

Abstract
Chronic alcohol abuse is associated with brain damage in a sex‐specific fashion, but the mechanisms involved are poorly described and remain controversial. Previous results have suggested that astrocyte gene expression is influenced by ethanol intoxication and during abstinence in vivo. Here, bioinformatic analysis of astrocyte‐enriched ethanol‐regulated genes in vivo revealed ubiquitin pathways as an ethanol target, but with sexually dimorphic cytokine signaling and changes associated with brain aging in females and not males. Consistent with this result, astrocyte activation was observed after exposure in female but not male animals, with reduced S100β levels in the anterior cingulate cortex and increased GFAP + cells in the hippocampus. In primary culture, the direct effects of chronic ethanol exposure followed by recovery on sex‐specific astrocyte function were examined. Male astrocyte responses were consistent with astrocyte deactivation with reduced GFAP expression during ethanol exposure. In contrast, female astrocytes exhibited increased expression of Tnf, reduced expression of the neuroprotective cytokine Tgfb1, disrupted bioenergetics and reduced excitatory amino acid uptake following exposure or recovery. These results indicate widespread astrocyte dysfunction in ethanol‐exposed females and suggest a mechanism that may underlie increased vulnerability to ethanol‐induced neurotoxicity in females.
Keywords: alcohol, astrocyte, glutamate uptake, sexually dimorphic, tumor necrosis factor
Introduction
Chronic alcohol abuse is a major public health problem and is frequently associated with cognitive impairment, neurotoxicity and brain damage among many other detrimental health consequences. For example, about half of the nearly 20 million alcoholics in the United States develop neuropsychological difficulties. Alcoholic dementia, associated with global severe amnesia and intellectual impairment, is the second‐leading cause of adult dementia in the United States and accounts for 10% of all cases. Ethanol‐induced brain damage includes brain volume reduction, loss of cortical neurons 61, increased microglia 46, and in some cases reactive astrogliosis and glial scar formation 13, 37, 74. While most studies have focused on direct effects on neurons, the role played by astrocytes, the most abundant cell type in the brain 12, is very poorly understood.
Reactive astrogliosis and glial scar formation occurs in a variety of settings, such as after stroke (ischemia), significant trauma and in neurodegenerative diseases, suggesting astrocytes are critically involved with some forms of brain insult. Astrocytes support neuronal health through structural and metabolic support, including maintenance of glutamate homeostasis. Neurotoxicity as a consequence of chronic alcohol abuse is postulated to result from decreased inhibitory GABAergic function concomitant with increased excitatory glutamatergic function as a consequence of neuroadaptation, resulting in oxidative stress, inflammation and excitotoxic brain damage during withdrawal 96. Astrocytes protect neurons from glutamate excitotoxicity and oxidative stress by removing glutamate from the extracellular space 17 via sodium‐dependent excitatory amino acid transporters (EAATs). Two of the five glutamate transporters, GLAST (human equivalent EAAT1) and GLT‐1 (EAAT2), are expressed predominantly by astrocytes and are responsible for the bulk of glutamate uptake in the brain 22. Elevated extracellular levels of glutamate result in inhibition of glutathione production 109 and persistent activation of glutamate receptors including N‐methyl‐D‐aspartate (NMDA) receptors, which may be a particularly important mediator of ethanol‐induced brain damage 41, 96. High concentrations of glutamate in the extracellular space can also lead to excitotoxic neuronal damage 64 mediated at least in part by accumulation of nitric oxide 106. Previous studies have demonstrated that acute exposure of unsexed primary hippocampal astrocytes to ethanol (15 minutes; 1–200 mM) evoked a dose‐dependent increase in glutamate secretion 103 and increased glial fibrillary acidic protein (GFAP) levels 39. Although changes in glutamate signaling have been observed in alcoholics during detoxification and rodents during withdrawal 47, the specific role of astrocytes in mediating the effects of chronic alcohol exposure in the central nervous system (CNS) is relatively unexplored.
Sexually dimorphic responses to alcohol exposure are becoming better characterized [see 121], with women more vulnerable to EtOH‐induced organ damage in the periphery. Females have higher rates or greater severity of cardiomyopathy, peripheral neuropathy, some types of cancer and liver cirrhosis 5, 59. Increased vulnerability to ethanol‐induced brain damage in the prefrontal cortex in females has been observed in some studies 44, 118. Other studies have not confirmed female vulnerability, but have noted different “brain morphological deficits” between males and females supporting the notion that neurodamage occurs via different mechanisms depending on sex 94. Nevertheless, increased vulnerability to EtOH‐induced neurotoxicity in females remains controversial [reviewed in 51] with additional studies needed to support or refute this hypothesis. This finding is buttressed by the observation that chronic ethanol exposure is associated with a strongly sexually dimorphic transcriptional response 44, 118, 119. In those studies, significantly regulated transcripts included several genes that are exclusively or predominantly expressed in astrocytes, suggesting that a component of the neuroadapative response in the brain after ethanol exposure reflects changes in astrocyte gene expression that are distinct between males and females. In the astrocyte population, limited characterization of sex differences has been described both in vivo and in primary astrocyte cultures harvested from male and female neonates. Females, but not males, exhibit increased astrocyte activation that is uncorrelated with infarct size in a model of ischemic stroke 26. Using sex‐specific primary cultures, differences in inflammatory response and sensitivity to stressors have previously been observed in primary astrocytes harvested from neonates 7, 72, 104. Such sexually dimorphic responses in astrocytes appear as a consequence of organizational gonadal steroid effects 122, also known as hormone imprinting, that result from a testosterone surge in male pups in utero just before birth and in the neonate shortly after birth 56, 127. Organizational effects are long lived or permanent, and are not dependent on subsequent exposure to hormone. Thus, male cells are different from female cells even in the neonatal animal. However, the specific consequences of chronic alcohol exposure and withdrawal on sex‐specific astrocyte function have not been characterized, either in cultures or in vivo.
In addition to playing a key role in glutamate homeostasis, recent work has also recognized the contribution of astrocytes to inflammatory signaling in the CNS 24. For example, evidence suggests astrocytes play a key role in development of excitatory autoimmune encephalomyelitis, an immune‐based model of the neurodegenerative disease multiple sclerosis 76. Astrocytes exhibit polarization toward pro‐ or anti‐inflammatory phenotypes similar to those exhibited by microglia (the resident immune cells of the CNS) and macrophages, and may regulate responding in microglia, oligodendrocytes and the adaptive immune system 55, 75. Importantly, astrocytes can produce potentially neurotoxic inflammatory mediators including tumor necrosis factor (TNF) 55. We have recently shown that following chronic intoxication in vivo, females exhibit a pro‐inflammatory response in the medial prefrontal cortex that includes increased Tnf expression, while males show a pattern consistent with immune suppression 118, 119. Associated with this pro‐inflammatory tone, females also exhibit increased neurotoxicity 44, 118. Combined, our studies suggest the possibility that sexually dimorphic ethanol‐induced neurotoxicity in the frontal cortex may be mediated in part by astrocytes both via induction and modulation of inflammatory signaling and through changes in astrocyte function including dysregulation of glutamate homeostasis. To date, these possibilities have not been carefully evaluated.
Thus, given evidence that vulnerability to ethanol‐induced brain damage is sex specific, that astrocytes can influence inflammatory signaling and glutamate levels, and that astrocyte function and response can be sexually dimorphic, the purpose of this study was to elucidate the effects of chronic ethanol intoxication specifically on astrocyte function in males and females. The hypothesis tested in these studies was that in females, astrocyte dysfunction is a critical component of the response to ethanol that may underlie subsequent ethanol‐induced neurodegeneration. Studies were performed in both males and females to characterize changes in astrocyte expression and function following chronic intoxication and withdrawal/recovery in vivo and in in vitro sex‐specific primary astrocyte cultures. The results demonstrate that females but not males exhibit enhanced astrocyte reactivity and gene expression changes in vivo and in astrocyte cultures that are consistent with increased excitatory glutamatergic and inflammatory signaling, altered bioenergetic homeostasis, and disrupted ubiquitin regulation following intoxication. The dysfunction seen in astrocytes exposed to chronic ethanol identifies several mechanisms by which astrocyte responses in females could create a permissive and vulnerable environment within the brain to enhance damage.
Methods
Animal subjects
For in vivo examination of astrocyte activation, male and female mice from both independently derived replicates of the selectively bred withdrawal seizure‐prone (WSP) and withdrawal seizure‐resistant (WSR) mice (provided by J. Crabbe, Portland, OR) were used. For array studies, female control mice were 69 ± 2 days old, female EtOH mice were 70 ± 2 days old, male control mice were 71 ± 1 days old, and male EtOH mice were 70 ± 1 days old at the start of the experiment. For immunohistochemical studies, female control mice were 74 ± 2 days old, female EtOH mice were 72 ± 2 days old, male control mice were 73 ± 2 days old, and male EtOH mice were 72 ± 3 days old at the start of the experiment. There were no significant differences between ages in any of these conditions. Breeding pairs of male and female B6D2F1 mice were purchased from Jackson Laboratory West for the isolation of primary astrocytes from B6D2F2 pups. Mice were maintained under a light/dark cycle of 0600–1800 light, with water and Purina Lab Diet chow available ad libitum. Room temperatures were maintained at 22 ± 1°C. Animal procedures were approved by the Institutional Animal Care and Use Committee at the Portland Oregon VA Health Care System and followed US National Institutes of Health animal welfare guidelines.
Chronic ethanol exposure
Mice were exposed to ethanol continuously for 72 h in vapor inhalation chambers as previously described 44, 119. This paradigm has been characterized as a vulnerability model [see 119], with a single chronic exposure to high intoxication, followed by synchronized withdrawal. Briefly, on day 1, ethanol animals were weighed, injected i.p. with ethanol (20% v/v) at 1.5 g/kg with 1 mmol/kg pyrazole (Pyz) to reduce variations in blood ethanol concentration (BEC), and placed into vapor inhalation chambers. On all days for control (Con) animals and days 2 and 3 for ethanol animals, mice were weighed, given i.p. injections of Pyz dissolved in 0.9% saline, and placed in vapor inhalation chambers. A saline‐only control was not included because previous analysis using an unbiased PCR differential display screening method that included WSR mice failed to identify significant differences in gene expression across a broad spectrum of genes between saline and Pyz‐treated animals in this paradigm 107. Ethanol‐exposed animals had tail blood drawn on days 2, 3 and 4 of the study for BEC determination via gas chromatography as previously described 14. BECs were in the highly intoxicated range for all groups and were not different between females and males [Ps > 0.05; BECs: female WSP = 2.31 ± 0.06 mg/mL (231 ± 60 mg/dL), male WSP = 2.21 ± 0.11 mg/mL, female WSR 2.22 ± 0.04 mg/mL, male WSR = 2.11 ± 0.11 mg/mL].
Astrocyte bioinformatics analysis
Gene profiling and bioinformatic analyses were performed on tissue harvested from medial prefrontal cortex at peak withdrawal following chronic intoxication as previously described 44, 119. Profiling analysis identified astrocytes as a component of the response and thus a cellular target of ethanol 44, 119. To characterize expression differences associated specifically with astrocytes and identify the biological pathways altered by chronic ethanol, all ethanol‐regulated genes from intoxication and peak withdrawal time points were compared using the “Astrocyte All” list as previously described 119. Briefly, the astrocyte list was compiled from a variety of sources 19, 29, 87, 88, where each publication identified genes with enriched expression in astrocyte cells. The “Astrocyte All” list was compared with our list of ethanol‐regulated genes to identify those genes expressed predominantly or exclusively in astrocytes. The gene lists were converted to mouse Entrez GeneID numbers and uploaded to Ingenuity Pathway Analysis (IPA, Qiagen, Redwood City, CA, USA) software for analysis. IPA was used to identify functional association networks, constructed to identify interactions or relationships between ethanol‐regulated genes enriched in astrocytes at early time points that may not have formal gene ontology annotations.
Brain histology
Following 5 days of ethanol withdrawal (corresponding to peak damage) or control treatment, animals were anesthetized with mouse cocktail [ketamine (7.5 mg/mL), xylazine (0.75 mg/mL) and acepromazine (0.15 mg/mL)], and perfused via the left ventricle with ice cold 0.9% saline followed by 4% paraformaldehyde in 0.1 M phosphate buffered solution (PBS, pH 7.3). This time point was chosen to allow animals to completely progress through the EtOH withdrawal period 44, 84 thought to be important for the neurotoxic effects of EtOH 112 and because it fits within the previously reported window of kainic acid neurotoxicity 52 which may exhibit mechanisms common to EtOH‐induced damage 78. Brains were removed and stored in 4% paraformaldehyde overnight at 4°C. Fixed brains were paraffin embedded and 6 μm sagittal sections were collected. Astrocyte activation was assessed by changes in cell morphology and altered expression of the astrocyte marker GFAP or S100β 111. Four adjacent brain sections collected in series were stained following the standard procedures for GFAP (Dako, Carpinteria, CA, USA; polyclonal rabbit anti‐GFAP) at 1:500 dilution or S100β (Abcam, Cambridge, UK) at 1:5000 dilution overnight at 4°C. Secondary detection of GFAP used Alexafluor 555 conjugated goat anti‐rabbit antiserum (Invitrogen) and counterstaining with 4′,6‐diamidino‐2‐phenylindole (DAPI). Staining was observed by indirect fluorescence microscopy using a Leica DM5000B microscope. GFAP+ cells were counted using the threshold function in Metamorph Premier Image Analysis software (v7.7.7, Molecular Devices, Downingtown, PA, USA). Staining intensity was evaluated as GFAP intensity per cell and found to be unchanged following EtOH (Ps > 0.05). The density of GFAP+ cells was determined by dividing the counts by the area of each region of interest (cells/mm2). Further, there were no differences in hippocampal areas for any of the subregions examined (Ps > 0.05). Detection of S100β used the standard diaminobenzidine method (Vector Laboratories Inc., Burlingame, CA, USA). S100β intensity analysis was carried out using Metamorph Premier Image Analysis software after thresholding using the integrated intensity module. Cell count (normalized to area) and morphology was determined using the Outgrowth module.
