

Abstract
The genotype (methionine, M or valine, V) at polymorphic codon 129 of the PRNP gene and the type (1 or 2) of abnormal prion protein in the brain are the major determinants of the clinicopathological features of sporadic Creutzfeldt–Jakob disease (CJD), thus providing molecular basis for classification of sporadic CJD, that is, MM1, MM2, MV1, MV2, VV1 or VV2. In addition to these “pure” cases, “mixed” cases presenting mixed neuropathological and biochemical features have also been recognized. The most frequently observed mixed form is the co‐occurrence of MM1 and MM2, namely MM1+2. However, it has remained elusive whether MM1+2 could be a causative origin of dura mater graft‐associated CJD (dCJD), one of the largest subgroups of iatrogenic CJD. To test this possibility, we performed transmission experiments of MM1+2 prions and a systematic neuropathological examination of dCJD patients in the present study. The transmission properties of the MM1+2 prions were identical to those of MM1 prions because MM2 prions lacked transmissibility. In addition, the neuropathological characteristics of MM2 were totally absent in dCJD patients examined. These results suggest that MM1+2 can be a causative origin of dCJD and causes neuropathological phenotype similar to that of MM1.
Keywords: Creutzfeldt–Jakob disease, prion
Introduction
Creutzfeldt–Jakob disease (CJD), a fatal transmissible neurodegenerative disease, is caused by an abnormal infectious isoform of prion protein (PrPSc), which is generated by a conformational change of the normal cellular isoform (PrPC) 31. The conformational conversion of PrPC occurs due to either one of three causes: spontaneous conversion in sporadic CJD (sCJD), pathogenic mutations of the PRNP gene in genetic CJD, or PrPSc infection in iatrogenic CJD and variant CJD.
The genotype (methionine, M or valine, V) at polymorphic codon 129 of the PRNP gene and the type (1 or 2) of PrPSc in the brain are the major determinants of the clinicopathological features of sCJD and permit molecular classification of sCJD, that is, MM1, MM2, MV1, MV2, VV1 or VV2 26. The PrPSc types 1 and 2 are distinguishable according to the size of the proteinase K‐resistant core of PrPSc (21 and 19 kDa, respectively), reflecting differences in the proteinase K‐cleavage site (at residues 82 and 97, respectively) 27. In addition, MM2 cases can be further divided into two subgroups, cortical form (MM2C) or thalamic form (MM2T), based on the neuropathological characteristics 26. Similarly, MV2 cases can also be divided into two neuropathologically distinct subgroups, cortical form (MV2C) or kuru plaque form (MV2K) 28. On the other hand, the MM1 and MV1 subgroups are indistinguishable in clinicopathological, biochemical and transmission properties, and are merged into one subgroup as MM/MV1 4, 26. Similarly, MM2C and MV2C are also indistinguishable and are merged into one subgroup as MM/MV2C. As a consequence, six subgroups are recognized in the current classification of sCJD, that is, MM/MV1, MM/MV2C, MM2T, MV2K, VV1 and VV2 30. In addition to these “pure” subgroups, “mixed” cases presenting mixed neuropathological features and more than one PrPSc type in the same brain have also been reported 5, 26, 28, 32, 33, 35. The most frequently observed mixed form is the co‐occurrence of MM/MV1 and MM/MV2C, denoted as MM/MV1+2C 28.
Dura mater graft‐associated CJD (dCJD) is an iatrogenic CJD caused by prion‐contaminated dura mater grafts obtained from human cadavers undiagnosed as CJD. There are two distinct subgroups in dCJD that show distinct clinicopathological, biochemical and transmission properties, reflecting their different causative origins. Plaque‐type dCJD is caused by the transmission of sCJD MV2K or VV2 and shows characteristic clinicopathological and biochemical features, that is, widespread PrP amyloid plaques (kuru plaques) and the accumulation of intermediate type PrPSc showing intermediate electrophoretic mobility between types 1 and 2 PrPSc 16, 18, 19, 23, 37. On the other hand, non‐plaque‐type dCJD has been considered to be caused by the transmission of sCJD MM/MV1 and shows clinicopathological and biochemical features indistinguishable from those of sCJD MM/MV1, that is, diffuse synaptic type PrP deposition and type 1 PrPSc accumulation.
