

Abstract
The polyglutamine (polyQ) diseases are a group of genetically and clinically heterogeneous neurodegenerative diseases, characterized by the expansion of polyQ sequences in unrelated disease proteins, which form different types of neuronal aggregates. The aim of this study was to characterize the aggregation pathology in the brainstem of spinocerebellar ataxia type 2 (SCA2) and 3 (SCA3) patients. For good recognition of neurodegeneration and rare aggregates, we employed 100 µm PEG embedded brainstem sections, which were immunostained with the 1C2 antibody, targeted at polyQ expansions, or with an antibody against p62, a reliable marker of protein aggregates. Brainstem areas were scored semiquantitatively for neurodegeneration, severity of granular cytoplasmic staining (GCS) and frequency of neuronal nuclear inclusions (NNI). SCA2 and SCA3 tissue exhibited the same aggregate types and similar staining patterns. Several brainstem areas showed statistically significant differences between disease groups, whereby SCA2 showed more severe GCS and SCA3 showed more numerous NNI. We observed a positive correlation between GCS severity and neurodegeneration in SCA2 and SCA3 and an inverse correlation between the frequency of NNI and neurodegeneration in SCA3. Although their respective disease proteins are unrelated, SCA2 and SCA3 showed the same aggregate types. Apparently, the polyQ sequence alone is sufficient as a driver of protein aggregation. This is then modified by protein context and intrinsic properties of neuronal populations. The severity of GCS was the best predictor of neurodegeneration in both disorders, while the inverse correlation of neurodegeneration and NNI in SCA3 tissue implies a protective role of these aggregates.
Keywords: neurodegeneration, p62, polyglutamine, protein aggregation disease, spinocerebellar ataxia
Introduction
The polyglutamine (polyQ) diseases are a group of clinically and genetically heterogeneous, progressive, neurodegenerative diseases, encompassing the autosomal dominant spinocerebellar ataxia (SCA) types 1–3, 6, 7 and 17, dentatorubropallidoluysian atrophy, Huntington's disease (HD) as well as the X‐linked, recessive spinobulbar muscular atrophy 10, 23, 26. They are characterized by the unstable expansion of polymorphic poly‐CAG sequences in otherwise unrelated disease genes beyond a disease‐specific threshold, which is then translated into an expanded polyQ sequence within the specific disease proteins 13, 23, 26, 43. The pathological alterations of the protein result in the formation of microscopically visible intraneuronal aggregates. These are mainly comprised of the disease protein, are present in specific brain areas and may result in the manifestation of corresponding clinical symptoms caused by the progressive neurodegeneration 13, 23, 43. However, the exact links between the manifestation of the neuronal protein aggregates and neurodegeneration are still unclear and subject to research.
This study focused on two of the more prevalent polyQ diseases, SCA2 and SCA3. SCA2 is one of the most frequent polyQ diseases, and is especially prevalent in the Holguín province of Cuba 22, 32. It is caused by a CAG expansion in the ATXN2 gene, encoding the ataxin‐2 protein, which is widely expressed in neuronal and non‐neuronal tissue, primarily in the perikarya 12, 18. The function of this protein has not been resolved, but a role in RNA processing was reported and ataxin‐2 is known to associate with stress granules 18, 21, 42. Post‐mortem studies in SCA2 demonstrated serious degenerative changes in brainstem, mesencephalon and thalamus, coupled with severe involvement of the cerebellum 29, 31, 36. The clinical picture of SCA2 is characterized by ataxia, dysphagia, dysarthria, oculomotor dysfunctions with a pronounced and early slowing of saccades, somatosensory deficits and muscle cramps in the early stages, coupled with bradykinesia, rigidity, executive dysfunctions and cognitive decline in the late disease stages 29, 36.
SCA3, also one of the most prevalent polyglutamine diseases worldwide, is caused by an expansion in the ATXN3 gene which encodes the ataxin‐3 protein. Ataxin‐3 primarily acts as a deubiquitinating protein in the ubiquitin‐proteasome pathway, where it can regulate the stability and activity of its protein substrates 10, 32, 44. The disease is characterized by widespread neurodegeneration, involving brainstem and thalamus, with only moderate cerebellar involvement 29, 31, 36. The spectrum of clinical symptoms includes ataxia of limb, stance and trunk, pyramidal and extrapyramidal signs, dysarthria, oculomotor dysfunctions, dysphagia, sensory and motor neuropathy, sensory deficits and amyotrophy 10, 29, 36.
There are several types of aggregates in the polyQ diseases. The well described neuronal nuclear inclusion bodies (NNI) are located within neuronal nuclei, are typically rounded, condensed and clearly demarked 1, 9, 11, 16, 20, 25, 27, 46. Additionally, granular cytoplasmic staining (GCS), representing small to medium sized, granular inclusions located in the perinuclear neuronal cytoplasm, has been reported in several polyQ expansion disorders 14, 25, 33, 35. These granular aggregates are in all likelihood the earliest microscopically visible type of aggregates during polyQ pathogenesis 35. Also frequently present are diffuse or finely granular aggregates in the neuronal nucleus and condensed aggregates in axonal processes which up to now have been described only in SCA3 and HD 9, 14, 34, 35.
Although all of these polyQ aggregates have been known for several years, their actual impact on neuronal survival is still elusive. While it was initially assumed that NNI were harmful to the neuron, it was not possible to correlate their presence in a given brain area to the severity of the neurodegeneration in that area. Furthermore recent studies imply a protective function of NNI 2, 5, 6, 28. The presence of axonal aggregates correlated quite well with the neurodegeneration in SCA3 tissue, but their relative scarcity makes it unlikely that they alone represent the harmful aggregate species 34. A progressive aggregation process was proposed in SCA3 and HD, where the aggregation process progresses from GCS to diffuse nuclear aggregates to NNI, and later leading to a possibly detrimental disruption of the cellular protein quality control 35. However, none of the different stages of the model unequivocally matched the pattern of neurodegeneration 37.
Several antibodies are useful to identify the polyQ aggregates in research as well as in routine neuropathology. The 1C2 antibody, initially targeted at the polyQ sequence of the TATA box binding protein with length of 38+ glutamine units has proven to be a very reliable tool in the identification of all types of polyQ aggregates 14, 19, 27, 40. Additionally, antibodies against the protein p62 are frequently used in analyses of polyQ expansion disorders and other neurodegenerative diseases, where they reliably stain different types of protein aggregates. In polyQ expansion disorders, p62 can be found primarily associated with NNI as well as axonal aggregates 17, 30, 34, 37. The presence of p62 at the disease protein aggregates also carries functional implication, because the protein is known to act as a shuttle protein in autophago‐lysosomal and proteasomal degradation pathways and can also contribute to active aggregation of toxic or misfolded disease proteins that cannot be degraded conventionally 3, 4, 24, 45.
To deepen the understanding of the aggregation processes underlying polyQ expansion disorders, we studied the distribution of specific polyQ aggregate types, that is, GCS and NNI, in brainstem tissue sections of SCA2 and SCA3 patients, using the 1C2 and p62 antibodies. Additionally, we aimed to elucidate if the severity of polyQ aggregation correlates to disease and/or brain region, and whether either aggregate type is correlated with neurodegeneration. GCS and NNI were present in both diseases, with SCA2 tissue exhibiting more severe GCS and SCA3 tissue showing more NNI. This pattern varied according to brain regions, but the relative severity of the aggregation pathology of a given brain area was quite similar across the patient groups. Statistical analysis showed a significant positive correlation between GCS and neurodegeneration in both diseases and an inverse correlation between NNI and neurodegeneration in SCA3 only.