Primary astrocyte cultures
Primary cortical type 1 astrocyte cultures were prepared from mouse cortex from male and female B6D2F2 pups between postnatal days 1–3 as described 71. Type 1 astrocytes are the predominant phenotype derived from neonate animals and exhibit a star‐shaped, protoplasmic phenotype and are localized in the gray matter 58, 100. Type 2 astrocytes are fibrous and derived from white matter, type 3 astrocytes are derived from Muller cells in the retina, while type 4 astrocytes (Bergmann glia) are derived from the molecular layer of the cerebellum. WSR and WSP models were originally chosen for these studies based on their withdrawal phenotype differences, with the expectation that this phenotype would be an important factor underlying genetic responses to ethanol intoxication and/or withdrawal. This hypothesis was not supported during the immediate withdrawal period and instead sex, not withdrawal phenotype, was found to exert the greatest influence on responses to chronic ethanol intoxication and early withdrawal 44, 118, 119. Because sex was the most important influence, primary cultures were instead derived using B6D2F2 animals. This allowed us to generalize effects of withdrawal phenotype and emphasize sex‐specific influences driving responses to ethanol. This particular line was chosen based on the observation that both C57/BL6 (B6) and DBA/2J (D2) mice were important contributors to the HS/Ibg progenitor heterogenous stock used to derive the WSR and WSP selected lines 27. Male and female pups were provisionally separated based on genital papilla size/anogenital distances 40 that was confirmed by genotyping for male‐specific sex‐determining region Y (Sry) [modified from 77] using Gapdh as an internal control. Brains were harvested and after the removal of the meninges and the isolation of cortical tissue, the samples were dissociated in 2 mg/mL Trypsin (Sigma, St. Louis, MO, USA) in the presence of 0.006% DNase I (Sigma) at 37°C for 3 minutes. Digestive enzyme activity was stopped using Trypsin Inhibitor (Invitrogen, Grand Island, NY, USA) on ice for 10 minutes, then cells from individual animals were strained through a cell strainer by gravity and pelleted at 1600 × g for 10 minutes. Plates (24‐well) were seeded at a density of 1.5 × 105 cells/cm2 and incubated at 37°C in 5% CO2 in high glucose Dulbecco's modified eagle medium (DMEM) supplemented with 10% heat‐inactivated fetal calf serum, and 100 U/mL penicillin/streptomycin. Contaminating microglial cells were detached from the astrocyte monolayer by shaking and with media replacement. Media was entirely changed a day or two after seeding, then changed twice weekly with fresh media during growth until confluence. Cultures in most cases were confluent 10–14 days after seeding. After another 7 days, chronic ethanol treatments were initiated in highly post‐confluent cultures at approximately day 21.
Chronic ethanol exposure and recovery in astrocyte cultures
To evaluate the effect of chronic ethanol exposure on astrocyte function, differentiated post‐confluent cultures were treated either with or without ethanol (100 mM final concentration) for 4 days similar to chronic EtOH exposures described previously 21. Thus, cultures were subjected to a highly intoxicating concentration of ethanol (461 mg/dL) consistent with what has been observed in emergency room patients and is in the expected range of concentrations observed in chronic alcoholics and binge drinkers 1, 92. Both ethanol and control cultures were placed in a sealed bag that also contained a reservoir of ethanol liquid at the same concentration, that is, 100 mM ethanol, or water. Ethanol concentrations in the media were tested over the chronic exposure time period using a colorimetric assay (QuantiChrom Ethanol Assay Kit, BioAssay Systems). Other cultures were allowed to progress through a “recovery” period of an additional 5 days after removal of the ethanol from the media. The term recovery is used here to distinguish between the more complex in vivo withdrawal associated with neuronal hyperexcitability and the in vitro exposure that occurs in nearly homogenous astrocyte cultures.
LDH assay
Lactate dehydrogenase (LDH) activity was measured to examine toxicity during the culture period and following chronic ethanol treatment using the LDH Cytotoxicity Assay kit from Cayman Chemical (Ann Arbor, MI, USA). All samples of media were diluted 1:10 for the assay procedure. LDH activity was assessed in: cultures grown in the sealed bag vs. cultures in the incubator, at the end of the 4 days of chronic ethanol exposure period, and again during the recovery period. Data for recovery were summed from determinations at the 3‐day media change time point and at the 5‐day time point. Intracellular LDH activity was assessed in cell lysate collected using 0.1% Triton X‐100. The LDH assessment was made using the manufacturer's standard protocol using 100 μL of media. Measurements were taken at 490 nm.
MTT assay
Changes in cell proliferation and viability were determined using a Vybrant 3‐(4,5‐dimethylthiazolyl‐2)‐2,5‐diphenyltetrazolium bromide (MTT) Cell Proliferation Assay kit from Invitrogen/Molecular Probes (Eugene, OR, USA). Cells were seeded in 96‐well plates at a density of 1.5 × 105 cells/cm2. MTT activity was measured in cultures grown in the sealed bag vs. cultures in the incubator and in cultures during chronic ethanol exposure. MTT was added to the cells according to the manufacturer's protocol and incubated for ∼6 h in a humidified 37°C incubator. Dimethyl sulfoxide (DMSO) was added to produce a formazen product for ∼16 h and read at 490 nm.
RNA isolation and quantitative PCR (qPCR) analysis
RNA isolation from astrocyte cultures was carried out following manufacturer's recommendations for the RNA Stat‐60 reagent (Tel‐Test, Inc. Friendswood, TX, USA) with modifications as previously described 43, 44. Contaminating genomic DNA was eliminated by digestion with RNase‐free DNase followed by Zymo‐spin column purification as described in the manufacturer's protocol (Zymo Research, Orange, CA, USA) and verified by qPCR. RNA integrity was assessed using a 1% agarose gel stained with SYBR Gold (Life Technologies, Grand Island, NY, USA) and quantified using Quant‐iT RiboGreen (Life Technologies). Expression differences in steady‐state mRNA levels in male and female cultures were determined using qRT‐PCR analysis as previously described 44. Briefly, qRT‐PCR was performed with the iCycler IQ Real Time PCR detection system (Bio‐Rad Laboratories, Inc., Hercules, CA, USA) using a one‐step QuantiTect SYBR Green RT‐PCR kit (Qiagen, Valencia, CA, USA). Relative expression of the RT‐PCR product was determined using the comparative ΔΔCt method, after normalizing expression to total RNA measured with RiboGreen 43. Primers were purchased, pre‐designed and validated from Qiagen or Bio‐Rad Laboratories, Inc.
Protein collection and Western blot
Protein was harvested from 6‐well dishes as whole cell lysates. Briefly, the cell layer was washed twice with sterile PBS then lysed in radioimmunoprecipitation assay buffer (RIPA) containing protease inhibitors (50 mM Tris–HCl, pH 7.4, 150 mM NaCl, 1 mM PMSF, 1 mM EDTA, 5 μg/mL aprotinin, 5 μg/mL leupeptin, 1% Triton X‐100, 1% sodium deoxycholate, 0.1% SDS). Lysates were collected after scraping and frozen at −20°C until protein quantification using the Pierce bicinchoninic acid assay (BCA) Protein Assay Kit. (Rockford, IL, USA). Western blot was performed using a previously described method 120. Antibodies for GFAP were obtained from Dako (Houston, TX, USA) and beta‐actin from Abcam (Cambridge, MA, USA). Blots for both GFAP (1:50 000) and beta‐actin (1:3000) primary antibodies were incubated overnight in Tris‐buffered saline with Tween‐20 (TBST). Bound antibodies were visualized using enhanced chemiluminescence (ECL) detection (Amersham Corp., Arlington Heights, IL, USA). Quantification of protein was determined in the linear response range of film using Optiquant software.
Glutamate transporter assay
EAAT activity was monitored by uptake of [3H]D‐aspartate 65 at the exposure 3 and recovery time points 5. Briefly, cells were rinsed with uptake buffer (135 mM NaCl, 5 mM KCL, 0.6 mM MgSO4, 1 mM D‐glucose, 10 mM HEPES, pH 7.5) for 5 minutes at 37°C. Uptake buffer was removed and replaced with uptake buffer containing 50 nM [3H]D‐aspartate for 5 minutes. Buffer was removed, cells rinsed in cold uptake buffer, and the cell layer was lysed in a 0.1% Triton X‐100 solution. Lysate was collected, transferred to scintillation vials containing scintillation fluid, and counted on a scintillation counter. Protein concentrations were determined in aliquots by the BCA method. Activity was determined as pmol/mg/min.
Lactate assay
Lactate measurements were conducted in culture media to characterize bioenergetics using the Lactate Plus Lactate Meter and Lactate Plus Lactate Test Strips (Nova Biomedical, Waltham, MA, USA). For each sample, a new test strip was placed in the meter and approximately 1–2 μL of culture media diluted 1:4 in PBS (pH 7.4) was assayed. Meter calibration was verified using lactate control solutions provided by the manufacturer.
Chemicals
Ethanol (ethyl alcohol, absolute, 200 proof) for use in chemical assays was purchased from AAPER Alcohol and Chemical (Shelbyville, KY, USA) and Pharmco Products, Inc. (Brookfield, CT, USA) that does not contain benzene for use in primary cultures. [3H]D‐aspartate with a specific activity of 12.9 Ci/mmol was purchased from Perkin Elmer Life Sciences, Inc. (Boston, MA, USA).
Statistical analyses
Data are expressed as mean ± standard error of the mean (SEM) and are normalized to fold of corresponding male control. Results were analyzed using a priori Student's t‐test for control vs. ethanol treatments (either exposure or recovery) using Microsoft Excel (Microsoft Corporation, Redmond, WA, USA) or Prism software (GraphPad Software Inc., San Diego, CA, USA). Power analyses were carried out by calculation of effect size based on the current statistics (http://www.uccs.edu/lbecker/), followed by calculation of sample size (http://www.danielsoper.com/statcalc3/default.aspx) with power = 0.80.
Results
Although astrocytes have been identified as an ethanol target following chronic ethanol intoxication and withdrawal in both rodents and humans 44, 69, 83, 119, there is little characterization of the specific consequences of chronic ethanol exposure on astrocyte function. Our previous gene expression analysis of ethanol effects in the medial prefrontal cortex had identified robust sexually dimorphic responses during a time course of intoxication, withdrawal (8 h post ethanol) and abstinence (21 days), particularly at the early time points (intoxication and withdrawal) 119. In the work presented here, we have extended our previous analyses 44, 119 to specifically focus on the effect of chronic intoxication or withdrawal on astrocytes in vivo in males and females. Ethanol‐regulated genes were identified in males and females in the medial prefrontal cortex harvested from WSR and WSP mice at the intoxication (0 h) and withdrawal (8 h) time points that encompass the early response to chronic ethanol exposure [see 119]. This set of genes was then compared with a database consisting of genes expressed predominantly or exclusively in astrocytes [similar to 83] to generate a list of astrocyte‐enriched genes regulated in vivo by chronic ethanol exposure and withdrawal in males vs. females. Ethanol‐regulated astrocyte genes in males and females are listed in Table 1. Notably, very few genes were regulated in both females and males, indicating that astrocytes are a target of ethanol effects but are differentially responsive depending on sex. These sets of ethanol‐regulated astrocyte‐enriched genes were then analyzed using IPA to characterize underlying biological pathways and networks that were statistically overrepresented in males vs. females. As shown in Figure 1, the top ethanol‐regulated network in males and in females was centered on hubs related to ubiquitin signaling in both sexes. However, with the exception of Tor3a (down‐regulated in females, mixed regulation in males), the interacting molecules and regulated genes were unique between males and females, revealing a distinct biological response following chronic intoxication and withdrawal. In females, network genes were generally up‐regulated and functionally are involved in cell cycle regulation, differentiation and aging. These results were consistent with our previous bioinformatic analysis of the complete set of regulated genes in this vulnerability paradigm, with a repair response observed in males but ethanol‐induced cell damage and inflammatory signaling in females 119.
Table 1.