Although sCJD MM/MV1+2C accounts for 28% of all sCJD cases in European countries where the causative dura mater grafts were manufactured 28, it has remained elusive whether this sCJD subgroup could be a causative origin of dCJD. To test this possibility, we performed experimental transmission of sCJD MM1+2C and systematic neuropathological examination of dCJD patients in the present study.
Materials and Methods
Patients
CJD cases included in this study were patients with clinically, genetically and histopathologically proven sCJD (five cases) and dCJD (45 cases). Brain tissues were obtained at autopsy from CJD patients after receiving informed consent for research use. The diagnosis of CJD, histopathological type and PrPSc type were confirmed by PrP immunohistochemistry and Western blot analysis 13, 33. The genotype and the absence of mutations in the open reading frame of the PRNP gene were determined by sequence analysis as described 14. According to Parchi's classification 30, the sCJD cases were classified as follows: MM1, two cases; MM2C, one case; MM1+2C, two cases. These patients showed the typical phenotypes of each sCJD subgroup in the clinicopathological and biochemical examinations. All dCJD cases examined were 129M/M homozygotes.
Transmission experiments
The production of knock‐in mice expressing human PrP with one of the codon 129 genotypes (Ki‐Hu129M/M, Ki‐Hu129M/V or Ki‐Hu129V/V), knock‐in mice expressing human PrP with lysine homozygosity at another polymorphic codon 219 (Ki‐Hu219K/K), and knock‐in mice expressing human/mouse chimeric PrP (Ki‐ChM) has been reported previously 2, 10, 15. The expression levels of human PrP in the brains of these knock‐in mice were identical to the level observed in wild‐type mouse. Intracerebral inoculation of 10% brain homogenates from the sCJD MM1+2C, MM1 or MM2C patients was performed as described 34. The inoculated mice were sacrificed at a predefined clinical endpoint, or at accidental death. One hemisphere of the brain was fixed in 10% buffered formalin for immunohistochemistry, and the other hemisphere was immediately frozen for Western blotting or serial passage. Incubation times are expressed as mean ± SEM. These experiments were approved by the Institutional Animal Care and Use Committee of Tohoku University, and performed in strict accordance with the Regulations for Animal Experiments and Related Activities at Tohoku University.
Immunohistochemistry
Formalin‐fixed brain tissues were treated with formic acid (99% for human tissues or 60% for mouse tissues) for 1 h to inactivate the infectivity, and embedded in paraffin. Tissue sections were pretreated by hydrolytic autoclaving before PrP immunohistochemistry 13. The anti‐PrP monoclonal antibody 3F4 (Signet, Dedham, MA, USA) was used as the primary antibody for human sections, and anti‐PrP antiserum PrP‐N 12 was used as the primary antibody for mouse sections. Goat‐anti‐mouse immunoglobulin polyclonal antibody labeled with the peroxidase‐conjugated dextran polymer, EnVision+ (Dako) and anti‐rabbit EnVision+ were used as the secondary antibodies.
Western blotting
PrPSc was obtained from the cerebral cortex after tissue homogenization, collagenase digestion and Sarkosyl‐NaCl extraction 9. For deglycosylation of PrPSc, samples were digested with PNGaseF (New England Biolabs, Ipswich, MA, USA) 16. Protein samples were subjected to sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS‐PAGE) and Western blotting 2. PrPSc type‐specific polyclonal antibodies [designated as Tohoku 1 (T1) or Tohoku 2 (T2)] 17 or anti‐PrP monoclonal antibody 3F4 were used as the primary antibodies. The type 1 PrPSc‐specific antibody T1 reacts with epitopes located between residues 82 and 96 of human PrP, which can be retained in type 1 PrPSc but not in type 2 PrPSc after proteinase K‐digestion 17. The type 2 PrPSc‐specific antibody T2 is a proteolytic cleavage site‐specific antibody and can specifically react with the N‐terminally cleaved type 2 PrPSc after proteinase K‐digestion 17. Anti‐rabbit EnVision+ and anti‐mouse EnVision+ were used as the secondary antibodies. The signal intensities of the Western blots were quantified with Quantity One software using an imaging device, VersaDoc 5000 (Bio‐Rad Laboratories, Hercules, CA, USA).
