

Abstract
There is a perfusion deficit in Alzheimer's disease (AD), commencing in the precuneus and spreading to other parts of the cerebral cortex. The deficit anticipates the development of dementia, contributes to brain damage, and is caused by both functional and structural abnormalities of the cerebral vasculature. Most of the abnormalities are probably secondary to the accumulation of Aβ but the consequent hypoperfusion may, in turn, increase Aβ production. In the early stages of disease, abnormalities that cause vasoconstriction predominate. These include cholinergic vascular denervation, inhibition of endothelial nitric oxide synthase, increased production of endothelin‐1 production and possibly also of angiotensin II. Patients with AD also have an increased prevalence of structural disease of cerebral microvessels, particularly CAA and capillary damage, and particularly in the later stages of disease these are likely to make an important contribution to the cerebral hypoperfusion. The metabolic abnormalities that cause early vascular dysfunction offer several targets for therapeutic intervention. However, for intervention to be effective it probably needs to be early. Prolonged cerebral hypoperfusion may induce compensatory circulatory changes that are themselves damaging, including hypertension and small vessel disease. This has implications for the use of antihypertensive drugs once there is accumulation of Aβ within the brain.
Keywords: Alzheimer's disease, amyloid‐β peptide, capillary damage, cerebral amyloid angiopathy, cholinergic innervation, endothelin‐1, endothelial nitric oxide synthase, hypoperfusion, ischemia, renin‐angiotensin system, vascular dysfunction
Introduction
A major contributor to the energy deficit in Alzheimer's disease (AD) is the reduction in cerebral perfusion that results from dysfunction and structural abnormalities of the cerebral vasculature. As discussed elsewhere in this mini‐symposium, there are also other contributors to the energy deficit in AD, including extracranial abnormalities that affect cerebral blood supply and intracellular disturbances of mitochondrial function, but those are not covered in the present review.
Within this review, we consider three questions:
Is cerebral hypoperfusion in AD caused by reduced demand or reduced supply?
What are the mechanisms of the hypoperfusion?
What are the therapeutic implications?
Reduced Demand or Reduced Supply?
The relative contributions of falling metabolic demand and reduction in blood supply vary according to the stage of disease. In preclinical and early AD there is evidence from multiple studies employing a range of methods that the cerebral hypoperfusion is pathological rather than physiological, that is, the decline in perfusion exceeds the reduction in metabolic demand, and causes tissue damage. Metabolic demand may, in fact, be increased during the earliest stages of amyloid accumulation 11, 27.
Some of the data comes from imaging studies using arterial spin‐labeled perfusion magnetic resonance imaging (ASL‐MRI) or 2‐deoxy‐2‐(18F)fluoro‐D‐glucose positron emission tomography (FDG‐PET) (there is a near‐perfect topographical correspondence between changes in ASL‐MRI and FDG‐PET in AD—see, eg, reference 24). In people with mutations that cause autosomal dominant forms of AD, in whom the timing of onset of the AD is highly predictable, a reduction in glucose uptake is demonstrable by FDG‐PET at least 10 years before the onset of clinical disease and before there is detectable atrophy 11. The decline shows a consistent pattern of topographical progression, starting in the precuneus (medial parietal cortex) and extending along the cingulate gyrus, lateral part of the parietal lobe and anterior part of the occipital lobe, then into the rest of the cerebrum.
A similar pattern of progression of hypoperfusion was demonstrated by ASL‐MRI in sporadic AD 12. In patients with MCI the hypoperfusion was most pronounced in the precuneus and posterior cingulate cortex but also involved lateral parietal, occipital and frontal cortex. Perfusion declined significantly in the temporal cortex only when patients became demented and did not fall in the hippocampus. Similar findings were reported in earlier studies on people with MCI 5, 30 and in healthy carriers of the APOE ε4 allele 65, a strong genetic risk factor for AD. This stereotypical distribution of hypoperfusion does not bear an obvious relationship to the distribution of cerebral atrophy, which correlates with that of neurofibrillary tangle pathology, commencing in the inferomedial part of the temporal lobes before spreading to other parts of the cerebrum 11. The distribution of hypoperfusion/reduced glucose uptake correlates much more closely with that of the preceding accumulation of amyloid (see, eg, references 11, 27).
We recently used a biochemical approach to assess the adequacy of perfusion and oxygenation of the precuneus in early AD. We compared the levels of two myelin proteins, myelin‐associated glycoprotein (MAG), which is highly susceptible to reduced tissue oxygenation, and proteolipid protein‐1 (PLP1), which is relatively resistant 8, 9. Both myelin proteins are synthesized in the oligodendrocyte cell body and require energy‐dependent transport to reach their sites of insertion into the myelin sheath (Figure 1). PLP1 is distributed throughout the myelin sheath whereas MAG is inserted only far from the cell body, in the adaxonal loop of myelin, the first part of the sheath to degenerate when blood supply is insufficient to meet the energy demands of the oligodendrocyte. Both myelin proteins are very stable under post‐mortem conditions 9, and as they have half‐lives of several months 47, 136, a decline in the MAG:PLP1 ratio in post‐mortem brain tissue reflects a pathological reduction in ante‐mortem perfusion over a relatively long period prior to death. We showed that the MAG:PLP1 ratio, was reduced in the precuneus by approximately 50% in Braak stage III–IV disease 75, indicating that even in early AD there is a disparity between oligodendrocyte energy demand and supply in the first region of brain to show hypoperfusion.
Figure 1.
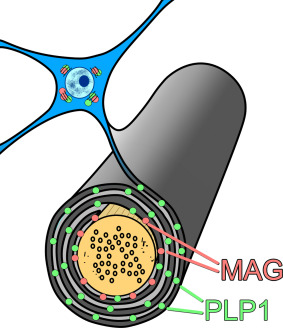
Schematic illustration of the distribution of MAG (pink dots) and PLP1 (green dots) in the myelin sheath. PLP1 is distributed throughout the myelin sheath whereas MAG is inserted only far from the cell body, in the adaxonal loop of myelin, the first part of the sheath to degenerate when blood supply is insufficient to meet the energy demands of the oligodendrocyte. As MAG and PLP1 are stable post mortem and have half‐lives of several months, a decline in MAG:PLP1 in post‐mortem brain tissue reflects a hypoperfusion‐related energy deficit over a relatively long period prior to death. Image adapted from reference 71.
Tarumi et al 131 used near‐infrared spectroscopy to compare the tissue oxygenation index (a measure of the saturation of hemoglobin by oxygen) in frontal cortex of people with amnestic MCI and age‐matched controls. In amnestic MCI patients the tissue oxygenation index was significantly reduced, both at rest and after a sit–stand maneuver, indicating increased oxygen extraction. Had hypoperfusion been a response to reduced metabolic demand rather than a pathological reduction in blood supply, the tissue oxygenation index would have been higher rather than lower.
Cerebral hypoperfusion predicts the development of dementia in patients with MCI and the rate of cognitive decline in patients with AD 14, 16, 22. The fact that the hypoperfusion actually damages the brain, even in preclinical or early disease, is well demonstrated on imaging of cerebral white matter in people with mutations that cause autosomal dominant forms of AD. A recent study showed that people with such mutations had a greater volume of white matter hyperintensities (WMH) several years before clinical disease, indicating that the hypoperfusion was severe enough to cause tissue damage 67. The increase in WMH was most pronounced in the parietal and occipital lobes; as noted above, this corresponds approximately to the distribution of early amyloid accumulation and reduced perfusion in the overlying cerebral cortex.
The relationship between cortical amyloid and WMH is likely to be relevant to the pathogenesis of the white matter hypoperfusion (see below). Other evidence for a relationship between amyloid burden and WMH comes from an MRI study of 150 cognitively normal people by Scott et al 122. The authors found that amyloid burden, as assessed by measuring Aβ42 level in the CSF, was an independent predictor of total WMH volume. Lee et al 67 found some correlation between WMH and the presence of microbleeds, suggesting a contribution from cerebral amyloid angiopathy. However, the increase in WMH remained significant after controlling for presence of microbleeds, which were calculated to account for 21% of the association between AD mutation status and WMH.
In later AD, it is likely that the decline in perfusion continues to exceed the reduction in metabolic demand but less so than in early disease. Several functional MRI studies have demonstrated an increase in the regional oxygen extraction fraction (rOEF) in the cerebral cortex and white matter in AD 83, 84, 137, indicating a continued pathological reduction in blood supply. Indeed, in the series of Toghi et al 137, rOEF was higher in the cerebral cortex of patients with AD than in those with vascular dementia (in the white matter the increase was more marked in patients with vascular dementia).
Our own studies found the MAG:PLP1 ratio to be reduced in the cerebral cortex in late, as well as early AD 75, 134. However, within the precuneus, the ratio was not as markedly decreased in brain tissue from patients with Braak stage V–VI disease as in those with earlier (Braak stage III–IV) disease and not significantly so in comparison with Braak stage 0–II disease (Figure 2). We interpreted this lessening of the perfusion deficit as being likely to reflect falling metabolic demand with increasing synaptic and neuronal damage.
