

Abstract
Maturation in central nervous system embryonal tumors is an uncommon phenomenon that is mainly reported in the context of specific histological subgroups of medulloblastoma. In this report we describe two cases of histological maturation in patients with supratentorial primitive neuroectodermal tumor with strikingly different outcomes. We discuss the potential impact of such findings on treatment and outcome.
Keywords: ETANTR, ETMR, maturation, prognosis, sPNET
Introduction
Differentiation/maturation of central nervous system (CNS) embryonal tumors has occasionally been reported spontaneously or following therapy. The meaning of this pathological finding in such aggressive tumors, however, is uncertain. In this report, we describe two patients with histologic evidence of tumor maturation following chemotherapy—one diagnosed with a supratentorial primitive neuroectodermal tumor (sPNET NOS), and the other with embryonal tumor with abundant neuropil and true rosettes (ETANTR). In addition to the review of the cases reported in the literature (Table 1) 2, 3, 5, 6, 8, 9, 12, 19, we discuss the potential impact of such finding on prognosis and therapeutic approach.
Table 1.
Patient characteristics of the different cases of maturation of sPNET and medulloblastoma published in the literature. Abbreviations: M = male; F = female; PR = partial resection; STR = subtotal resection; NTR = near total resection; GTR = gross total resection; MBEN = medulloblastoma with extensive nodularity; MB = medulloblastoma; PNET = primitive neuroectodermal tumor; CT = chemotherapy; CSI = craniospinal irradiation; HDC = high‐dose chemotherapy; RT = radiation; VPA = valproic acid; DOD = dead of disease; NA = not available; ETANTR = embryonal tumor with abundant neuropil and true rosettes; ETMR = embryonal tumor with multilayered rosettes
| Sex/age at diagnosis | Tumor location | Initial surgery | First histology diagnosis | Initial treatment | Time of first relapse/progression from diagnosis | Second and repeat histology diagnosis | Treatment for first relapse | Outcome from diagnosing | |
|---|---|---|---|---|---|---|---|---|---|
| Warzok et al (1983) 19 | M/6 months | Infratentorial | PR | Cerebellar neuroblastoma (MBEN) | CT | 5 years 1/2 | Ganglioglioma | None | ALIVE |
| de Chadarévian et al (1987) 5 | F/2.5 years | Infratentorial | STR | Cerebellar neuroblastoma (MBEN) | CSI | 4 years (resection for residual) | Ganglioneuroma | None | ALIVE |
| Geyer et al (1992) 8 | F/16 months | Infratentorial | NTR | Cerebellar neuroblastoma (MBEN) | CT | 18 months | Ganglioglioma | None | ALIVE 4 years |
| Cai et al (2000) 2 | M/2 years | Infratentorial | GTR | Desmoplastic MB | CT | 8 months | Neuronal maturation | CSI |
DOD Third relapse Classic Mb |
| Cai et al (2000) | M/14 years | Infratentorial | STR | MB with extensive desmoplasia | CSI+ CT | 8 years (resection for residual) | Mature neuronal neoplasm with extensive calcification | None | ALIVE 11 years |
| Chelliah et al (2010) 3 | M/22 months | Infratentorial | GTR | MB with extensive nodularity (MBEN) | CSI+CT | 10 years† | Gangliocytoma | Unknown | Unknown |
| Driever et al (2004) 6 | M/5 years | Left parietal lobe | GTR | Undifferentiated PNET with focal neuronal differentiation | CSI+ HDC | 9 months | Astrocytic differentiation | VPA, CT | ALIVE, 31 months |
| Horbinski et al (2011) 9 | Newborn | Left frontotemporo parietal lobe | STR | High‐grade neoplasm with astrocytic and PNET like features | 0 | 13 years | Low‐grade tumor predominantly astrocytic + minor component of ganglionic cell | 0 | NA |
| Patient 1 | F/11 months | Supratentorial/ infratentorial | STR | sPNET/MB | HDC | 8 months | Residual neoplasm with advanced neuronal and astrocytic component | NA | ALIVE 30 months |
| Patient 2 | F/21 months | Left parieto occipital lobe | GTR | sPNET, secondary revised as ETANTR/ETMR | HDC | 4 months | ETANTR/ETMR with neuronal maturation, astrocytic component (ganglioglioma like) | Surgery, focal RT |
DOD Second relapse 21 months from diagnosis |
†Second progression.