Results
The clinical features of the two patients with sCJD MM1+2C are summarized in Table 1. Histopathological examination of the brain revealed the coexistence of MM/MV1 pathology, that is, diffuse synaptic type PrP deposition, and MM/MV2C pathology (ie, large confluent vacuoles and perivacuolar PrP deposition) (Figure 1A–C). Western blot analysis of PrPSc using a conventional anti‐PrP antibody 3F4, type 1 PrPSc‐specific antibody T1 or type 2 PrPSc‐specific antibody T2 showed the coexistence of types 1 and 2 PrPSc in both patients (Figure 1D). To compare the relative amounts of the type 1 or type 2 PrPSc between the patients, the signal intensities of the T1‐ or T2‐reactive PrPSc were normalized by those of the 3F4‐reactive PrPSc. The type 1 : type 2 ratios in case #1 and case #2 were 90:10 and 55:45, respectively. We then inoculated intracerebrally the brain homogenates from the two sCJD MM1+2C cases into PrP‐humanized knock‐in mice. These knock‐in mice expressed human PrP with either one of the codon 129 genotypes (Ki‐Hu129M/M, Ki‐Hu129M/V or Ki‐Hu129V/V), human PrP with the 219K/K genotype (Ki‐Hu219K/K) or human/mouse chimeric PrP (Ki‐ChM) 2, 10, 15. The mean incubation periods and attack rates of sCJD MM1+2C prion‐inoculated mice were similar to those of MM1 prion‐inoculated mice (Table 2). In particular, Ki‐ChM mice showed remarkably short incubation times after challenge with sCJD MM1+2C prions, similar to those after challenge with MM1 prions 34. By contrast, sCJD MM2C prions could not be transmitted to any PrP‐humanized knock‐in mouse lines examined, as reported previously 18. Indeed, although experimental coinfection with distinct prion strains can result in prolonged incubation periods, referred to as “prion strain interference” 3, 7, the coexisting MM2C prions did not interfere with the transmission of MM1 prions even in the mice inoculated with MM1+2C case #2 materials, which contained equal amounts of type 2 PrPSc and type 1 PrPSc. Moreover, the neuropathological features were also identical between the sCJD MM1+2C prion‐inoculated mice and the MM1 prion‐inoculated mice, regardless of the PRNP genotype of mice (Figure 2A). All MM1+2C prion‐inoculated mice showed only diffuse synaptic type PrP deposition, and the neuropathological characteristics of MM2C such as large confluent vacuoles or perivacuolar PrP deposition were not observed. Moreover, Western blot analysis of PrPSc in the brain revealed that all sCJD MM1+2C prion‐inoculated mice produced only type 1 PrPSc regardless of the PRNP genotype (Figure 2B). Even in Western blot analysis using type 2 PrPSc‐specific antibody T2, no PrPSc signal was detected in the MM1+2C prion‐inoculated mice and the MM1 prion‐inoculated mice. The sizes of type 1 PrPSc produced in the sCJD prion‐inoculated mice seemed to be slightly smaller than those in the human cases (inocula). Similar downward size shift of PrPSc after transmission has been recognized in a transmission experiment using a different PrP‐humanized knock‐in mouse line 22. However, we reported previously that type 1 PrPSc produced in the sCJD prion‐inoculated mice retained epitopes for the type 1 PrPSc‐specific antibody T1 (residues 82–96) or for another type 1 PrPSc‐specific antibody POM2 (residues 83–89) 17. In addition, we confirmed that type 2 PrPSc produced in the sCJD prion‐inoculated knock‐in mice could be detected by the N‐terminal cleavage site‐specific antibody T2 similar to type 2 PrPSc of human sCJD cases 17, 18, suggesting that the PrPSc type‐specific N‐terminal cleavage by proteinase K is not altered in these knock‐in mice. Therefore, although the sizes of PrPSc produced in the sCJD prion‐inoculated mice can slightly differ from those of human counterpart, it does not affect the typing of PrPSc that was determined by differences in the N‐terminal proteolytic cleavage site. Whether the size shift can relate to C‐terminal cleavage of PrPSc or to posttranslational modification of PrP, for example, glycosylphosphatidylinositol anchor, should be examined in the future. Taken together, the neuropathological and biochemical analyses revealed that MM2C prions were not transmitted to the sCJD MM1+2C prion‐inoculated mice.
Table 1.