Patients and Methods
In this study, we analyzed the brainstems of 5 SCA2 and 6 SCA3 patients which were clinically and genetically characterized at the University Clinics of Groningen, Enschede, Düsseldorf, Tübingen and Budapest, along with sections from 4 control brains (Table 1). The investigation was approved by the ethical board of the JW Goethe University Medical School, where the research was performed.
Table 1.
Patients. Data from SCA2 patients, SCA3 patients and control individuals involved in this study. List of patient number, age at death, gender, diagnosis and CAG repeats, if available. Control patients were without history of neurodegenerative or psychiatric diseases. n.d.: not determined.
| Patient number | Age at death | Gender | Diagnosis | CAG‐repeats |
|---|---|---|---|---|
| 1 | 26 | F | SCA2 | 52/n.d. |
| 2 | 51 | M | SCA2 | 39/20 |
| 3 | 52 | M | SCA2 | 35/33 |
| 4 | 55 | M | SCA2 | 40/22 |
| 5 | 88 | F | SCA2 | 36/22 |
| 6 | 45 | M | SCA3 | 69/n.d. |
| 7 | 52 | M | SCA3 | 69/14 |
| 8 | 62 | F | SCA3 | 75/20 |
| 9 | 63 | M | SCA3 | n.d. |
| 10 | 73 | M | SCA3 | 65/28 |
| 11 | 85 | M | SCA3 | 66/21 |
| 12 | 52 | F | Control | n.d. |
| 13 | 59 | M | Control | n.d. |
| 14 | 62 | F | Control | n.d. |
| 15 | 84 | M | Control | n.d. |
The presence of NNI and GCS was analyzed in select 100‐µm‐thick PEG embedded sections through the pons, the pontomedullary junction area and the medulla oblongata 38. For the visualization of general polyQ aggregation, we used the 1C2 antibody (1:3200, Merck‐Millipore, Darmstadt, Germany) 40. We furthermore used the sc‐25575 rabbit‐anti‐p62 antibody (1:100, Santa Cruz, Dallas, TX), which is known to detect several types of pathological protein aggregates and exhibits low affinity to granular cytoplasmic staining 3, 35, 45. After de‐waxing, antibody retrieval was performed with Tris/HCl pH9.0 (Carl Roth, Karlsruhe, Germany) at 90–95°C for 30 minutes for both the 1C2 and the p62 antibodies. For the 1C2 antibody the sections were additionally treated with 99% formic acid (Sigma–Aldrich, St Louis, MO) for 10 minutes at room temperature. Consecutively, the sections were incubated with the primary antibody at 4°C over night, followed by treatment with a biotinylated secondary antibody (1:200, Linaris, Dossenheim, Germany) for 30 minutes at room temperature and the AB‐complex (Linaris, Dossenheim, Germany) again for 30 minutes at room temperature. For visualization of a positive immunoreaction, the sections were incubated with diaminobenzidine (Merck‐Millipore, Darmstadt, Germany) and H2O2 (Carl Roth, Karlsruhe, Germany) for 15 minutes at room temperature. After dehydration, the sections were mounted.
To assess the severity of the neurodegeneration, we used 100‐µm‐thick PEG embedded sections, stained with the pigment‐Nissl histochemical staining method. After de‐waxing, Darrow‐Red (Merck‐Millipore, Darmstadt, Germany) was used as a basophilic pigment to stain Nissl material, while aldehyde‐fuchsin (Serva, Heidelberg, Germany) was employed to highlight lipofuscin 7.
The severity and extent of GCS type aggregation pathology was scored semiquantitatively as follows: 0 = no discernable aggregation in the investigated brainstem area; 1 = light granular staining of neuronal perikarya; 2 = moderate to intense granular staining of the neuronal perikarya; 3 = intense labeling of the neuronal perikarya and proximal neurites (Supporting Information Figure S1D–F,J,K). The severity and extent of NNI was scored as follows: 0 = no discernable NNI in the brain area investigated; 1 = very rare NNI; 2 = easily discernable NNI in a minority of neurons; 3 = discernable NNI in a majority of neurons (Supporting Information Figure S1A–C,G–I). The severity of the neurodegeneration was scored as follows: 0 = no discernable neuronal loss; 1 = slight reduction of neuronal density; 2 = severe reduction of neural density; 3 = complete or almost complete neuronal loss.
We created a contingency table for each brainstem region investigated including the frequency distribution of the severity of the aggregation pathology in our SCA2 and SCA3 patients. Each of these region‐specific contingency tables was subjected to Fisher‐Freeman‐Halton's exact test to assess whether there were significant differences between the SCA2 and SCA3 patient groups regarding the proportions of the four grades of the aggregation pathology. Kendall's rank correlation coefficient tau was calculated separately for our SCA2 or SCA3 patients to determine whether the severity of the neuronal aggregations pathology (ie, GCS or NNI) was correlated with the extent of brainstem neurodegeneration. To test for inter‐rater reliability, a second investigator (M. Fredrich) assessed the sections blinded to genotype, and a weighted kappa‐score was calculated (BiAS for Windows, version 11.0, Epsilon, Darmstadt, Germany).
Results
Antibody‐specific staining patterns
The 1C2 and the anti‐p62 antibodies yielded different staining patterns, similar to earlier observations in 5‐µm‐thick paraffin sections (35). The 1C2 antibody stained both GCS and NNI equally well (Figure 1A,C,E,G). Because of the strong intensity of the cytoplasmic 1C2 labeling in thick sections, the neuronal nucleus was sometimes partially obscured and NNI were difficult to identify (Figure 1A). Conversely, the anti‐p62 antibody labeled NNI clearly and intensely (Figure 1F,H), while only rarely showing GCS (Figure 1B,D). The latter was mostly the case in areas with very severe GCS type aggregation pathology that could be ascertained in the corresponding 1C2 stained sections (Figure 1A,B). Control cases displayed neither 1C2 nor p62 positive aggregates, but did display a light, diffuse cytoplasmic p62 staining. Both staining types were consistently observed in SCA2 and SCA3 patient tissue, with GCS and NNI frequently occurring in the same neurons. Overall, the 1C2 antibody is superior in staining GCS, while at least in thick sections, p62 is preferable for assessing NNI.
Figure 1.