Figure 2.
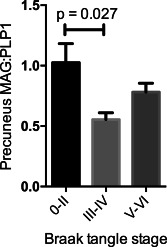
Bar chart showing decline of MAG:PLP1 in the precuneus in AD. The decline is most marked in early disease (Braak tangle stage III–IV). The ratio may rise in late disease as a consequence of falling metabolic demand. Reproduced from reference 75.
Mechanisms: Metabolic Vascular Dysfunction
Under normal circumstances, cerebral perfusion is tightly regulated to match the supply of oxygenated blood to metabolic requirements, both of the brain as a whole (through autoregulation—the maintenance of relatively constant blood flow despite changes in perfusion pressure) and of the individual regions within it (through neurovascular coupling) 25, 106. This regulation is effected through multiple neurogenic, myogenic and metabolic pathways. In AD, the activity of several pathways that regulate intracerebral vascular tone and influence neurovascular coupling is abnormally altered. Most of the alterations promote vasoconstriction, acting on smooth muscle cells in the tunica media of arterioles, on pericytes in capillaries or on both types of cell, and reduce tissue oxygenation. Other abnormalities allow inappropriate local vasodilatation, diverting blood away from regions of higher metabolic demand. Both types of alteration have the potential to affect neurological function and, if sustained, to cause permanent damage.
Cerebral Cortex
Cholinergic innervation
Arterioles in the cerebral cortex are innervated by cholinergic nerves, originating in the nucleus basalis of Meynert 139, 142. Stimulation of neurons in the nucleus basalis (see reference 26 for review) or of muscarinic receptors in isolated arterioles 48 causes vasodilatation, partly mediated by stimulation of the production of nitric oxide (NO) 151. Tong and Hamel 139 found a reduction in the cholinergic innervation of cortical blood vessels in AD, mirroring a general loss of cholinergic nerve terminals from the cerebral cortex and in keeping with the loss of neurons from the nucleus basalis from an early stage of disease (see references 31, 68 for review). Cholinergic deafferentation reduces blood flow in the cerebral cortex, as was demonstrated after targeted ablation of cholinergic neurons by administration of 192 IgG‐saporin 144. Although interpretation of the findings is complicated by possible effects of cholinergic denervation on neuronal activity and metabolic demand, it seems likely that reduced stimulation of muscarinic receptors in the walls of cortical arterioles contributes to the hypoperfusion of the cortex in AD, and possible beneficial effects of cholinesterase inhibitors in AD may relate partly to augmented cerebral perfusion 13, 26. It should be noted that AD is also associated with alterations in a range of other neurotransmitters that have direct or indirect effects on vascular contractility, including glutamate, γ‐aminobutyric acid, noradrenaline (norepinephrine), serotonin and dopamine 28, 34, 44, 63, 113.
Amyloid‐β peptide (Aβ)
Perhaps not surprisingly, several of the processes that mediate vascular dysfunction in AD are probably initiated by the accumulation of Aβ. Topical application of Aβ40 or Aβ42 to isolated arteries causes vasoconstriction, Aβ40 being more potent than Aβ42 in this regard 29. The vasoconstriction can be reduced by free radical scavengers and cyclo‐oxygenase inhibitors 99, 112, 135, 140. Aβ40 enhances the constriction induced by endothelin‐1 (EDN1) and reduces the vasodilatation produced by NO 99, 101.
Aβ40 also induces vasoconstriction in vivo, as demonstrated by its application to mouse cortex 89. This can be prevented by administering free radical scavengers or by an M35Nle amino acid substitution in Aβ40 which interferes with its ability to generate reactive oxygen species 89, 90. In a series of studies on mice overexpressing Aβ‐precursor protein (APP), Iadecola et al showed that elevated endogenous Aβ also caused vasoconstriction from an early age (2 months in Tg2576 mice, well before plaque formation), impaired autoregulation and interfered with neurovascular coupling (in this case the functional hyperemia of the barrel cortex that is normally induced by whisker stimulation) 86, 87, 88, 90. Shin et al 125 confirmed that Tg2576 mice had an attenuated hyperemic response to hypercapnia and whisker stimulation but were unable to demonstrate this until the mice had reached the age of 9 months, that is, after commencement of vascular deposition of Aβ. The authors suggested that vascular deposition of Aβ was a prerequisite for the vascular dysfunction. It is also possible that the development of vascular dysfunction in these mice simply depends on the concentration of soluble Aβ, which increases with age, although it is not clear why the two research groups found so marked a difference in the timing of onset of the dysfunction. The abnormal cerebral vasoconstriction in Tg2576 mice requires the production of free radicals by nicotinamide adenine dinucleotide phosphate oxidase (NADPH oxidase), as shown by studies in which NADPH oxidase was either inhibited 103, or inactivated by deletion of Nox2 104. These findings are in keeping with the studies on isolated arteries, described above.
For simplicity, the various processes that contribute to hypoperfusion in AD are considered under separate headings in the present review. However, as noted below, the different pathways overlap and interact substantially—particularly insofar as they involve Aβ, for example, in upregulating the production of EDN1 by endothelin‐converting enzymes‐1 and −2 (ECE1 and ECE2) and that of angiotensin II (Ang II) by angiotensin‐converting enzyme (ACE), in reducing NO production by endothelial cells, in binding to and sequestering vascular endothelial growth factor (VEGF) in plaques, and in blocking VEGF receptor 2 (VEGFR‐2) signaling in endothelial cells. There is also interaction between the endothelin and renin‐angiotensin systems (reviewed in reference 76); Aβ and the cholinergic systems (see references 55, 94, 147 for review); and the cholinergic system and VEGF production 53.
Endothelin system
EDN1 is a potent vasoconstrictor and its concentration is increased significantly in cerebral cortex from patients with AD 75, 97, 134. The increase is demonstrable from an early stage of disease, including within the precuneus 75, the first region in which blood flow declines (see above). Paradoxically, the gene that encodes EDN1 is also upregulated by hypoxia 127. The extent to which EDN1 is increased in AD correlates with the severity of cortical hypoperfusion/tissue hypoxia, as measured by the decline in the MAG:PLP1 ratio.
Both Aβ40 and Aβ42 are capable of increasing EDN1 production in vitro: Aβ40 through upregulation of ECE1 in endothelial cells 97, 98, and Aβ42 through upregulation of ECE2 in neurons 96. In cortex from patients with AD the concentration of EDN1 correlates closely with that of Aβ42 but bears no relationship to that of Aβ40 75, suggesting that the increase in EDN1 and decrease in tissue oxygenation are caused, at least in part, by Aβ42‐mediated neuronal upregulation of ECE2. Aβ is a physiological substrate of both ECEs 36, 37, 38 and the upregulation of ECE2 and consequent sustained overproduction of EDN1 in AD may simply be an unfortunate side effect of the parenchymal accumulation of substrate in the form of Aβ42 76. The lack of association between Aβ40 and EDN1 (or MAG:PLP1) does not discount a role for Aβ40 in the vascular dysfunction of AD, but such a role is likely to be predominantly episodic: interfering with autoregulation and neurovascular coupling rather than causing sustained hypoperfusion. Palmer et al 98 showed that the enhanced release of EDN1 that follows the addition of Aβ40 to human cerebrovascular endothelial cells in vitro could be prevented by the addition of superoxide dismutase, potentially linking upregulation of ECE1 with the observations of Niwa et al 89, 90 on free‐radical mediated vasoconstriction, and suggesting that the episodic cerebral vasoconstriction induced by Aβ40 results from a free radical‐mediated increase in endothelial ECE‐1 activity and EDN1 production.
Angiotensin
Increased production of the vasoconstrictor Ang II may contribute to hypoperfusion of the frontal cortex, where ACE activity 74, 77, 78 and Ang II level (unpublished observations) are elevated in AD, perhaps in response to the accumulation of Aβ42. Miners et al 78 showed that ACE activity in SH‐SY5Y neuroblastoma cells was upregulated by aggregated Aβ42 (but not Aβ40 or freshly solubilized Aβ42). The relationship between Aβ42, ACE activity, Ang II production and hypoperfusion is, however, less clear‐cut than that between Aβ42, ECE1 activity, EDN1 production and hypoperfusion, in that neither ACE activity nor Ang II level was increased in precuneus from patients with AD 75.
Like so many of the dysregulated pathways that are the focus of this review, the renin‐angiotensin system has a complex interrelationship with other vasoregulatory processes. ACE cleaves 141 and probably thereby limits the duration of action of the vasodilator bradykinin, the production of which is likely to be elevated in AD, as a result of increased activity of plasma kallikrein 4. Ang II was reported to increase EDN1 production in endothelial 32 and vascular adventitial fibroblasts 1, probably by inducing transcription of the preproendothelin‐1 gene 117 and by a mechanism involving NADPH oxidase 2, thereby contributing to EDN1‐mediated hypoperfusion. Conversely, hypoxia was shown to upregulate the expression and activation of ACE 62.