Clinical Case Report
Patient 1
An 11‐month‐old girl was assessed to the emergency room for a 2‐week history of head tilt and decrease mobility of her left side. The initial head computed tomography and subsequent magnetic resonance imaging (MRI) disclosed a large midline mass involving the infra and supratentorial level associated with obstructive hydrocephalus (Figure 1A). She underwent an initial partial surgical resection of the infratentorial portion of her mass (Figure 1B). The pathologic diagnosis was in keeping with a primitive neuroectodermal tumor (see pathologic description later ). Postoperatively, she had significant truncal ataxia and persistent left‐sided weakness. The MRI of the spine was normal, but the cerebrospinal fluid (CSF) was positive for malignant cells.
Figure 1.
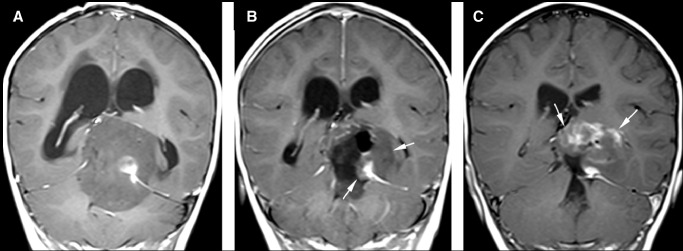
Magnetic resonance (MR) images of patient 1. Post‐Gadolinium sagittal MR images of the initial scan (A) demonstrate a large mass lesion with supra‐ and infratentorial involvement. The mass is predominantly hypointense with a small area of marked enhancement. On the post‐Gadolinium sagittal MR images obtained on postoperative day 2 (B), residual tumor (arrows) is seen. Post‐Gadolinium sagittal MR images obtained after completion of induction chemotherapy (C) show interval increase in size of the residual tumor (arrows) with more extensive and higher degree of enhancement.
The patient was enrolled in the Children's Oncology Group high‐risk infant PNET protocol consisting of three cycles of induction chemotherapy (methotrexate, vincristine, cisplatinum, cyclophosphamide) with a plan for consolidation with high‐dose chemotherapy and stem cell rescue. Following completion of induction chemotherapy, repeated MRI demonstrated increased in size of the remaining supratentorial mass (Figure 1C), therefore the patient went off study. Attempt for a gross total resection was elected in order to offer consolidation in minimal residual disease. The subtotal resection was performed, revealing the presence of differentiated ganglioglioma‐like tissue (described later), but the surgery was complicated by infarction of left posterior cerebellar artery territory.
The patient underwent three cycles of high‐dose chemotherapy (Cb, Thiotepa) followed by stem cell rescue. At completion of therapy, the residual tumor was stable in size on MRI.
Adjuvant focal radiation was discussed, but given the significant neurological impairment of the patient and the change in the histological features of the tumor, the decision was made to postpone its use until evidence of progression. Follow‐up imaging at 30 months post‐therapy showed a residual mass unchanged in size.
Pathological findings, Patient 1
Primary surgical resection
The original tumor was composed of sheets of small round blue cells characterized by hyperchromatic oval to angulated nuclei, scant cytoplasm, little morphologic differentiation, and frequent mitotic figures (Figure 2, upper panels). A few small foci of neuropil with cells showing larger nuclei, visible nucleoli, and increased cytoplasm were seen, consistent with focal neuronal differentiation; however, most of the tumor was primitive in morphology. Rare Homer–Wright rosettes were present, but there were no multilayered rosettes. The tumor cells were positive for Ini1, and were negative for epithelial membrane antigen (EMA) and Lin28. Scattered tumor cells were positive for glial fibrillary acidic protein (GFAP). With respect to differentiation, the tumor cells labeled strongly and diffusely positive for the neural stem/progenitor marker Nestin and the synaptic vesicle protein Synaptophysin. Scattered cells were positive for the neuronal marker protein gene protein 9.5 (PGP9.5), and rare cells were positive for the more mature neuronal marker NeuN. In keeping with the numerous mitoses, Ki‐67 showed a high proliferative index. Fluorescence in situ hybridization (FISH) for N‐myc, c‐myc and chr 19q13.42 showed no amplification. FISH for chromosome 17p and 17q showed no imbalances such as iso17q or 17p‐. The lesion was classified as a CNS‐PNET, NOS.