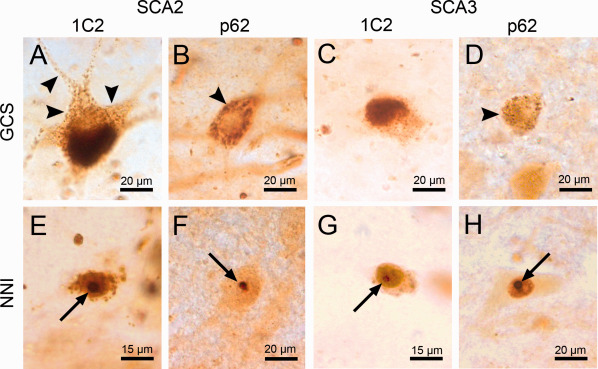
Aggregates in SCA2 and SCA 3 patient tissue (A‐D). GCS in brainstem neurons in SCA2 (A,B) and SCA3 (C,D) patient tissue, stained with the 1C2 (A,C) and p62 (B,D) antibodies. Note the extension of GCS into proximal neurites in SCA2 (A, arrowhead). (E–H) NNI in brainstem neurons of SCA2 (E,F) and SCA3 (G,H) stained with the 1C2 (E,G) and p62 antibodies (F,H) (arrows). A,C,E,G: 1C2 staining; B,D,F,H: anti‐p62 staining; 100 μm PEG embedded sections.
Mapping of aggregates in SCA2 patient tissue
Brainstem tissue sections from the SCA2 patient group exhibited overall strong GCS type aggregation pathology. The granular aggregates were primarily located in the neuronal perinuclear cytoplasm and often extended into the proximal neurites (Figure 1A). GCS was present in all brainstem areas investigated and it was very severe in the motor trigeminal, abducens, facial, ambiguus, reticulotegmental, vestibular, pontine, sensor trigeminal, great raphe, dorsal motor vagal and lateral reticular nuclei as well as the superior and inferior olives (Tables 2, 3, 4) (Figures 2E, 3A,E and 4A). NNI were also present in SCA2 tissue but were notably scarce, and were only present in significant numbers in the pontine and arcuate nuclei (Tables 2, 3, 4). Several nuclei were devoid of NNI, e.g. the lateral reticular and solitary nuclei (Tables 2, 3, 4).
Table 2.
Aggregation pathology of the upper pons. Medians of the severity of different aggregate types and neurodegeneration in brainstem nuclei of the upper pons, listed for SCA2 patients only (SCA2), SCA3 patients only (SCA3) or for the whole patient group. SCA2 and SCA3 groups were tested against each other with Fisher‐Freeman‐Haltons exact test for statistically significant differences in aggregation severity (GCS with 1C2, NNI with p62), with P‐values listed for each area and aggregation type.
| Area | SCA2 | SCA3 | All | SCA2 vs. SCA3, exact test | ||||
|---|---|---|---|---|---|---|---|---|
| 1C2 | p62 | 1C2 | p62 | 1C2 | p62 | |||
| Pontine reticular formation (oral) | GCS | 2 | 1 | 1.5 | 0 | 2 | 0.5 | GCS (1C2): P = 0.2857 |
| NNI | 0.5 | 1 | 1.5 | 1 | 1 | 1 | NNI(p62): P = 0.4667 | |
| ND | 0 | 0 | 0 | |||||
| Pontine reticular formation (caudal) | GCS | 2 | 1 | 1 | 0 | 2 | 1 | GCS (1C2): P = 0.0606 |
| NNI | 1 | 0 | 2 | 2 | 1 | 1 | NNI(p62): P = 0.0152 | |
| ND | 0 | 0 | 0 | |||||
| Locus coeruleus | GCS | 2 | 0.5 | 2 | 0 | 2 | 0 | GCS (1C2): P = 0.5455 |
| NNI | 0.5 | 1 | 0.5 | 1 | 0.5 | 1 | NNI(p62): P = 0.4000 | |
| ND | 1 | 1 | 1 | |||||
| Lateral parabrachial ncl. | GCS | 2 | 0 | 2 | 0 | 2 | 0 | GCS (1C2): P = 0.1818 |
| NNI | 1 | 0 | 1 | 1 | 1 | 1 | NNI(p62): P = 0.0411 | |
| ND | 0 | 0 | 0 | |||||
| Medial parabrachial ncl. | GCS | 2 | 1 | 1 | 0 | 2 | 1 | GCS (1C2): P = 0.1775 |
| NNI | 0 | 1 | 1.5 | 1 | 1 | 1 | NNI(p62): P = 0.2121 | |
| ND | 0 | 0 | 0 | |||||
| Reticulotegmental nucleus | GCS | 3 | 1 | 2 | 0 | 2 | 1 | GCS (1C2): P = 0.0333 |
| NNI | 1 | 1 | 1.5 | 1 | 1 | 1 | NNI(p62): P = 0.3030 | |
| ND | 2 | 2 | 2 | |||||
| Pontine nuclei | GCS | 3 | 1 | 2 | 0 | 2 | 0 | GCS (1C2): P = 0.0281 |
| NNI | 2 | 2 | 3 | 3 | 3 | 3 | NNI(p62): P = 0.0152 | |
| ND | 1 | 0.5 | 1 | |||||
| Superior vestibular nucleus | GCS | 3 | 1 | 2 | 0 | 2 | 0 | GCS (1C2): P = 0.1061 |
| NNI | 1 | 1 | 1 | 1 | 1 | 1 | NNI(p62): P = 1.0000 | |
| ND | 1 | 1 | 1 | |||||
| Motor trigeminal nucleus | GCS | 3 | 2 | 3 | 0 | 3 | 0 | GCS (1C2): P = 1.0000 |
| NNI | 1 | 0 | 1 | 1 | 1 | 1 | NNI(p62): P = 0.0476 | |
| ND | 2 | 2 | 2 | |||||
| Sensor trigeminal nucleus | GCS | 2 | 0 | 1 | 0 | 2 | 0 | GCS (1C2): P = 1.0000 |
| NNI | 1 | 0 | 1 | 1 | 1 | 1 | NNI(p62): P = 0.0151 | |
| ND | 1 | 1 | 1 | |||||
Table 3.
Aggregation pathology of the pontomedullary junction area. Medians of the severity of different aggregation types and neurodegeneration in brainstem nuclei of the pontomedullary junction area, listed for SCA2 patients only (SCA2), SCA3 patients only (SCA3) or for the whole patient group (all). SCA2 and SCA3 groups were tested against each other with Fisher‐Freeman‐Haltons exact test for statistically significant differences in aggregation severity (GCS with 1C2, NNI with p62), with P‐values listed for each area and aggregation type.