Endothelial nitric oxide synthase (eNOS)
NO, a potent vasodilator, is synthesized within the endothelium by eNOS, the activity of which plays an important role in local regulation of the cerebral microcirculation 151. eNOS is activated by a wide range of stimuli, including acetylcholine 40, bradykinin, oxidative stress, shear stress and hypoxia. Both Aβ40 and Aβ42 inhibit eNOS activity. Aβ40 was reported to do so through a mechanism that depends on protein kinase C 46, and Aβ42 through interfering with Akt/GSK‐3β signaling and a mechanism involving interaction of eNOS with heat shock protein 90 64, 128. Mice partially deficient in eNOS develop cognitive impairment associated with a range of neuropathological abnormalities, including cerebral amyloid angiopathy (CAA) and disruption of the blood–brain barrier (BBB), largely confined to the temporoparietal and retrosplenial granular cortex and hippocampus 129. Jeynes and Provias 52 reported a significant negative correlation between the number of eNOS‐immunolabeled capillaries and the density of neurofibrillary tangles and Aβ plaques in sections of temporal and calcarine cortex in AD.
White Matter
As noted above, most of the studies on mechanisms of vascular dysfunction in AD have used rodent models and have focused on cerebral cortex. Whilst several of the local metabolic abnormalities that contribute to hypoperfusion of the cerebral cortex may also apply in the white matter, our studies have highlighted differences that are relevant to our understanding of the pathogenesis of ischemic white matter damage in AD and have implications for treatment.
Whereas we found the concentration of EDN1 to be elevated approximately twofold in the cerebral cortex in AD, presumably in response to Aβ42‐induced upregulation of ECE2, EDN1 level was significantly reduced in the underlying white matter 8, 75. This reduction occurred in association with a modest decline in MAG and in MAG:PLP1 in the white matter 9 (in keeping with other evidence of ischemic white matter damage in AD, eg, on neuroimaging, as discussed above). The relationship between MAG:PLP1 and EDN1 in the white matter was the converse of that in the cortex: in the cortex MAG:PLP1 and EDN1 correlated negatively and in the white matter they correlated positively 75. White matter hypoperfusion in AD is not therefore caused by increased white matter EDN1, which falls as would be expected physiologically in response to hypoperfusion. However, MAG:PLP1 in the white matter did correlate positively with the concentration of EDN1 in the overlying cortex, suggesting that hypoperfusion of the white matter in AD results partly from vasoconstriction of perforating arterioles as they traverse the cortex (Figure 3) 23, 75. This mechanism of white matter hypoperfusion is likely to be relevant to other Aβ‐dependent processes that increase vasoconstriction within the cortex, including cholinergic denervation, Ang II production and reduction in activity of eNOS.
Figure 3.
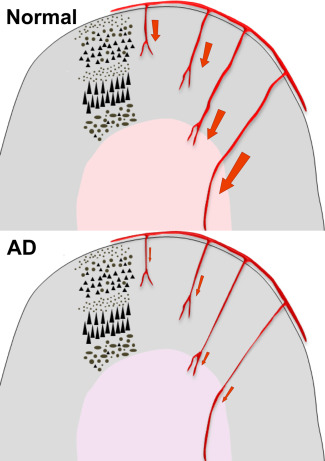
Perforating arterioles that arise from meningeal branches of the major cerebral arteries supply both the cerebral cortex and the underlying white matter. Excessive vasoconstriction within the cerebral cortex in AD affects not only cortical arterioles but also perforating arterioles that traverse the cortex. Thus vasoconstriction within the cerebral cortex contributes to hypoperfusion of the white matter even if arterioles in the white matter are not themselves constricted. Modified from reference 23.
Mechanisms: Structural Abnormalities of the Cerebral Vasculature
The abnormalities described above affect vascular function but are not associated with long‐lasting structural alterations. As recently reviewed 71, patients with AD also have an increased prevalence of structural disease of cerebral microvessels, particularly CAA and capillary damage. They may also have more severe non‐amyloid small vessel disease (SVD) than elderly people without AD but most of the cited evidence is indirect, based on the identification of white matter abnormalities on neuroimaging, and as likely to have resulted from CAA or capillary damage as from SVD. An MRI‐based study of regional cerebrovascular resistance (CVRi) found this to be increased in several regions of brain that are not affected by CAA and have a predilection for SVD, including thalamus and caudate nucleus 85, but the pathological substrate of the increased CVRi remains to be demonstrated. For further consideration of the possible association of SVD with AD, see reference 71.
Cerebral Amyloid Angiopathy
Most patients with AD have CAA, in some series over 90%, compared with about 30% in elderly controls 21, 42, 69, 72, 73, 143, 146. In many cases the CAA is relatively mild, affecting only occasional arterioles in the leptomeninges, but some patients have widespread involvement of cortical and meningeal arterioles, as well as deposition of Aβ in the adventitia of meningeal venules. In patients with AD, possession of APOE ε4 is a risk factor for more‐severe CAA 21, 111, 121 and is strongly associated with capillary CAA 6, 69, 133 (in controls, arteriolar Aβ amyloid angiopathy is more strongly associated with APOE ε2). In the majority of patients CAA is restricted to the cerebral cortex and overlying leptomeninges but it may also involve the cerebellum (particularly the meningeal vessels) and occasionally the brain stem. Aβ may also accumulate in the walls of capillaries, sometimes extensively so. Capillary CAA predominantly involves the entorhinal and occipital cortex but can be present in other parts of the neocortex and is often, but not always, associated with severe arteriolar CAA.
CAA has several adverse effects on cerebral perfusion. Perhaps the most widely recognized are cerebral micro‐hemorrhages and larger lobar hemorrhages, but there is also extensive documentation of ischemic abnormalities (predominantly cortical microinfarcts) 3, 18, 41, 43, 93, 138, some of which are caused by local thrombosis, some by the marked narrowing of severely affected blood vessels, and some probably by impaired neurovascular coupling. Evidence of neurovascular decoupling in human patients comes from MRI studies of occipital vascular reactivity in response to visual stimulation in patients with probable CAA 35, 107, 126. Peca et al 107 also found that impaired neurovascular coupling, as evidenced by lower functional MRI responses to visual stimuli, was associated with more microbleeds and a higher volume of white matter lesions, linking impairment of neurovascular coupling with severity of tissue damage.
Capillary Damage
The capillary bed constitutes much the largest part of the cerebral vasculature and is also the most important in terms of metabolic homeostasis. Yet the contribution of capillary damage to hypoperfusion in AD has been somewhat neglected, perhaps because of the small size and inconspicuous histological appearance of individual capillaries.
Despite the hypoperfusion, the density of capillaries in the cerebral cortex is unchanged or reduced in AD 8, 15, 17, 45, 60, 134, and more of them show degenerative changes in AD than in age‐matched controls 7, 20, 51, 123. Both endothelial cells and pericytes are affected, their degeneration eventually leaving residual “string” vessels consisting solely of tubes of collagen. These degenerative changes occur despite a significant increase in the concentration of VEGF in AD 54, 75, 130, 134, which would be expected to promote angiogenesis, with the formation of new capillaries 92, 150. Several factors may contribute to this lack of angiogenic response. Aβ peptides have direct anti‐angiogenic activity 99, 100, 102 and also bind to VEGF receptor 2, blocking VEGF signaling. In addition, Aβ within plaques binds and thereby potentially sequesters VEGF, interfering with its biological availability 105, 148.
At the level of the capillary bed, degeneration of pericytes has emerged as a key contributor to hypoperfusion. Changes in the contractile activity of pericytes modulate capillary caliber and cerebral blood flow and probably play an important role in neurovascular coupling 10, 49, 108. Dore‐Duffy et al reported that pericytes in primary cultures express both EDN1 and its two receptors (EDNRA and EDNRB) 33. The authors also provided in vivo evidence that EDN1 (which is elevated in AD—see above) contributes to the regulation of capillary perfusion through binding to EDNRA receptors in pericytes. Experimental traumatic brain injury in mice caused an increase in the number of smooth muscle actin‐positive pericytes around capillaries, a rise in capillary EDN1, and reduced capillary diameter. These changes could be prevented by administration of an EDNRA antagonist. Pericytes are also important for maintenance of the BBB 10.
A series of studies by Zlokovic et al have shown that loss of pericytes exacerbates multiple pathological processes in AD 145, including hypoperfusion and disruption of the BBB 10, 123, accumulation of Aβ40 and Aβ42, tau pathology and neuronal loss 120. Montagne et al 80 quantified BBB permeability in the hippocampus of young and older adult volunteers by dynamic contrast‐enhanced MRI, and showed the degree of increase in permeability to correlate with the CSF:plasma albumin ratio (a marker of BBB breakdown) and the CSF concentration of soluble platelet‐derived growth factor receptor β (a marker of pericyte injury). Both BBB permeability and pericyte injury were more pronounced in participants with MCI than in older individuals who were cognitively normal.
