

Abstract
Huntington's disease (HD) is caused by a CAG‐repeat encoding a polyglutamine (polyQ) tract in the huntingtin protein. There is plenty of evidence of polyQ‐driven toxicity. However, CAG repeat RNA‐driven alteration of splicing has recently been proposed in analogy to CUG‐repeat diseases. Here we review the reported alteration of the CAG‐repeat associated splicing factor SRSF6 in brains of HD patients and mouse models and how this correlates with altered splicing of, at least, two microtubule‐associated proteins in HD, namely MAPT (tau) and MAP2. Regarding tau, altered splicing of exon 10 has been reported, along with increased levels and 4R/3R‐tau ratio and detection of tau in a new nuclear rod‐shaped histopathological hallmark termed tau nuclear rod (TNR) or tau nuclear indentation (TNI). These findings, together with an attenuation of HD phenotype in R6/1 mice with tau deficiency and subsequent studies showing increased phosphorylation in mouse models and increased levels in CSF of patients, has led to proposing HD as a tauopathy. Regarding MAP2, an increase in its juvenile form and a decrease in total MAP2 together with redistribution from dendrites to soma is observed in HD patients, which may contribute to the dendritic atrophy in HD. Furthermore, MAP2 positive structures filling nuclear indentations have occasionally been found and co‐localized with tau. Therefore, altered MAP function with imbalance in tau/MAP2 content could contribute to HD striatal atrophy and dysfunction. Besides, TNIs might be indicative of such MAP abnormalities. TNIs are also found in early pathology Alzheimer's disease and in tauopathy mice over‐expressing mutant 4R‐tau. This indicates that tau alteration is sufficient for TNI detection, which becomes a marker of increased total tau and/or altered 4R/3R‐tau ratio and reporter of pathology‐associated nuclear indentations. Altogether, these recent studies suggest that correcting the SRSF6‐driven missplicing and/or microtubule‐associated imbalance might be of therapeutic value in HD.
Keywords: Huntington's disease, MAP2, splicing, SRSF6, tau, tauopathy, TNR (tau nuclear rod), TNI (tau‐immunopositive nuclear indentation)
INTRODUCTION
Huntington's disease (HD) is a strictly inherited autosomal‐dominant neurodegenerative disorder that affects 5–10 in 100 000 individuals 53. It is characterized by a marked degeneration of the striatum and a widespread cortical atrophy leading to involuntary movements such as chorea and tremor that typically appear in midlife. The disease then progresses to dementia and leads to death of the patient 15–20 years after onset 28. Currently, there is no curative treatment for HD.
The mutation causing HD is a CAG repeat expansion in exon 1 of the huntingtin gene (HTT) 29. HD thus belongs to the group of developmental and degenerative diseases that are caused by expansion of unstable repeats 21. More precisely, it is the paradigm of the subgroup of neurodegenerative diseases caused by a gain of function of translated CAG repeats encoding polyglutamine (polyQ) tracts in the corresponding proteins. This subgroup also includes dentatorubralpallidoluysian atrophy (DRPLA), spinal and bulbar muscular atrophy (SBMA) and spinocerebellar ataxia 1, 2, 3, 6, 7 and 17 (SCA1/2/3/6/7/17).
There is plenty of evidence supporting toxicity of the expanded polyQ‐containing proteins as they interfere with multiple essential functions of the cell such as transcription—by sequestering multiple transcription factors with PolyQ domains 39, mitochondrial function—by interacting with various protein complexes in the external mitochondrial membrane 59 or the proteostasis mechanisms such as the ubiquitin proteasome system 43 and autophagy 33, among others. Here we will pay particular attention to those pathogenic mechanisms of mutant huntingtin related to alteration of the neuronal cytoskeleton.
Thanks to their specialized cytoskeleton, neurons maintain their complex morphology with two types of cytoplasmic extensions, axons and dendrites. Neural transmission occurs through these processes and changes in neuronal morphology may affect neuronal function and induce pathology. The cytoskeleton is composed of three main components: the microtubules, the microfilaments and the intermediate filaments. Microtubules are very dynamic tubulin polymers that in neurons become stabilized in specific directions to maintain the axonal and dendritic cytoplasmic extensions 40. The proteins that serve to stabilize microtubules are the microtubule‐associated proteins (or MAPs) MAP1A, MAP1B, MAP2 and tau 38. In support of the role of these proteins in maintaining the integrity of the axonal and dendritic cytoplasmic extensions, an asymmetric distribution of MAPs 38 is seen in mature neurons 10 with tau being preferentially localized in axons 3. Anterograde and retrograde transport of cargoes along neuronal projections takes place along microtubules by the action of the kinesins and the dynein–dynactin motor proteins 24. A classic pathogenic mechanism of expanded PolyQ‐Htt is the interference with transport machinery along microtubule cytoskeleton, by interaction of mutant huntingtin with motor proteins that transport cargoes along microtubules such as dynein–dynactin 22, 36.