EtOH‐regulated astrocyte‐enriched genes. Abbreviations: MPE = male WSP ethanol; MPA = male WSP control; MRE = male WSR ethanol; MRA = male WSR control; FPE = female WSP ethanol; FPA = female WSP control; FRE = female WSR ethanol; FRA = female WSR control
| GeneID | ACC | UGCluster | Name | Symbol | 0 h MPE‐MPA | 0 h MRE‐MRA | 0 h FPE‐FPA | 0 h FRE‐FRA | 8 h MPE‐MPA | 8 h MRE‐MRA | 8 h FPE‐FPA | 8 h FRE‐FRA |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 66278 | AI426191 | Mm.27900 | RIKEN cDNA 1810013D10 gene | 1810013D10Rik | 0.148 | −0.132 | −0.805 | −0.83 | −0.545 | 0.188 | −0.02 | −0.097 |
| 75572 | AI327338 | Mm.390319 | Acylphosphatase 2, muscle type | Acyp2 | −0.176 | −0.177 | −1.149 | −0.591 | −0.026 | −0.328 | −0.249 | 0.02 |
| 27360 | AI324178 | Mm.426080 | Adducin 3 (gamma) | Add3 | 0.114 | 0.338 | −0.323 | −0.252 | 0.168 | −0.547 | 0.19 | 0.166 |
| 27360 | AI449373 | Mm.426080 | Adducin 3 (gamma) | Add3 | 0.114 | 0.338 | −0.323 | −0.252 | 0.168 | −0.547 | 0.19 | 0.166 |
| 52231 | AI465332 | Mm.259326 | Ankyrin repeat and zinc finger domain containing 1 | Ankzf1 | −0.268 | 0.04 | −1.468 | −0.406 | −0.375 | −0.683 | 0.676 | 0.519 |
| 52231 | AI425962 | Mm.259326 | Ankyrin repeat and zinc finger domain containing 1 | Ankzf1 | −0.268 | 0.04 | −1.468 | −0.406 | −0.375 | −0.683 | 0.676 | 0.519 |
| 11886 | AI413346 | Mm.22547 | N‐acylsphingosine amidohydrolase 1 | Asah1 | 0.096 | 0.452 | −0.317 | −1.029 | 0.064 | 0.217 | 0.081 | −0.231 |
| 65973 | AI451660 | Mm.222206 | Aspartate‐beta‐hydroxylase | Asph | 0.248 | −0.344 | −0.699 | 0.265 | 0.3 | −0.28 | 0.62 | 0.861 |
| 12419 | AI893653 | Mm.262059 | Chromobox homolog 5 (Drosophila HP1a) | Cbx5 | 0.039 | −0.016 | −0.677 | −0.293 | −0.405 | 0.212 | −0.025 | −0.059 |
| 12419 | AI450379 | Mm.262059 | Chromobox homolog 5 (Drosophila HP1a) | Cbx5 | 0.039 | −0.016 | −0.677 | −0.293 | −0.405 | 0.212 | −0.025 | −0.059 |
| 12212 | AI452330 | Mm.42223 | Cysteine‐rich hydrophobic domain 1 | Chic1 | −0.109 | −0.125 | 0.633 | −0.148 | −0.283 | 0.257 | −0.014 | 0.164 |
| 70568 | AI451962 | Mm.38390 | Copine III | Cpne3 | −0.151 | −0.052 | 0.303 | 0.826 | 0.506 | 0.273 | 0.254 | −0.011 |
| 13198 | AI323295 | Mm.110220 | DNA‐damage inducible transcript 3 | Ddit3 | −0.088 | −0.103 | −0.445 | 0.469 | −0.014 | 0.102 | 0.343 | −0.219 |
| 68187 | AI465387 | Mm.87130 | Family with sequence similarity 135, member A | Fam135a | −0.361 | −0.079 | 1.259 | −0.879 | 0.094 | 0.122 | 0.122 | 0.31 |
| 14221 | AI465262 | Mm.29730 | Four jointed box 1 (Drosophila) | Fjx1 | 0.039 | −0.037 | −0.383 | 0.526 | −0.009 | −0.03 | 0.079 | −0.263 |
| 233899 | AI415639 | Mm.25220 | Predicted gene 166 | Gm166 | −0.193 | −0.051 | 0.018 | 1.146 | 0.135 | 0.083 | 0.045 | 0.25 |
| 215798 | AI449247 | Mm.440142 | G‐protein coupled receptor 126 | Gpr126 | 0.053 | −0.141 | 1.529 | 0.223 | 0.267 | −0.233 | 0.516 | −0.114 |
| 53379 | AI449004 | Mm.155896 | Heterogenous nuclear ribonucleoprotein A2/B1 | Hnrnpa2b1 | −0.268 | 0.392 | −0.491 | −0.25 | 0.356 | 0.09 | 0.237 | 0.001 |
| 16848 | AI451068 | Mm.12834 | LFNG O‐fucosylpeptide 3‐beta‐N‐acetylglucosaminyltransferase | Lfng | −0.354 | 0.13 | −0.23 | 1.416 | −0.34 | −0.115 | 0.06 | 0.296 |
| 16905 | AI464505 | Mm.243014 | Lamin A | Lmna | −0.156 | 0.034 | −0.39 | 0.029 | −0.017 | −0.323 | 0.33 | 0.257 |
| 11426 | AI449659 | Mm.402299 | Microtubule‐actin cross‐linking factor 1 | Macf1 | −0.419 | 0.428 | −0.474 | 0.733 | 0.237 | −0.176 | 0.222 | −0.088 |
| 69900 | AI415185 | Mm.348004 | Major facilitator superfamily domain containing 11 | Mfsd11 | 0.14 | −0.429 | −0.233 | −0.469 | −0.19 | 0.197 | 0.066 | −0.306 |
| 171580 | AI447388 | Mm.290431 | Microtubule associated monoxygenase, calponin and LIM domain containing 1 | Mical1 | −0.224 | 0.066 | 0.074 | −0.073 | −0.016 | 0.049 | −0.317 | −0.384 |
| 382793 | AI428081 | Mm.296770 | Metaxin 3 | Mtx3 | −0.053 | 0.478 | −0.011 | 0.646 | −0.454 | −0.13 | 0.326 | −0.389 |
| 210622 | AI414397 | Mm.28649 | Peptidase domain containing associated with muscle regeneration 1 | Pamr1 | −0.028 | −0.319 | −0.069 | −0.062 | −0.523 | 0.011 | −0.1 | 0.125 |
| 235587 | AI451081 | Mm.273659 | Poly (ADP‐ribose) polymerase family, member 3 | Parp3 | 0.389 | −0.144 | 0.181 | 0.517 | −0.128 | 0.114 | 0.767 | −0.359 |
| 18578 | AI452258 | Mm.20181 | Phosphodiesterase 4B, cAMP specific | Pde4b | −0.249 | −0.402 | 0.603 | 0.836 | 0.106 | 0.294 | −0.017 | 0.111 |
| 18933 | AI447573 | Mm.288642 | Paired related homeobox 1 | Prrx1 | 0.091 | −0.044 | −0.157 | 0.196 | 0.075 | 0.143 | 0.034 | −0.177 |
| 19659 | AI893623 | Mm.279741 | Retinol binding protein 1, cellular | Rbp1 | 0.306 | 0.179 | −0.14 | −0.426 | −0.184 | −0.265 | 0.493 | −0.192 |
| 58887 | AI425994 | Mm.219183 | Replication initiator 1 | Repin1 | 0.109 | −0.041 | −0.011 | −0.103 | 0.087 | −0.384 | 0.218 | 0.476 |
| 244058 | AI429605 | Mm.333943 | RGM domain family, member A | Rgma | −0.185 | −0.451 | 0.191 | −0.201 | −0.376 | 0.172 | −0.332 | −0.058 |
| 11853 | AI324259 | Mm.262 | Ras homolog gene family, member C | Rhoc | 0.024 | 0.159 | 0.251 | 1.453 | −0.035 | 0.087 | 0.264 | −0.202 |
| 20516 | AI415416 | Mm.323901 | Solute carrier family 20, member 2 | Slc20a2 | 0.448 | −0.393 | 0.133 | 0.644 | 0.312 | 0.074 | −0.368 | 0.478 |
| 66500 | AI451495 | Mm.34550 | Solute carrier family 30 (zinc transporter), member 7 | Slc30a7 | 0.104 | −0.135 | −0.454 | −0.115 | 0.022 | −0.303 | 0.553 | 0.426 |
| 20751 | AI894147 | Mm.28393 | Sepiapterin reductase | Spr | 0.4 | 0.032 | −0.86 | −0.581 | 0.207 | −0.543 | 1.028 | 0.38 |
| 107513 | AI452176 | Mm.426670 | Signal sequence receptor, alpha | Ssr1 | −0.178 | −0.12 | −0.899 | −0.129 | 0.041 | 0.008 | 0.342 | 0.506 |
| 272589 | AI451204 | Mm.34483 | Tubulin folding cofactor E‐like | Tbcel | −0.337 | 0.398 | −0.066 | −0.182 | 0.173 | −0.415 | −0.085 | −0.073 |
| 76355 | AI325957 | Mm.251537 | TDP‐glucose 4,6‐dehydratase | Tgds | 0.311 | 0.103 | −0.578 | 0.146 | 0.357 | −0.306 | −0.028 | 0.213 |
| 68059 | AI448035 | Mm.275191 | Transmembrane 9 superfamily member 2 | Tm9sf2 | 0.052 | 0.012 | −0.793 | −1.04 | −0.342 | −0.256 | 0.422 | 0.024 |
| 30935 | AI429495 | Mm.206737 | Torsin family 3, member A | Tor3a | −0.087 | −0.003 | −0.844 | 0.215 | −0.353 | 0.275 | 0.138 | 0.104 |
| 66637 | AI450184 | Mm.379120 | TRNA splicing endonuclease 15 homolog (Saccharomyces cerevisiae) | Tsen15 | −0.311 | −0.087 | −0.443 | −0.602 | −0.459 | −0.241 | 0.065 | 0.694 |
| 67120 | AI426949 | Mm.275710 | Tetratricopeptide repeat domain 14 | Ttc14 | −0.021 | 0.01 | −0.87 | −0.08 | −0.352 | −0.702 | 0.518 | 0.317 |
| 72114 | AI428386 | Mm.3774 | Zinc finger, BED domain containing 3 | Zbed3 | −0.072 | 0.175 | −1.058 | 0.225 | −0.844 | −0.294 | 0.443 | −0.361 |
| 233410 | AI447270 | Mm.29525 | Zinc finger protein 592 | Zfp592 | −0.46 | 0.091 | −0.493 | −1.945 | −0.094 | 0.302 | −0.256 | −0.007 |
Fold change in gene expression, with significantly regulated genes highlighted in bold at 0 h (immediately after chronic EtOH) and 8 h (peak withdrawal).
Figure 1.
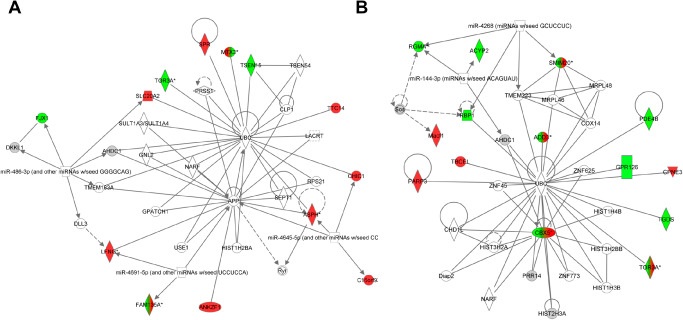
The top bioinformatic network for male and female mice derived from analysis of ethanol‐regulated astrocyte‐enriched genes. A. The top female network (P ≈ 10−32; see Supporting Information Table S1) of ethanol‐regulated astrocyte‐enriched genes centered on ubiquitin (UBC) and amyloid precursor protein (APP). B. The top male network (P ≈ 10−38) of ethanol‐regulated astrocyte‐enriched genes centered on ubiquitin (UBC). Of particular interest is that the only overlapping, regulated gene between the two networks is TOR3A (Torsin 3A). All other regulated genes are unique between the two networks indicating an almost completely sexually dimorphic response to ethanol. Solid lines represent direct interactions while dashed lines represent indirect interactions between genes. Genes in red are up‐regulated in response to ethanol. Genes in green are down‐regulated in response to ethanol. Genes colored in both red and green indicate mixed regulation.
The effect of chronic ethanol intoxication on astrocyte activation in vivo was examined using immunofluorescence for the traditional astrocyte activation marker GFAP 34. Visual examination of labeling intensity indicated that GFAP+ labeling in the CNS was localized to the hippocampus and white matter in the corpus callosum. This result is consistent with the reported low/undetectable expression of GFAP outside of these regions in mice 50. Following immunolabeling, the number of GFAP+ cells was assessed in the hippocampus and hippocampal subregions including the dentate gyrus (DG) and hilus, CA 1–2, and CA3 (Figure 2) of WSR mice. By using WSR mice, astrocyte responses were examined in the absence of ethanol‐withdrawal‐induced seizure activity. Ethanol exposure led to an increase in GFAP+ cells in females in the DG and hilus (P < 0.01) and CA 1–2 subregions (P < 0.05), but ethanol had no significant effect on the number of GFAP+ cells in any subregion in males (P > 0.05). Similar results were observed in WSP mice (data not shown). Thus, chronic exposure and withdrawal induced a significant increase in astrocyte activation in several hippocampal subregions, but only in females. Because of the very low levels of GFAP staining outside of the hippocampus, astrocyte activation in the medial prefrontal cortex was then characterized via immunolabeling of the calcium binding protein S100β, another marker associated with astrocyte reactivity 18. Analysis focused on the anterior cingulate cortex (ACC), a subregion of the medial prefrontal cortex previously identified as damaged in females following ethanol exposure 118. ACC volumes did not differ between control and alcohol‐exposed animals (all Ps > 0.05). The number of S100β+ cells was reduced in males but not females following ethanol exposure (P < 0.05; Figure 3). Males also had a trend toward lower mean cell body area (P = 0.07, with 5 additional samples per group required to fully test the hypothesis based on power analysis). In contrast, staining intensity was reduced in females following ethanol exposure (P < 0.05), with no difference in males.
Figure 2.