| Area | SCA2 | SCA3 | All | SCA2 vs. SCA3, exact test | ||||
|---|---|---|---|---|---|---|---|---|
| 1C2 | p62 | 1C2 | p62 | 1C2 | p62 | |||
| Superior olive | GCS | 3 | 1 | 2 | 0 | 3 | 1 | GCS (1C2): P = 0.0152 |
| NNI | 0 | 1 | 0 | 0 | 0 | 0 | NNI(p62): P = 0.0606 | |
| ND | 2 | 2 | 2 | |||||
| Abducens nucleus | GCS | 3 | 1 | 3 | 0 | 3 | 0.5 | GCS (1C2): P = 1.0000 |
| NNI | 0.5 | 1 | 1 | 1 | 1 | 1 | NNI(p62): P = 1.0000 | |
| ND | 1 | 3 | 2 | |||||
| Nucleus raphe magnus | GCS | 3 | 1 | 2 | 0 | 2 | 0 | GCS (1C2): P = 0.0152 |
| NNI | 1 | 1 | 2 | 2.5 | 2 | 2 | NNI(p62): P = 0.1126 | |
| ND | 1 | 0 | 0 | |||||
| Gigantocellular reticular nucleus | GCS | 2 | 1 | 2 | 0 | 3 | 0 | GCS (1C2): P = 0.2424 |
| NNI | 1 | 1 | 3 | 2 | 1 | 2 | NNI(p62): P = 0.0368 | |
| ND | 1 | 1 | 1 | |||||
| Facialis nucleus | GCS | 3 | 1 | 3 | 0 | 3 | 0 | GCS (1C2): P = 0.4545 |
| NNI | 1 | 0 | 1 | 1 | 1 | 1 | NNI(p62): P = 0.0152 | |
| ND | 2 | 2 | 2 | |||||
| Medial vestibular nucleus | GCS | 3 | 1 | 2 | 0 | 2 | 0 | GCS (1C2): P = 0.1061 |
| NNI | 1 | 1 | 1 | 1 | 1 | 1 | NNI(p62): P = 1.0000 | |
| ND | 1 | 2 | 1 | |||||
| Lateral vestibular nucleus | GCS | 3 | 1 | 2 | 0 | 2 | 1 | GCS (1C2): P = 0.0476 |
| NNI | 0 | 1 | 1 | 1 | 0 | 1 | NNI(p62): P = 1.0000 | |
| ND | 1.5 | 2.5 | 2 | |||||
| Inferior vestibular nucleus | GCS | 3 | 1 | 2 | 0 | 2 | 0 | GCS (1C2): P = 0.0736 |
| NNI | 0 | 1 | 1.5 | 1 | 1 | 1 | NNI(p62): P = 0.2121 | |
| ND | 1 | 1 | 1 | |||||
| Prepositus hypoglossus ncl. | GCS | 2 | 1 | 2 | 0 | 2 | 0 | GCS (1C2): P = 0.4545 |
| NNI | 1 | 1 | 2 | 2.5 | 2 | 2 | NNI(p62): P = 0.0303 | |
| ND | 0 | 1.5 | 1 | |||||
| Arcuate nucleus | GCS | 3 | 1 | 2 | 0 | 2 | 0 | GCS (1C2): P = 0.0281 |
| NNI | 1.5 | 2 | 3 | 3 | 2 | 3 | NNI(p62): P = 0.0152 | |
| ND | 0 | 0 | 0 | |||||
Table 4.
Aggregation pathology in the medulla oblongata. Medians of the severity of different aggregate types and neurodegeneration in brainstem nuclei of the medulla oblongata, listed for SCA2 patients only (SCA2), SCA3 patients only (SCA3) or for the whole patient group (all). SCA2 and SCA3 groups were tested against each other with Fisher‐Freeman‐Haltons exact test for statistically significant differences in aggregation severity (GCS with 1C2, NNI with p62), with P values listed for each area and aggregation type.
| Area | SCA2 | SCA3 | All | SCA2 vs. SCA3 exact test | ||||
|---|---|---|---|---|---|---|---|---|
| 1C2 | p62 | 1C2 | p62 | 1C2 | p62 | |||
| Nucleus hypoglossus | GCS | 3 | 2 | 3 | 0 | 3 | 1 | GCS (1C2): P = 1.0000 |
| NNI | 1 | 1 | 1 | 1 | 1 | 1 | NNI(p62): P = 0.1818 | |
| ND | 1 | 2 | 2 | |||||
| Dorsal motor vagus | GCS | 3 | 1 | 2 | 0 | 2 | 0 | GCS (1C2): P = 0.0152 |
| NNI | 1 | 1 | 2 | 2 | 1 | 2 | NNI(p62): P = 0.0260 | |
| ND | 1 | 1.5 | 1 | |||||
| Spinal trigeminal nucleus | GCS | 2 | 1 | 1 | 0 | 1 | 0 | GCS (1C2): P = 0.0022 |
| NNI | 0 | 1 | 1 | 1 | 1 | 1 | NNI(p62): P = 0.1818 | |
| ND | 1 | 1 | 1 | |||||
| Lateral reticular nucleus | GCS | 3 | 1 | 2 | 0 | 2 | 0 | GCS (1C2): P = 0.0022 |
| NNI | 0.5 | 0 | 1 | 1.5 | 1 | 1 | NNI(p62): P = 0.0584 | |
| ND | 2 | 2 | 2 | |||||
| Inferior olive | GCS | 3 | 1 | 2 | 0 | 2 | 1 | GCS (1C2): P = 0.0152 |
| NNI | 1 | 1 | 2 | 2 | 1.5 | 1 | NNI(p62): P = 0.0152 | |
| ND | 2 | 1 | 1 | |||||
| Solitary nucleus | GCS | 2 | 0 | 1.5 | 0 | 1 | 0 | GCS (1C2): P = 1.0000 |
| NNI | 0 | 0 | 1 | 1 | 1 | 1 | NNI(p62): P = 0.1061 | |
| ND | 1 | 0 | 0 | |||||
| Ambiguus nucleus | GCS | 3 | 1 | 3 | 0 | 3 | 0 | GCS (1C2): P = 1.0000 |
| NNI | 0 | 1 | 1 | 0.5 | 0.5 | 1 | NNI(p62): P = 1.0000 | |
| ND | 1 | 1 | 1 | |||||
| Gracile nucleus | GCS | 2 | 1 | 2 | 0 | 2 | 0 | GCS (1C2): P = 1.0000 |
| NNI | 0 | 1 | 1 | 1 | 1 | 1 | NNI(p62): P = 1.0000 | |
| ND | 2 | 2 | 2 | |||||
| Cuneate nucleus | GCS | 2 | 1 | 1 | 0 | 2 | 0 | GCS (1C2): P = 0.1775 |
| NNI | 0 | 1 | 1 | 1 | 1 | 1 | NNI(p62): P = 1.0000 | |
| ND | 1 | 2 | 2 | |||||
| External cuneate nucleus | GCS | 2 | 1 | 1.5 | 0 | 2 | 0 | GCS (1C2): P = 0.1818 |
| NNI | 0 | 1 | 1 | 1 | 1 | 1 | NNI(p62): P = 1.0000 | |
| ND | 2 | 2 | 2 | |||||
Figure 2.
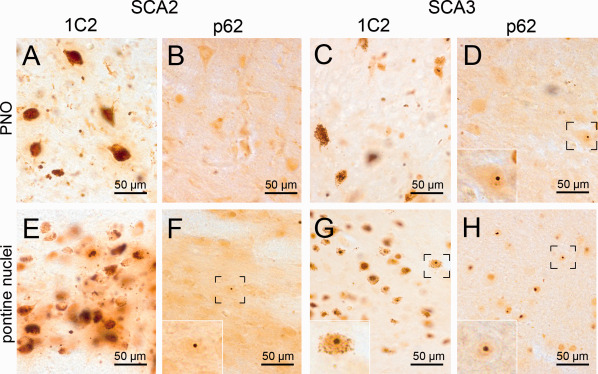
Aggregates in the pontine reticular formation and the pontine nuclei (A–D). The oral pontine reticular formation (PNO) shows moderate GCS in SCA2 (A) and SCA3 (C), whereby the staining of SCA2 tissue is slightly more intense. NNI were rare (D, insert). (E–H) The basal pontine nuclei show moderate GCS in both SCA2 (E) and SCA3 (G). NNI were very frequent in the pontine nuclei, especially in SCA3 (H). A,C,E,G: 1C2 staining; B,D,F,H: p62 staining; 100 µm PEG embedded sections.