In this review we will elaborate on impaired alternative splicing as a recently identified pathogenic mechanism in HD 35 that in turn affects microtubule‐associated proteins such as tau and MAP2, thus impinging on their loss function and, in the case of tau, a toxic gain of function as well that has led to considering HD as a tauopathy.
THE SPLICING FACTOR SRSF6 IS ALTERED IN HD
Probably because of the mentioned strong evidence supporting the toxicity of expanded PolyQ, the potential toxicity of the expanded CAG mRNA has often been partially overlooked (see Chapter by E. Martí in this mini‐symposium for extensive review on RNA bases of HD pathology). However, other trinucleotide repeat expansion disorders, some of them with equivalent neuropathology and symptoms, are caused by expansions in non‐coding regions of their transcripts 9. These include the CAG repeat disorder (SCA12) and several CUG repeat disorders such as Huntington's disease‐like‐2 (HDL‐2) and myotonic dystrophy 1 (DM1) 9, 42. Although toxicity could still arise from translation of polyaminoacids—in some cases PolyQ itself—because of the repeat‐associated non‐ATG mediated initiation of translation 7, for these diseases with non‐exonic expansions there is clear evidence of expanded repeat containing mRNAs being toxic. This is best characterized in the case of DM1 for which a key element in the pathogenesis is the binding of splicing factors by the mutant CUG transcript, thus leading to alternative splicing aberrations in multiple genes 44.
Alternative precursor‐mRNA splicing is a key mechanism for regulating gene expression in mammals. Alternative splicing patterns are regulated by specialized RNA‐binding proteins that alter spliceosome assembly at specific splice sites. These include splicing regulators such as CUGBP1 or MBNL1/2 proteins, the SR‐proteins and the hnRNPs 19. As mentioned, CUG repeats sequester specific splicing factors and, intriguingly, CAG repeats have recently been shown to mimic CUG repeats in the misregulation of alternative splicing 41, while the splicing factor SRSF6 (also known as SRp55) was recently bioinformatically predicted by Bates et al 48 to bind CAG repeats according to the 6‐mer YRCRKM (T/C,A/G,C,A/G,G/T,A/C). This consensus has been refined and extended to a 9‐mer sequence out of which the last eight nucleotides show a perfect match with CAGCAGCA sequence 32.
In this regard, it has recently been reported that the splicing factor SRSF6 is altered in brain of HD patients and mouse models 15. SRSF6 protein levels are increased in striatum of HD patients with the appearance of a higher molecular weight band that could reflect a rise in its phosphorylation status. In fact, a phospho‐SR antibody showed increased levels at 55 kDa, the predicted molecular weight for phospho‐SRSF6, both in HD patient striatum 15 and cortex 6 and in the R6/1 mouse model of the disease 15. When looking at its cellular localization, it has been found that SRSF6 accumulates into nuclear inclusion bodies in R6/1 mice, where SRSF6 and Htt co‐localize. In the case of human HD striatum, SRSF6 also accumulates in Htt‐inclusion bodies and, in addition, other nuclear and cytoplasmic accumulations with a variety of shapes have been found, showing inclusion‐, foci‐, thread‐ and rod‐like aggregates (Figure 1).
Figure 1.
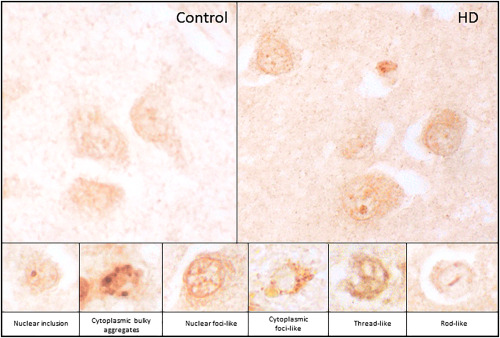
Immunohistochemistry with SRSF6 antibody in human striatum from a control (upper left panel) and a HD (upper right panel) case. Magnified images (lower panels) show the different shapes that SRSF6 depositions can adopt in HD.
In line with this, two SRSF6‐mediated splicing alterations have been described that may be important for the pathogenesis of HD, one in the HTT gene itself, giving rise to a highly toxic N‐terminal form of mutant HTT 48. More recently, we and others have reported the neurodegeneration causing‐missplicing of exon 10 of the tau (MAPT) gene in HD patients 15, 54, which led us to propose HD as a secondary tauopathy.