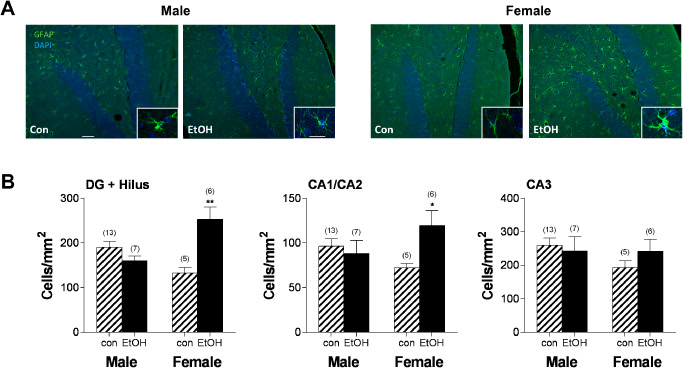
Ethanol ‐induced astrocyte activation in females but not males. A. Representative images of GFAP staining in the dentate gyrus and hilus of the hippocampus in male control (Con) and ethanol (EtOH), and female Con and EtOH sections following a 5‐day withdrawal period at 20× magnification. Insets illustrate the morphology and intensity of staining following ethanol exposure in male and female mice at 63× magnification. Scale bars 50 μm, inset 5 μm. B. Quantitation of the density of GFAP‐labeled cells in the dentate gyrus (DG) and hilus, CA1 and CA2, and the CA3 subregions of the hippocampus. Females exhibit increased density of GFAP‐labeled cells in the DG and hilus, as well as CA1/CA2 subregions. Striped bars indicate control groups. Solid black bars indicate ethanol‐exposed groups. Numbers in parentheses indicate the n for each group. *P < 0.05; **P < 0.01.
Figure 3.
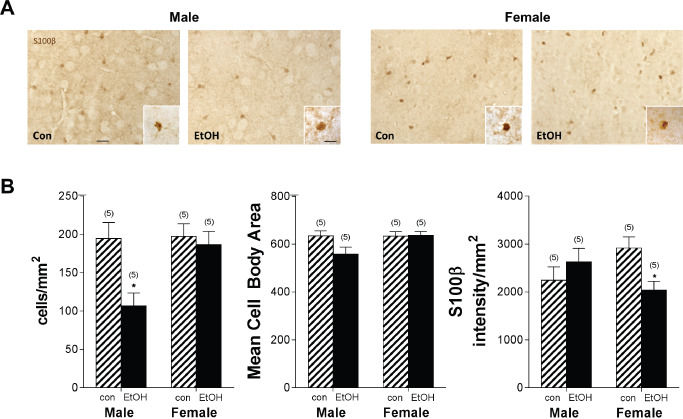
Ethanol‐induced astrocyte dysfunction in the anterior cingulate cortex. Following chronic ethanol exposure, S100β immunohistochemistry was used to assess astrocyte expression and morphology. A. Representative images of S100β staining in the ACC of the prefrontal cortex in male control (Con) and ethanol (EtOH), and female Con and ethanol sections following a 5‐day withdrawal period at 20× magnification. Insets illustrate the morphology and intensity of staining following ethanol exposure at 63× magnification. Scale bars 50 μm, inset 5 μm. B. Males exhibit a reduction in S100β+ cells following ethanol exposure. Females exhibited reduced S100β staining intensity following ethanol exposure. Striped bars indicate control groups. Solid black bars indicate ethanol‐exposed groups. Numbers in parentheses indicate the n for each group. *P < 0.05.
Given sexually dimorphic astrocyte responses following chronic ethanol in vivo, we next analyzed the direct effects of chronic ethanol exposure on astrocyte function. For this analysis, a primary astrocyte culture model system employing chronic ethanol exposure was developed. Sex‐specific cultures were derived from cortical tissue obtained from individual pups (Figure 4A). Following repeated shaking to remove microglial cells early in the culture period, nearly all cells exhibited GFAP labeling in post‐confluent mature cultures (21 days; Figure 4B). The sex of each individual culture was assessed using the Sry marker which is only present in males (Figure 4C). Chronic ethanol exposure and recovery were modeled using this paradigm, with cultures subjected to control (Con) or 100 mM chronic ethanol exposure (Exp) conditions for 4 days, then allowed to progress through a recovery period of 5 additional days in culture in the absence of ethanol. This concentration of ethanol is consistent with the high levels observed in chronic alcoholics admitted for detoxification programs 1, 92. The 5‐day recovery time point was chosen because it is consistent with neurotoxicity following excitatory insult 90, and because our previous data indicated sexually dimorphic neurotoxicity following chronic ethanol intoxication at this time 44, 119. One potential concern with ethanol treatment is evaporation from the media, resulting in inconsistent ethanol concentrations over time. To address this concern, cultures were placed in plastic bags containing a reservoir of ethanol at the target concentration. Control cultures were placed in plastic bags containing a water reservoir. Notably, cell viability (measured by MTT) was unaltered by incubation in the bag for 4 days compared with normal control cultures that were not incubated in the bag (Figure 4D). Using this procedure, ethanol concentrations in the media remained near the target range throughout the exposure period (98.1 ± 1.8 mM at the end of the 4‐day exposure period).
Figure 4.
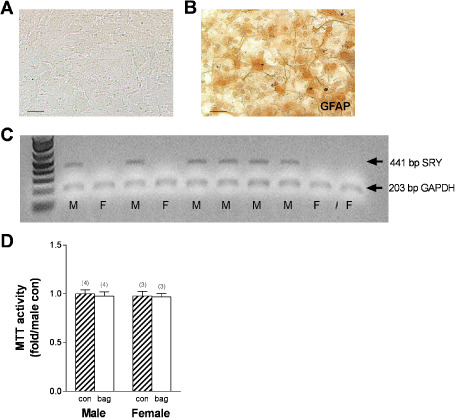
Sex‐specific mature astrocytic primary cultures. A. Representative phase contrast image of cell growth after approximately 21 days in cultures, scale bar = 5 μm. B. Representative GFAP labeling of primary cultures showing nearly universal GFAP expression, scale bar = 5 μm. C. Agarose gel showing SRY positive and negative cultures following amplification. D. Astrocyte viability was assessed using MTT assays after incubation in a standard freezer bag (numbers in parentheses indicate the n for each group). Striped bars indicate control astrocyte cultures. Solid white bars indicate bag‐incubated astrocyte cultures.
Potential toxicity from chronic ethanol treatment was carefully evaluated using two independent assays; one assessed cellular MTT for viability changes, while the other determined cytotoxicity via leakage of LDH into the media. As shown in Figure 5A, ethanol exposure did not induce markers of cell death as indicated by the lack of increase in either MTT or LDH in culture. In contrast, ethanol exposure surprisingly led to a reduction in media LDH activity in females (P < 0.05). Upon loss of membrane integrity, LDH is released from the cell and extracellular levels increase. Thus, reduced extracellular LDH is not indicative of toxicity, but instead may indicate altered lactate homeostasis and changes in bioenergetics. During recovery, there was no difference in either MTT activity or media or cellular LDH (Figure 5B). Blood EtOH concentrations as high as 130 mM have been observed in emergency room patients, with a median concentration of approximately 50 mM 92. Thus, 100 mM EtOH is in the range of concentrations observed in alcoholics and was not toxic to the astrocyte cultures as indicated by the MTT and LDH results. Future studies will incorporate dose–response curves to determine if lower EtOH concentrations induce similar dysfunction in female astrocyte cultures. Thus, this paradigm subjected primary cultures of highly enriched astrocytes from each sex to consistent high levels of chronic ethanol without inducing toxicity, providing a reliable model for assessing the direct effect of ethanol exposure on astrocyte function, without the contribution of changes in other cell types and/or circulating hormones as would be seen in vivo.
Figure 5.
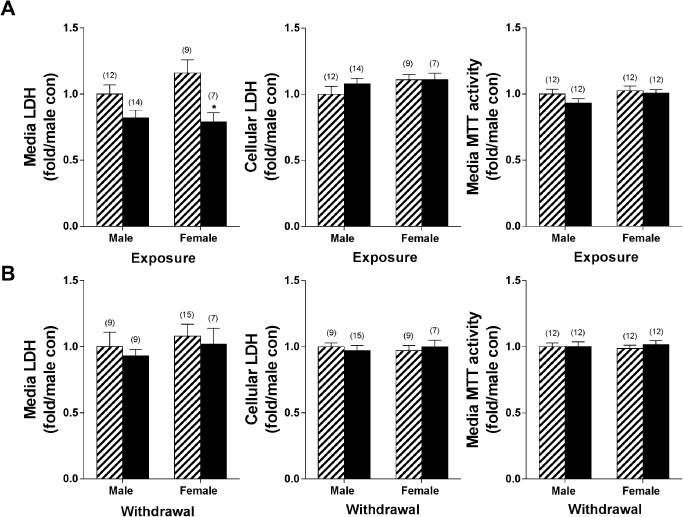
Lack of effects of ethanol on astrocyte viability. Astrocyte viability in highly confluent cultures was assessed using MTT assays after (A) chronic ethanol exposure and after (B) recovery. No significant differences were observed in any of the conditions tested. Lactate dehydrogenase activity (LDH) was also used as a measure of cytotoxicity, with leakage of LDH into the media an indicator of compromised membrane integrity. Ethanol exposure reduced media LDH concentrations, and had no effect during recovery (numbers in parentheses indicate the n for each group). Striped bars indicate control groups. Solid black bars indicate ethanol‐exposed groups. *P < 0.05.
Using this approach, the effects of ethanol exposure on astrocyte gene expression were examined with a focus on three overarching aspects of astrocyte function: astrocyte markers 125, proteins important in glutamate homeostasis and cytokine/chemokine expression. Analysis was performed after chronic exposure and following recovery. As shown in Figure 6A, ethanol exposure trended toward reduced S100β expression in females (P = 0.08, with 10–13 additional samples per group required to fully test the hypothesis based on power analysis). The direct effects of ethanol on molecules involved in glutamate homeostasis were then examined, as one mechanism of alcohol‐induced neurotoxicity previously proposed is hyperexcitability resulting from increased glutamate signaling 96. Eaat2 expression was significantly increased in males following ethanol exposure (P < 0.05), but Eaat1 and glutamine synthetase (GluI) levels were unchanged. Finally, the effect of chronic ethanol exposure on cytokine and chemokine expression was determined, as another potential mechanism of neurotoxicity is induction of inflammatory signaling by alcohol resulting in activation of cell death pathways either directly by cytokines such as Tnf, or following positive feedback and enhancement of inflammatory signaling. Interestingly, the only transcript that was significantly up‐regulated in female cultures was Tnf, which was significantly induced following chronic ethanol (P < 0.05; Figure 6C). Alcohol exposure did not influence expression of fibroblast growth factor 2 (Fgf2), metabotropic glutamate receptor 1 (Grm1; mGluR1), Grik2 (ionotropic glutamate receptor; kainate 2) or interleukin‐1β (Supporting information Figures S1 and S2). Thus, despite a general lack of effect on gene expression in astrocytes during exposure, alcohol directly induced expression of the inflammatory cytokine Tnf in female cultures, which could have wide‐ranging effects on neurotoxicity, astrocyte cell viability, blood–brain barrier (BBB) integrity and downstream inflammatory signaling.
Figure 6.
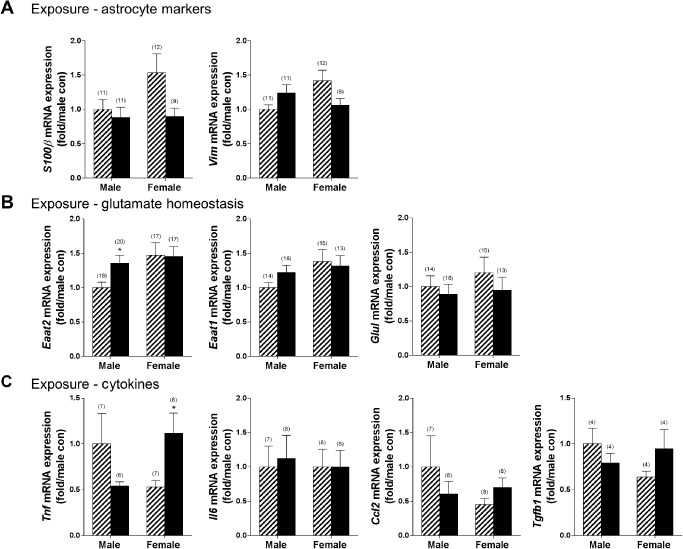
Induction of Tnf expression in female mice following chronic ethanol. The effect of 4 days of ethanol exposure (100 mm) was assessed in sex‐specific primary astrocyte cultures using qRT‐PCR. A. No changes were observed in expression of the astrocyte markers S100β and Vim (vimentin). B. Males exhibited increased expression of Eaat2, but no other differences were noted. C. Females exhibited increased expression of Tnf following ethanol exposure. Striped bars indicate control groups. Solid black bars indicate ethanol‐exposed groups. Numbers in parentheses indicate the n for each group. *P < 0.05.
Next, the influence of subsequent recovery on gene expression was assessed. Astrocyte markers indicated that males exhibited a significant reduction in vimentin expression (P < 0.05), with no other effects of ethanol (Figure 7A). Recovery did not influence Eaat expression, but did lead to a reduction in GluI (P < 0.05) in males (Figure 7B). Expression of cytokines and chemokines identified reduced Tgfb1 expression in females (Figure 7C; P < 0.05) with no other differences observed.
Figure 7.