Figure 3.
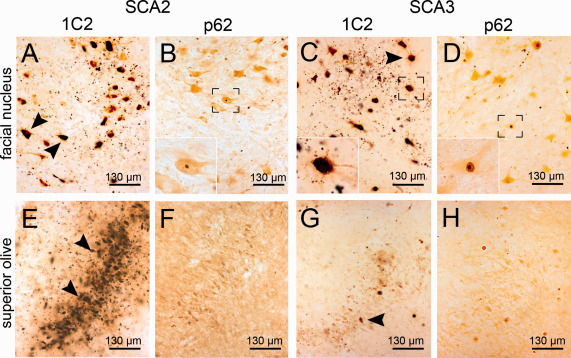
Aggregates in the superior olive and the facial nucleus (A‐D). The facial nucleus of SCA2 and SCA3 cases shows severe GCS (A,C) (arrowheads), which extends into the proximal neuronal processes in both SCA2 and SCA3. NNI were rare but present (B,D, inserts). (E–H) The superior olive shows severe GCS in SCA2 (A) and moderate staining in SCA3 (G). NNI were usually rare or completely absent (F,H). A,C,E,G: 1C2 staining; B,D,F,H: p62 staining; 100 μm PEG embedded sections.
Figure 4.
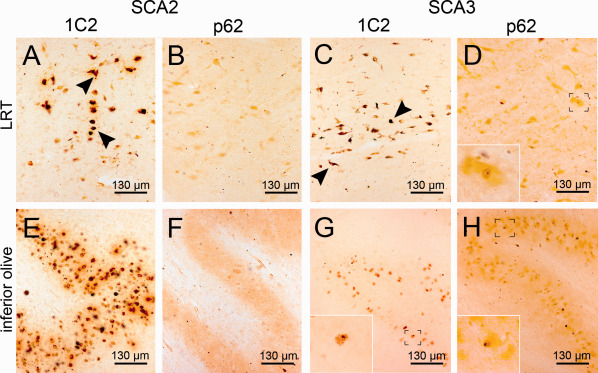
Aggregates in the lateral reticular nucleus and the inferior olive (A‐D). The lateral reticular nucleus (LRT) of the medulla oblongata displays severe GCS in a SCA2 and a SCA3 case (A,C) (arrowheads), while showing almost no NNI in the SCA2 patient (B) and only a singular NNI in SCA3 (D, insert). (E–H) The inferior olive shows intense GCS in SCA2 (E) and only moderate GCS in SCA3 case (G), while discernable NNI are almost absent in SCA2 but frequently present in SCA3 (G,H). A,C,E,G: 1C2 staining; B,D,F,H: p62 staining; 100 μm PEG embedded sections.
Within our SCA2 patient cohort, two cases were of special notice. Patient 1 had a notably large pathological CAG expansion in one of her ataxin‐2 alleles, showed severe volume loss and neurodegenerative changes in her brainstem and had markedly earlier age of onset (6 years) and death (25 years) than the other patients of our SCA2 cohort. Patient 1 showed very intense GCS and markedly more NNI in several brainstem nuclei than other SCA2 patients (Supporting Information Tables S1–S3). Patient 3 notably carried a mildly expanded allele (35Q) and another intermediate length allele (33Q) (Table 1). Additionally, because the patient died at an early disease stage caused by prostate cancer, the donated brain is one of the very few early stage SCA2 brains available for histopathological analysis. The patient brain showed macroscopically visible atrophy of the cerebellum as well as moderate neurodegenerative changes that are typical for SCA2 15. The brainstem sections showed GCS type aggregation that was comparable to other, more advanced SCA2 cases. NNI were also present and only slightly less frequent than in the other SCA2 cases (Supporting Information Tables S1–S3). Despite the comparatively short expansions, aggregates were readily recognizable by the 1C2 antibody.
Mapping of aggregates in SCA3 patient tissue
SCA3 patient tissue exhibited GCS type aggregates in all brainstem areas investigated. The granular aggregates were mainly confined to the perinuclear cytoplasm, while staining of proximal neurites was rare (Tables 2, 3, 4) (Figure 1). GCS was most abundant in the motor nuclei of the brainstem and the superior olive (Figure 3C,G). Labeling of GCS with p62 was notably scarce (Figure 1D). NNI were frequently present in all brain areas investigated, and most numerous in the caudal pontine reticular formation, the pontine nuclei, gigantocellular reticular nucleus, great raphe nucleus, arcuate nucleus and dorsal motor vagal nucleus (Tables 2, 3, 4) (Figure 2G,H).
Comparison of the distribution of different aggregate types in SCA2 and SCA3
To confirm the reproducibility of the analysis, after the initial assessment by K. Seidel, a second rater (M. Fredrich) who was blinded to the genotype, independently scored the regional aggregation pathologies. We performed an inter‐rater comparison between both data sets, which yielded a linear weighted kappa score of 0.728. The comparison of aggregation patterns in both disease groups revealed a complex picture. When assessing GCS with the 1C2 antibody, 11 out of 30 brain areas showed a significantly more intense staining in SCA2 than in SCA3 (Fisher‐Freeman‐Halton's exact test; P ≤ 0.05), while no area exhibited significantly more severe GCS type aggregation in SCA3 than in SCA2 on average (Tables 2, 3, 4). When assessing NNI with the p62 antibody, 11 out of 30 brain areas showed significantly more NNI in SCA3 than in SCA2 (Fisher‐Freeman‐Halton's exact test; P ≤ 0.05). Likewise, SCA2 brain tissue did not show significantly more NNI on average than corresponding SCA3 sections (Tables 2, 3, 4). Other brain areas did not display statistically significant differences between both disease groups. Also, several similarities in both patient groups were notable: The pontine and arcuate nuclei always displayed the highest relative amount of NNI (Tables 2, 3, 4) (Figure 2E–H). Furthermore, the brainstem motor nuclei always displayed relatively severe GCS in both patient groups, coupled with a comparatively low number of NNI (Tables 2, 3, 4) (Figure 3A–D).
Correlation between different aggregate types and neurodegeneration
The severity of the GCS type aggregation pathology, visualized with the 1C2 antibody, showed a significant positive correlation with the severity of neurodegeneration in the SCA2 (Kendall's tau test; tau = 0.425; P < 0.05) and SCA3 patient cohorts (Kendall's tau = 0.304; P < 0.05). In addition, in the SCA3 patient cohort only, neurodegeneration was inversely correlated with the frequency of p62 immunostained NNI (Kendall's tau = −0.330; P < 0.05).
Discussion
Aggregations of the disease protein are a longstanding hallmark of polyQ expansion disorders. NNI were first considered to be a toxic aggregate species, however later investigations in model organisms assumed a more protective role for these aggregates 2, 27. GCS type aggregates are notably smaller, less conspicuous and more difficult to stain and have only been reported recently 14, 35. They are also likely the first aggregate to appear in polyQ disorders, but their role in neuropathology is completely unclear 35, 37. Several antibodies that are capable of identifying these aggregates are currently used in routine diagnostics as well as in research. However, different antibodies do not necessarily produce the same staining results. We compared the 1C2 antibody, which reacts with expanded polyQ repeats and has been shown to provide good results in polyQ disease research and diagnostics 40, and an antibody against the p62 protein, which is known to stain several different types of polyQ and non‐polyQ protein aggregates 3, 4, 24, 45. We used these antibodies to map the distribution of aggregates in the brainstems of SCA2 and SCA3 patients and compared the different staining patterns of the disease groups with each other and with the neuronal loss in the affected areas.