HD AS A MISSPLICING TAUOPATHY
Tauopathies are a group of neurodegenerative diseases characterized by altered metabolism and deposition of the neuronal microtubule‐associated protein tau, including Alzheimer's disease, progressive supranuclear palsy (PSP), Pick's disease and forms of frontotemporal lobar degeneration (FTLD) among others 2, 34. Tau modifications in tauopathies include phosphorylation and truncation that may decrease or increase their affinity to bind with and stabilize microtubules, thus causing loss or excess of function. A toxic gain of function has also been attributed to tau deposits either in the form of the small micro‐aggregates and the bigger filamentous tangles or spheroid Pick bodies. The discovery of mutations in MAPT as causative of familial FTLD‐tau evidenced that tau alteration is sufficient to cause neurodegenerative disease 30. Apart from point mutations, FTLD‐tau can also be caused by silent and intronic mutations that affect the alternative splicing of MAPT exon 10 (Figure 2A), which generates tau isoforms with either three or four tubulin‐binding repeats (3R‐ and 4R‐tau), thus demonstrating that an imbalance in the 4R/3R‐tau ratio is pathological per se.
Figure 2.
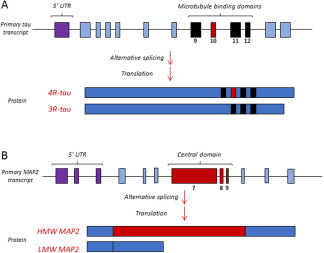
Schematic view of the alternative splicing of tau exon 10 (A) and MAP2 central domain (B). Regarding tau, alternative splicing of exon 10 will define the isoforms with 3 or 4 microtubule binding domains (3R and 4R tau). In the case of MAP2, alternative splicing of (HMW) or low (LMW) molecular weight, being the HMW MAP2 characteristic of adult neurons and the LMW MAP2 a juvenile form.
SRSF6 is one of the splicing factors that clearly affect alternative splicing of MAPT exon 10 20, 55, 60. When the ratio of 4R‐ and 3R‐tau mRNA was analyzed in the brain of HD patients, an alteration in the splicing of tau exon 10 was observed, producing an imbalance in tau mRNA isoforms with an increase in the tau‐4R mRNA isoforms and a decrease in the tau‐3R isoforms in the cortex and striatum of HD patients 15. With 3R‐tau and 4R‐tau specific antibodies, an equivalent increase in the 4R/3R ratio was observed at the protein level and, furthermore, it was found that the total amount of tau is increased in cortex of HD patients 15. Imbalance in favor of 4R‐tau at the RNA and protein levels and increased total tau protein content were observed also in brain of two HD mouse models, namely R6/1 and HD94 mice.
This, together with the detection of tau accumulation in the forms of rods that seemed to span the neuronal nuclei and which were initially termed Tau Nuclear Rods (TNRs), as well as the attenuation of HD phenotype of R6/1 mice in tau deficient genetic background 15 led to the proposal that HD should be considered as a tauopathy. Four independent studies conducted in human HD brains and in mouse models appeared shortly thereafter and corroborated the 4R/3R imbalance and the presence of TNRs in human HD brains 54 and reported further alteration of tau in HD such as increased phosphorylation in mouse models 4, 26 and increased levels in CSF of HD patients 56. An excellent, comprehensive review of all these recent studies pointing to HD as a tauopathy has recently been published 25. This review also summarizes the previous clinical studies exploring tau pathology comorbidity in HD brains based on analysis of tau neurofibrillary pathology with antibodies that mainly detect hyperphosphorylated tau 31, 51 as well as the increased detection of tau in CSF of HD patients 8, with levels that correlate with those of mutant huntingtin in CSF 56. In addition and more recently, CSF total tau concentration has been reported to predict clinical phenotype in HD 45. The review by Gratuze et al 25 also summarizes the studies reporting increased tau phosphorylation in various mouse models of HD such as R6/1, R6/2, Q175 and KI140 mice in relation to the level and activity of tau‐kinases (GSK3, CDK5, ERK1, ERK2, MAPK1, CamKII) and tau‐phosphatases (PP1, PP2 and PP2B), suggesting a crucial role of the previously known decrease of PP2B (also known as calcineurin) in HD 58 in the observed increased tau phosphorylation. This also fits well with the reported decreased levels and activity of GSK3, one of the main tau kinases, in striatum and cortex of HD brains 16, 37. The level and haplotype of tau has also been related to the dementia in HD 26, 54.
In summary, altered tau exon 10 splicing, increased total tau levels and 4R/3R‐tau protein ratio in brains and increased tau correlating with mutant huntingtin levels in CSF have been found in HD patients. Furthermore, HD phenotype attenuation under tau deficiency, increased total tau levels, increased tau phosphorylation and altered tau‐kinases and tau‐phosphatases have been found in HD mouse models. Altogether, it seems reasonable to consider HD as a secondary tauopathy.