Therapeutic Implications
There is therefore overwhelming evidence of a wide range of functional and structural abnormalities of the cerebral microvasculature in AD, that contribute to hypoperfusion and the resulting energy deficit as well as to other aspects of the disease. Implications for therapy are both specific and general. Specific implications concern the potential for targeting of particular pathways or receptors to ameliorate the vascular abnormalities—particularly those that are not the result of structural changes. General implications relate to the timing and broader consequences of intervention.
Several of the pathways implicated in abnormal microvascular function in AD are potentially amenable to treatment. The cholinergic system is, of course, already routinely targeted in AD patients through the administration of cholinesterase inhibitors. These drugs improve cerebral perfusion in mild to moderate disease 19, and several studies found evidence of an association between cognitive response and cerebral blood flow 13, 26, 124. The potential for intervention in the renin‐angiotensin system has been extensively reviewed 56, 57, 58, 59 and the effects on cognition and cerebral blood flow of losartan, an angiotensin receptor antagonist, are currently being tested in a multicenter UK clinical trial 115.
Another potential target is the endothelin system. Bosentan, a non‐selective EDNR antagonist 119, improves pulmonary blood flow and exercise tolerance in patients with pulmonary hypertension, another disease in which there is elevated production of EDN1 114, 118. Bosentan also preserves endothelium‐dependent aortic and carotid vasodilatation in Tg2576 mice 39. Selective EDNRA receptor antagonists such as zibotentan 50, 81 offer theoretical advantages, in that they target the predominant type of EDN1 receptor responsible for mediating vasoconstriction in both smooth muscle cells of cerebral arterioles and pericytes that surround capillaries 33, 91. For discussion see reference 95.
Interventions aimed at reversing functional abnormalities of the vasculature in AD have the potential not only to improve symptoms but also to slow the progression of disease. Hypoperfusion probably increases the production of Aβ42, thereby accelerating the progression of disease. Simulation of neuronal ischemia in vitro, or experimental cerebral hypoperfusion in animal models increases Aβ42 production through multiple mechanisms (reviewed in references 70, 71), including upregulation of amyloid‐β precursor protein and β‐secretase and possibly reduced neprilysin‐mediated degradation. Indirect evidence of a hypoperfusion‐induced increase in Aβ comes from observations in patients who had survived a recent cardiac arrest 149 or diffuse traumatic brain injury with cerebral edema (and, therefore, almost certainly hypoperfusion) 79. Both groups had elevated serum Aβ42 over several days. In the patients with diffuse traumatic brain injury, Aβ42 was also monitored in the CSF where the level declined, arguing against non‐specific leakage of Aβ from damaged brain tissue as the explanation for the rising level in the serum.
The timing of intervention is likely to be critical, as prolonged hypoperfusion causes permanent brain damage (as discussed above), and as the disease progresses the balance tends to shift from metabolic to structural vascular dysfunction (Figure 4). It seems possible too that prolonged cerebral hypoperfusion may induce compensatory changes in the circulation that are themselves damaging, including hypertension and SVD. Mid‐life hypertension is significantly associated with AD 109, 110. Several clinical studies have reported an association between hypertension before the age of 65 years (particularly if there is elevation of diastolic blood pressure) and later development of AD 61, 66, 82. Hypertension in cognitively normal adults with at least 1 APOE ε4 allele was associated with increased binding of the Aβ tracer F18–labeled florbetapir 116. The conventional interpretation is that hypertension increases the risk of developing AD by promoting the accumulation of Aβ. However, another possible explanation is that hypertension is a physiological response to tonic cerebral vasoconstriction induced by mid‐life accumulation of Aβ; a means of maintaining cerebral perfusion. There is experimental evidence that this is the case, in that cerebroventricular infusion of Aβ was shown to cause a progressive, highly significant rise in blood pressure in rats 132. This has obvious implications for autoregulation and for the treatment of hypertension once Aβ has begun to accumulate, and suggests that blood pressure in such patients should be lowered only cautiously, ideally with monitoring of the effects on cerebral perfusion.
Figure 4.
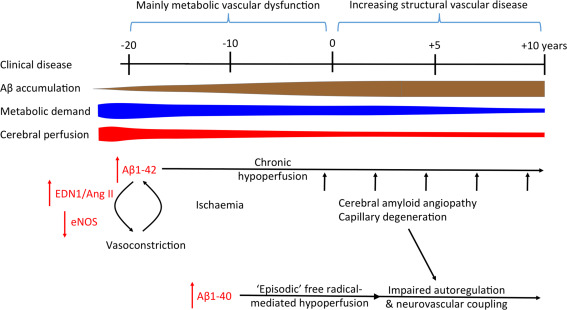
Shift from purely metabolic to structural vascular dysfunction over the course of AD (in relation to the onset of clinical disease, at 0 years). After a brief period of increased metabolic demand and cerebral blood flow, the progressive accumulation of Aβ in early (preclinical) stages of AD drives several metabolic pathways that lead to excessive vasoconstriction and reduced cerebral perfusion. Cerebral perfusion declines faster than metabolic demand. Aβ42‐induced metabolic processes may be more important in driving chronic hypoperfusion, and Aβ40‐induced processes in impairing vascular responsiveness. As the disease progresses, capillary damage and, in many patients, CAA, become increasingly important contributors to both chronic hypoperfusion and abnormalities of autoregulation and neurovascular coupling.
Acknowledgments
This work was supported by Alzheimer's Research UK (ART‐PG2011‐1 and ARUK‐PG2015‐11). The South West Dementia Brain Bank is part of the Brains for Dementia Research program, jointly funded by Alzheimer's Research UK and Alzheimer's Society, and is supported by BRACE (Bristol Research into Alzheimer's and Care of the Elderly) and the Medical Research Council.
References
- 1. An SJ, Boyd R, Wang Y, Qiu X, Wang HD (2006) Endothelin‐1 expression in vascular adventitial fibroblasts. Am J Physiol Heart Circ Physiol 290:H700–H708. [DOI] [PubMed] [Google Scholar]
- 2. An SJ, Boyd R, Zhu M, Chapman A, Pimentel DR, Wang HD (2007) NADPH oxidase mediates angiotensin II‐induced endothelin‐1 expression in vascular adventitial fibroblasts. Cardiovasc Res 75:702–709. [DOI] [PubMed] [Google Scholar]
- 3. Arvanitakis Z, Capuano AW, Leurgans SE, Buchman AS, Bennett DA, Schneider JA (2016) The relationship of cerebral vessel pathology to brain microinfarcts. Brain Pathol doi: 10.1111/bpa.12365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ashby EL, Love S, Kehoe PG (2012) Assessment of activation of the plasma kallikrein‐kinin system in frontal and temporal cortex in Alzheimer's disease and vascular dementia. Neurobiol Aging 33:1345–1355. [DOI] [PubMed] [Google Scholar]
- 5. Asllani I, Habeck C, Scarmeas N, Borogovac A, Brown TR, Stern Y (2008) Multivariate and univariate analysis of continuous arterial spin labeling perfusion MRI in Alzheimer's disease. J Cereb Blood Flow Metab 28:725–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Attems J, Jellinger KA (2004) Only cerebral capillary amyloid angiopathy correlates with Alzheimer pathology–a pilot study. Acta Neuropathol 107:83–90. [DOI] [PubMed] [Google Scholar]
- 7. Baloyannis SJ, Baloyannis IS (2012) The vascular factor in Alzheimer's disease: a study in Golgi technique and electron microscopy. J Neurol Sci 322:117–121. [DOI] [PubMed] [Google Scholar]