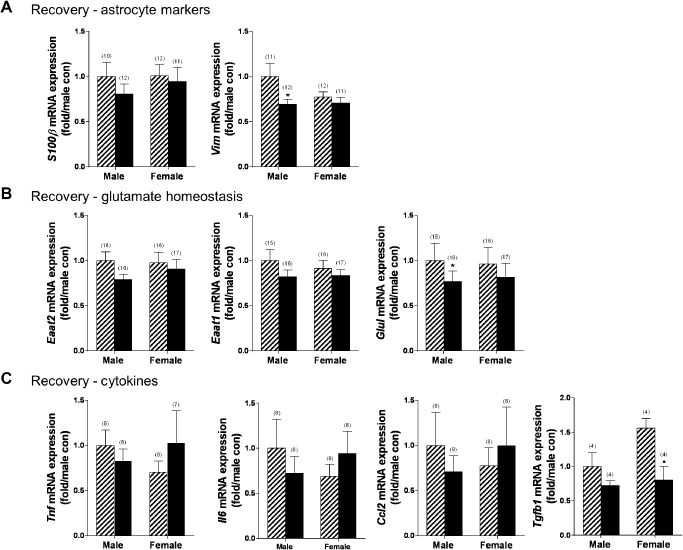
Reduced T gfb1 expression in female mice after recovery from chronic ethanol. The effect of ethanol exposure (4 days, 100 mm) and subsequent recovery (5 days) were assessed in sex‐specific astrocyte cultures using qPCR. A. Males exhibited reduced expression of the astrocyte marker vimentin. B. Males also had reduced expression of glutamine synthetase (glui). C. Females exhibited reduced expression of the cytokine T gfb1. Striped bars indicate control groups. Solid black bars indicate ethanol‐exposed groups. Numbers in parentheses indicate the n for each group. *P < 0.05.
Given evidence of bioenergetic alterations associated with chronic ethanol in astrocytes as evidenced by altered LDH activity during exposure and reduced glutamine synthetase during recovery, lactate concentrations in the media were examined. Lactate levels in males were unchanged during ethanol exposure (Con: 10.4 ± 0.5 mM; ethanol: 10.1 ± 0.8 mM; n = 3–5; P > 0.05) whereas lactate levels in females trended toward an increase following ethanol exposure (Con: 10.7 ± 0.3 mM; ethanol: 11.5 ± 0.3 mM; n = 4/group; P = 0.07; with 2 additional samples per group needed to fully test the hypothesis based on power analysis). Thus, although LDH activity in the media was reduced in females, lactate levels were elevated.
The number of GFAP+ cells in females in the hippocampus was increased following ethanol, therefore, the cell‐specific effects of ethanol on GFAP protein levels in highly confluent primary astrocyte cultures were determined to evaluate whether the increased numbers of cells represent potential proliferation or increased cellular expression. Western blotting was used to examine GFAP levels following ethanol exposure and recovery (Figure 8A). In males, GFAP levels were significantly reduced following ethanol exposure, but returned to control levels during recovery. In females, GFAP levels were unaltered during ethanol exposure or during recovery. Thus, males, but not females, exhibit reduced GFAP levels as a result of ethanol exposure in the culture model.
Figure 8.
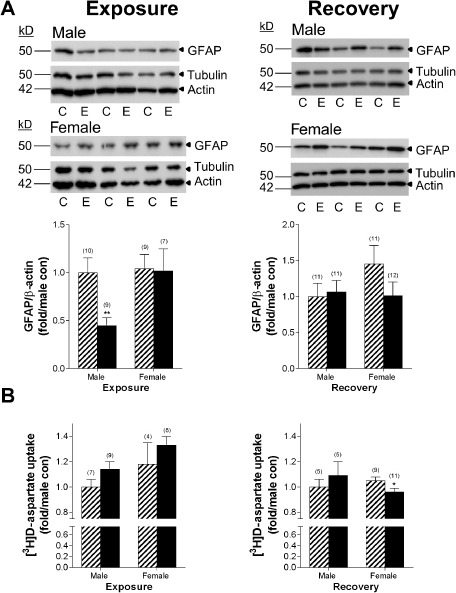
Sexually dimorphic astrocyte response: deactivation of astrocytes in males following ethanol but loss of glutamate uptake in females. A. Representative Western blots are shown depicting GFAP, tubulin and actin labeling both following chronic ethanol exposure and during recovery in males and females. Control (C); ethanol (E). Expression of the marker of astrocyte activation, GFAP, was assessed via Western blotting. Primary astrocytes derived from male mice exhibited a reduction in GFAP expression following ethanol exposure, with no changes observed in females. Numbers in parentheses indicate the n for each group. B. EAAT uptake was unchanged during exposure, but was reduced in females during recovery. Striped bars indicate control groups. Solid black bars indicate ethanol‐exposed groups. Numbers in parentheses indicate the n for each group. *P < 0.05, **P < 0.01.
Finally, the functional impact of ethanol exposure on astrocytic glutamate uptake was examined. These studies were undertaken given ethanol effects on Eaat2 expression in males, previous work showing alcohol‐induced neurotoxicity in females, but not males, and evidence of sexually dimorphic astrocyte responses as determined by GFAP+ cells in vivo and reduced S100β levels in vivo and in primary cultures. Glutamate uptake capacity was characterized using [3H]D‐aspartate, a substrate for both EAAT1 and EAAT2 transporters 57. Neither ethanol exposure nor recovery impacted [3H]D‐aspartate uptake in male cultures. By contrast, [3H]D‐aspartate uptake was significantly reduced in female cultures during the recovery period following ethanol exposure (Figure 8B; P < 0.05). Dysregulation of glutamate uptake in female cultures, which would result in reduced removal of glutamate from the extracellular space following ethanol exposure, reflects changes in glutamate homeostasis that would exert important effects on neuronal health in vivo.
Discussion
This study identified astrocytes as an ethanol target in the medial prefrontal cortex and hippocampus, with distinct sex differences following chronic ethanol exposure and withdrawal/recovery. Increased astrocyte reactivity was observed in female but not male mice following chronic ethanol intoxication. Astrocytes were prevalent in the ACC in both males and females, but only males also had a reduction in S100β+ cells in the ACC following ethanol. In astrocyte‐enriched cultures, female but not male astrocytes exhibited increased expression of the pro‐inflammatory cytokine Tnf, reduced expression of the neuroprotective cytokine Tgfb1, increased media lactate concentration (trend) and reduced glutamate uptake capacity. Based on these results and evidence from the literature, we propose a novel model of sex‐specific astrocyte dysfunction associated with chronic ethanol exposure (Figure 9). Thus, in female astrocytes, ethanol induces increased inflammatory signaling in the presence of a reduced neuroprotective response, reduced glutamate clearance and altered lactate homeostasis. As shown in this model, the downstream effects of such cellular dysfunction are inflammation, oxidative stress, compromised BBB integrity, demyelination, impaired bioenergetic homeostasis and increased neuronal hyperexcitability which lead to neurotoxicity and neurodegeneration in females 11, 24, 85, 119. These cellular mechanisms are not mutually exclusive as studies suggest interactions between TNF signaling, glutamate homeostasis 95, 113, BBB integrity 70 and myelin degradation 124. Similarly, while TNF signaling is detrimental to CNS homeostasis, transforming growth factor‐β (TGF‐β) is strongly neuroprotective as signaling acts to reduce inflammation, excitotoxicity and apoptosis, and to enhance neuronal regeneration [for review, see 31]. The trend toward increased lactate release in females is also intriguing, as lactate levels are increased during several types of brain injury and are thought to both indicate and enhance tissue damage 63, 102. Thus, alcohol‐induced astrocyte dysfunction in females is predicted to accelerate alcohol‐induced neurotoxicity in a sexually dimorphic fashion. Although neurodamage is observed in both male and female chronic alcoholics, the mechanisms and pathways for damage are distinct and therefore represent opportunities for novel intervention development and targeted sex‐specific therapies.
Figure 9.
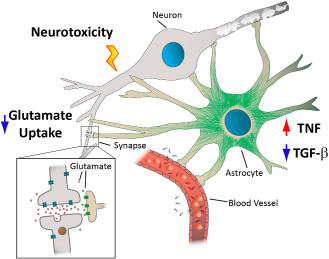
Model of ethanol‐induced astrocyte dysfunction in females. Ethanol targets several critical aspects of brain homeostasis which are maintained by proper astrocyte function. Primary astrocyte cultures from females exhibited increased expression of the inflammatory cytokine T nf, reduced expression of the neuroprotective cytokine T gfb1 and a trend toward increased lactate production following ethanol exposure. TNF can directly induce cell death via interaction with “death” receptors, and can serve to amplify inflammatory signaling pathways leading to neuroinflammation and oxidative stress. Increased T nf and reduced T gfb1 could also compromise blood–brain barrier (BBB) integrity leading to extravasation of pathogens and danger signals and induce damage to endothelial cells and oligodendrocytes. Alcoholics exhibit significant white matter shrinkage, consistent with disruption of proper myelination which is dependent on normal astrocyte function. Alcohol also led to a reduction in astrocyte‐mediated uptake of excitatory amino acids in females, who are already prone to excitotoxic damage due to reduced TGF‐β expression.
Bioinformatic analysis of expression differences in astrocyte‐enriched genes in vivo demonstrated a sexually dimorphic response to ethanol. Although ubiquitin signaling was a network hub in astrocyte genes from both sexes, distinct interacting gene sets were identified between males and females. Ethanol‐induced expression differences in this top network observed in astrocytes from females were consistent with a pro‐inflammatory tone, cellular dysfunction and a cytotoxic response in the prefrontal cortex. For example, up‐regulation of the nitric oxide synthase cofactor sepiapterin reductase (Spr) in females would result in excess amounts of tetrahydrobiopterin that has been associated with age‐related brain dysfunction in the cerebral cortex 80 and cognitive impairments. Also suggestive of brain dysfunction, amyloid precursor protein (App) was a secondary hub identified in females in the ubiquitin network of genes. Cleavage of amyloid precursor protein (APP) produces amyloid β (Aβ), the hallmark protein observed in Alzheimer's disease. The presence of APP in the female gene expression network is intriguing, as APP serves important trophic, cell survival and cell proliferation functions 115. Aβ can also activate transcription of stress genes via NF‐κB (nuclear factor kappa‐light‐chain‐enhancer of activated B cells) 62 and we have previously reported sexually dimorphic NF‐κB signaling in females during withdrawal 118, 119. Dysregulated ubiquitination contributes to the development of Alzheimer's disease and other neurodegenerative disorders [for review, see 8], and is associated with demyelination and white matter degeneration 123. Myelin degradation has been associated with ethanol abuse 2, 28, 68, but to our knowledge males and females have not been examined separately. Though complex, TNF production has been associated with enhanced demyelination 66, while Tgfb1 expression reduces demyelination 33. Metaxin 3 (Mtx3) in the ubiquitin network was also dysregulated in female astrocytes and plays an important role in TNF‐induced cell death 89. By contrast in males, the ubiquitin gene network is suggestive of a protective environment in the CNS in this model. For example, reduced expression of the retinol binding protein 1 (Rbp1) would increase free concentrations of retinoids. Retinoids reduce the production of pro‐inflammatory cytokines and chemokines by astrocytes and microglia 110. Down‐regulation of phosphodiesterase 4B, cAMP specific (Pde4b) could improve BBB function and reduce inflammatory responses 73. Finally, withdrawal may activate repair mechanisms in males, as poly(ADP‐ribose) polymerase 3 (Parp3, also known as Artd3), an important player in the cellular response to cytotoxic double‐strand breaks in DNA 15, is up‐regulated in males. The identification of sexually dimorphic mechanisms of cell death will be important for improved understanding of our previous observation of ethanol‐induced neurodamage in female but not male mice in our vulnerability model 44, 118. Unfortunately, most clinical studies are underpowered to adequately examine both sexes, thus careful analysis of female‐specific biological responses to chronic ethanol in the CNS is poorly characterized 108.
Although the specific mechanism(s) that underlie alcoholic neurodegeneration are complex and likely differ depending on the insult, brain region, genetic background and sex [for review, see 42], it is well established that normal astrocyte function is critical for neuronal health. Astrocytes are important regulators of glutamate signaling and homeostasis, can produce inflammatory and supportive cytokine mediators, aid in the maintenance of the BBB 24, 85, and influence myelination 54, 85. Severe brain injury is associated with an increase in the number of reactive astrocytes (particularly in the hippocampus), changes in astrocyte morphology and gene expression including induction of GFAP, and increased lactate production 10. In the studies presented here, females exhibited an increase in the density of GFAP+ cells in the hippocampus (in particular in DG and hilus and CA1/2) following ethanol exposure and withdrawal. Increased GFAP immunofluorescence can reflect rearrangement of cytoskeletal proteins including F‐actin depolymerization 86, and may reduce astrocyte motility by providing structural stability to astrocytic processes. Activation of astrocytes observed in the hippocampus in females is consistent with altered function that could contribute to neurodegeneration in frontal regions, given known circuitry associated with the reinforcing effects of alcohol 35 and alcohol withdrawal 38, 42. GFAP levels outside of the hippocampus in mice are difficult to assess 50 and are too low to quantify in the ACC using immunohistochemical methods. However, we observed a reduction in the intensity of the calcium binding protein S100β in the ACC in ethanol‐exposed female mice consistent with the reduced expression observed in ethanol‐exposed mature female astrocyte cultures. Decreased S100β expression in females is significant because this molecule binds GFAP and inhibits its phosphorylation 125. Forced reduction of S100β levels using siRNA interventions leads to loss of motility and adoption of a more stellate and potentially reactive phenotype 18.