MAP2 SPLICING IS ALTERED IN HD
An emerging concept in splicing factor physiology is the possibility that these proteins regulate multiple functionally related genes as entire gene expression programs 1, 23. The closest homolog to tau is the microtubule‐binding protein MAP2 11. We recently hypothesized that MAP2 might be target of SRSF6 and altered in HD 6.
As mentioned, MAP2 is a neuronal cytoskeleton regulator and the main microtubule‐associated protein in dendrites is MAP2. Interestingly, dendritic alteration of striatal medium spiny neurons is one of the earliest morphological abnormalities in HD 18. Different MAP2 isoforms are generated by alternative splicing (Figure 2B). The low‐molecular weight isoforms of MAP2 (LMW‐MAP2) are the juvenile forms resulting from exclusion of the sequence encoded by exons E7‐E9. LMW‐MAP2 is down‐regulated after the early stages of neuronal development, when E7‐E9 exon‐including high‐molecular weight isoforms (HMW‐MAP2) are favored. Alternative splicing of MAP2 is critical for its subcellular distribution. HMW isoforms are restricted to dendrites and cell bodies 50, whereas LMW isoforms are more widely distributed across the different neuronal compartments 14. This subcellular localization of MAP2 in dendrites and cell bodies suggests a complementary function with tau that, as mentioned, is the main microtubule‐associated protein expressed specifically in axons 11, 47.
To explore whether SRSF6 indeed affects MAP2 alternative splicing, SRSF6 knockdown experiments were performed in neuroblastoma cells 6. These experiments confirmed that splicing of MAP2 E7‐E9 exons is affected by SRSF6. Accordingly, an imbalance in LMW and HMW MAP2 mRNA isoforms was detected in striatum of HD patients in favor of the juvenile LMW forms, together with a decrease in total MAP2 mRNA. This was accompanied by a global decrease in total MAP2 protein. In good agreement, the predominant dendritic MAP2 staining normally observed in striatal neuropil of control subjects is absent in HD cases. In these, MAP2‐immunoreactivity is faint and restricted to neuronal cell bodies, often showing a sharp boundary at the base of dendrites. This study, therefore, highlights the importance of splicing alteration for the cytoskeleton alterations in HD, and suggests that MAP2 alteration contributes to dendritic atrophy.
Another observation from the analysis of the MAP2 staining in control and HD subjects is occasional detection of filamentous MAP2 structures that appear to extend along the nucleus of neurons both in controls and HD subjects. As for tau‐immunopositive nuclear indentations (TNIs) in HD neurons, confocal sections show that these MAP2 positive structures fill nuclear indentations and they even co‐localize. Since these MAP2 positive rod‐like deposits appear in control and HD patients in a similar number with total levels of MAP2 being severely decreased in the striatum of HD, it is difficult to infer whether they are enriched in the patients or not.
In any case, the question arises of whether alteration of microtubule dynamics as a consequence of altered composition of two MAPs, tau and MAP2, would result in the formation of these rods spanning the nuclear space.
ARE NUCLEAR INDENTATIONS RELATED TO CYTOSKELETON ABNORMALITIES IN HD NEURONS?
When we first reported the nuclear rod‐shaped tau immunostainings in neurons of cortex and striatum of HD brains 15 and in hippocampus of early Braak stage AD brains we coined the term tau nuclear rods (TNRs) 15, as they appear to traverse the nuclear space 15, 54. However, immunoelectron microscopy and confocal analysis of tau immunofluorescence with fluorescent nuclear counterstaining revealed that TNRs in brains of HD patients in fact occupy narrow cytoplasmic extensions that fill infoldings of the nuclear envelope 15, so we will here refer to this histological hallmark as TNIs.
Interestingly, rod‐shaped indentations of the nuclear envelope are also known to be common in neurons 27, 49 and their incidence has been reported to change as a consequence of the synaptic activity of the neurons 57. Most importantly for our subject, increased incidence of nuclear indentations has been known for decades in striatum of HD brain and more recently also in related CAG triplet repeat disorder patients 5, 46, 52 as well as cell and mouse models 12, 13, 61.
We reasoned that TNI formation in neurons of HD patients was likely secondary to tau alterations as TNI detection in HD correlates with an increase in total tau, particularly of the isoforms with four tubulin‐binding repeats (4R‐tau). In order to test this hypothesis, tau levels and localization in the P301S mouse model, which overexpress 4R‐tau with a FTLD point mutation, were assessed 17. As expected, total tau levels were dramatically increased in these animals in all analyzed regions. When looking for TNIs with various tau antibodies, they were found profusely, preferentially with non‐phosphorylated tau antibodies. Surprisingly, an increase in nuclear indentation incidence was not found, suggesting that tau alteration in P301S mice does not cause per se an increment in the amount of nuclear indentation. On the other hand, tau alteration in P301S mice is indeed enough to detect TNIs, which could become, in histopathological analysis of human as well as mouse brain tissue, a suitable indicator of neurons with pathology‐associated nuclear indentations, increased total tau and/or altered 4R/3R‐tau ratio.