Figure 2.
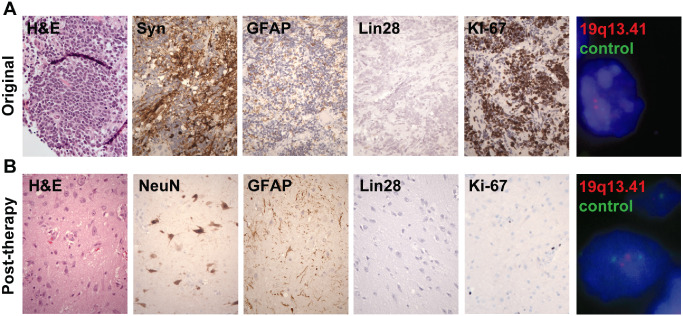
Pathology specimens of patient 1. A. Original resection specimen consisting of densely cellular primitive neuroectodermal tumor. B. Specimen post‐chemotherapy at second‐look surgery composed entirely of less cellular ganglioglioma‐like tissue including large ganglion cells and astrocytes. (400×)
Second‐look surgery
The second resection specimen consisted entirely of vaguely nodular, ganglioglioma‐like tissue containing numerous haphazardly oriented and dysplastic appearing ganglion cells embedded in abundant neuropil matrix (Figure 2, lower panels). Among the larger, more mature‐appearing neurons, there were scattered astrocytes and smaller cells with round nuclei. Additionally, some fragments showed a desmoplastic reaction around the vasculature. There were no microcystic changes, calcifications, eosinophilic granular bodies, Rosenthal fibers or perivascular lymphocytic cuffs. Mitotic figures were not identified, and no residual primitive “blastoma”‐like component was identified in any of the tissue examined. The astrocytes strongly expressed GFAP and vimentin. The neuronal forms frequently expressed PGP9.5 and NeuN. Both the large ganglion cells as well as some smaller cells with round nuclei stained with Neu‐N, supporting the predominant ganglion cell component and a minor neurocytic component. Nestin staining was weak and focal. Ki‐67 labeling was low. Compared with the primary tumor, the increased abundance of neuropil, morphologic neuronal changes, decreased nestin expression, increased Neu‐N expression and decreased Ki‐67 labeling all suggest that maturation of residual tumor had occurred.
Patient 2
A 21‐month‐old girl presented with a 3‐week history of progressive right‐sided weakness and limping, and 1 week of nausea and vomiting. MRI of the brain revealed a large left frontoparietal tumor with cystic and solid component associated with mass effect, midline shift and uncal herniation (Figure 3A). She underwent an urgent surgical resection, reported as gross total resection on postoperative MRI. The pathologic findings of this primitive neuroectodermal tumor are described later. Staging work‐up including spinal MRI and CSF cytology was negative for metastasis.
Figure 3.
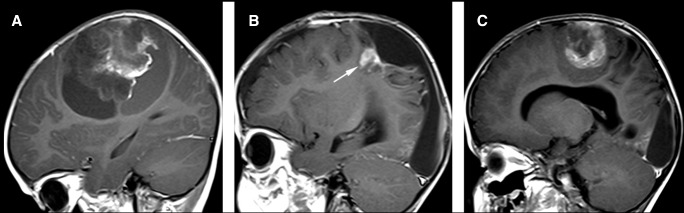
Magnetic resonance images (MRI) of patient 2. (A) On the initial MRI, l. Post‐Gadolinium sagittal T1‐weighted image shows patchy mild enhancement of the solid component. On the MRI performed prior to the second craniotomy (B), a round recurrent tumor is present markedly enhancing on post‐contrast T1 (arrows). (C) Before the third craniotomy, a large recurrent tumor is present demonstrating patchy mild enhancement on post‐contrast T1. The MR features of the recurrent tumor are similar to those of the solid portion of the primary tumor.