Summary of clinical features of the sCJD MM1+2C cases
| Case #1 | Case #2 | |
|---|---|---|
| Gender | Male | Female |
| Age at onset (years) | 68 | 66 |
| Initial symptom | Progressive dementia | Progressive dementia |
| Myoclonus (months)* | 1 | 6 |
| Akinetic mutism (months)* | 2 | 10 |
| PSWCs on EEG (months)* | 1 | 10 |
| Duration of illness (months) | 2 | 44 |
*Duration until the appearance of myoclonus, akinetic mutism or PSWCs on EEG from onset.
Abbreviations: PSWC = periodic sharp‐wave complex; EEG = electroencephalogram.
Figure 1.
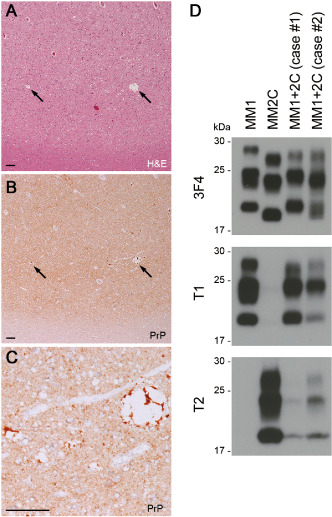
Neuropathological and biochemical properties of the sCJD MM1+2C cases. A. Histopathological analysis showed scattered large confluent vacuoles (arrows) in addition to spongiform changes in the cerebral cortex (H&E; frontal cortex of case #1). B. Immunohistochemical analysis of PrP revealed the coexistence of perivacuolar PrP deposition (arrows) and synaptic type PrP deposition (anti‐PrP antibody 3F4; frontal cortex of case #1). C. Higher magnification of perivacuolar PrP deposition in panel B. Scale bars: 100 μm. D. Western blot analysis using a conventional anti‐PrP antibody 3F4, type 1 PrPSc‐specific antibody T1 or type 2 PrPSc‐specific antibody T2 revealed the co‐occurrence of types 1 and 2 PrPSc in the sCJD MM1+2C cases. The amounts loaded per lane were as follows: MM1, 280 μg (wet weight of brain tissue); MM2C, 100 μg; MM1+2C (case #1), 250 μg; or MM1+2C (case #2), 125 μg.
Table 2.
Transmission of sCJD MM1+2C, MM1 or MM2C to PrP‐humanized knock‐in mice
| Inoculum (ID) | Mouse line (human PRNP genotypes at codons 129 and 219) | ||||
|---|---|---|---|---|---|
| Ki‐Hu129M/M (M/M, E/E) | Ki‐Hu129M/V (M/V, E/E) | Ki‐Hu129V/V (V/V, E/E) | Ki‐Hu219K/K (M/M, K/K) | Ki‐ChM | |
| MM1+2C (case #1) | 615 ± 8 (6/6)a | ND | 717 ± 44 (6/6) | 647 ± 49 (5/5) | 162 ± 13 (6/6) |
| MM1+2C (case #2) | 580 ± 8 (5/5) | 595 ± 6 (4/4) | 588 ± 11 (4/4) | 594 ± 10 (5/5) | 160 ± 9 (4/4) |
| MM1 (H3)b | 467 ± 24 (8/8) | 490 ± 26 (5/5) | 774 ± 32 (6/6) | 573 ± 52 (5/5) | 142 ± 3 (5/5) |
| MM1 (NR) | 531 ± 47 (5/5) | 508 ± 55 (6/6) | 588 ± 3 (5/5) | 527 ± 31 (6/6) | 154 ± 5 (6/6) |
| MM2Cc | — (0/7) | — (0/7) | — (0/7) | — (0/8) | — (0/6) |
Mean incubation periods (days ± SEM) and attack rates (number of mice positive for PrP accumulation in immunohistochemical analysis/number of inoculated mice).
None of the sCJD MM2C prion‐inoculated mice had developed disease by the end of their life span. Transmission data of Ki‐Hu129M/M, Ki‐Hu129M/V and Ki‐Hu129V/V have been reported previously 18.
Abbreviation: ND = not done.
Figure 2.
