behav Immun 36:9–14. [DOI] [PubMed] [Google Scholar]
- 19. Carlson C, Estergard W, Oh J, Suhy J, Jack CR Jr, Siemers E, Barakos J (2011) Prevalence of asymptomatic vasogenic edema in pretreatment Alzheimer's disease study cohorts from phase 3 trials of semagacestat and solanezumab. Alzheimers Dement 7:396–401. [DOI] [PubMed] [Google Scholar]
- 20. Chabriat H, Joutel A, Dichgans M, Tournier‐Lasserve E, Bousser MG (2009) CADASIL. Lancet Neurol 8:643–653. [DOI] [PubMed] [Google Scholar]
- 21. Charidimou A, Meegahage R, Fox Z, Peeters A, Vandermeeren Y, Laloux P et al (2013) Enlarged perivascular spaces as a marker of underlying arteriopathy in intracerebral haemorrhage: a multicentre MRI cohort study. J Neurol Neurosurg Psychiat 84:624–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Charidimou A, Jäger RH, Peeters A, Vandermeeren Y, Laloux P, Baron JC, Weering DJ (2014) White matter perivascular spaces are related to cortical superficial siderosis in cerebral amyloid angiopathy. Stroke 45:2930–2935. [DOI] [PubMed] [Google Scholar]
- 23. Christov A, Ottman J, Hamdheydari L, Grammas P (2008) Structural changes in Alzheimer's disease brain microvessels. Curr Alzheimer Res 5:392–395. [DOI] [PubMed] [Google Scholar]
- 24. Clifford PM, Siu G, Kosciuk M, Levin EC, Venkataraman V, D'Andrea MR, Nagele RG (2008) Alpha7 nicotinic acetylcholine receptor expression by vascular smooth muscle cells facilitates the deposition of Abeta peptides and promotes cerebrovascular amyloid angiopathy. Brain Res 1234:158–171. [DOI] [PubMed] [Google Scholar]
- 25. Craggs LJ, Hagel C, Kuhlenbaeumer G, Borjesson‐Hanson A, Andersen O, Viitanen M et al (2013) Quantitative vascular pathology and phenotyping familial and sporadic cerebral small vessel diseases. Brain Pathol 23:547–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cribbs DH, Ghochikyan A, Vasilevko V, Tran M, Petrushina I, Sadzikava N et al (2003) Adjuvant‐dependent modulation of Th1 and Th2 responses to immunization with beta‐amyloid. Int Immunol 15:505–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Cumurciuc R, Guichard JP, Reizine D, Gray F, Bousser MG, Chabriat H (2006) Dilation of Virchow‐Robin spaces in CADASIL. Eur J Neurol 13:187–190. [DOI] [PubMed] [Google Scholar]
- 28. Deane R, Bell RD, Sagare A, Zlokovic BV (2009) Clearance of amyloid‐beta peptide across the blood‐brain barrier: implication for therapies in Alzheimer's disease. CNS Neurol Disord Drug Targets 8:16–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Debette S, Markus HS (2010) The clinical importance of white matter hyperintensities on brain magnetic resonance imaging: systematic review and meta‐analysis. BMJ 341:c3666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Doubal FN, MacLullich AM, Ferguson KJ, Dennis MS, Wardlaw JM (2010) Enlarged perivascular spaces on MRI are a feature of cerebral small vessel disease. Stroke 41:450–454. [DOI] [PubMed] [Google Scholar]
- 31. Dubroca C, Lacombe P, Domenga V, Maciazek J, Levy B, Tournier‐Lasserve E et al (2005) Impaired vascular mechanotransduction in a transgenic mouse model of CADASIL arteriopathy. Stroke 36:113–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Duering M, Csanadi E, Gesierich B, Jouvent E, Herve D, Seiler S et al (2013) Incident lacunes preferentially localize to the edge of white matter hyperintensities: insights into the pathophysiology of cerebral small vessel disease. Brain 136:2717–2726. [DOI] [PubMed] [Google Scholar]
- 33. Duvernoy HM, Delon S, Vannson JL (1981) Cortical blood vessels of the human brain. Brain Res Bull 7:519–579. [DOI] [PubMed] [Google Scholar]