The patient was treated according to the infant malignant brain tumor CCG 99703 with three cycles of induction chemotherapy. Prior to consolidation, a repeat MRI confirmed the absence of residual disease. The patient then underwent high‐dose chemotherapy with stem cell rescue (three cycles of Cb, Thiotepa). The end‐of‐therapy imaging disclosed a 1‐cm diameter nodular mass suspicious for recurrence (Figure 3B). Repeat spinal MRI and CSF cytology were negative for metastasis.
Second‐look surgery was undertaken with the plan to consolidate with focal radiation. The recurrent tumor showed features of maturation described further, later. Given parental concerns regarding radiation‐induced morbidity and the maturation changes described in the recurrent tumor sample, focal radiation was postponed until further relapse. Meanwhile the child was prescribed oral tamoxifen and cis retinoic acid as maturating agents.
The patient returned 2 months later with new onset of left‐sided limping. The MRI confirmed a 5.5 × 4.0 × 2.0‐cm enhancing mass within the surgical cavity (Figure 3C). A near total resection was achieved and focal radiation was undertaken (54 Gy). While the child was asymptomatic, the follow‐up MRI at 2 months post‐radiation disclosed recurrent disease both within and outside the field of radiation and the patient went on palliative therapy. She received two cycles of oral VP16 and then dichloroacetate orally and naturopathic medication at home for 11 months before dying of disease 21 months from diagnosis.
Pathological findings, Patient 2
Primary surgical resection
The tumor was a primitive neuroectodermal tumor with high cellularity and moderate to marked anaplasia (Figure 4A). The tumor was composed nearly entirely of sheets of poorly differentiated neuroepithelial cells with hyperchromatic nuclei and minimal cytoplasm, punctuated by frequent zones of spontaneous micronecrosis. Rare true (but not multilayered) rosettes were noted among the expanses of primitive tumor. Minimal areas of neuropil formation containing scattered cells with small amounts of cytoplasm were seen. Immunohistochemistry showed that the tumor was positive for Ini1 and negative for EMA. A subset of cells was positive for GFAP. Nestin was diffuse and strongly positive, as was synaptophysin. PGP9.5 showed scattered positive cells. Exceedingly rare cells were positive for NeuN. Lin28 immunohistochemistry was strongly and diffusely positive in the tumor.
Figure 4.
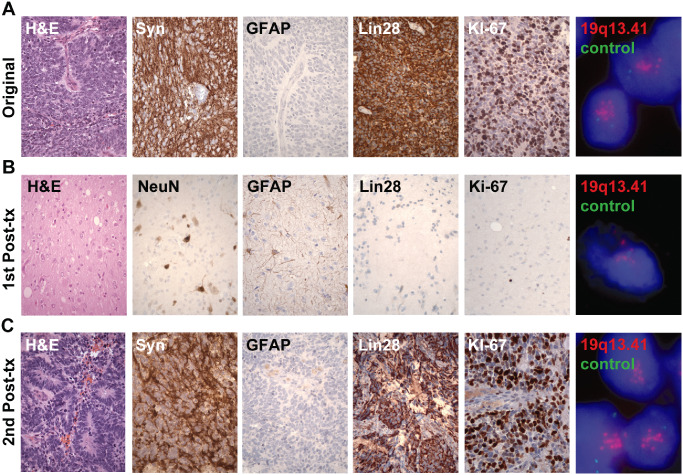
Pathology specimens of patient 2. A. Original resection specimen showing densely cellular primitive neuroectodermal tumor. B. Specimen at first recurrence post chemotherapy showing decreased cellularity and maturation of cellular elements into ganglioglioma‐like tissue. C. Specimen at second recurrence shows high cellularity and abundant multilayered rosettes. (400×)
FISH for N‐myc and c‐myc showed no amplification. Analysis of the 19q13.41 region by FISH was found to be positive for amplification with homogeneously staining regions (HSRs). Although neuropil was minimal and multilayered rosettes were not identified, in light of the 19q13.41 amplification and Lin28 positivity, the features were considered consistent with embryonal tumor with abundant neuropil and true rosettes (ETANTR), alternatively called embryonal tumor with multilayered rosettes (ETMR).