CONCLUSIONS
Alternative splicing alteration is a well‐established pathogenic mechanism in CUG triplet repeat disorders because expanded CUG RNA sequences form hairpins able to sequester specific splicing factors such as MBNL. Expanded CAG sequences have been shown to form similar hairpins and to bind splicing factors. SRSF6 is one such factors, as its consensus binding motif fits well with the expanded CAG sequence. SRSF6 has been found to be profoundly altered in striatum and cortex of HD patient brains and mouse models and two neurodegeneration‐associated missplicing events have been reported that can be explained by the alteration of SRSF6 in HD. One such missplicing event affects the first intron in HTT gene and results in the formation of highly toxic exon 1 encoded short forms of mutant huntingtin. The other missplicing event affects exon 10 of the MAPT gene and results in an imbalance of tau isoforms with three or four microtubule binding motifs in favor of the latter; such imbalance is sufficient to cause neurodegeneration as evidenced in FTLD‐affected families with intronic and silent mutations flanking exon 10 splicing sites. This neurodegeneration causing 4R/3R‐tau imbalance correlates with an increase in total tau levels in brains of HD patients and mouse models and a possible explanation might be increased half‐life of 4R‐tau over 3R‐tau, although this has not been explored. Another microtubule‐associated protein, MAP2, has recently been reported to be a target of SRSF6 and to be misspliced in striatum of HD patients. More precisely, the juvenile form of MAP2 lacking exons 7–9 is favored in HD brains. This correlates with a redistribution of MAP2 from the dendrites to the neuronal soma and with a decrease in total MAP2 levels that might be related to the early dendritic arborization abnormalities observed in striatal neurons of HD patients. Therefore, toxic gain of function of MAPS such as the increased 4R/3R‐tau ratio, possible excess function because of the increased total tau, decreased function such as reduced total MAP2 or an imbalance in tau/MAP2 content in the neuron are likely contributors to HD striatal atrophy and dysfunction (Figure 3). A new histopathological marker, the rod‐like tau positive nuclear indentations (TNIs), is found to be dramatically increased in brain of HD patients and may be a reporter of such MAP abnormalities in HD brains. Interestingly, this histopathological hallmark is not restricted to HD as it is also found in hippocampal neurons of early neuropathological stages of AD cases and in tauopathy mice over‐expressing 4R‐tau with FTLD P301S mutation. This indicates that tau alteration is sufficient for detection of TNIs that may be possible indicators of increased total tau and/or increased 4R/3R‐tau ratio in the affected neurons, in addition to being an efficient way to monitor pathology‐associated nuclear indentations. Summing up, these recent studies suggest that mending altered splicing events driven by SRSF6 could be of therapeutic value for HD, as well as for correcting the microtubule‐associated imbalance.
Figure 3.
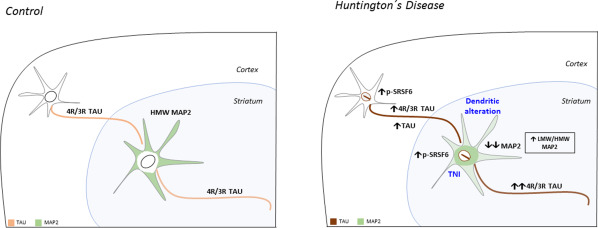
Schematic representation of the cytoskeleton abnormalities related to SRSF6 splicing in HD. In control conditions, 4R/3R tau will be balanced between both isoforms and MAP2 predominant isoform will be the HMW MAP2, located mainly in dendrites. In HD, the increase in p‐SRSF6 levels both in cortex and striatum of HD patients produces an imbalance in the 4R/3R tau ratio in favor of 4R‐tau and an increase in total tau in cortex. In addition, MAP2 splicing is altered in favor of the LMW MAP2 isoforms with a redistribution from the dendrites to the soma. A global decrease in the levels of MAP2 is also found. This alterations correlate with dendritic atrophy and the appearance of TNIs contributing to cytoskeleton abnormalities in HD neurons.
ACKNOWLEDGEMENTS
This work was supported by Centro de Investigación Biomédica en Red de Enfermedades Neurodegenerativas (CiberNed‐PI2015‐2/06‐3), by Spanish Ministery of Economy and Competitiveness (MINECO, SAF2015‐65371‐R) and by Fundación BBVA.