S100β exhibits a complex expression pattern that varies with astrocyte maturation and environmental influences. Expression of S100β identifies mature astrocytes from neural stem cells 98; however, S100β also regulates astrocyte shape and movement 18. Levels of S100β can also change with insult. Some have suggested that female astrocytes remain partially undifferentiated, thereby allowing greater plasticity 81. Future studies will unravel the complex, sexually dimorphic regulation of astrocytic maturation, morphology and cytoskeletal changes induced by ethanol. Findings of increased gliosis in alcoholics are not universal, and may be dependent on the extent and nature of the abuse, the brain region examined, the genetic background of the population and/or the gender of the alcoholic. In fact, chronic ethanol exposure has been associated with reduced GFAP levels in some situations, as shown in the orbitofrontal cortex in young (predominantly male) uncomplicated alcoholics and in rats chronically exposed for longer than a few weeks 79. These results are consistent with the Western blot findings of reduced GFAP protein from the male primary cultures we observed during ethanol exposure. It should be noted that our quantitation methods for GFAP and S100β may be unable to differentiate between changes in cell size (ie, swelling) and increases in cell number. However, there was no indication of increased GFAP+ cell area in female EtOH‐treated mice, indicating a greater likelihood of an increase in cell number (data not shown). Regardless, significant changes in astrocyte cytoskeleton were observed in females following EtOH exposure. Thus, the development of astrogliosis and the potential for astrocyte dysfunction in the setting of chronic alcohol abuse is incompletely understood. Our results suggest that female alcoholics are at greater risk for the development of gliosis, but to our knowledge females have not been specifically examined. Additional studies comparing males and females are warranted as astrocytes exert widespread influence on CNS health and have been suggested as a therapeutic target for ameliorating the negative health consequences of drugs of abuse 45.
Neurotoxicity following chronic ethanol abuse may result from decreased inhibitory tone concomitant with increased excitatory glutamatergic signaling due to neuroadaptative responses to the CNS depressant effects of alcohol. In our vulnerability model, we have observed alcohol‐induced neurotoxicity in female but not male mice 44, 118. Glutamate is the major excitatory neurotransmitter in the CNS, and high concentrations of glutamate in the extracellular space can precipitate a cascade of cellular events that lead to excitotoxicity, elevated oxidative stress, glutathione depletion and cell death 9, 48. Astrocytes are primarily responsible for terminating glutamate signaling at the synapse, which is critical for normal neurotransmission and also to protect neurons from hyperexcitability 30. Notably, female but not male astrocytes exhibited reduced EAAT capacity 5 days after recovery. Reduced transport in females is consistent with increased Tnf expression and elevated extracellular lactate levels, which are both associated with impaired glutamate transport in other contexts 20, 60. In addition, reduced Tgfb1 expression in females is associated with increased vulnerability to excitotoxicity 23. Lactate is a component of a complex energy homeostasis maintained in the CNS by astrocytes and neurons that may exert both oxidative and metabolic stress following ethanol exposure in females 49. Also notable is that alcoholics (not analyzed separately for males vs. females) can exhibit pronounced increases in blood lactate levels during withdrawal 97. Female mice also had disrupted S100β expression that has been associated with alterations in glutamate homeostasis 116, 117. Although changes in glutamate levels have been observed in alcoholics during detoxification 47, females were not specifically examined and cellular mechanisms underlying glutamate dysfunction are likely to be sexually dimorphic. Increased extracellular glutamate in vivo as a consequence of reduced uptake could lead to sexually dimorphic hyperexcitability in postsynaptic neurons with oxidative stress and subsequent toxicity in brain regions where projections end. Again, a detailed comparison between male and female alcoholics in brain region‐specific damage or the extent of damage has not been reported. In addition, reduced glutamate uptake in female astrocytes would lead to a reduction in Na+/K+‐ATPase activity 99 and reduced glycolysis 93. This result is also consistent with sexually dimorphic astrocyte dysfunction observed in patients with temporal lobe epilepsy, with females showing more pronounced intracortical gliosis 32. In male astrocyte cultures, although Eaat2 mRNA levels were increased, there was no corresponding increase in glutamate uptake with exposure. This may be a consequence of two offsetting phenomena: increased Eaat2 expression but reduced GFAP protein levels. Loss of GFAP protein results in blockade of EAAT2 trafficking to the cell surface 53. Combined, the studies presented here suggest that glutamate‐mediated neurodamage due to sexually dimorphic astrocyte dysfunction may be a central consequence of alcohol abuse associated with neurotoxicity, particularly in females.
While the importance of astrocytes in glutamate homeostasis in the CNS is clear, it is less well appreciated that astrocytes are also involved in inflammatory signaling and produce cytokines and chemokines similar to microglia, the resident macrophage‐like CNS immune cells 114. The immunological consequences of ethanol abuse are complex, inducing immunosuppression in some organs, and increased infection rates, but also inflammation and cell death in other target organs 25. In the brain, astrocytes respond to “danger molecules” such as bacterially derived lipopolysaccharide (LPS) via toll‐like receptor 4 (TLR4) activation 114. Guerri's group has examined the influence of TLR4 knockout on several aspects of ethanol‐induced dysfunction, establishing contributions of TLR4 to ethanol‐induced neurotoxicity, cognitive dysfunction, neuroinflammation and myelin damage 2, 3, 4, 82, 91. The identification of increased expression of Tnf as a direct consequence of ethanol exposure in female but not male astrocytes is intriguing, as TNF alone is capable of inducing neuronal cell death 113, 126, but can also be a mechanism to induce an even more potent and damaging inflammatory response 16. Others using unsexed (ie, mixed) astrocyte cultures have found that ethanol exposure reduces TNF concentration following 7 days in culture, which is consistent with down‐regulation of Tnf in males 105. Interestingly, lactate induces Tnf expression in astroglial cultures, with the trend toward increased lactate in female cultures suggesting one potential mechanism of Tnf induction 6. In models of chronic ethanol abuse, Tnf activation has been associated with activation of the nucleotide‐binding oligomerization domain, leucine‐rich repeat and pyrin domain containing peptide (NLRP3) inflammasome 67 in females, identifying another pathway of potential toxicity due to ethanol. In contrast, the reduced Tgfb1 expression observed in female astrocytes is also detrimental and is consistent with a vulnerability to damage in females, especially in models of excitotoxicity and ischemia 101. Thus, astrocyte dysfunction as characterized by induction of Tnf and suppression of Tgfb1 may contribute directly to the previously identified female vulnerability to ethanol‐induced neurotoxicity, or due to the downstream effects of signaling pathways such as via altered glutamate homeostasis 44, 119.
Studies presented here indicate that sexually dimorphic responses to chronic ethanol exposure and recovery are indeed observed in sex‐specific astrocyte cultures. It is important to note that in this paradigm, primary astrocyte cultures do not fully recapitulate the effects of ethanol exposure in the CNS, as evidenced by the increased Il6 expression observed in females in the prefrontal cortex in vivo 119, but expression was unchanged in female astrocyte cultures following chronic ethanol treatment. This is in contrast to the elevation of Tnf in females, which was observed both in vivo and in the culture model. Such differences may be due to the post‐confluent nature of the cultures, to cellular changes due to adherence to the culture dish, or finally to the absence of other cell types or hormonal changes which may provide signals that affect astrocyte function. A caveat to the culture model is that astrocytes from both sexes would not be exposed to circulating hormones which are protective under certain conditions like stroke. Our previous data suggest that levels of estradiol drop following ethanol exposure in both sexes, thus minimizing any influence these factors might have on the observed results 36. Nevertheless, these studies do identify direct effects of chronic alcohol exposure in the astrocyte population that are fundamentally sexually dimorphic, and independent of other influences.
In conclusion, sexually dimorphic astrocyte dysfunction is observed following chronic ethanol intoxication in vivo and in sex‐specific primary astrocyte cultures, with female astrocytes demonstrating a pervasive dysfunction including induction of pro‐inflammatory signaling, suppression of the neuroprotective cytokine Tgfb1, dysregulation of EAAT function, changes in bioenergetics, and expression differences consistent with neurotoxicity and cognitive impairment. The results presented here and summarized in our model emphasize the crucial role played by astrocytes in CNS homeostasis and implicate astrocyte dysregulation as an important contributor to the heightened vulnerability to ethanol‐induced neurotoxicity observed in female alcoholics. Sex differences in astrocyte function suggest that females may be more sensitive to certain types of insults, including alcohol intoxication. Understanding these sex‐specific differences in astrocyte function will be critical for developing more efficacious treatments for alcohol‐induced damage during withdrawal, maintenance of sobriety and also for other neurodegenerative disorders.
Conflict of Interest
The authors declare that they have no conflict of interest.
Supporting information
Figure S1. Gene expression following chronic EtOH.
Figure S2. Gene expression following recovery from chronic EtOH.
Table S1. Female and male ethanol‐regulated networks.
Acknowledgments
This work was supported by Merit Review Award #BX001172 (KMW) and Career Development Award #BX001294 (CJW) from the United States (U.S.) Department of Veterans Affairs Biomedical Laboratory Research and Development and from the NIH/NIAAA [R01AA021468 and R21AA018420 (KMW)]. Additionally, this material is the result of work supported with resources and the use of facilities at the VA Portland Health Care System (KMW) and the Research Career Scientist Program (KMW). We acknowledge support from NIAAA for the Portland Alcohol Research Center (P60AA010760) and for the maintenance of colonies of WSR and WSP mice (R24AA020245) used in the present studies. Contents do not necessarily represent the views of the U.S. Department of Veterans Affairs or the United States Government.
References
- 1. Adachi J, Mizoi Y, Fukunaga T, Ogawa Y, Ueno Y, Imamichi H (1991) Degrees of alcohol intoxication in 117 hospitalized cases. J Stud Alcohol 52:448–453. [DOI] [PubMed] [Google Scholar]
- 2. Alfonso‐Loeches S, Pascual M, Gomez‐Pinedo U, Pascual‐Lucas M, Renau‐Piqueras J, Guerri C (2012) Toll‐like receptor 4 participates in the myelin disruptions associated with chronic alcohol abuse. Glia 60:948–964. [DOI] [PubMed] [Google Scholar]
- 3. Alfonso‐Loeches S, Pascual M, Guerri C (2013) Gender differences in alcohol‐induced neurotoxicity and brain damage. Toxicology 311:27–34. [DOI] [PubMed] [Google Scholar]
- 4. Alfonso‐Loeches S, Pascual‐Lucas M, Blanco AM, Sanchez‐Vera I, Guerri C (2010) Pivotal role of TLR4 receptors in alcohol‐induced neuroinflammation and brain damage. J Neurosci 30:8285–8295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ammendola A, Gemini D, Iannaccone S, Argenzio F, Ciccone G, Ammendola E et al (2000) Gender and peripheral neuropathy in chronic alcoholism: a clinical‐electroneurographic study. Alcohol Alcohol 35:368–371. [DOI] [PubMed] [Google Scholar]
- 6. Andersson AK, Ronnback L, Hansson E (2005) Lactate induces tumour necrosis factor‐alpha, interleukin‐6 and interleukin‐1beta release in microglial‐ and astroglial‐enriched primary cultures. J Neurochem 93:1327–1333. [DOI] [PubMed] [Google Scholar]
- 7. Astiz M, Acaz‐Fonseca E, Garcia‐Segura LM (2014) Sex differences and effects of estrogenic compounds on the expression of inflammatory molecules by astrocytes exposed to the insecticide dimethoate. Neurotox Res 25:271–285. [DOI] [PubMed] [Google Scholar]
- 8. Atkin G, Paulson H (2014) Ubiquitin pathways in neurodegenerative disease. Front Mol Neurosci 7:63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Atlante A, Calissano P, Bobba A, Giannattasio S, Marra E, Passarella S (2001) Glutamate neurotoxicity, oxidative stress and mitochondria. FEBS Lett 497:1–5. [DOI] [PubMed] [Google Scholar]