First recurrence
Compared with the sample from initial diagnosis, the sample obtained at first post‐therapy recurrence appeared much less cellular, and more mature. The tumor was now composed of cells containing larger nuclei with visible nucleoli and variable amounts of cytoplasm, within a background of abundant neuropil (Figure 4B). Nuclear atypia was moderate, but no mitotic figures were identified. No areas of densely cellular, small round blue cell tumor were identified. There were no rosettes. Although bland areas of partially calcifying geographic necrosis were present, there were no areas of spontaneous tumor micronecrosis as found in the original tumor. Overall, the recurrent tumor histologically resembled a low‐grade glioneuronal tumor. Consistent with the ganglion cell morphology, Neu‐N was positive in many of the larger cells. GFAP was also positive in a subset of cells, supporting a glial component. Nestin showed weak/focal positivity. Lin28 was largely negative except for scattered cells with weak staining that was only slightly more intense than background. The Ki‐67 proliferative index was low. FISH for chr19q13.41 showed a mix of amplified and non‐amplified cells, with the amplified cells displaying HSRs.
Second recurrence
The tumor sample from the second recurrence was histologically similar to the patient's original tumor without any of the differentiation/maturation pattern seen at the time of the first recurrence. In addition, the second recurrence tumor displayed numerous perivascular pseudorosettes as well as frequent multilayered “ependymoblastic” true rosettes (Figure 4C). Like the original tumor, only a minor component displayed somewhat larger cells and neuropil. Nestin and Lin28 expression, which were both high in the original tumor, but had decreased or were absent at first recurrence, were again highly expressed in the second recurrence. Like both the original and the first recurrence, FISH for chr19q13.42 in the second recurrence showed amplification with HSRs. The final pathology diagnosis was recurrent ETANTR.
Selected histologic, immunohistochemical, and molecular features of the tumors from both patients 1 and 2 are summarized in Table 2.
Table 2.
Histologic, immunohistochemical, and molecular comparison between patient 1 and patient 2. ND = not done.
| Patient | Tumor | Ini1 IHC | Lin28 IHC | c‐Myc FISH | N‐Myc FISH | chr 17 FISH | 19q13.41 FISH |
|---|---|---|---|---|---|---|---|
| 1 | Primary | Positive | Negative | Not amplified | Not amplified | No imbalance | Not amplified |
| 1 | First recurrence | Positive | Negative | ND | ND | ND | Not amplified |
| 2 | Primary | Positive | Strong, diffuse | Not amplified | Not amplified | No imbalance | Amplified |
| 2 | First recurrence | Positive | Weak, rare cells | ND | ND | ND | Mix of non‐amplified and amplified |
| 2 | Second recurrence | Positive | Strong, diffuse | ND | ND | ND | Amplified |
Discussion
Maturation or differentiation of CNS embryonal tumors has been reported to occur spontaneously or following treatment but these pathological changes remain rare and therefore their interpretation and possible impact on patient outcome remain unknown. Initial descriptions of maturation in embryonal CNS tumors were reported in infratentorial medulloblastoma variant, essentially in the previously called cerebellar neuroblastoma 5, 8, 19 currently designated as medulloblastoma with extensive nodularity (MBEN) 1, 14. Subsequent case of maturation post‐therapy have been reported other variants such as desmoplastic or melanocytic meduloblastoma 2, 3, 12. All but one of the cases reported in the literature had a favorable outcome without further therapy beyond the re‐resection (Table 1). It is possible that the one patient with poor outcome may have had another type of aggressive medulloblastoma or PNET such as ETANTR, since that case report was from an era that predated much of what is now known on the molecular classification of embryonal tumors. Our report of two cases expands the tumor types that may undergo treatment‐induced maturation to include supratentorial embryonal tumors, and shows that maturation may occur even in well documented cases of the highly aggressive ETANTR.
Whether the presence of a maturation/differentiation process can be viewed as a favourable indicator of outcome in supratentorial PNET remains unclear. Only 2 cases of maturation in PNET‐like or malignant glioneuronal tumors have been reported to date, one following treatment with valproic acid and one spontaneous without any treatment following surgery 6, 9 (Table 1). The rarity of the report of maturation/differentiation at the supratentorial level also limits our understanding of the natural history of this phenomenon. Whether these findings are transient changes induced by the chemotherapy or whether they can represent the ultimate stage of evolution of the tumor, as described in neuroblastoma spontaneously or after treatment with retinoic acid, cannot be predicted 4, 17. In both our patients, the presence tumor maturation/differentiation weighed on the decision not to proceed to adjuvant radiation treatment. For the first patient, no further therapy was administered and she is currently 3 years off therapy without evidence of growth of her residual mass. In our second patient, the observation of maturation was only transient and did not prevent further progression of the tumor, which was eventually characterized as an ETANTR/ETMR.