REFERENCES
- 1. Anko ML (2014) Regulation of gene expression programmes by serine‐arginine rich splicing factors. Semin Cell Dev Biol 32:11–21. [DOI] [PubMed] [Google Scholar]
- 2. Avila J, Lucas JJ, Perez M, Hernandez F (2004) Role of tau protein in both physiological and pathological conditions. Physiol Rev 84:361– 384. [DOI] [PubMed] [Google Scholar]
- 3. Binder LI, Frankfurter A, Rebhun LI (1985) The distribution of tau in the mammalian central nervous system. J Cell Biol 101:1371–1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Blum D, Herrera F, Francelle L, Mendes T, Basquin M et al (2015) Mutant huntingtin alters Tau phosphorylation and subcellular distribution. Hum Mol Genet 24:76–85. [DOI] [PubMed] [Google Scholar]
- 5. Bots GT, Bruyn GW (1981) Neuropathological changes of the nucleus accumbens in Huntington's chorea. Acta Neuropathol 55:21–22. [DOI] [PubMed] [Google Scholar]
- 6. Cabrera JR, Lucas JJ (2016) MAP2 splicing is altered in Huntington's disease. Brain Pathol, 2016 Apr 21. doi: 10.1111/bpa.12387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cleary JD, Ranum LP (2014) Repeat associated non‐ATG (RAN) translation: new starts in microsatellite expansion disorders. Curr Opin Genet Dev 26:6–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Constantinescu R, Romer M, Zetterberg H, Rosengren L, Kieburtz K (2011) Increased levels of total tau protein in the cerebrospinal fluid in Huntington's disease. Parkinsonism Relat Disord 17:714–715. [DOI] [PubMed] [Google Scholar]
- 9. Cooper TA (2009) Molecular biology. Neutralizing toxic RNA. Science 325:272–273. [DOI] [PubMed] [Google Scholar]
- 10. Craig AM, Banker G (1994) Neuronal polarity. Annu Rev Neurosci 17:267–310. [DOI] [PubMed] [Google Scholar]
- 11. Dehmelt L, Halpain S (2005) The MAP2/Tau family of microtubule‐associated proteins. Genome Biol 6:204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Diaz‐Hernandez M, Hernandez F, Martin‐Aparicio E, Gomez‐Ramos P, Moran MA et al (2003) Neuronal induction of the immunoproteasome in Huntington's disease. J Neurosci 23:11653–11661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Evert BO, Wullner U, Schulz JB, Weller M, Groscurth P et al (1999) High level expression of expanded full‐length ataxin‐3 in vitro causes cell death and formation of intranuclear inclusions in neuronal cells. Hum Mol Genet 8:1169–1176. [DOI] [PubMed] [Google Scholar]
- 14. Ferhat L, Represa A, Ferhat W, Ben‐Ari Y, Khrestchatisky M (1998) MAP2d mRNA is expressed in identified neuronal populations in the developing and adult rat brain and its subcellular distribution differs from that of MAP2b in hippocampal neurones. Eur J Neurosci 10:161–171. [DOI] [PubMed] [Google Scholar]
- 15. Fernandez‐Nogales M, Cabrera JR, Santos‐Galindo M, Hoozemans JJ, Ferrer I et al (2014) Huntington's disease is a four‐repeat tauopathy with tau nuclear rods. Nat Med 20:881–885. [DOI] [PubMed] [Google Scholar]
- 16. Fernandez‐Nogales M, Hernandez F, Miguez A, Alberch J, Gines S et al (2015) Decreased glycogen synthase kinase‐3 levels and activity contribute to Huntington's disease. Hum Mol Genet 24:5040–5052. [DOI] [PubMed] [Google Scholar]
- 17. Fernandez‐Nogales M, Santos‐Galindo M, Merchan‐Rubira J, Hoozemans J, Rabano A et al (2016) Tau‐positive nuclear indentations in P301S tauopathy mice. Brain Pathol, 2016 Jun 24. doi: 10.1111/bpa.12407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ferrante RJ, Kowall NW, Richardson EP Jr. (1991) Proliferative and degenerative changes in striatal spiny neurons in Huntington's disease: a combined study using the section‐Golgi method and calbindin D28k immunocytochemistry. J Neurosci 11:3877–3887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Fu XD, Ares M Jr. (2014) Context‐dependent control of alternative splicing by RNA‐binding proteins. Nat Rev Genet 15:689–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gao QS, Memmott J, Lafyatis R, Stamm S, Screaton G, Andreadis A (2000) Complex regulation of tau exon 10, whose missplicing causes frontotemporal dementia. J Neurochem 74:490–500. [DOI] [PubMed] [Google Scholar]