The 1C2 antibody showed clear and consistent staining of both GCS and NNI. In contrast, the p62 antibody showed a marked preference for NNI and only rarely stained GCS. The superposition of structures in 100µm thick, PEG embedded sections not only facilitated the recognition of comparatively scarce aggregates that could not be detected in thin sections, but also enabled us to reliably grade the severity of the aggregation pathology as well as the accompanying neurodegeneration. However, particularly strong GCS often obscured the neuronal nucleus, which impeded the identification of NNI. We did not encounter this problem in p62 stained sections. Furthermore, although the 1C2 antibody was generated to target polyQ sequences of 38+ glutamine units 19, 40, it is known to react with shorter sequences 39. In our own patient cohort, a SCA2 patient with a short polyQ expansion of 35 repeats still contained GCS and NNI that were clearly labeled by the 1C2 antibody.
We were able to confirm GCS in neurons of all brainstem areas in our SCA2 and SCA3 patient cohort. Additionally, the presence of NNI could be verified in a majority of brainstem nuclei for both disorders. GCS regularly extended into the proximal neurites in SCA2 tissue, while it was mostly confined to the perikarya in SCA3 except in the most severely affected regions. The severity of the aggregation pathology varied strongly depending on brain area. In both diseases the pontine and arcuate nuclei reliably exhibited the highest relative frequency of NNI, while the brainstem motor nuclei (eg, trigeminal, abducens, facial and hypoglossal nuclei) consistently showed a relatively high amount of GCS type staining and very few NNI (Tables 2, 3, 4). Additionally, several of the brain areas investigated showed significant differences between the two disease groups. In these cases the SCA2 patient group showed more severe GCS type staining on average, while SCA3 patient tissue contained more NNI.
Despite the fact that ataxin‐2 and ataxin‐3 have no sequence homology except for the common feature of a polyQ sequence, SCA2 and SCA3 exhibit the same basic aggregate types. It can thus be assumed that the expansion of this polyQ sequence alone is sufficient to cause the observed proteinopathy 43, 47. The differences regarding the severity of the protein aggregation pathology between different brain areas, as well as the partial variations between SCA2 and SCA3 patient groups, suggest that the common mechanisms of polyQ mediated aggregation are modified by both disease protein context (eg, the primarily cytoplasmic localization of ataxin‐2) and intrinsic neuronal properties (eg, the capability of neuronal populations to handle aggregating protein) 35, 37, 43, 47.
Comparing the disease protein aggregation with neuronal degeneration yielded a significant positive correlation between the severity of GCS and neuronal loss in both SCA2 and SCA3 patients. Our data imply that on average, the severity of GCS serves as the most reliable predictor of neuronal survival. There are several lines of research indicating that distributed cytoplasmic polyQ protein can interfere with vital cell functions such as formation of the endoplasmatic reticulum and, interestingly, DNA repair 8, 41. However, because of the limitations of human post‐mortem material we are unable to ascertain whether GCS itself represents the primary toxic aggregate type or if it is merely an indicator of underlying or downstream pathological events.
Additionally, our data from SCA3 patient material showed an inverse correlation between the presence of NNI and neurodegeneration. These post‐mortem results are in accordance with theories that ascribe a primarily protective role to the NNI, and imply that neuronal populations, such as the brainstem motor nuclei, that fail to form NNI, cope less well with polyQ protein aggregation and are subject to earlier and/or more severe neurodegeneration 2, 5, 6, 27. This is further supported by the fact that NNI are immunopositive for p62, a protein that is known to be involved in active protein aggregation, and by the decreased survival rate of neurons that are incapable of forming NNI in model organisms 2, 6, 45. The formation of the NNI may represent an attempt by an affected neuron to assemble the dispersed, toxic disease protein into more condensed structures, thereby reducing the reactive surface area of the toxic protein granules. However, we failed to find a similar statistically significant correlation in SCA2 tissue, possibly caused by the low overall frequency of NNI outside of the pontine and arcuate brain nuclei. Furthermore, because sometimes individual brain areas of single patients could deviate from this, other factors that were not investigated in this study (eg, axonal aggregates, protein quality control systems, trophic factors, cellular metabolism) likely have further detrimental or protective effects on neuronal populations.
Outlook
Our investigation of the protein aggregation in SCA2 and SCA3 showed highly similar aggregate types in both patient groups, hinting at common mechanisms of protein pathology in the polyQ expansion disorders which are modified by disease protein context and brain area. Further investigations of additional polyQ expansion disorders (eg, SCA1, 6, 7) could ascertain the existence of these common mechanisms. Our investigation also hinted at a positive correlation between GCS type staining and neurodegeneration. In contrast NNI seemed to assume a protective role, at least in SCA3 tissue. The exact neuropathological and neuroprotective pathways underlying these phenomena need to be elucidated, which would not only add to our understanding of polyQ neuropathology, but might even open avenues to therapy.
Supporting information
Additional Supporting Information may be found in the online version of this article at the publisher's web‐site:
Figure S1. Exemplary staining types. (A–C) shows mild, marked and severe 1C2 positive NNI type aggregation pathology, (D–F) shows mild, marked and severe 1C2 positive GCS type aggregation pathology, (G–I) shows mild, marked or severe p62 positive NNI type aggregation pathology, (J,K) shows mild or marked p62 positive GCS type pathology. Arrows indicate neurons carrying archetypical aggregates. Severe GCS type aggregation was not visualized with the p62 antibody and is thus not represented on this figure (NA).
Table S1. Detailed scoring of the aggregation pathology of the upper pons. Assessment of the severity of polyQ aggregation pathology in different brain areas of the upper pons of SCA2 and SCA3 patients. Aggregation (GCS, NNI) and neurodegeneration (ND) severity are listed as: 0: non discernable; 1: mild; 2: moderate; 3: severe?: neuronal nuclei could not be assessed because severe NNI;/: indicates that the tissue sections was unavailable.
Table S2. Detailed scoring of the aggregation pathology of the pontomedullary junction area. Assessment of the severity of polyQ aggregation pathology in different brain areas of the pontomedullary junction area of SCA2 and SCA3 patients. Aggregation and neurodegeneration severity are listed as: 0: non discernable; 1: mild; 2: moderate; 3: severe.?: neuronal nuclei could not be assessed because severe NNI;/: indicates that the tissue sections was unavailable.
Table S3. Detailed scoring of the aggregation pathology of the medulla oblongata. Assessment of the severity of GCS and NNI aggregation pathology in different brain areas of the medulla oblongata of SCA2 and SCA3 patients. Aggregation and neurodegeneration severity are listed as: 0: non discernable; 1: mild; 2: moderate; 3: severe.?: neuronal nuclei could not be assessed because severe NNI;/: indicates that the tissue sections was unavailable.