- 8. Barker R, Ashby EL, Wellington D, Barrow VM, Palmer JC, Kehoe PG et al (2014) Pathophysiology of white matter perfusion in Alzheimer's disease and vascular dementia. Brain 137:1524–1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Barker R, Wellington D, Esiri MM, Love S (2013) Assessing white matter ischemic damage in dementia patients by measurement of myelin proteins. J Cereb Blood Flow Metab 33:1050–1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bell RD, Winkler EA, Sagare AP, Singh I, LaRue B, Deane R, Zlokovic BV (2010) Pericytes control key neurovascular functions and neuronal phenotype in the adult brain and during brain aging. Neuron 68:409–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Benzinger TL, Blazey T, Jack CR, Jr. , Koeppe RA, Su Y, Xiong C et al (2013) Regional variability of imaging biomarkers in autosomal dominant Alzheimer's disease. Proc Natl Acad Sci U S A 110:E4502–E4509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Binnewijzend MA, Kuijer JP, Benedictus MR, van der Flier WM, Wink AM, Wattjes MP et al (2013) Cerebral blood flow measured with 3D pseudocontinuous arterial spin‐labeling MR imaging in Alzheimer disease and mild cognitive impairment: a marker for disease severity. Radiology 267:221–230. [DOI] [PubMed] [Google Scholar]
- 13. Blin J, Ivanoiu A, Coppens A, De Volder A, Labar D, Michel C, Laterre EC (1997) Cholinergic neurotransmission has different effects on cerebral glucose consumption and blood flow in young normals, aged normals, and Alzheimer's disease patients. Neuroimage 6:335–343. [DOI] [PubMed] [Google Scholar]
- 14. Borroni B, Perani D, Broli M, Colciaghi F, Garibotto V, Paghera B et al (2005) Pre‐clinical diagnosis of Alzheimer disease combining platelet amyloid precursor protein ratio and rCBF spect analysis. J Neurol 252:1359–1362. [DOI] [PubMed] [Google Scholar]
- 15. Bouras C, Kovari E, Herrmann FR, Rivara CB, Bailey TL, von Gunten A et al (2006) Stereologic analysis of microvascular morphology in the elderly: Alzheimer disease pathology and cognitive status. J Neuropathol Exp Neurol 65:235–244. [DOI] [PubMed] [Google Scholar]
- 16. Brown DR, Hunter R, Wyper DJ, Patterson J, Kelly RC, Montaldi D, McCullouch J (1996) Longitudinal changes in cognitive function and regional cerebral function in Alzheimer's disease: a SPECT blood flow study. J Psychiatr Res 30:109–126. [DOI] [PubMed] [Google Scholar]
- 17. Buee L, Hof PR, Bouras C, Delacourte A, Perl DP, Morrison JH, Fillit HM (1994) Pathological alterations of the cerebral microvasculature in Alzheimer's disease and related dementing disorders. Acta Neuropathol 87:469–480. [DOI] [PubMed] [Google Scholar]
- 18. Cadavid D, Mena H, Koeller K, Frommelt RA (2000) Cerebral β amyloid angiopathy is a risk factor for cerebral ischemic infarction. A case control study in human brain biopsies. J Neuropathol Exp Neurol 59:768–773. [DOI] [PubMed] [Google Scholar]
- 19. Ceravolo R, Volterrani D, Tognoni G, Dell'Agnello G, Manca G, Kiferle L et al (2004) Cerebral perfusional effects of cholinesterase inhibitors in Alzheimer disease. Clin Neuropharmacol 27:166–170. [DOI] [PubMed] [Google Scholar]
- 20. Challa VR, Thore CR, Moody DM, Anstrom JA, Brown WR (2004) Increase of white matter string vessels in Alzheimer's disease. J Alzheimers Dis 6:379–383. [DOI] [PubMed] [Google Scholar]
- 21. Chalmers K, Wilcock GK, Love S (2003) APOE ε4 influences the pathological phenotype of Alzheimer's disease by favouring cerebrovascular over parenchymal accumulation of A beta protein. Neuropathol Appl Neurobiol 29:231–238. [DOI] [PubMed] [Google Scholar]
- 22. Chao LL, Buckley ST, Kornak J, Schuff N, Madison C, Yaffe K et al (2010) ASL perfusion MRI predicts cognitive decline and conversion from MCI to dementia. Alzheimer Dis Assoc Disord 24:19–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Charidimou A, Pantoni L, Love S (2016) The concept of sporadic cerebral small vessel disease: a road map on key definitions and current concepts. Int J Stroke 11:6–18. [DOI] [PubMed] [Google Scholar]
- 24. Chen Y, Wolk DA, Reddin JS, Korczykowski M, Martinez PM, Musiek ES et al (2011) Voxel‐level comparison of arterial spin‐labeled perfusion MRI and FDG‐PET in Alzheimer disease. Neurology 77:1977–1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cipolla MJ (2009) Control of cerebral blood flow. In: The Cerebral Circulation, Chapter 5, pp. 41–52. Morgan & Claypool Life Sciences: San Rafael, CA. [PubMed] [Google Scholar]
- 26. Claassen JA, Jansen RW (2006) Cholinergically mediated augmentation of cerebral perfusion in Alzheimer's disease and related cognitive disorders: the cholinergic‐vascular hypothesis. J Gerontol a Biol Sci Med Sci 61:267–271. [DOI] [PubMed] [Google Scholar]
- 27. Cohen AD, Price JC, Weissfeld LA, James J, Rosario BL, Bi W et al (2009) Basal cerebral metabolism may modulate the cognitive effects of Abeta in mild cognitive impairment: an example of brain reserve. J Neurosci 29:14770–14778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Cohen Z, Bonvento G, Lacombe P, Hamel E (1996) Serotonin in the regulation of brain microcirculation. Prog Neurobiol 50:335–362. [DOI] [PubMed] [Google Scholar]
- 29. Crawford F, Suo Z, Fang C, Mullan M (1998) Characteristics of the in vitro vasoactivity of β‐amyloid peptides. Exp Neurol 150:159–168. [DOI] [PubMed] [Google Scholar]
- 30. Dai W, Lopez OL, Carmichael OT, Becker JT, Kuller LH, Gach HM (2009) Mild cognitive impairment and alzheimer disease: patterns of altered cerebral blood flow at MR imaging. Radiology 250:856–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Davies P, Maloney AJ (1976) Selective loss of central cholinergic neurons in Alzheimer's disease. Lancet 2:1403. [DOI] [PubMed] [Google Scholar]
- 32. Dohi Y, Hahn AW, Boulanger CM, Buhler FR, Luscher TF (1992) Endothelin stimulated by angiotensin II augments contractility of spontaneously hypertensive rat resistance arteries. Hypertension 19:131–137. [DOI] [PubMed] [Google Scholar]
- 33. Dore‐Duffy P, Wang S, Mehedi A, Katyshev V, Cleary K, Tapper A et al (2011) Pericyte‐mediated vasoconstriction underlies TBI‐induced hypoperfusion. Neurol Res 33:176–186. [DOI] [PubMed] [Google Scholar]
- 34. Drake CT, Iadecola C (2007) The role of neuronal signaling in controlling cerebral blood flow. Brain Lang 102:141–152. [DOI] [PubMed] [Google Scholar]
- 35. Dumas A, Dierksen GA, Gurol ME, Halpin A, Martinez‐Ramirez S, Schwab K et al (2012) Functional magnetic resonance imaging detection of vascular reactivity in cerebral amyloid angiopathy. Ann Neurol 72:76–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Eckman EA, Adams SK, Troendle FJ, Stodola BA, Kahn MA, Fauq AH et al (2006) Regulation of steady‐state β‐amyloid levels in the brain by neprilysin and endothelin‐converting enzyme but not angiotensin‐converting enzyme. J Biol Chem 281:30471–30478. [DOI] [PubMed] [Google Scholar]
- 37. Eckman EA, Reed DK, Eckman CB (2001) Degradation of the Alzheimer's amyloid β peptide by endothelin‐converting enzyme. J Biol Chem 276:24540–24548. [DOI] [PubMed] [Google Scholar]
- 38. Eckman EA, Watson M, Marlow L, Sambamurti K, Eckman CB (2003) Alzheimer's disease β‐amyloid peptide is increased in mice deficient in endothelin‐converting enzyme. J Biol Chem 278:2081–2084. [DOI] [PubMed] [Google Scholar]
- 39. Elesber AA, Bonetti PO, Woodrum JE, Zhu XY, Lerman LO, Younkin SG, Lerman A (2006) Bosentan preserves endothelial function in mice overexpressing APP. Neurobiol Aging 27:446–450. [DOI] [PubMed] [Google Scholar]
- 40. Elhusseiny A, Hamel E (2000) Muscarinic–but not nicotinic–acetylcholine receptors mediate a nitric oxide‐dependent dilation in brain cortical arterioles: a possible role for the M5 receptor subtype. J Cereb Blood Flow Metab 20:298–305. [DOI] [PubMed] [Google Scholar]