- 34. Dziewulska D, Lewandowska E (2012) Pericytes as a new target for pathological processes in CADASIL. Neuropathology 32:515–521. [DOI] [PubMed] [Google Scholar]
- 35. Fan R, Defilippis K, Van Nostrand WE (2007) Induction of complement proteins in a mouse model of cerebral microvascular Abeta deposition. J Neuroinflammation 4:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Fazekas F, Kleinert R, Offenbacher H, Schmidt R, Kleinert G, Payer F et al (1993) Pathologic correlates of incidental MRI white matter signal hyperintensities. Neurology 43:1683–1689. [DOI] [PubMed] [Google Scholar]
- 37. Fernandez‐Vizarra P, Lopez‐Franco O, Mallavia B, Higuera‐Matas A, Lopez‐Parra V, Ortiz‐Muñoz G et al (2012) Immunoglobulin G Fc receptor deficiency prevents Alzheimer‐like pathology and cognitive impairment in mice. Brain 135:2826–2837. [DOI] [PubMed] [Google Scholar]
- 38. Fernando MS, Simpson JE, Matthews F, Brayne C, Lewis CE, Barber R et al (2006) White matter lesions in an unselected cohort of the elderly: molecular pathology suggests origin from chronic hypoperfusion injury. Stroke 37:1391–1398. [DOI] [PubMed] [Google Scholar]
- 39. Ferrer I, Boada Rovira M, Sánchez Guerra ML, Rey MJ, Costa‐Jussá F (2004) Neuropathology and pathogenesis of encephalitis following amyloid‐beta immunization in Alzheimer's disease. Brain Pathol 14:11–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Frackowiak J, Zoltowska A, Wisniewski HM (1994) Non‐fibrillar beta‐amyloid protein is associated with smooth muscle cells of vessel walls in Alzheimer disease. J Neuropathol Exp Neurol 53:637–645. [DOI] [PubMed] [Google Scholar]
- 41. Gilman S, Koller M, Black RS, Jenkins L, Griffith SG, Fox NC et al (2005) Clinical effects of Abeta immunization (AN1792) in patients with AD in an interrupted trial. Neurology 64:1553–1562. [DOI] [PubMed] [Google Scholar]
- 42. Goldman EE (1913) Vitalfarbung am Zentralnervensystem. Abh Preuss Akad Wiss Phys‐Math K1 1:1–60. [Google Scholar]
- 43. Gorter JW (1999) Major bleeding during anticoagulation after cerebral ischemia: patterns and risk factors. Stroke Prevention In Reversible Ischemia Trial (SPIRIT). European Atrial Fibrillation Trial (EAFT) study groups. Neurology 53:1319–1327. [DOI] [PubMed] [Google Scholar]
- 44. Gouw AA, Seewann A, van der Flier WM, Barkhof F, Rozemuller AM, Scheltens P, Geurts JJ (2011) Heterogeneity of small vessel disease: a systematic review of MRI and histopathology correlations. J Neurol Neurosurg Psychiatry 82:126–135. [DOI] [PubMed] [Google Scholar]
- 45. Gregoire SM, Charidimou A, Gadapa N et al (2011) Acute ischaemic brain lesions in intracerebral haemorrhage: multicentre cross‐sectional magnetic resonance imaging study. Brain 134:2376–2386. [DOI] [PubMed] [Google Scholar]
- 46. Gurol ME, Irizarry MC, Smith EE et al (2006) Plasma beta‐amyloid and white matter lesions in AD, MCI, and cerebral amyloid angiopathy. Neurology 66:23–29. [DOI] [PubMed] [Google Scholar]
- 47. Gurol ME, Viswanathan A, Gidicsin C et al (2013) Cerebral amyloid angiopathy burden associated with leukoaraiosis: a positron emission tomography/magnetic resonance imaging study. Ann Neurol 73:529–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hachinski VC, Potter P, Merskey H (1986) Leuko‐araiosis: an ancient term for a new problem. Can J Neurol Sci 13:533–534. [DOI] [PubMed] [Google Scholar]
- 49. Hawkes CA, Hartig W, Kacza J, Schliebs R, Weller RO, Nicoll JA, Carare RO (2011) Perivascular drainage of solutes is impaired in the ageing mouse brain and in the presence of cerebral amyloid angiopathy. Acta Neuropathol 121:431–443. [DOI] [PubMed] [Google Scholar]