- 10. Bambrick L, Kristian T, Fiskum G (2004) Astrocyte mitochondrial mechanisms of ischemic brain injury and neuroprotection. Neurochem Res 29:601–608. [DOI] [PubMed] [Google Scholar]
- 11. Barnett SC, Linington C (2013) Myelination: do astrocytes play a role? Neuroscientist 19:442–450. [DOI] [PubMed] [Google Scholar]
- 12. Bass NH, Hess HH, Pope A, Thalheimer C (1971) Quantitative cytoarchitectonic distribution of neurons, glia, and DNA in rat cerebral cortex. J Comp Neurol 143:481–490. [DOI] [PubMed] [Google Scholar]
- 13. Baydas G, Tuzcu M (2005) Protective effects of melatonin against ethanol‐induced reactive gliosis in hippocampus and cortex of young and aged rats. Exp Neurol 194:175–181. [DOI] [PubMed] [Google Scholar]
- 14. Beadles‐Bohling AS, Wiren KM (2006) Anticonvulsive effects of kappa‐opioid receptor modulation in an animal model of ethanol withdrawal. Genes Brain Behav 5:483–496. [DOI] [PubMed] [Google Scholar]
- 15. Beck C, Boehler C, Guirouilh Barbat J, Bonnet ME, Illuzzi G, Ronde P et al (2014) PARP3 affects the relative contribution of homologous recombination and nonhomologous end‐joining pathways. Nucleic Acids Res 42:5616–5632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bradley JR (2008) TNF‐mediated inflammatory disease. J Pathol 214:149–160. [DOI] [PubMed] [Google Scholar]
- 17. Brambilla L, Martorana F, Rossi D (2013) Astrocyte signaling and neurodegeneration: new insights into CNS disorders. Prion 7:28–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Brozzi F, Arcuri C, Giambanco I, Donato R (2009) S100B protein regulates astrocyte shape and migration via interaction with src kinase: implications for astrocyte development, activation, and tumor growth. J Biol Chem 284:8797–8811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cahoy JD, Emery B, Kaushal A, Foo LC, Zamanian JL, Christopherson KS et al (2008) A transcriptome database for astrocytes, neurons, and oligodendrocytes: a new resource for understanding brain development and function. J Neurosci 28:264–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Carmen J, Rothstein JD, Kerr DA (2009) Tumor necrosis factor‐alpha modulates glutamate transport in the CNS and is a critical determinant of outcome from viral encephalomyelitis. Brain Res 1263:143–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chandler LJ, Newsom H, Sumners C, Crews F (1993) Chronic ethanol exposure potentiates NMDA excitotoxicity in cerebral cortical neurons. J Neurochem 60:1578–1581. [DOI] [PubMed] [Google Scholar]
- 22. Chaudhry FA, Lehre KP, van Lookeren Campagne M, Ottersen OP, Danbolt NC, Storm‐Mathisen J (1995) Glutamate transporters in glial plasma membranes: highly differentiated localizations revealed by quantitative ultrastructural immunocytochemistry. Neuron 15:711–720. [DOI] [PubMed] [Google Scholar]
- 23. Chen NF, Huang SY, Chen WF, Chen CH, Lu CH, Chen CL et al (2013) TGF‐beta1 attenuates spinal neuroinflammation and the excitatory amino acid system in rats with neuropathic pain. J Pain 14:1671–1685. [DOI] [PubMed] [Google Scholar]
- 24. Claycomb KI, Johnson KM, Winokur PN, Sacino AV, Crocker SJ (2013) Astrocyte regulation of CNS inflammation and remyelination. Brain Sci 3:1109–1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cook RT (1998) Alcohol abuse, alcoholism, and damage to the immune system–a review. Alcohol Clin Exp Res 22:1927–1942. [PubMed] [Google Scholar]
- 26. Cordeau P Jr, Lalancette‐Hebert M, Weng YC, Kriz J (2008) Live imaging of neuroinflammation reveals sex and estrogen effects on astrocyte response to ischemic injury. Stroke 39:935–942. [DOI] [PubMed] [Google Scholar]
- 27. Crabbe JC, Kosobud A, Young ER, Tam BR, McSwigan JD (1985) Bidirectional selection for susceptibility to ethanol withdrawal seizures in Mus musculus. Behav Genet 15:521–536. [DOI] [PubMed] [Google Scholar]
- 28. de la Monte SM, Kril JJ (2014) Human alcohol‐related neuropathology. Acta Neuropathol 127:71–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Daginakatte GC, Gadzinski A, Emnett RJ, Stark JL, Gonzales ER, Yan P et al (2008) Expression profiling identifies a molecular signature of reactive astrocytes stimulated by cyclic AMP or proinflammatory cytokines. Exp Neurol 210:261–267. [DOI] [PubMed] [Google Scholar]
- 30. Danbolt NC (2001) Glutamate uptake. Prog Neurobiol 65:1–105. [DOI] [PubMed] [Google Scholar]
- 31. Dobolyi A, Vincze C, Pal G, Lovas G (2012) The neuroprotective functions of transforming growth factor Beta proteins. Int J Mol Sci 13:8219–8258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Doherty MJ, Rostad SW, Kraemer DL, Vossler DG, Haltiner AM (2007) Neocortical gliosis in temporal lobe epilepsy: gender‐based differences. Epilepsia 48:1455–1459. [DOI] [PubMed] [Google Scholar]
- 33. Drescher KM, Murray PD, Lin X, Carlino JA, Rodriguez M (2000) TGF‐beta 2 reduces demyelination, virus antigen expression, and macrophage recruitment in a viral model of multiple sclerosis. J Immunol 164:3207–3213. [DOI] [PubMed] [Google Scholar]
- 34. Eng LF (1985) Glial fibrillary acidic protein (GFAP): the major protein of glial intermediate filaments in differentiated astrocytes. J Neuroimmunol 8:203–214. [DOI] [PubMed] [Google Scholar]
- 35. Feduccia AA, Chatterjee S, Bartlett SE (2012) Neuronal nicotinic acetylcholine receptors: neuroplastic changes underlying alcohol and nicotine addictions. Front Mol Neurosci 5:83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Forquer MR, Hashimoto JG, Roberts ML, Wiren KM (2011) Elevated testosterone in females reveals a robust sex difference in altered androgen levels during chronic alcohol withdrawal. Alcohol 45:161–171. [DOI] [PubMed] [Google Scholar]
- 37. Franke H (1995) Influence of chronic alcohol treatment on the GFAP‐immunoreactivity in astrocytes of the hippocampus in rats. Acta Histochem 97:263–271. [DOI] [PubMed] [Google Scholar]
- 38. Godsil BP, Kiss JP, Spedding M, Jay TM (2013) The hippocampal‐prefrontal pathway: the weak link in psychiatric disorders? Eur Neuropsychopharmacol 23:1165–1181. [DOI] [PubMed] [Google Scholar]
- 39. Gonzalez A, Pariente JA, Salido GM (2007) Ethanol stimulates ROS generation by mitochondria through Ca2+ mobilization and increases GFAP content in rat hippocampal astrocytes. Brain Res 1178:28–37. [DOI] [PubMed] [Google Scholar]
- 40. Greenham LW, Greenham V (1977) Sexing mouse pups. Lab Anim 11:181–184. [DOI] [PubMed] [Google Scholar]
- 41. Hardingham GE, Fukunaga Y, Bading H (2002) Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut‐off and cell death pathways. Nat Neurosci 5:405–414. [DOI] [PubMed] [Google Scholar]
- 42. Harper C (2009) The neuropathology of alcohol‐related brain damage. Alcohol Alcohol 44:136–140. [DOI] [PubMed] [Google Scholar]
- 43. Hashimoto JG, Beadles‐Bohling AS, Wiren KM (2004) Comparison of RiboGreen and 18S rRNA quantitation for normalizing real‐time RT‐PCR expression analysis. Biotechniques 36:54–56, 8‐60. [DOI] [PubMed] [Google Scholar]
- 44. Hashimoto JG, Wiren KM (2008) Neurotoxic consequences of chronic alcohol withdrawal: expression profiling reveals importance of gender over withdrawal severity. Neuropsychopharmacology 33:1084–1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Haydon PG, Blendy J, Moss SJ, Rob Jackson F (2009) Astrocytic control of synaptic transmission and plasticity: a target for drugs of abuse? Neuropharmacology 56 (Suppl. 1):83–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. He J, Crews FT (2008) Increased MCP‐1 and microglia in various regions of the human alcoholic brain. Exp Neurol 210:349–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hermann D, Weber‐Fahr W, Sartorius A, Hoerst M, Frischknecht U, Tunc‐Skarka N et al (2012) Translational magnetic resonance spectroscopy reveals excessive central glutamate levels during alcohol withdrawal in humans and rats. Biol Psychiatry 71:1015–1021. [DOI] [PubMed] [Google Scholar]
- 48. Herrera F, Sainz RM, Mayo JC, Martin V, Antolin I, Rodriguez C (2001) Glutamate induces oxidative stress not mediated by glutamate receptors or cystine transporters: protective effect of melatonin and other antioxidants. J Pineal Res 31:356–362. [DOI] [PubMed] [Google Scholar]
- 49. Hertz L (2004) The astrocyte‐neuron lactate shuttle: a challenge of a challenge. J Cereb Blood Flow Metab 24:1241–1248. [DOI] [PubMed] [Google Scholar]
- 50. Hinkle DA, Baldwin SA, Scheff SW, Wise PM (1997) GFAP and S100beta expression in the cortex and hippocampus in response to mild cortical contusion. J Neurotrauma 14:729–738. [DOI] [PubMed] [Google Scholar]
- 51. Hommer DW (2003) Male and female sensitivity to alcohol‐induced brain damage. Alcohol Res Health 27:181–185. [PMC free article] [PubMed] [Google Scholar]
- 52. Hopkins KJ, Wang G, Schmued LC (2000) Temporal progression of kainic acid induced neuronal and myelin degeneration in the rat forebrain. Brain Res 864:69–80. [DOI] [PubMed] [Google Scholar]
- 53. Hughes EG, Maguire JL, McMinn MT, Scholz RE, Sutherland ML (2004) Loss of glial fibrillary acidic protein results in decreased glutamate transport and inhibition of PKA‐induced EAAT2 cell surface trafficking. Brain Res Mol Brain Res 124:114–123. [DOI] [PubMed] [Google Scholar]
- 54. Ishibashi T, Dakin KA, Stevens B, Lee PR, Kozlov SV, Stewart CL, Fields RD (2006) Astrocytes promote myelination in response to electrical impulses. Neuron 49:823–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Jang E, Kim JH, Lee S, Kim JH, Seo JW, Jin M et al (2013) Phenotypic polarization of activated astrocytes: the critical role of lipocalin‐2 in the classical inflammatory activation of astrocytes. J Immunol 191:5204–5219. [DOI] [PubMed] [Google Scholar]
- 56. Jansson JO, Ekberg S, Isaksson O, Mode A, Gustafsson JA (1985) Imprinting of growth hormone secretion, body growth, and hepatic steroid metabolism by neonatal testosterone. Endocrinology 117:1881–1889. [DOI] [PubMed] [Google Scholar]
- 57. Jensen AA, Brauner‐Osborne H (2004) Pharmacological characterization of human excitatory amino acid transporters EAAT1, EAAT2 and EAAT3 in a fluorescence‐based membrane potential assay. Biochem Pharmacol 67:2115–2127. [DOI] [PubMed] [Google Scholar]
- 58. Kimelberg HK, Nedergaard M (2010) Functions of astrocytes and their potential as therapeutic targets. Neurotherapeutics 7:338–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Kovacs EJ, Messingham KA (2002) Influence of alcohol and gender on immune response. Alcohol Res Health 26:257–263. [PMC free article] [PubMed] [Google Scholar]
- 60. Koyama Y, Kimura Y, Hashimoto H, Matsuda T, Baba A (2000) L‐lactate inhibits L‐cystine/L‐glutamate exchange transport and decreases glutathione content in rat cultured astrocytes. J Neurosci Res 59:685–691. [DOI] [PubMed] [Google Scholar]
- 61. Kril JJ, Halliday GM, Svoboda MD, Cartwright H (1997) The cerebral cortex is damaged in chronic alcoholics. Neuroscience 79:983–998. [DOI] [PubMed] [Google Scholar]
- 62. Kuner P, Schubenel R, Hertel C (1998) Beta‐amyloid binds to p57NTR and activates NFkappaB in human neuroblastoma cells. J Neurosci Res 54:798–804. [DOI] [PubMed] [Google Scholar]
- 63. Lama S, Auer RN, Tyson R, Gallagher CN, Tomanek B, Sutherland GR (2014) Lactate storm marks cerebral metabolism following brain trauma. J Biol Chem 289:20200–20208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Lau A, Tymianski M (2010) Glutamate receptors, neurotoxicity and neurodegeneration. Pflugers Arch 460:525–542. [DOI] [PubMed] [Google Scholar]
- 65. Lau CL, Beart PM, O'Shea RD (2010) Transportable and non‐transportable inhibitors of L‐glutamate uptake produce astrocytic stellation and increase EAAT2 cell surface expression. Neurochem Res 35:735–742. [DOI] [PubMed] [Google Scholar]
- 66. Liefner M, Siebert H, Sachse T, Michel U, Kollias G, Bruck W (2000) The role of TNF‐alpha during Wallerian degeneration. J Neuroimmunol 108:147–152. [DOI] [PubMed] [Google Scholar]
- 67. Lippai D, Bala S, Petrasek J, Csak T, Levin I, Kurt‐Jones EA, Szabo G (2013) Alcohol‐induced IL‐1beta in the brain is mediated by NLRP3/ASC inflammasome activation that amplifies neuroinflammation. J Leukoc Biol 94:171–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Liu J, Lewohl JM, Dodd PR, Randall PK, Harris RA, Mayfield RD (2004) Gene expression profiling of individual cases reveals consistent transcriptional changes in alcoholic human brain. J Neurochem 90:1050–1058. [DOI] [PubMed] [Google Scholar]
- 69. Liu J, Lewohl JM, Harris RA, Dodd PR, Mayfield RD (2007) Altered gene expression profiles in the frontal cortex of cirrhotic alcoholics. Alcohol Clin Exp Res 31:1460–1466. [DOI] [PubMed] [Google Scholar]
- 70. Liu J, Marino MW, Wong G, Grail D, Dunn A, Bettadapura J et al (1998) TNF is a potent anti‐inflammatory cytokine in autoimmune‐mediated demyelination. Nat Med 4:78–83. [DOI] [PubMed] [Google Scholar]
- 71. Liu M, Hurn PD, Roselli CE, Alkayed NJ (2007) Role of. J Cereb Blood Flow Metab 27. [DOI] [PubMed] [Google Scholar]