- 21. Gatchel JR, Zoghbi HY (2005) Diseases of unstable repeat expansion: mechanisms and common principles. Nat Rev Genet 6:743–755. [DOI] [PubMed] [Google Scholar]
- 22. Gauthier LR, Charrin BC, Borrell‐Pages M, Dompierre JP, Rangone H et al (2004) Huntingtin controls neurotrophic support and survival of neurons by enhancing BDNF vesicular transport along microtubules. Cell 118:127–138. [DOI] [PubMed] [Google Scholar]
- 23. Germann S, Gratadou L, Dutertre M, Auboeuf D (2012) Splicing programs and cancer. J Nucleic Acids 2012:269570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Goldstein LS, Yang Z (2000) Microtubule‐based transport systems in neurons: the roles of kinesins and dyneins. Annu Rev Neurosci 23:39– 71. [DOI] [PubMed] [Google Scholar]
- 25. Gratuze M, Cisbani G, Cicchetti F, Planel E (2016) Is Huntington's disease a tauopathy? Brain 139:1014–1025. [DOI] [PubMed] [Google Scholar]
- 26. Gratuze M, Noel A, Julien C, Cisbani G, Milot‐Rousseau P et al (2015) Tau hyperphosphorylation and deregulation of calcineurin in mouse models of Huntington's disease. Hum Mol Genet 24:86–99. [DOI] [PubMed] [Google Scholar]
- 27. Graveland GA, DiFiglia M (1985) The frequency and distribution of medium‐sized neurons with indented nuclei in the primate and rodent neostriatum. Brain Res 327:307–311. [DOI] [PubMed] [Google Scholar]
- 28. Harper PS (1992) The epidemiology of Huntington's disease. Hum Genet 89:365–376. [DOI] [PubMed] [Google Scholar]
- 29. HDCRG (1993) A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. The Huntington's Disease Collaborative Research Group. Cell 72:971–983. [DOI] [PubMed] [Google Scholar]
- 30. Hutton M, Lendon CL, Rizzu P, Baker M, Froelich S et al (1998) Association of missense and 5'‐splice‐site mutations in tau with the inherited dementia FTDP‐17. Nature 393:702–705. [DOI] [PubMed] [Google Scholar]
- 31. Jellinger KA (1998) Alzheimer‐type lesions in Huntington's disease. J Neural Transm (Vienna) 105:787–799. [DOI] [PubMed] [Google Scholar]
- 32. Jensen MA, Wilkinson JE, Krainer AR (2014) Splicing factor SRSF6 promotes hyperplasia of sensitized skin. Nat Struct Mol Biol 21:189–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Jimenez‐Sanchez M, Thomson F, Zavodszky E, Rubinsztein DC (2012) Autophagy and polyglutamine diseases. Prog Neurobiol 97:67–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lee VM, Goedert M, Trojanowski JQ (2001) Neurodegenerative tauopathies. Annu Rev Neurosci 24:1121–1159. [DOI] [PubMed] [Google Scholar]
- 35. Lewis S (2014) Neurodegenerative disease: faulty splicing in Huntington's disease. Nat Rev Neurosci 15:564. [Google Scholar]
- 36. Li SH, Gutekunst CA, Hersch SM, Li XJ (1998) Interaction of huntingtin‐associated protein with dynactin P150Glued. J Neurosci 18:1261–1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lim NK, Hung LW, Pang TY, McLean CA, Liddell JR et al (2014) Localized changes to glycogen synthase kinase‐3 and collapsin response mediator protein‐2 in the Huntington's disease affected brain. Hum Mol Genet 23:4051–4063. [DOI] [PubMed] [Google Scholar]
- 38. Matus A (1988) Microtubule‐associated proteins: their potential role in determining neuronal morphology. Annu Rev Neurosci 11:29–44. [DOI] [PubMed] [Google Scholar]
- 39. McCampbell A, Fischbeck KH (2001) Polyglutamine and CBP: fatal attraction? Nat Med 7:528–530. [DOI] [PubMed] [Google Scholar]
- 40. Mitchison T, Kirschner M (1988) Cytoskeletal dynamics and nerve growth. Neuron 1:761–772. [DOI] [PubMed] [Google Scholar]
- 41. Mykowska A, Sobczak K, Wojciechowska M, Kozlowski P, Krzyzosiak WJ (2011) CAG repeats mimic CUG repeats in the misregulation of alternative splicing. Nucleic Acids Res 39:8938–8951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Orr HT, Zoghbi HY (2007) Trinucleotide repeat disorders. Annu Rev Neurosci 30:575–621. [DOI] [PubMed] [Google Scholar]
- 43. Ortega Z, Lucas JJ (2014) Ubiquitin‐proteasome system involvement in Huntington's disease. Front Mol Neurosci 7:77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ranum LP, Cooper TA (2006) RNA‐mediated neuromuscular disorders. Annu Rev Neurosci 29:259–277. [DOI] [PubMed] [Google Scholar]