- 50. Hawkes CA, Sullivan PM, Hands S, Weller RO, Nicoll JA, Carare RO (2012) Disruption of arterial perivascular drainage of amyloid‐beta from the brains of mice expressing the human APOE epsilon4 allele. PLoS ONE 7:e41636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Hawkes CA, Gatherer M, Sharp MM, Dorr A, Yuen HM, Kalaria R et al (2013) Regional differences in the morphological and functional effects of aging on cerebral basement membranes and perivascular drainage of amyloid‐beta from the mouse brain. Aging Cell 12:224–236. [DOI] [PubMed] [Google Scholar]
- 52. Head E, Barrett EG, Murphy MP et al (2006) Immunization with fibrillar Abeta(1–42) in young and aged canines: antibody generation and characteristics, and effects on CSF and brain Abeta. Vaccine 24:2824–2834. [DOI] [PubMed] [Google Scholar]
- 53. Herzig MC, Van Nostrand WE, Jucker M (2006) Mechanism of cerebral beta‐amyloid angiopathy: murine and cellular models. Brain Pathol 16:40–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Hurford R, Charidimou A, Fox Z, Cipolotti L, Jager R, Werring DJ (2014) MRI‐visible perivascular spaces: relationship to cognition and small vessel disease MRI markers in ischaemic stroke and TIA. J Neurol Neurosurg Psychiatry 85:522–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Hutchings M, Weller RO (1986) Anatomical relationships of the pia mater to cerebral blood vessels in man. J Neurosurg 65:316–325. [DOI] [PubMed] [Google Scholar]
- 56. Ihara M, Polvikoski TM, Hall R, Slade JY, Perry RH, Oakley AE et al (2010) Quantification of myelin loss in frontal lobe white matter in vascular dementia, Alzheimer's disease, and dementia with Lewy bodies. Acta Neuropathol 119:579–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Iliff JJ, Wang M, Liao Y, Plogg BA, Peng W, Gundersen GA et al (2012) A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid beta. Sci Transl Med 4:147ra11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Iliff JJ, Wang M, Zeppenfeld DM, Venkataraman A, Plog BA, Liao Y et al (2013) Cerebral arterial pulsation drives paravascular CSF‐interstitial fluid exchange in the murine brain. J Neurosci 33:18190–18199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Johanson CE, Duncan JA III, Klinge PM, Brinker T, Stopa EG, Silverberg GD (2008) Multiplicity of cerebrospinal fluid functions: new challenges in health and disease. Cerebrospinal Fluid Res 5:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Johnston M, Zakharov A, Papaiconomou C, Salmasi G, Armstrong D (2004) Evidence of connections between cerebrospinal fluid and nasal lymphatic vessels in humans, non‐human primates and other mammalian species. Cerebrospinal Fluid Res 1:2–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Joutel A, Monet‐Lepretre M, Gosele C, Baron‐Menguy C, Hammes A, Schmidt S et al (2010) Cerebrovascular dysfunction and microcirculation rarefaction precede white matter lesions in a mouse genetic model of cerebral ischemic small vessel disease. J Clin Invest 120:433–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Jouvent E, Mangin JF, Porcher R, Viswanathan A, O'Sullivan M, Guichard JP et al (2008) Cortical changes in cerebral small vessel diseases: a 3D MRI study of cortical morphology in CADASIL. Brain 131:2201–2208. [DOI] [PubMed] [Google Scholar]
- 63. Kalaria RN (2002) Small vessel disease and Alzheimer's dementia: pathological considerations. Cerebrovasc Dis 13(Suppl. 2):48–52. [DOI] [PubMed] [Google Scholar]
- 64. Kaufer D, Gandy S (2009) APOE ε4 and bapineuzumab: infusing pharmacogenomics into Alzheimer disease therapeutics. Neurology 73:2052–2053. [DOI] [PubMed] [Google Scholar]