Supratentorial PNETs, although historically viewed as the medulloblastoma counterpart at the supratentorial level, are progressively being dissected into more discrete clinicopathologic entities with the advancement of molecular characterization. Some of them, like the group of PNET associated with chr19q13.41 amplification and histology of ETANTR/ETMR, have an aggressive natural history and dismal prognosis compare to others 10, 13, 16.
Thus, in sPNET, such maturation/differentiation findings must be interpreted in the context of the molecular characterization of these embryonal tumors, as our cases illustrate. We suggest that all primitive neuroectodermal tumors should be tested for chr19q13.41–42 amplification and/or Lin28 immunopositivity—regardless of the abundance/paucity of neuropil and presence/absence of multilayered true rosettes—to rule out the diagnosis of ETANTR. With respect to 19q13.41 FISH versus Lin28 IHC, however, some interesting differences were highlighted by our second case. The chr19q13.41 amplification by FISH tracked well with the patient's eventual poor outcome; the persistent population of amplified cells was detected within the histologically mature‐appearing tumor tissue in this patient who eventually succumbed to disease. In contrast, Lin28 immunopositivity varied more with the apparent histologic differentiation; Lin28 labeled the first and third specimens diffusely and strongly, while the recurrence that displayed maturation showed only weak/equivocal staining in scattered cells. Nevertheless, Lin28 IHC is still useful in that its positivity, albeit focal, may have suggested the cells' ongoing malignant potential. Our findings underscore the diagnostic utility and prognostic significance of these markers, and support the findings that both the chr19q13.41 amplification and Lin28 IHC are strong diagnostic markers of these aggressive lesions 1, 11, 13, 16 We also suggest that in cases of significant residual disease following therapy, second look surgery should be strongly considered to rule out not only persistent disease but also the possibility of residual matured tissue, before recommending further adjuvant therapy. When confronted with maturation/differentiation on repeated histology, close monitoring is certainly warranted.
In light of these findings of maturation, one may wonder about the role in PNET of maturating agents such as 13‐cis retinoic acid which has been associated with long term survival benefit in high risk neuroblastoma 15, 18. Our first case highlights the possibility of doing well with stable disease after histologic maturation. In contrast, our second case showed transient maturation, and the tumor eventually recurred in its original primitive form with fatal outcome. Nevertheless, while ETANTR have a very aggressive behavior and the prognosis is currently dismal, the possibility of inducing maturation may be a pathway for further exploration to improve their current outcomes. Indeed, although our patient did relapse after maturation, her overall survival (21 months) was longer than most patients with ETANTR, which typically have overall survival less than 1 year 7, 13. Only with a concerted effort to further document the phenomenon of spontaneous and post‐therapy maturation in PNETs, combined with molecular testing for specific variants of PNET such as ETANTR/ETMR, will we be able to better understand its biologic and prognostic significance.
None of the authors have conflict of interest to declare.