Acknowledgments
Supported by grants from the ADCA‐Vereniging Nederland (Hoek van Holland, the Netherlands), the deutsche Heredo‐Ataxie‐Gesellschaft (Stuttgart, Germany), the Stiftung Hoffnung (Köln, Germany), and the Dr. Senckenberg Stiftung (Frankfurt/Main, Germany). The skilful assistance of D. von Meltzer (secretary) and I. Szasz (graphics) is thankfully acknowledged.
References
- 1. Ansorge O, Giunti P, Michalik A, Van BC, Harding B, Wood N et al (2004) Ataxin‐7 aggregation and ubiquitination in infantile SCA7 with 180 CAG repeats. Ann Neurol 56:448–452. [DOI] [PubMed] [Google Scholar]
- 2. Arrasate M, Mitra S, Schweitzer ES, Segal MR, Finkbeiner S (2004) Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature 431:805–810. [DOI] [PubMed] [Google Scholar]
- 3. Bjorkoy G, Lamark T, Brech A, Outzen H, Perander M, Overvatn A et al (2005) p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin‐induced cell death. J Cell Biol 171:603–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bjorkoy G, Lamark T, Johansen T (2006) p62/SQSTM1: a missing link between protein aggregates and the autophagy machinery. Autophagy 2:138–139. [DOI] [PubMed] [Google Scholar]
- 5. Bodner RA, Outeiro TF, Altmann S, Maxwell MM, Cho SH, Hyman BT et al (2006) Pharmacological promotion of inclusion formation: a therapeutic approach for Huntington's and Parkinson's diseases. Proc Natl Acad Sci U S A 103:4246–4251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bowman AB, Yoo SY, Dantuma NP, Zoghbi HY (2005) Neuronal dysfunction in a polyglutamine disease model occurs in the absence of ubiquitin‐proteasome system impairment and inversely correlates with the degree of nuclear inclusion formation. Hum Mol Genet 14:679–691. [DOI] [PubMed] [Google Scholar]
- 7. Braak H, Rub U, Del TK (2003) Involvement of precerebellar nuclei in multiple system atrophy. Neuropathol Appl Neurobiol 29:60–76. [DOI] [PubMed] [Google Scholar]
- 8. Chatterjee A, Saha S, Chakraborty A, Silva‐Fernandes A, Mandal SM, Neves‐Carvalho A et al (2015) The role of the mammalian DNA end‐processing enzyme polynucleotide kinase 3′‐phosphatase in spinocerebellar ataxia type 3 pathogenesis. PLoS Genet 11:e1004749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. DiFiglia M, Sapp E, Chase KO, Davies SW, Bates GP, Vonsattel JP et al (1997) Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science 277:1990–1993. [DOI] [PubMed] [Google Scholar]
- 10. Durr A (2010) Autosomal dominant cerebellar ataxias: polyglutamine expansions and beyond. Lancet Neurol 9:885–894. [DOI] [PubMed] [Google Scholar]
- 11. Duyckaerts C, Durr A, Cancel G, Brice A (1999) Nuclear inclusions in spinocerebellar ataxia type 1. Acta Neuropathol 97:201–207. [DOI] [PubMed] [Google Scholar]
- 12. Gispert S, Twells R, Orozco G, Brice A, Weber J, Heredero L et al (1993) Chromosomal assignment of the second locus for autosomal dominant cerebellar ataxia (SCA2) to chromosome 12q23‐24.1. Nat Genet 4:295–299. [DOI] [PubMed] [Google Scholar]
- 13. Hands SL, Wyttenbach A (2010) Neurotoxic protein oligomerisation associated with polyglutamine diseases. Acta Neuropathol 120:419–437. [DOI] [PubMed] [Google Scholar]
- 14. Hayashi M, Kobayashi K, Furuta H (2003) Immunohistochemical study of neuronal intranuclear and cytoplasmic inclusions in Machado‐Joseph disease. Psychiatry Clin Neurosci 57:205–213. [DOI] [PubMed] [Google Scholar]
- 15. Hoche F, Baliko L, den DW, Steinecker K, Bartos L, Safrany E et al (2011) Spinocerebellar ataxia type 2 (SCA2): Identification of early brain degeneration in one monozygous twin in the initial disease stage. Cerebellum 10:245–253. [DOI] [PubMed] [Google Scholar]
- 16. Koyano S, Uchihara T, Fujigasaki H, Nakamura A, Yagishita S, Iwabuchi K (1999) Neuronal intranuclear inclusions in spinocerebellar ataxia type 2: triple‐labeling immunofluorescent study. Neurosci Lett 273:117–120. [DOI] [PubMed] [Google Scholar]
- 17. Kuusisto E, Kauppinen T, Alafuzoff I (2008) Use of p62/SQSTM1 antibodies for neuropathological diagnosis. Neuropathol Appl Neurobiol 34:169–180. [DOI] [PubMed] [Google Scholar]
- 18. Lastres‐Becker I, Rub U, Auburger G (2008) Spinocerebellar ataxia 2 (SCA2). Cerebellum 7:115–124. [DOI] [PubMed] [Google Scholar]
- 19. Lescure A, Lutz Y, Eberhard D, Jacq X, Krol A, Grummt I et al (1994) The N‐terminal domain of the human TATA‐binding protein plays a role in transcription from TATA‐containing RNA polymerase II and III promoters. EMBO J 13:1166–1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Li M, Nakagomi Y, Kobayashi Y, Merry DE, Tanaka F, Doyu M et al (1998) Nonneural nuclear inclusions of androgen receptor protein in spinal and bulbar muscular atrophy. Am J Pathol 153:695–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Nonis D, Schmidt MH, van de Loo S, Eich F, Dikic I, Nowock J et al (2008) Ataxin‐2 associates with the endocytosis complex and affects EGF receptor trafficking. Cell Signal 20:1725–1739. [DOI] [PubMed] [Google Scholar]
- 22. Orozco DG, Nodarse FA, Cordoves SR, Auburger G (1990) Autosomal dominant cerebellar ataxia: clinical analysis of 263 patients from a homogeneous population in Holguin, Cuba. Neurology 40:1369–1375. [DOI] [PubMed] [Google Scholar]
- 23. Orr HT, Zoghbi HY (2007) Trinucleotide repeat disorders. Annu Rev Neurosci 30:575–621. [DOI] [PubMed] [Google Scholar]
- 24. Paine MG, Babu JR, Seibenhener ML, Wooten MW (2005) Evidence for p62 aggregate formation: role in cell survival. FEBS Lett 579:5029–5034. [DOI] [PubMed] [Google Scholar]
- 25. Pang JT, Giunti P, Chamberlain S, An SF, Vitaliani R, Scaravilli T et al (2002) Neuronal intranuclear inclusions in SCA2: a genetic, morphological and immunohistochemical study of two cases. Brain 125:656–663. [DOI] [PubMed] [Google Scholar]
- 26. Paulson HL (1999) Protein fate in neurodegenerative proteinopathies: polyglutamine diseases join the (mis)fold. Am J Hum Genet 64:339–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Paulson HL, Perez MK, Trottier Y, Trojanowski JQ, Subramony SH, Das SS et al (1997) Intranuclear inclusions of expanded polyglutamine protein in spinocerebellar ataxia type 3. Neuron 19:333–344. [DOI] [PubMed] [Google Scholar]