- 65. Kerchner GA, Boxer AL (2010) Bapineuzumab. Expert Opin Biol Ther 10:1121–1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Kida S, Pantazis A, Weller RO (1993) CSF drains directly from the subarachnoid space into nasal lymphatics in the rat. Anatomy, histology and immunological significance. Neuropathol Appl Neurobiol 19:480–488. [DOI] [PubMed] [Google Scholar]
- 67. Kovari E, Gold G, Herrmann FR, Canuto A, Hof PR, Bouras C, Giannakopoulos P (2007) Cortical microinfarcts and demyelination affect cognition in cases at high risk for dementia. Neurology 68:927–931. [DOI] [PubMed] [Google Scholar]
- 68. Laman JD, Weller RO (2013) Drainage of cells and soluble antigen from the CNS to regional lymph nodes. J Neuroimmune Pharmacol 8:840–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Launer LJ (2004) Epidemiology of white matter lesions. Top Magn Reson Imaging 15:365–367. [DOI] [PubMed] [Google Scholar]
- 70. Lister KJ, James WG, Hickey MJ (2007) Immune complexes mediate rapid alterations in microvascular permeability: roles for neutrophils, complement, and platelets. Microcirculation 14:709–722. [DOI] [PubMed] [Google Scholar]
- 71. Lue LF, Kuo YM, Roher AE, Brachova L, Shen Y, Sue L et al (1999) Soluble amyloid beta peptide concentration as a predictor of synaptic change in Alzheimer's disease. Am J Pathol 155:853–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Martinez‐Ramirez S, Pontes‐Neto OM, Dumas AP et al (2013) Topography of dilated perivascular spaces in subjects from a memory clinic cohort. Neurology 80:1551–1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Masliah E, Hansen L, Adame A, Crews L, Bard F, Lee C et al (2005) Abeta vaccination effects on plaque pathology in the absence of encephalitis in Alzheimer disease. Neurology 64:129–131. [DOI] [PubMed] [Google Scholar]
- 74. McCarron MO, Nicoll JA (1998) Recurrent hemorrhage in cerebral amyloid angiopathy. Neurology 51:924–925. [DOI] [PubMed] [Google Scholar]
- 75. McCarron MO, Nicoll JA (2004) Cerebral amyloid angiopathy and thrombolysis‐related intracerebral haemorrhage. Lancet Neurol 3:484–492. [DOI] [PubMed] [Google Scholar]
- 76. McLean CA, Cherny RA, Fraser FW, Fuller SJ, Smith MJ, Beyreuther K et al (1999) Soluble pool of Abeta amyloid as a determinant of severity of neurodegeneration in Alzheimer's disease. Ann Neurol 46:860–866. [DOI] [PubMed] [Google Scholar]
- 77. Miao Q, Paloneva T, Tuominen S, Poyhonen M, Tuisku S, Viitanen M, Kalimo H (2004) Fibrosis and stenosis of the long penetrating cerebral arteries: the cause of the white matter pathology in cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy. Brain Pathol 14:358–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Murata T, Handa H, Mori K, Nakano Y (1981) The significance of periventricular lucency on computed tomography: experimental study with canine hydrocephalus. Neuroradiol 20:221–227. [DOI] [PubMed] [Google Scholar]
- 79. Nicoll JA, Wilkinson D, Holmes C, Steart P, Markham H, Weller RO (2003) Neuropathology of human Alzheimer disease after immunization with amyloid‐beta peptide: a case report. Nat Med 9:448–452. [DOI] [PubMed] [Google Scholar]
- 80. Okamoto Y, Yamamoto T, Kalaria RN, Senzaki H, Maki T, Hase Y et al (2012) Cerebral hypoperfusion accelerates cerebral amyloid angiopathy and promotes cortical microinfarcts. Acta Neuropathol 123:381–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Okeda R, Arima K, Kawai M (2002) Arterial changes in cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL) in relation to pathogenesis of diffuse myelin loss of cerebral white matter: examination of cerebral medullary arteries by reconstruction of serial sections of an autopsy case. Stroke 33:2565–2569. [DOI] [PubMed] [Google Scholar]