- 45. Rodrigues FB, Byrne L, McColgan P, Robertson N, Tabrizi SJ et al (2016) Cerebrospinal fluid total tau concentration predicts clinical phenotype in Huntington's disease. J Neurochem, 2016 Jun 25. doi: 10.1111/jnc.13719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Roos RA, Bots GT (1983) Nuclear membrane indentations in Huntington's chorea. J Neurol Sci 61:37–47. [DOI] [PubMed] [Google Scholar]
- 47. Sanchez C, Diaz‐Nido J, Avila J (2000) Phosphorylation of microtubule‐associated protein 2 (MAP2) and its relevance for the regulation of the neuronal cytoskeleton function. Prog Neurobiol 61:133–168. [DOI] [PubMed] [Google Scholar]
- 48. Sathasivam K, Neueder A, Gipson TA, Landles C, Benjamin AC et al (2013) Aberrant splicing of HTT generates the pathogenic exon 1 protein in Huntington disease. Proc Natl Acad Sci U S A 110:2366–2370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Seress L, Ribak CE (1992) Ultrastructural features of primate granule cell bodies show important differences from those of rats: axosomatic synapses, somatic spines and infolded nuclei. Brain Res 569:353–357. [DOI] [PubMed] [Google Scholar]
- 50. Shafit‐Zagardo B, Kalcheva N, Dickson D, Davies P, Kress Y (1997) Distribution and subcellular localization of high‐molecular‐weight microtubule‐associated protein‐2 expressing exon 8 in brain and spinal cord. J Neurochem 68:862–873. [DOI] [PubMed] [Google Scholar]
- 51. Singhrao SK, Thomas P, Wood JD, MacMillan JC, Neal JW et al (1998) Huntingtin protein colocalizes with lesions of neurodegenerative diseases: an investigation in Huntington's, Alzheimer's, and Pick's diseases. Exp Neurol 150:213–222. [DOI] [PubMed] [Google Scholar]
- 52. Takahashi H, Egawa S, Piao YS, Hayashi S, Yamada M et al (2001) Neuronal nuclear alterations in dentatorubral‐pallidoluysian atrophy: ultrastructural and morphometric studies of the cerebellar granule cells. Brain Res 919:12–19. [DOI] [PubMed] [Google Scholar]
- 53. Vonsattel JP, DiFiglia M (1998) Huntington disease. J Neuropathol Exp Neurol 57:369–384. [DOI] [PubMed] [Google Scholar]
- 54. Vuono R, Winder‐Rhodes S, de Silva R, Cisbani G, Drouin‐Ouellet J et al (2015) The role of tau in the pathological process and clinical expression of Huntington's disease. Brain 138:1907–1918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Wang Y, Wang J, Gao L, Lafyatis R, Stamm S, Andreadis A (2005) Tau exons 2 and 10, which are misregulated in neurodegenerative diseases, are partly regulated by silencers which bind a SRp30c.SRp55 complex that either recruits or antagonizes htra2beta1. J Biol Chem 280:14230–14239. [DOI] [PubMed] [Google Scholar]
- 56. Wild EJ, Boggio R, Langbehn D, Robertson N, Haider S et al (2015) Quantification of mutant huntingtin protein in cerebrospinal fluid from Huntington's disease patients. J Clin Invest 125:1979–1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Wittmann M, Queisser G, Eder A, Wiegert JS, Bengtson CP et al (2009) Synaptic activity induces dramatic changes in the geometry of the cell nucleus: interplay between nuclear structure, histone H3 phosphorylation, and nuclear calcium signaling. J Neurosci 29:14687–14700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Xifro X, Giralt A, Saavedra A, Garcia‐Martinez JM, Diaz‐Hernandez M et al (2009) Reduced calcineurin protein levels and activity in exon‐1 mouse models of Huntington's disease: role in excitotoxicity. Neurobiol Dis 36:461–469. [DOI] [PubMed] [Google Scholar]
- 59. Yano H, Baranov SV, Baranova OV, Kim J, Pan Y et al (2014) Inhibition of mitochondrial protein import by mutant huntingtin. Nat Neurosci 17:822–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Yin X, Jin N, Gu J, Shi J, Zhou J et al (2012) Dual‐specificity tyrosine phosphorylation‐regulated kinase 1A (Dyrk1A) modulates serine/arginine‐rich protein 55 (SRp55)‐promoted Tau exon 10 inclusion. J Biol Chem 287:30497–30506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Zander C, Takahashi J, El Hachimi KH, Fujigasaki H, Albanese V et al (2001) Similarities between spinocerebellar ataxia type 7 (SCA7) cell models and human brain: proteins recruited in inclusions and activation of caspase‐3. Hum Mol Genet 10:2569–2579. [DOI] [PubMed] [Google Scholar]