- 28. Rub U, de Vos RA, Brunt ER, Sebesteny T, Schols L, Auburger G et al (2006) Spinocerebellar ataxia type 3 (SCA3): thalamic neurodegeneration occurs independently from thalamic ataxin‐3 immunopositive neuronal intranuclear inclusions. Brain Pathol 16:218–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Rub U, Schols L, Paulson H, Auburger G, Kermer P, Jen JC et al (2013) Clinical features, neurogenetics and neuropathology of the polyglutamine spinocerebellar ataxias type 1, 2, 3, 6 and 7. Prog Neurobiol. 104:38–66. [DOI] [PubMed] [Google Scholar]
- 30. Rub U, Seidel K, Vonsattel J, Lange H, Eisenmenger W, Gotz M et al (2014) Huntington's disease (HD): neurodegeneration of Brodmann's primary visual area 17 (BA17). Brain Pathol. 25:701–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Scherzed W, Brunt ER, Heinsen H, de Vos RA, Seidel K, Burk K et al (2012) Pathoanatomy of cerebellar degeneration in spinocerebellar ataxia type 2 (SCA2) and type 3 (SCA3). Cerebellum 11:749–760. [DOI] [PubMed] [Google Scholar]
- 32. Schols L, Bauer P, Schmidt T, Schulte T, Riess O (2004) Autosomal dominant cerebellar ataxias: clinical features, genetics, and pathogenesis. Lancet Neurol 3:291–304. [DOI] [PubMed] [Google Scholar]
- 33. Seidel K, Brunt ER, de Vos RA, Dijk F, van der Want HJ, Rub U et al (2009) The p62 antibody reveals various cytoplasmic protein aggregates in spinocerebellar ataxia type 6. Clin Neuropathol 28:344. [DOI] [PubMed] [Google Scholar]
- 34. Seidel K, den Dunnen WF, Schultz C, Paulson H, Frank S, de Vos RA et al (2010) Axonal inclusions in spinocerebellar ataxia type 3. Acta Neuropathol 120:449–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Seidel K, Meister M, Dugbartey GJ, Zijlstra MP, Vinet J, Brunt ER et al (2012) Cellular protein quality control and the evolution of aggregates in spinocerebellar ataxia type 3 (SCA3). Neuropathol Appl Neurobiol 38:548–558. [DOI] [PubMed] [Google Scholar]
- 36. Seidel K, Siswanto S, Brunt ER, den DW, Korf HW, Rub U (2012) Brain pathology of spinocerebellar ataxias. Acta Neuropathol 124:1–21. [DOI] [PubMed] [Google Scholar]
- 37. Seidel K, Siswanto S, Fredrich M, Bouzrou M, Brunt ER, van Leeuwen FW et al (2015) Polyglutamine aggregation in Huntington's disease and spinocerebellar ataxia type 3: similar mechanisms in aggregate formation. Neuropathol Appl Neurobiol 42:153–166. [DOI] [PubMed] [Google Scholar]
- 38. Smithson KG, MacVicar BA, Hatton GI (1983) Polyethylene glycol embedding: a technique compatible with immunocytochemistry, enzyme histochemistry, histofluorescence and intracellular staining. J Neurosci Methods 7:27–41. [DOI] [PubMed] [Google Scholar]
- 39. Tanaka M, Morishima I, Akagi T, Hashikawa T, Nukina N (2001) Intra‐ and intermolecular beta‐pleated sheet formation in glutamine‐repeat inserted myoglobin as a model for polyglutamine diseases. J Biol Chem 276:45470–45475. [DOI] [PubMed] [Google Scholar]
- 40. Trottier Y, Lutz Y, Stevanin G, Imbert G, Devys D, Cancel G et al (1995) Polyglutamine expansion as a pathological epitope in Huntington's disease and four dominant cerebellar ataxias. Nature 378:403–406. [DOI] [PubMed] [Google Scholar]
- 41. Ueda M, Li S, Itoh M, Hayakawa‐Yano Y, Wang MX, Hayakawa M et al (2014) Polyglutamine expansion disturbs the endoplasmic reticulum formation, leading to caspase‐7 activation through Bax. Biochem Biophys Res Commun 443:1232–1238. [DOI] [PubMed] [Google Scholar]
- 42. van de Loo S, Eich F, Nonis D, Auburger G, Nowock J (2009) Ataxin‐2 associates with rough endoplasmic reticulum. Exp Neurol 215:110–118. [DOI] [PubMed] [Google Scholar]
- 43. Williams AJ, Paulson HL (2008) Polyglutamine neurodegeneration: protein misfolding revisited. Trends Neurosci 31:521–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Winborn BJ, Travis SM, Todi SV, Scaglione KM, Xu P, Williams AJ et al (2008) The deubiquitinating enzyme ataxin‐3, a polyglutamine disease protein, edits Lys63 linkages in mixed linkage ubiquitin chains. J Biol Chem 283:26436–26443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wooten MW, Hu X, Babu JR, Seibenhener ML, Geetha T, Paine MG et al (2006) Signaling, polyubiquitination, trafficking, and inclusions: sequestosome 1/p62's role in neurodegenerative disease. J Biomed Biotechnol 2006:62079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Yamada M (2010) Dentatorubral‐pallidoluysian atrophy (DRPLA). Neuropathology 39:453–457. [DOI] [PubMed] [Google Scholar]
- 47. Yamada M, Sato T, Tsuji S, Takahashi H (2008) CAG repeat disorder models and human neuropathology: similarities and differences. Acta Neuropathol 115:71–86. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting Information may be found in the online version of this article at the publisher's web‐site:
Figure S1. Exemplary staining types. (A–C) shows mild, marked and severe 1C2 positive NNI type aggregation pathology, (D–F) shows mild, marked and severe 1C2 positive GCS type aggregation pathology, (G–I) shows mild, marked or severe p62 positive NNI type aggregation pathology, (J,K) shows mild or marked p62 positive GCS type pathology. Arrows indicate neurons carrying archetypical aggregates. Severe GCS type aggregation was not visualized with the p62 antibody and is thus not represented on this figure (NA).
Table S1. Detailed scoring of the aggregation pathology of the upper pons. Assessment of the severity of polyQ aggregation pathology in different brain areas of the upper pons of SCA2 and SCA3 patients. Aggregation (GCS, NNI) and neurodegeneration (ND) severity are listed as: 0: non discernable; 1: mild; 2: moderate; 3: severe?: neuronal nuclei could not be assessed because severe NNI;/: indicates that the tissue sections was unavailable.
Table S2. Detailed scoring of the aggregation pathology of the pontomedullary junction area. Assessment of the severity of polyQ aggregation pathology in different brain areas of the pontomedullary junction area of SCA2 and SCA3 patients. Aggregation and neurodegeneration severity are listed as: 0: non discernable; 1: mild; 2: moderate; 3: severe.?: neuronal nuclei could not be assessed because severe NNI;/: indicates that the tissue sections was unavailable.
Table S3. Detailed scoring of the aggregation pathology of the medulla oblongata. Assessment of the severity of GCS and NNI aggregation pathology in different brain areas of the medulla oblongata of SCA2 and SCA3 patients. Aggregation and neurodegeneration severity are listed as: 0: non discernable; 1: mild; 2: moderate; 3: severe.?: neuronal nuclei could not be assessed because severe NNI;/: indicates that the tissue sections was unavailable.