- 82. O'Sullivan M (2010) Imaging small vessel disease: lesion topography, networks, and cognitive deficits investigated with MRI. Stroke 41:S154–S158. [DOI] [PubMed] [Google Scholar]
- 83. O'Sullivan M, Lythgoe DJ, Pereira AC et al (2002) Patterns of cerebral blood flow reduction in patients with ischemic leukoaraiosis. Neurology 59:321–326. [DOI] [PubMed] [Google Scholar]
- 84. Patankar TF, Mitra D, Varma A, Snowden J, Neary D, Jackson A (2005) Dilatation of the Virchow‐Robin space is a sensitive indicator of cerebral microvascular disease: study in elderly patients with dementia. Am J Neuroradiol 26:1512–1520. [PMC free article] [PubMed] [Google Scholar]
- 85. Patton RL, Kalback WM, Esh CL, Kokjohn TA, Van Vickle GD, Luehrs DC et al (2006) Amyloid‐β peptide remnants in AN‐1792‐immunized Alzheimer's disease patients: a biochemical analysis. Am J Pathol 169:1048–1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Petrushina I, Ghochikyan A, Mktrichyan M, Mamikonyan G, Movsesyan N, Davtyan H et al (2007) Alzheimer's disease peptide epitope vaccine reduces insoluble but not soluble/oligomeric Abeta species in amyloid precursor protein transgenic mice. J Neurosci 27:12721–12731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Phillips MJ, Needham M, Weller RO (1997) Role of cervical lymph nodes in autoimmune encephalomyelitis in the Lewis rat. J Pathol 182:457–464. [DOI] [PubMed] [Google Scholar]
- 88. Pollock H, Hutchings M, Weller RO, Zhang ET (1997) Perivascular spaces in the basal ganglia of the human brain: their relationship to lacunes. J Anat 191:337–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Rennels ML, Gregory TF, Blaumanis OR, Fujimoto K, Grady PA (1985) Evidence for a “paravascular” fluid circulation in the mammalian central nervous system, provided by the rapid distribution of tracer protein throughout the brain from the subarachnoid space. Brain Res 326:47–63. [DOI] [PubMed] [Google Scholar]
- 90. Revesz T, Ghiso J, Lashley T, Plant G, Rostagno A, Frangione B, Holton JL (2003) Cerebral amyloid angiopathies: a pathologic, biochemical, and genetic view. J Neuropathol Exp Neurol 62:885–898. [DOI] [PubMed] [Google Scholar]
- 91. Revesz T, Holton JL, Lashley T, Plant G, Frangione B, Rostagno A, Ghiso J (2009) Genetics and molecular pathogenesis of sporadic and hereditary cerebral amyloid angiopathies. Acta Neuropathol 118:115–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Roher AE, Kuo Y‐M, Esh C, Knebel C, Weiss N, Kalback W et al (2003) Cortical and leptomeningeal cerebrovascular amyloid and white matter pathology in Alzheimer's disease. Mol Med 9:112–122. [PMC free article] [PubMed] [Google Scholar]
- 93. Roman GC (1991) White matter lesions and normal‐pressure hydrocephalus: Binswanger disease or Hakim syndrome? Am J Neuroradiol 12:40–41. [PMC free article] [PubMed] [Google Scholar]
- 94. Salloway S, Gur T, Berzin T, Zipser B, Correia S, Hovanesian V et al (2002) Effect of APOE genotype on microvascular basement membrane in Alzheimer's disease. J Neurol Sci 203–204:183–187. [DOI] [PubMed] [Google Scholar]
- 95. Salzman KL, Osborn AG, House P, Jinkins JR, Ditchfield A, Cooper JA, Weller RO (2005) Giant tumefactive perivascular spaces. Am J Neuroradiol 26:298–305. [PMC free article] [PubMed] [Google Scholar]
- 96. Samarasekera N, Smith C, Al‐Shahi Salman R (2011) The association between cerebral amyloid angiopathy and intracerebral haemorrhage: systematic review and meta‐analysis. J Neurol Neurosurg Psychiatry 83:275–281. [DOI] [PubMed] [Google Scholar]