- 72. Liu M, Oyarzabal EA, Yang R, Murphy SJ, Hurn PD (2008) A novel method for assessing sex‐specific and genotype‐specific response to injury in astrocyte culture. J Neurosci Methods 171:214–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Ma H, Shi J, Wang C, Guo L, Gong Y, Li J et al (2014) Blockade of PDE4B limits lung vascular permeability and lung inflammation in LPS‐induced acute lung injury. Biochem Biophys Res Commun 450:1560–1567. [DOI] [PubMed] [Google Scholar]
- 74. Maeda J, Higuchi M, Inaji M, Ji B, Haneda E, Okauchi T et al (2007) Phase‐dependent roles of reactive microglia and astrocytes in nervous system injury as delineated by imaging of peripheral benzodiazepine receptor. Brain Res 1157:100–111. [DOI] [PubMed] [Google Scholar]
- 75. Mayo L, Quintana FJ, Weiner HL (2012) The innate immune system in demyelinating disease. Immunol Rev 248:170–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Mayo L, Trauger SA, Blain M, Nadeau M, Patel B, Alvarez JI et al (2014) Regulation of astrocyte activation by glycolipids drives chronic CNS inflammation. Nat Med 20:1147–1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. McClive PJ, Sinclair AH (2001) Rapid DNA extraction and PCR‐sexing of mouse embryos. Mol Reprod Dev 60:225–226. [DOI] [PubMed] [Google Scholar]
- 78. McGeer EG, Jakubovic A, Singh EA (1980) Ethanol, baclofen, and kainic acid neurotoxicity. Exp Neurol 69:359–364. [DOI] [PubMed] [Google Scholar]
- 79. Miguel‐Hidalgo JJ, Waltzer R, Whittom AA, Austin MC, Rajkowska G, Stockmeier CA (2010) Glial and glutamatergic markers in depression, alcoholism, and their comorbidity. J Affect Disord 127:230–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Miyajima M, Minoshima M, Tanaka M, Nishimura R, Hishioka N, Numata T et al (2013) Increase in tetrahydrobiopterin concentration with aging in the cerebral cortex of the senescence‐accelerated mouse prone 10 strain caused by abnormal regulation of tetrahydrobiopterin biosynthesis. Biogerontology 14:491–501. [DOI] [PubMed] [Google Scholar]
- 81. Mong JA, Kurzweil RL, Davis AM, Rocca MS, McCarthy MM (1996) Evidence for sexual differentiation of glia in rat brain. Horm Behav 30:553–562. [DOI] [PubMed] [Google Scholar]
- 82. Montesinos J, Pascual M, Pla A, Maldonado C, Rodriguez‐Arias M, Minarro J, Guerri C (2015) TLR4 elimination prevents synaptic and myelin alterations and long‐term cognitive dysfunctions in adolescent mice with intermittent ethanol treatment. Brain Behav Immun 45:233–244. [DOI] [PubMed] [Google Scholar]
- 83. Mulligan MK, Rhodes JS, Crabbe JC, Mayfield RD, Harris RA, Ponomarev I (2011) Molecular profiles of drinking alcohol to intoxication in C57BL/6J mice. Alcohol Clin Exp Res 35:659–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Nakajima W, Ishida A, Lange MS, Gabrielson KL, Wilson MA, Martin LJ et al (2000) Apoptosis has a prolonged role in the neurodegeneration after hypoxic ischemia in the newborn rat. J Neurosci 20:7994–8004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Nash B, Thomson CE, Linington C, Arthur AT, McClure JD, McBride MW, Barnett SC (2011) Functional duality of astrocytes in myelination. J Neurosci 31:13028–13038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Nath V, Reneau JC, Dertien JS, Agrawal RG, Guerra I, Bhakta Y et al (2012) An in vitro model for studying the effects of continuous ethanol exposure on N‐methyl‐D‐aspartate receptor function. Alcohol 46:3–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Obayashi S, Tabunoki H, Kim SU, Satoh J (2009) Gene expression profiling of human neural progenitor cells following the serum‐induced astrocyte differentiation. Cell Mol Neurobiol 29:423–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Oldham MC, Konopka G, Iwamoto K, Langfelder P, Kato T, Horvath S, Geschwind DH (2008) Functional organization of the transcriptome in human brain. Nat Neurosci 11:1271–1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Ono K, Wang X, Kim SO, Armstrong LC, Bornstein P, Han J (2010) Metaxin deficiency alters mitochondrial membrane permeability and leads to resistance to TNF‐induced cell killing. Protein Cell 1:161–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Panegyres PK (1998) The effects of excitotoxicity on the expression of the amyloid precursor protein gene in the brain and its modulation by neuroprotective agents. J Neural Transm 105:463–478. [DOI] [PubMed] [Google Scholar]
- 91. Pascual M, Balino P, Alfonso‐Loeches S, Aragon CM, Guerri C (2011) Impact of TLR4 on behavioral and cognitive dysfunctions associated with alcohol‐induced neuroinflammatory damage. Brain Behav Immun 25 (Suppl. 1):S80–S91. [DOI] [PubMed] [Google Scholar]
- 92. Pelissier F, Lauque D, Charpentier S, Franchitto N (2014) Blood alcohol concentration in intoxicated patients seen in the emergency department: does it influence discharge decisions? J Stud Alcohol Drugs 75:937–944. [DOI] [PubMed] [Google Scholar]
- 93. Pellerin L, Magistretti PJ (1994) Glutamate uptake into astrocytes stimulates aerobic glycolysis: a mechanism coupling neuronal activity to glucose utilization. Proc Natl Acad Sci USA 91:10625–10629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Pfefferbaum A, Rosenbloom M, Deshmukh A, Sullivan E (2001) Sex differences in the effects of alcohol on brain structure. Am J Psychiatry 158:188–197. [DOI] [PubMed] [Google Scholar]
- 95. Pickering M, Cumiskey D, O'Connor JJ (2005) Actions of TNF‐alpha on glutamatergic synaptic transmission in the central nervous system. Exp Physiol 90:663–670. [DOI] [PubMed] [Google Scholar]
- 96. Prendergast MA, Harris BR, Mullholland PJ, Blanchard JA 2nd, Gibson DA, Holley RC, Littleton JM (2004) Hippocampal CA1 region neurodegeneration produced by ethanol withdrawal requires activation of intrinsic polysynaptic hippocampal pathways and function of N‐methyl‐D‐aspartate receptors. Neuroscience 124:869–877. [DOI] [PubMed] [Google Scholar]
- 97. Pronko PS, Velichko MG, Moroz AR, Rubanovich NN (1997) Low‐molecular‐weight metabolites relevant to ethanol metabolism: correlation with alcohol withdrawal severity and utility for identification of alcoholics. Alcohol and alcoholism. 32:761–768. [DOI] [PubMed] [Google Scholar]
- 98. Raponi E, Agenes F, Delphin C, Assard N, Baudier J, Legraverend C, Deloulme JC (2007) S100B expression defines a state in which GFAP‐expressing cells lose their neural stem cell potential and acquire a more mature developmental stage. Glia 55:165–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Rose EM, Koo JC, Antflick JE, Ahmed SM, Angers S, Hampson DR (2009) Glutamate transporter coupling to Na,K‐ATPase. J Neurosci 29:8143–8155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Rosen CL, Bunge RP, Ard MD, Wood PM (1989) Type 1 astrocytes inhibit myelination by adult rat oligodendrocytes in vitro . J Neurosci. 9:3371–3379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Ruocco A, Nicole O, Docagne F, Ali C, Chazalviel L, Komesli S et al (1999) A transforming growth factor‐beta antagonist unmasks the neuroprotective role of this endogenous cytokine in excitotoxic and ischemic brain injury. J Cereb Blood Flow Metab 19:1345–1353. [DOI] [PubMed] [Google Scholar]
- 102. Sahuquillo J, Merino MA, Sanchez‐Guerrero A, Arikan F, Vidal‐Jorge M, Martinez‐Valverde T et al (2014) Lactate and the lactate‐to‐pyruvate molar ratio cannot be used as independent biomarkers for monitoring brain energetic metabolism: a microdialysis study in patients with traumatic brain injuries. PLoS ONE 9:e102540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Salazar M, Pariente JA, Salido GM, Gonzalez A (2008) Ethanol induces glutamate secretion by Ca2+ mobilization and ROS generation in rat hippocampal astrocytes. Neurochem Int 52:1061–1067. [DOI] [PubMed] [Google Scholar]
- 104. Santos‐Galindo M, Acaz‐Fonseca E, Bellini MJ, Garcia‐Segura LM (2011) Sex differences in the inflammatory response of primary astrocytes to lipopolysaccharide. Biol Sex Differ 2:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Sarc L, Wraber B, Lipnik‐Stangelj M (2011) Ethanol and acetaldehyde disturb TNF‐alpha and IL‐6 production in cultured astrocytes. Hum Exp Toxicol 30:1256–1265. [DOI] [PubMed] [Google Scholar]
- 106. Sattler R, Xiong Z, Lu WY, Hafner M, MacDonald JF, Tymianski M (1999) Specific coupling of NMDA receptor activation to nitric oxide neurotoxicity by PSD‐95 protein. Science 284:1845–1848. [DOI] [PubMed] [Google Scholar]
- 107. Schafer GL, Crabbe JC, Wiren KM (1998) Identification of neuroendocrine‐specific protein as an ethanol‐regulated gene with mRNA differential display. Mamm Genome 9:979–982. [DOI] [PubMed] [Google Scholar]
- 108. Schuckit MA, Smith TL, Trim RS, Kuperman S, Kramer J, Hesselbrock V et al (2012) Sex differences in how a low sensitivity to alcohol relates to later heavy drinking. Drug Alcohol Rev 31:871–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Shih AY, Erb H, Sun X, Toda S, Kalivas PW, Murphy TH (2006) Cystine/glutamate exchange modulates glutathione supply for neuroprotection from oxidative stress and cell proliferation. J Neurosci 26:10514–10523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Sodhi RK, Singh N (2014) Retinoids as potential targets for Alzheimer's disease. Pharmacol Biochem Behav 120:117–123. [DOI] [PubMed] [Google Scholar]
- 111. Sosunov AA, Wu X, Tsankova NM, Guilfoyle E, McKhann GM 2nd, Goldman JE (2014) Phenotypic heterogeneity and plasticity of isocortical and hippocampal astrocytes in the human brain. J Neurosci 34:2285–2298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Stepanyan TD, Farook JM, Kowalski A, Kaplan E, Barron S, Littleton JM (2008) Alcohol withdrawal‐induced hippocampal neurotoxicity in vitro and seizures in vivo are both reduced by memantine. Alcohol Clin Exp Res 32:2128–2135. [DOI] [PubMed] [Google Scholar]
- 113. Takeuchi H, Jin S, Wang J, Zhang G, Kawanokuchi J, Kuno R et al (2006) Tumor necrosis factor‐alpha induces neurotoxicity via glutamate release from hemichannels of activated microglia in an autocrine manner. J Biol Chem 281:21362–21368. [DOI] [PubMed] [Google Scholar]
- 114. Tarassishin L, Suh HS, Lee SC (2014) LPS and IL‐1 differentially activate mouse and human astrocytes: role of CD14. Glia 62:999–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Thinakaran G, Koo EH (2008) Amyloid precursor protein trafficking, processing, and function. J Biol Chem 283:29615–29619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Tramontina F, Leite MC, Goncalves D, Tramontina AC, Souza DF, Frizzo JK et al (2006) High glutamate decreases S100B secretion by a mechanism dependent on the glutamate transporter. Neurochem Res 31:815–820. [DOI] [PubMed] [Google Scholar]
- 117. Tramontina F, Tramontina AC, Souza DF, Leite MC, Gottfried C, Souza DO et al (2006) Glutamate uptake is stimulated by extracellular S100B in hippocampal astrocytes. Cell Mol Neurobiol 26:81–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Wilhelm CJ, Hashimoto JG, Roberts ML, Bloom SH, Beard DK, Wiren KM (2015) Females Uniquely vulnerable to alcohol‐induced neurotoxicity show altered glucocorticoid signaling. Brain Res 19:102–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Wilhelm CJ, Hashimoto JG, Roberts ML, Sonmez MK, Wiren KM (2014) Understanding the addiction cycle: a complex biology with distinct contributions of genotype vs. sex at each stage. Neuroscience 279C:168–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Wiren K, Keenan E, Zhang X, Ramsey B, Orwoll E (1999) Homologous androgen receptor up‐regulation in osteoblastic cells may be associated with enhanced functional androgen responsiveness. Endocrinology 140:3114–3124. [DOI] [PubMed] [Google Scholar]
- 121. Wiren KM (2013) Males and females are just different: sexually dimorphic responses to chronic ethanol exposure in hippocampal slice cultures. Neurosci Lett 550:1–5. [DOI] [PubMed] [Google Scholar]
- 122. Witt ED (2007) Puberty, hormones, and sex differences in alcohol abuse and dependence. Neurotoxicol Teratol 29:81–95. [DOI] [PubMed] [Google Scholar]
- 123. Zerbi V, Kleinnijenhuis M, Fang X, Jansen D, Veltien A, Van Asten J et al (2013) Gray and white matter degeneration revealed by diffusion in an Alzheimer mouse model. Neurobiol Aging 34:1440–1450. [DOI] [PubMed] [Google Scholar]
- 124. Zhao C, Ling Z, Newman MB, Bhatia A, Carvey PM (2007) TNF‐alpha knockout and minocycline treatment attenuates blood‐brain barrier leakage in MPTP‐treated mice. Neurobiol Dis 26:36–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Ziegler DR, Innocente CE, Leal RB, Rodnight R, Goncalves CA (1998) The S100B protein inhibits phosphorylation of GFAP and vimentin in a cytoskeletal fraction from immature rat hippocampus. Neurochem Res 23:1259–1263. [DOI] [PubMed] [Google Scholar]
- 126. Zou JY, Crews FT (2005) TNF alpha potentiates glutamate neurotoxicity by inhibiting glutamate uptake in organotypic brain slice cultures: neuroprotection by NF kappa B inhibition. Brain Res 1034:11–24. [DOI] [PubMed] [Google Scholar]
- 127. Zuloaga DG, McGivern RF, Handa RJ (2009) Organizational influence of the postnatal testosterone surge on the circadian rhythm of core body temperature of adult male rats. Brain Res 1268:68–75. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Gene expression following chronic EtOH.
Figure S2. Gene expression following recovery from chronic EtOH.
Table S1. Female and male ethanol‐regulated networks.