- 41. Ellis RJ, Olichney JM, Thal LJ, Mirra SS, Morris JC, Beekly D, Heyman A (1996) Cerebral amyloid angiopathy in the brains of patients with Alzheimer's disease: the CERAD experience, Part XV. Neurology 46:1592–1596. [DOI] [PubMed] [Google Scholar]
- 42. Esiri MM, Wilcock GK (1986) Cerebral amyloid angiopathy in dementia and old age. J Neurol Neurosurg Psychiatry 49:1221–1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Esiri MM, Wilcock GK, Morris JH (1997) Neuropathological assessment of the lesions of significance in vascular dementia. J Neurol Neurosurg Psychiatry 63:749–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Fergus A, Lee KS (1997) GABAergic regulation of cerebral microvascular tone in the rat. J Cereb Blood Flow Metab 17:992–1003. [DOI] [PubMed] [Google Scholar]
- 45. Fischer VW, Siddiqi A, Yusufaly Y (1990) Altered angioarchitecture in selected areas of brains with Alzheimer's disease. Acta Neuropathol 79:672–679. [DOI] [PubMed] [Google Scholar]
- 46. Gentile MT, Vecchione C, Maffei A, Aretini A, Marino G, Poulet R et al (2004) Mechanisms of soluble β‐amyloid impairment of endothelial function. J Biol Chem 279:48135–48142. [DOI] [PubMed] [Google Scholar]
- 47. Greer JM, Lees MB (2002) Myelin proteolipid protein–the first 50 years. Int J Biochem Cell Biol 34:211–215. [DOI] [PubMed] [Google Scholar]
- 48. Hamel E (2004) Cholinergic modulation of the cortical microvascular bed. Prog Brain Res 145:171–178. [DOI] [PubMed] [Google Scholar]
- 49. Hamilton NB, Attwell D, Hall CN (2010) Pericyte‐mediated regulation of capillary diameter: a component of neurovascular coupling in health and disease. Front Neuroenergetics 2:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Haque SU, Dashwood MR, Heetun M, Shiwen X, Farooqui N, Ramesh B et al (2013) Efficacy of the specific endothelin a receptor antagonist zibotentan (ZD4054) in colorectal cancer: a preclinical study. Mol Cancer Ther 12:1556–1567. [DOI] [PubMed] [Google Scholar]
- 51. Hunter JM, Kwan J, Malek‐Ahmadi M, Maarouf CL, Kokjohn TA, Belden C et al (2012) Morphological and pathological evolution of the brain microcirculation in aging and Alzheimer's disease. PLoS One 7:e36893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Jeynes B, Provias J (2009) Significant negative correlations between capillary expressed eNOS and Alzheimer lesion burden. Neurosci Lett 463:244–248. [DOI] [PubMed] [Google Scholar]
- 53. Kakinuma Y, Furihata M, Akiyama T, Arikawa M, Handa T, Katare RG, Sato T (2010) Donepezil, an acetylcholinesterase inhibitor against Alzheimer's dementia, promotes angiogenesis in an ischemic hindlimb model. J Mol Cell Cardiol 48:680–693. [DOI] [PubMed] [Google Scholar]
- 54. Kalaria RN, Cohen DL, Premkumar DR, Nag S, LaManna JC, Lust WD (1998) Vascular endothelial growth factor in Alzheimer's disease and experimental cerebral ischemia. Brain Res Mol Brain Res 62:101–105. [DOI] [PubMed] [Google Scholar]
- 55. Kar S, Quirion R (2004) Amyloid β peptides and central cholinergic neurons: functional interrelationship and relevance to Alzheimer's disease pathology. Prog Brain Res 145:261–274. [DOI] [PubMed] [Google Scholar]
- 56. Kehoe PG (2003) The renin‐angiotensin‐aldosterone system and Alzheimer s disease? J Renin Angiotensin Aldosterone Syst 4:80–93. [DOI] [PubMed] [Google Scholar]
- 57. Kehoe PG, Miners S, Love S (2009) Angiotensins in Alzheimer's disease ‐ friend or foe? Trends Neurosci 32:619–628. [DOI] [PubMed] [Google Scholar]
- 58. Kehoe PG, Passmore PA (2012) The renin‐angiotensin system and antihypertensive drugs in Alzheimer's disease: current standing of the angiotensin hypothesis? J Alzheimers Dis 30 Suppl 2:S251–S268. [DOI] [PubMed] [Google Scholar]
- 59. Kehoe PG, Wilcock GK (2007) Is inhibition of the renin‐angiotensin system a new treatment option for Alzheimer's disease? Lancet Neurol 6:373–378. [DOI] [PubMed] [Google Scholar]
- 60. Kitaguchi H, Ihara M, Saiki H, Takahashi R, Tomimoto H (2007) Capillary beds are decreased in Alzheimer's disease, but not in Binswanger's disease. Neurosci Lett 417:128–131. [DOI] [PubMed] [Google Scholar]
- 61. Kivipelto M, Helkala EL, Laakso MP, Hanninen T, Hallikainen M, Alhainen K et al (2002) Apolipoprotein E ε4 allele, elevated midlife total cholesterol level, and high midlife systolic blood pressure are independent risk factors for late‐life Alzheimer disease. Ann Intern Med 137:149–155. [DOI] [PubMed] [Google Scholar]
- 62. Krick S, Hanze J, Eul B, Savai R, Seay U, Grimminger F et al (2005) Hypoxia‐driven proliferation of human pulmonary artery fibroblasts: cross‐talk between HIF‐1α and an autocrine angiotensin system. FASEB J 19:857–859. [DOI] [PubMed] [Google Scholar]
- 63. Krimer LS, Muly EC, 3rd , Williams GV, Goldman‐Rakic PS (1998) Dopaminergic regulation of cerebral cortical microcirculation. Nat Neurosci 1:286–289. [DOI] [PubMed] [Google Scholar]
- 64. Lamoke F, Mazzone V, Persichini T, Maraschi A, Harris MB, Venema RC et al (2015) Amyloid β peptide‐induced inhibition of endothelial nitric oxide production involves oxidative stress‐mediated constitutive eNOS/HSP90 interaction and disruption of agonist‐mediated Akt activation. J Neuroinflammation 12:84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Langbaum JB, Chen K, Caselli RJ, Lee W, Reschke C, Bandy D et al (2010) Hypometabolism in Alzheimer‐affected brain regions in cognitively healthy Latino individuals carrying the apolipoprotein E ε4 allele. Arch Neurol 67:462–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Launer LJ, Ross GW, Petrovitch H, Masaki K, Foley D, White LR, Havlik RJ (2000) Midlife blood pressure and dementia: the Honolulu‐Asia aging study. Neurobiol Aging 21:49–55. [DOI] [PubMed] [Google Scholar]
- 67. Lee S, Viqar F, Zimmerman ME, Narkhede A, Tosto G, Benzinger TL et al (2016) White matter hyperintensities are a core feature of Alzheimer's disease: evidence from the Dominantly Inherited Alzheimer Network. Ann Neurol (in press). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Liu AK, Chang RC, Pearce RKB, Gentleman SM (2015) Nucleus basalis of Meynert revisited: anatomy, history and differential involvement in Alzheimer's and Parkinson's disease. Acta Neuropathol 129:527–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Love S, Chalmers K, Ince P, Esiri M, Attems J, Jellinger K et al (2014) Development, appraisal, validation and implementation of a consensus protocol for the assessment of cerebral amyloid angiopathy in post‐mortem brain tissue. Am J Neurodegener Dis 3:19–32. [PMC free article] [PubMed] [Google Scholar]
- 70. Love S, Miners JS (2015) White matter hypoperfusion and damage in dementia: post‐mortem assessment. Brain Pathol 25:99–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Love S, Miners JS (2016) Cerebrovascular disease in ageing and Alzheimer's disease. Acta Neuropathol 131:645–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Love S, Nicoll JA, Hughes A, Wilcock GK (2003) APOE and cerebral amyloid angiopathy in the elderly. Neuroreport 14:1535–1536. [DOI] [PubMed] [Google Scholar]
- 73. Masuda J, Tanaka K, Ueda K, Omae T (1988) Autopsy study of incidence and distribution of cerebral amyloid angiopathy in Hisayama, Japan. Stroke 19:205–210. [DOI] [PubMed] [Google Scholar]
- 74. Miners JS, Ashby E, Van Helmond Z, Chalmers KA, Palmer LE, Love S, Kehoe PG (2008) Angiotensin‐converting enzyme (ACE) levels and activity in Alzheimer's disease, and relationship of perivascular ACE‐1 to cerebral amyloid angiopathy. Neuropathol Appl Neurobiol 34:181–193. [DOI] [PubMed] [Google Scholar]