- 97. Schenk D, Barbour R, Dunn W, Gordon G, Grajeda H, Guido T et al (1999) Immunization with amyloid‐beta attenuates Alzheimer‐disease‐like pathology in the PDAPP mouse. Nature 400:173–177. [DOI] [PubMed] [Google Scholar]
- 98. Schley D, Carare‐Nnadi R, Please CP, Perry VH, Weller RO (2006) Mechanisms to explain the reverse perivascular transport of solutes out of the brain. J Theor Biol 238:962–974. [DOI] [PubMed] [Google Scholar]
- 99. Shimizu H, Ghazizadeh M, Sato S, Oguro T, Kawanami O (2009) Interaction between beta‐amyloid protein and heparan sulfate proteoglycans from the cerebral capillary basement membrane in Alzheimer's disease. J Clin Neurosci 16:277–282. [DOI] [PubMed] [Google Scholar]
- 100. Simpson JE, Fernando MS, Clark L, Ince PG, Matthews F, Forster G et al (2007) White matter lesions in an unselected cohort of the elderly: astrocytic, microglial and oligodendrocyte precursor cell responses. Neuropathol Appl Neurobiol 33:410–419. [DOI] [PubMed] [Google Scholar]
- 101. Sjoberg AP, Trouw LA, Blom AM (2009) Complement activation and inhibition: a delicate balance. Trends Immunol 30:83–90. [DOI] [PubMed] [Google Scholar]
- 102. Sperling R, Salloway S, Brooks DJ et al (2012) Amyloid‐related imaging abnormalities in patients with Alzheimer's disease treated with bapineuzumab: a retrospective analysis. Lancet Neurol 11:241–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Szentistvanyi I, Patlak CS, Ellis RA, Cserr HF (1984) Drainage of interstitial fluid from different regions of rat brain. Am J Physiol 246:F835–F844. [DOI] [PubMed] [Google Scholar]
- 104. Teeling JL, Carare RO, Glennie MJ, Perry VH (2012) Intracerebral immune complex formation induces inflammation in the brain that depends on Fc receptor interaction. Acta Neuropathol 124:479–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Thal DR, Rüb U, Orantes M, Braak H (2002) Phases of A beta‐deposition in the human brain and its relevance for the development of AD. Neurology 58:1791–1800. [DOI] [PubMed] [Google Scholar]
- 106. Thal DR, Larionov S, Abramowski D, Wiederhold KH, Van Dooren T, Yamaguchi H et al (2007) Occurrence and co‐localization of amyloid beta‐protein and apolipoprotein E in perivascular drainage channels of wild‐type and APP‐transgenic mice. Neurobiol Aging 28:1221–1230. [DOI] [PubMed] [Google Scholar]
- 107. Uspenskaia O, Liebetrau M, Herms J, Danek A, Hamann GF (2004) Aging is associated with increased collagen type IV accumulation in the basal lamina of human cerebral microvessels. BMC Neurosci 5:37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. van Zwam M, Huizinga R, Heijmans N, van Meurs M, Wierenga‐Wolf AF, Melief M‐J et al (2009) Surgical excision of CNS‐draining lymph nodes reduces relapse severity in chronic‐relapsing EAE. J Pathol 217:543–551. [DOI] [PubMed] [Google Scholar]
- 109. Vasilevko V, Xu F, Previti ML, Van Nostrand WE, Cribbs DH (2007) Experimental investigation of antibody‐mediated clearance mechanisms of amyloid‐beta in CNS of Tg‐SwDI transgenic mice. J Neurosci 27:13376–13383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Viswanathan A, Gray F, Bousser MG, Baudrimont M, Chabriat H (2006) Cortical neuronal apoptosis in CADASIL. Stroke 37:2690–2695. [DOI] [PubMed] [Google Scholar]
- 111. Wardlaw JM, Smith EE, Biessels GJ et al (2013) Neuroimaging standards for research into small vessel disease and its contribution to ageing and neurodegeneration. Lancet Neurol 12:822–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Weller RO (1998) Pathology of cerebrospinal fluid and interstitial fluid of the CNS: significance for Alzheimer disease, prion disorders and multiple sclerosis. J Neuropath Exper Neurol 57:885–894. [DOI] [PubMed] [Google Scholar]