References
- 1. Adesina AM, Hunter J (2010) Medulloblastoma. In: Atlas of Pediatric Brain Tumors, Adesina AM, Tihan T, Fuller CE, Poussaint TY (eds), pp. 75–94. Springer: New York. [Google Scholar]
- 2. Cai DX, Mafra M, Schmidt RE, Scheithauer BW, Park TS, Perry A (2000) Medulloblastomas with extensive posttherapy neuronal maturation. Report of two cases. J Neurosurg 93:330–334. [DOI] [PubMed] [Google Scholar]
- 3. Chelliah D, Mensah Sarfo‐Poku C, Stea BD, Gardetto J, Zumwalt J (2010) Medulloblastoma with extensive nodularity undergoing post‐therapeutic maturation to a gangliocytoma: a case report and literature review. Pediatr Neurosurg 46:381–384. [DOI] [PubMed] [Google Scholar]
- 4. Das S, Foley N, Bryan K, Watters KM, Bray I, Murphy DM et al (2010) MicroRNA mediates DNA demethylation events triggered by retinoic acid during neuroblastoma cell differentiation. Cancer Res 70:7874–7881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. de Chadarévian JP, Montes JL, O'Gorman AM, Freeman CR (1987) Maturation of cerebellar neuroblastoma into ganglioneuroma with melanosis. A histologic, immunocytochemical, and ultrastructural study. Cancer 59:69–76. [DOI] [PubMed] [Google Scholar]
- 6. Driever PH, Wagner S, Hofstädter F, Wolff JE (2004) Valproic acid induces differentiation of a supratentorial primitive neuroectodermal tumor. Pediatr Hematol Oncol 21:743–751. [DOI] [PubMed] [Google Scholar]
- 7. Gessi M, Giangaspero F, Lauriola L, Gardiman M, Scheithauer BW, Halliday W et al (2009) Embryonal tumors with abundant neuropil and true rosettes: a distinctive CNS primitive neuroectodermal tumor. Am J Surg Pathol 33:211–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Geyer JR, Schofield D, Berger M, Milstein J (1992) Differentiation of a primitive neuroectodermal tumor into a benign ganglioglioma. J Neurooncol 14:237–241. [DOI] [PubMed] [Google Scholar]
- 9. Horbinski C, Dillon D, Pittman T (2011) Low‐grade recurrence of a congenital high‐grade supratentorial tumor with astrocytic features in the absence of adjuvant therapy. Neuropathology 31:286–291. [DOI] [PubMed] [Google Scholar]
- 10. Korshunov A, Remke M, Gessi M, Ryzhova M, Hielscher T, Witt H et al (2010) Focal genomic amplification at 19q13.42 comprises a powerful diagnostic marker for embryonal tumors with ependymoblastic rosettes. Acta Neuropathol 120:253–260. [DOI] [PubMed] [Google Scholar]
- 11. Korshunov A, Ryzhova M, Jones DT, Northcott PA, van Sluis P, Volckmann R et al (2012) LIN28A immunoreactivity is a potent diagnostic marker of embryonal tumor with multilayered rosettes (ETMR). Acta Neuropathol 124:875–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kubota KC, Itoh T, Yamada Y, Yamaguchi S, Ishida Y, Nakasu Y et al (2009) Melanocytic medulloblastoma with ganglioneurocytomatous differentiation: a case report. Neuropathology 29:72–77. [DOI] [PubMed] [Google Scholar]
- 13. Li M, Lee KF, Lu Y, Clarke I, Shih D, Eberhart C et al (2009) Frequent amplification of a chr19q13.41 microRNA polycistron in aggressive primitive neuroectodermal brain tumors. Cancer Cell 16:533–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Burger PC, Jouvet A et al (2007) The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol 114:97–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Matthay KK, Reynolds CP, Seeger RC, Shimada H, Adkins ES, Haas‐Kogan D et al (2009) Long‐term results for children with high‐risk neuroblastoma treated on a randomized trial of myeloablative therapy followed by 13‐cis‐retinoic acid: a children's oncology group study. J Clin Oncol 27:1007–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Nobusawa S, Yokoo H, Hirato J, Kakita A, Takahashi H, Sugino T et al (2012) Analysis of chromosome 19q13.42 amplification in embryonal brain tumors with ependymoblastic multilayered rosettes. Brain Pathol 22:689–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Reynolds CP (2000) Differentiating agents in pediatric malignancies: retinoids in neuroblastoma. Curr Oncol Rep 2:511–518. [DOI] [PubMed] [Google Scholar]
- 18. Spiller SE, Ditzler SH, Pullar BJ, Olson JM (2008) Response of preclinical medulloblastoma models to combination therapy with 13‐cis retinoic acid and suberoylanilide hydroxamic acid (SAHA). J Neurooncol 87:133–141. [DOI] [PubMed] [Google Scholar]
- 19. Warzok R, Jaenisch W, Lang G (1983) Morphology and biology of cerebellar neuroblastomas. J Neurooncol 1:373–379. [DOI] [PubMed] [Google Scholar]