- 75. Miners JS, Palmer J, Love S (in press) Pathophysiology of hypoperfusion of the precuneus in early Alzheimer's disease. Brain Pathol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Miners JS, Palmer JC, Tayler H, Palmer LE, Ashby E, Kehoe PG, Love S (2014) Aβ degradation or cerebral perfusion? Divergent effects of multifunctional enzymes. Front Aging Neurosci 6:238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Miners JS, van Helmond Z, Raiker M, Love S, Kehoe PG (2010) ACE variants and association with brain Aβ levels in Alzheimer's disease. Am J Transl Res 3:73–80. [PMC free article] [PubMed] [Google Scholar]
- 78. Miners S, Ashby E, Baig S, Harrison R, Tayler H, Speedy E et al (2009) Angiotensin‐converting enzyme levels and activity in Alzheimer's disease: differences in brain and CSF ACE and association with ACE1 genotypes. Am J Transl Res 1:163–177. [PMC free article] [PubMed] [Google Scholar]
- 79. Mondello S, Buki A, Barzo P, Randall J, Provuncher G, Hanlon D et al (2014) CSF and plasma amyloid‐β temporal profiles and relationships with neurological status and mortality after severe traumatic brain injury. Sci Rep 4:6446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Montagne A, Barnes SR, Sweeney MD, Halliday MR, Sagare AP, Zhao Z et al (2015) Blood‐brain barrier breakdown in the aging human hippocampus. Neuron 85:296–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Morris CD, Rose A, Curwen J, Hughes AM, Wilson DJ, Webb DJ (2005) Specific inhibition of the endothelin A receptor with ZD4054: clinical and pre‐clinical evidence. Br J Cancer 92:2148–2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Morris MC, Scherr PA, Hebert LE, Glynn RJ, Bennett DA, Evans DA (2001) Association of incident Alzheimer disease and blood pressure measured from 13 years before to 2 years after diagnosis in a large community study. Arch Neurol 58:1640–1646. [DOI] [PubMed] [Google Scholar]
- 83. Nagata K, Kondoh Y, Atchison R, Sato M, Satoh Y, Watahiki Y et al (2000) Vascular and metabolic reserve in Alzheimer's disease. Neurobiol Aging 21:301–307. [DOI] [PubMed] [Google Scholar]
- 84. Nagata K, Sato M, Satoh Y, Watahiki Y, Kondoh Y, Sugawara M et al (2002) Hemodynamic aspects of Alzheimer's disease. Ann N Y Acad Sci 977:391–402. [DOI] [PubMed] [Google Scholar]
- 85. Nation DA, Wierenga CE, Clark LR, Dev SI, Stricker NH, Jak AJ et al (2013) Cortical and subcortical cerebrovascular resistance index in mild cognitive impairment and Alzheimer's disease. J Alzheimers Dis 36:689–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Niwa K, Carlson GA, Iadecola C (2000) Exogenous Aβ1‐40 reproduces cerebrovascular alterations resulting from amyloid precursor protein overexpression in mice. J Cereb Blood Flow Metab 20:1659–1668. [DOI] [PubMed] [Google Scholar]
- 87. Niwa K, Kazama K, Younkin L, Younkin SG, Carlson GA, Iadecola C (2002) Cerebrovascular autoregulation is profoundly impaired in mice overexpressing amyloid precursor protein. Am J Physiol Heart Circ Physiol 283:H315–H323. [DOI] [PubMed] [Google Scholar]
- 88. Niwa K, Kazama K, Younkin SG, Carlson GA, Iadecola C (2002) Alterations in cerebral blood flow and glucose utilization in mice overexpressing the amyloid precursor protein. Neurobiol Dis 9:61–68. [DOI] [PubMed] [Google Scholar]
- 89. Niwa K, Porter VA, Kazama K, Cornfield D, Carlson GA, Iadecola C (2001) Aβ‐peptides enhance vasoconstriction in cerebral circulation. Am J Physiol Heart Circ Physiol 281:H2417–H2424. [DOI] [PubMed] [Google Scholar]
- 90. Niwa K, Younkin L, Ebeling C, Turner SK, Westaway D, Younkin S et al (2000) Aβ1‐40‐related reduction in functional hyperemia in mouse neocortex during somatosensory activation. Proc Natl Acad Sci U S A 97:9735–9740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Noll G, Wenzel RR, Luscher TF (1996) Endothelin and endothelin antagonists: potential role in cardiovascular and renal disease. Mol Cell Biochem 157:259–267. [DOI] [PubMed] [Google Scholar]
- 92. Nor JE, Christensen J, Mooney DJ, Polverini PJ (1999) Vascular endothelial growth factor (VEGF)‐mediated angiogenesis is associated with enhanced endothelial cell survival and induction of Bcl‐2 expression. Am J Pathol 154:375–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Okazaki H, Reagan TJ, Campbell RJ (1979) Clinicopathologic studies of primary cerebral amyloid angiopathy. Mayo Clin Proc 54:22–31. [PubMed] [Google Scholar]
- 94. Pákáski M, Kálmán J (2008) Interactions between the amyloid and cholinergic mechanisms in Alzheimer's disease. Neurochem Int 53:103–111. [DOI] [PubMed] [Google Scholar]
- 95. Palmer J, Love S (2011) Endothelin receptor antagonists: potential in Alzheimer's disease. Pharmacol Res 63:525–531. [DOI] [PubMed] [Google Scholar]
- 96. Palmer JC, Baig S, Kehoe PG, Love S (2009) Endothelin‐converting enzyme‐2 is increased in Alzheimer's disease and up‐regulated by Aβ. Am J Pathol 175:262–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Palmer JC, Barker R, Kehoe PG, Love S (2012) Endothelin‐1 is elevated in Alzheimer's disease and upregulated by amyloid‐β. J Alzheimers Dis 29:853–861. [DOI] [PubMed] [Google Scholar]
- 98. Palmer JC, Tayler HM, Love S (2013) Endothelin‐converting enzyme‐1 activity, endothelin‐1 production, and free radical‐dependent vasoconstriction in Alzheimer's disease. J Alzheimers Dis 36:577–587. [DOI] [PubMed] [Google Scholar]
- 99. Paris D, Humphrey J, Quadros A, Patel N, Crescentini R, Crawford F, Mullan M (2003) Vasoactive effects of Aβ in isolated human cerebrovessels and in a transgenic mouse model of Alzheimer's disease: role of inflammation. Neurol Res 25:642–651. [DOI] [PubMed] [Google Scholar]
- 100. Paris D, Patel N, DelleDonne A, Quadros A, Smeed R, Mullan M (2004) Impaired angiogenesis in a transgenic mouse model of cerebral amyloidosis. Neurosci Lett 366:80–85. [DOI] [PubMed] [Google Scholar]
- 101. Paris D, Town T, Parker TA, Humphrey J, Mullan M (1998) Isoform‐specific vasoconstriction induced by apolipoprotein E and modulation of this effect by Alzheimer's β‐amyloid peptide. Neurosci Lett 256:73–76. [DOI] [PubMed] [Google Scholar]
- 102. Paris D, Townsend K, Quadros A, Humphrey J, Sun J, Brem S et al (2004) Inhibition of angiogenesis by Aβ peptides. Angiogenesis 7:75–85. [DOI] [PubMed] [Google Scholar]
- 103. Park L, Anrather J, Zhou P, Frys K, Pitstick R, Younkin S et al (2005) NADPH‐oxidase‐derived reactive oxygen species mediate the cerebrovascular dysfunction induced by the amyloid β peptide. J Neurosci 25:1769–1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Park L, Zhou P, Pitstick R, Capone C, Anrather J, Norris EH et al (2008) Nox2‐derived radicals contribute to neurovascular and behavioral dysfunction in mice overexpressing the amyloid precursor protein. Proc Natl Acad Sci U S A 105:1347–1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Patel NS, Mathura VS, Bachmeier C, Beaulieu‐Abdelahad D, Laporte V, Weeks O et al (2010) Alzheimer's β‐amyloid peptide blocks vascular endothelial growth factor mediated signaling via direct interaction with VEGFR‐2. J Neurochem 112:66–76. [DOI] [PubMed] [Google Scholar]
- 106. Paulson OB, Strandgaard S, Edvinsson L (1990) Cerebral autoregulation. Cerebrovasc Brain Metab Rev 2:161–192. [PubMed] [Google Scholar]
- 107. Peca S, McCreary CR, Donaldson E, Kumarpillai G, Shobha N, Sanchez K et al (2013) Neurovascular decoupling is associated with severity of cerebral amyloid angiopathy. Neurology 81:1659–1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Peppiatt CM, Howarth C, Mobbs P, Attwell D (2006) Bidirectional control of CNS capillary diameter by pericytes. Nature 443:700–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Power MC, Weuve J, Gagne JJ, McQueen MB, Viswanathan A, Blacker D Blood Pressure. The AlzRisk Database. Alzheimer Research Forum.