- 113. Weller RO (2005) Microscopic morphology and histology of the human meninges. Morphologie 89:22–34. [DOI] [PubMed] [Google Scholar]
- 114. Weller RO, Subash M, Preston SD, Mazanti I, Carare RO (2008) Perivascular drainage of amyloid‐beta peptides from the brain and its failure in cerebral amyloid angiopathy and Alzheimer's disease. Brain Pathol 18:253–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Weller RO, Boche D, Nicoll JA (2009) Microvasculature changes and cerebral amyloid angiopathy in Alzheimer's disease and their potential impact on therapy. Acta Neuropathol 118:87–102. [DOI] [PubMed] [Google Scholar]
- 116. Weller RO, Djuanda E, Yow HY, Carare RO (2009) Lymphatic drainage of the brain and the pathophysiology of neurological disease. Acta Neuropathol 117:1–14. [DOI] [PubMed] [Google Scholar]
- 117. Weller RO, Preston SD, Subash M, Carare RO (2009) Cerebral amyloid angiopathy in the aetiology and immunotherapy of Alzheimer disease. Alzheimers Res Ther 1:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Weller RO, Galea I, Carare RO, Minagar A (2010) Pathophysiology of the lymphatic drainage of the central nervous system: implications for pathogenesis and therapy of multiple sclerosis. Pathophysiology 17:295–306. [DOI] [PubMed] [Google Scholar]
- 119. Wilcock DM, Colton CA (2009) Immunotherapy, vascular pathology, and microhemorrhages in transgenic mice. CNS Neurol Disord Drug Targets 8:50–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Wisniewski HM, Wegiel J (1994) Beta‐amyloid formation by myocytes of leptomeningeal vessels. Acta Neuropathol 87:233–241. [DOI] [PubMed] [Google Scholar]
- 121. Wyss‐Coray T, Lin C, Sanan DA, Mucke L, Masliah E (2000) Chronic overproduction of transforming growth factor‐beta1 by astrocytes promotes Alzheimer's disease‐like microvascular degeneration in transgenic mice. Am J Pathol 156:139–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Yamamoto Y, Ihara M, Tham C, Low RW, Slade JY, Moss T et al (2009) Neuropathological correlates of temporal pole white matter hyperintensities in CADASIL. Stroke 40:2004–2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Yamamoto Y, Craggs L, Baumann M, Kalimo H, Kalaria RN (2011) Review: molecular genetics and pathology of hereditary small vessel diseases of the brain. Neuropathol Appl Neurobiol 37:94–113. [DOI] [PubMed] [Google Scholar]
- 124. Yao M, Herve D, Allili N, Jouvent E, Duering M, Dichgans M, Chabriat H (2012) NIHSS scores in ischemic small vessel disease: a study in CADASIL. Cerebrovasc Dis 34:419–423. [DOI] [PubMed] [Google Scholar]
- 125. Yao M, Herve D, Jouvent E, Duering M, Reyes S, Godin O et al (2014) Dilated perivascular spaces in small‐vessel disease: a study in CADASIL. Cerebrovasc Dis 37:155–163. [DOI] [PubMed] [Google Scholar]
- 126. Zarow C, Barron E, Chang Chui H, Perlmutter LS (1997) Vascular basement membrane pathology and Alzheimer's disease. Ann N Y Acad Sci 826:147–160. [DOI] [PubMed] [Google Scholar]
- 127. Zhang ET, Inman CB, Weller RO (1990) Interrelationships of the pia mater and the perivascular (Virchow‐Robin) spaces in the human cerebrum. J Anat 170:111–123. [PMC free article] [PubMed] [Google Scholar]
- 128. Zhang ET, Richards HK, Kida S, Weller RO (1992) Directional and compartmentalised drainage of interstitial fluid and cerebrospinal fluid from the rat brain. Acta Neuropathol 83:233–239. [DOI] [PubMed] [Google Scholar]