- 110. Power MC, Weuve J, Gagne JJ, McQueen MB, Viswanathan A, Blacker D (2011) The association between blood pressure and incident Alzheimer disease: a systematic review and meta‐analysis. Epidemiology 22:646–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Premkumar DR, Cohen DL, Hedera P, Friedland RP, Kalaria RN (1996) Apolipoprotein E‐ε4 alleles in cerebral amyloid angiopathy and cerebrovascular pathology associated with Alzheimer's disease. Am J Pathol 148:2083–2095. [PMC free article] [PubMed] [Google Scholar]
- 112. Price JM, Sutton ET, Hellermann A, Thomas T (1997) β‐amyloid induces cerebrovascular endothelial dysfunction in the rat brain. Neurol Res 19:534–538. [DOI] [PubMed] [Google Scholar]
- 113. Raichle ME, Hartman BK, Eichling JO, Sharpe LG (1975) Central noradrenergic regulation of cerebral blood flow and vascular permeability. Proc Natl Acad Sci U S A 72:3726–3730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Raja SG, Dreyfus GD (2008) Current status of bosentan for treatment of pulmonary hypertension. Ann Card Anaesth 11:6–14. [DOI] [PubMed] [Google Scholar]
- 115.Reducing pathology in Alzheimer's disease through angiotensin targeting. [DOI] [PubMed]
- 116. Rodrigue KM, Rieck JR, Kennedy KM, Devous MD, Sr. , Diaz‐Arrastia R, Park DC (2013) Risk factors for β‐amyloid deposition in healthy aging: vascular and genetic effects. JAMA Neurol 70:600–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Rossi GP, Sacchetto A, Cesari M, Pessina AC Interactions between endothelin‐1 and the renin‐angiotensin‐aldosterone system. Cardiovasc Res 43:300–307. [DOI] [PubMed] [Google Scholar]
- 118. Rubin LJ, Badesch DB, Barst RJ, Galie N, Black CM, Keogh A et al (2002) Bosentan therapy for pulmonary arterial hypertension. N Engl J Med 346:896–903. [DOI] [PubMed] [Google Scholar]
- 119. Rubin LJ, Roux S (2002) Bosentan: a dual endothelin receptor antagonist. Expert Opin Investig Drugs 11:991–1002. [DOI] [PubMed] [Google Scholar]
- 120. Sagare AP, Bell RD, Zhao Z, Ma Q, Winkler EA, Ramanathan A, Zlokovic BV (2013) Pericyte loss influences Alzheimer‐like neurodegeneration in mice. Nat Commun 4:2932. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 121. Schmechel DE, Saunders AM, Strittmatter WJ, Crain BJ, Hulette CM, Joo SH et al (1993) Increased amyloid β‐peptide deposition in cerebral cortex as a consequence of apolipoprotein E genotype in late‐onset Alzheimer disease. Proc Natl Acad Sci U S A 90:9649–9653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Scott JA, Braskie MN, Tosun D, Thompson PM, Weiner M, DeCarli C, Carmichael OT Alzheimer's Disease Neuroimaging I (2015) Cerebral amyloid and hypertension are independently associated with white matter lesions in elderly. Front Aging Neurosci 7:221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Sengillo JD, Winkler EA, Walker CT, Sullivan JS, Johnson M, Zlokovic BV (2013) Deficiency in mural vascular cells coincides with blood‐brain barrier disruption in Alzheimer's disease. Brain Pathol 23:303–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Shimizu S, Hanyu H, Iwamoto T, Koizumi K, Abe K (2006) SPECT follow‐up study of cerebral blood flow changes during Donepezil therapy in patients with Alzheimer's disease. J Neuroimaging 16:16–23. [DOI] [PubMed] [Google Scholar]
- 125. Shin HK, Jones PB, Garcia‐Alloza M, Borrelli L, Greenberg SM, Bacskai BJ et al (2007) Age‐dependent cerebrovascular dysfunction in a transgenic mouse model of cerebral amyloid angiopathy. Brain 130:2310–2319. [DOI] [PubMed] [Google Scholar]
- 126. Smith EE, Vijayappa M, Lima F, Delgado P, Wendell L, Rosand J, Greenberg SM (2008) Impaired visual evoked flow velocity response in cerebral amyloid angiopathy. Neurology 71:1424–1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Stow LR, Jacobs ME, Wingo CS, Cain BD (2011) Endothelin‐1 gene regulation. Faseb J 25:16–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Suhara T, Magrane J, Rosen K, Christensen R, Kim HS, Zheng B et al (2003) Aβ42 generation is toxic to endothelial cells and inhibits eNOS function through an Akt/GSK‐3beta signaling‐dependent mechanism. Neurobiol Aging 24:437–451. [DOI] [PubMed] [Google Scholar]
- 129. Tan XL, Xue YQ, Ma T, Wang X, Li JJ, Lan L et al (2015) Partial eNOS deficiency causes spontaneous thrombotic cerebral infarction, amyloid angiopathy and cognitive impairment. Mol Neurodegener 10:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Tarkowski E, Issa R, Sjogren M, Wallin A, Blennow K, Tarkowski A, Kumar P (2002) Increased intrathecal levels of the angiogenic factors VEGF and TGF‐β in Alzheimer's disease and vascular dementia. Neurobiol Aging 23:237–243. [DOI] [PubMed] [Google Scholar]
- 131. Tarumi T, Dunsky DI, Khan MA, Liu J, Hill C, Armstrong K et al (2014) Dynamic cerebral autoregulation and tissue oxygenation in amnestic mild cognitive impairment. J Alzheimers Dis 41:765–778. [DOI] [PubMed] [Google Scholar]
- 132. Tayler HM, Palmer JC, Thomas TL, Kehoe PG, Paton JFR, Love S (2014) Investigating the relationship between cerebral Αβ and systemic hypertension [Abstract]. Neuropathol Appl Neurobiol 40:41. [Google Scholar]
- 133. Thal DR, Ghebremedhin E, Rub U, Yamaguchi H, Del Tredici K, Braak H (2002) Two types of sporadic cerebral amyloid angiopathy. J Neuropathol Exp Neurol 61:282–293. [DOI] [PubMed] [Google Scholar]
- 134. Thomas T, Miners S, Love S (2015) Post‐mortem assessment of hypoperfusion of cerebral cortex in Alzheimer's disease and vascular dementia. Brain 138:1059–1069. [DOI] [PubMed] [Google Scholar]
- 135. Thomas T, Thomas G, McLendon C, Sutton T, Mullan M (1996) Amyloid‐mediated vasoactivity and vascular endothelial damage. Nature 380:168–171. [DOI] [PubMed] [Google Scholar]
- 136. Toews AD, White FV, Morell P (1988) Metabolism of functional groups modifying the CNS myelin‐associated glycoprotein. J Neurochem 51:1646–1650. [DOI] [PubMed] [Google Scholar]
- 137. Tohgi H, Yonezawa H, Takahashi S, Sato N, Kato E, Kudo M et al (1998) Cerebral blood flow and oxygen metabolism in senile dementia of Alzheimer's type and vascular dementia with deep white matter changes. Neuroradiology 40:131–137. [DOI] [PubMed] [Google Scholar]
- 138. Tomonaga M (1981) Cerebral amyloid angiopathy in the elderly. J Am Geriatr Soc 29:151–157. [DOI] [PubMed] [Google Scholar]
- 139. Tong XK, Hamel E (1999) Regional cholinergic denervation of cortical microvessels and nitric oxide synthase‐containing neurons in Alzheimer's disease. Neuroscience 92:163–175. [DOI] [PubMed] [Google Scholar]
- 140. Townsend KP, Obregon D, Quadros A, Patel N, Volmar C, Paris D, Mullan M (2002) Proinflammatory and vasoactive effects of Aβ in the cerebrovasculature. Ann N Y Acad Sci 977:65–76. [DOI] [PubMed] [Google Scholar]
- 141. Tschope C, Schultheiss HP, Walther T (2002) Multiple interactions between the renin‐angiotensin and the kallikrein‐kinin systems: role of ACE inhibition and AT1 receptor blockade. J Cardiovasc Pharmacol 39:478–487. [DOI] [PubMed] [Google Scholar]
- 142. Vaucher E, Hamel E (1995) Cholinergic basal forebrain neurons project to cortical microvessels in the rat: electron microscopic study with anterogradely transported Phaseolus vulgaris leucoagglutinin and choline acetyltransferase immunocytochemistry. J Neurosci 15:7427–7441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Vinters HV, Gilbert JJ (1983) Cerebral amyloid angiopathy: incidence and complications in the aging brain. II. The distribution of amyloid vascular changes. Stroke 14:924–928. [DOI] [PubMed] [Google Scholar]
- 144. Waite JJ, Holschneider DP, Scremin OU (1999) Selective immunotoxin‐induced cholinergic deafferentation alters blood flow distribution in the cerebral cortex. Brain Res 818:1–11. [DOI] [PubMed] [Google Scholar]
- 145. Winkler EA, Sagare AP, Zlokovic BV (2014) The pericyte: a forgotten cell type with important implications for Alzheimer's disease? Brain Pathol 24:371–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Yamada M, Tsukagoshi H, Otomo E, Hayakawa M (1987) Cerebral amyloid angiopathy in the aged. J Neurol 234:371–376. [DOI] [PubMed] [Google Scholar]
- 147. Yan Z, Feng J (2004) Alzheimer's disease: interactions between cholinergic functions and β‐amyloid. Curr Alzheimer Res 1:241–248. [DOI] [PubMed] [Google Scholar]
- 148. Yang SP, Bae DG, Kang HJ, Gwag BJ, Gho YS, Chae CB (2004) Co‐accumulation of vascular endothelial growth factor with β‐amyloid in the brain of patients with Alzheimer's disease. Neurobiol Aging 25:283–290. [DOI] [PubMed] [Google Scholar]
- 149. Zetterberg H, Mortberg E, Song L, Chang L, Provuncher GK, Patel PP et al (2011) Hypoxia due to cardiac arrest induces a time‐dependent increase in serum amyloid β levels in humans. PLoS One 6:e28263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Zhang ZG, Zhang L, Jiang Q, Zhang R, Davies K, Powers C et al (2000) VEGF enhances angiogenesis and promotes blood‐brain barrier leakage in the ischemic brain. J Clin Invest 106:829–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Zhu J, Song W, Li L, Fan X (2016) Endothelial nitric oxide synthase: a potential therapeutic target for cerebrovascular diseases. Mol Brain 9:30. [DOI] [PMC free article] [PubMed] [Google Scholar]