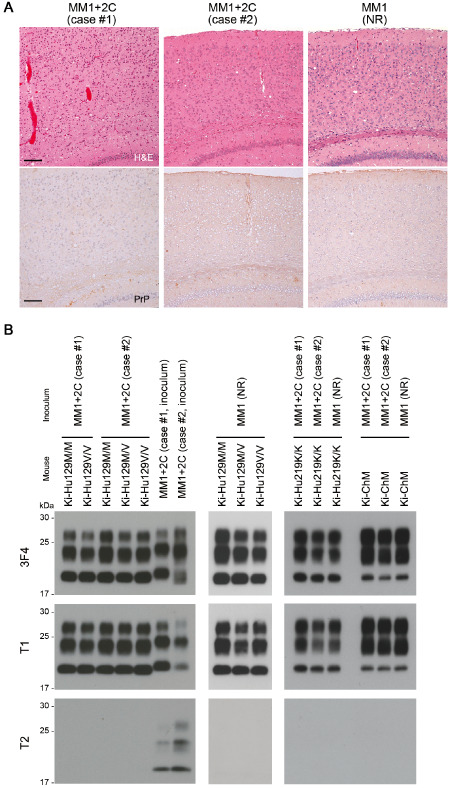
Neuropathological and biochemical properties of the PrP‐humanized mice inoculated with sCJD MM1+2C prions. A. Histopathological analysis of the brain of Ki‐Hu129M/M mice inoculated with sCJD MM1+2C prions (case #1 or case #2) or MM1 prions (NR). The MM1+2C prion‐inoculated mice showed only spongiform changes (H&E, upper panels) and synaptic type PrP deposition (anti‐PrP antibody PrP‐N, lower panels), similar to the MM1 prion‐inoculated mice. Large confluent vacuoles or perivacuolar PrP deposition were absent in all the MM1+2C prion‐inoculated mice. Scale bars: 100 μm. B. Western blot analysis using a conventional anti‐PrP antibody 3F4, type 1 PrPSc‐specific antibody T1 or type 2 PrPSc‐specific antibody T2. The MM1+2C prion‐inoculated mice produced only type 1 PrPSc regardless of the PRNP genotype. Type 2 PrPSc was not detected even in Western blot analysis using the T2 antibody.
To examine whether the transmission properties of sCJD MM/MV1+2C were identical to those of MM/MV1 in human iatrogenic CJD cases, we performed systematic neuropathological evaluation of 45 cases with dCJD, representing approximately one‐third of dCJD cases in Japan. Histopathological examination throughout the whole brain has an advantage over Western blot analysis to detect the focal coexistence of MM/MV2C prions with MM/MV1 prions 28. Among 45 dCJD cases, 29 showed diffuse synaptic type PrP deposition, and 16 showed widespread PrP amyloid plaques (kuru plaques) (Figure 3A). However, none of the dCJD cases showed sCJD MM/MV1+2C‐like pathology despite the fact that sCJD MM/MV1+2C accounts for 28% of all sCJD cases in European countries where the causative dura mater grafts were manufactured (Figure 3B) 28, suggesting that MM/MV2C prions are also not transmissible in human iatrogenic CJD cases.
Figure 3.
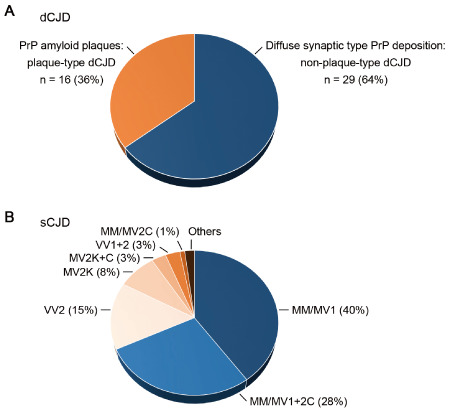
Neuropathological evaluation of 45 cases with dCJD. A. Histopathological examination throughout the whole brain of the dCJD patients revealed that 29 cases (64%) showed the diffuse synaptic type PrP deposition and 16 cases (36%) showed widespread PrP amyloid plaques. None of the dCJD patients had large confluent vacuoles or perivacuolar PrP deposition. B. Frequency of each sCJD subgroup in European countries 30.
Discussion
sCJD MM1+2C prion‐inoculated mice lacked the neuropathological and biochemical characteristics of MM2C prions. In addition, sCJD MM/MV1+2C‐like pathology was entirely absent in the dCJD cases in the systematic evaluation in the present study. These results suggest that sCJD MM/MV1+2C prions and MM/MV1 prions are identical in transmission properties both in animal experiments and in human iatrogenic CJD cases because MM/MV2C prions lack transmissibility. As transmission experiments using PrP‐humanized knock‐in mice have been so far conducted on very limited numbers of sCJD MM1+2C cases, further analyses with larger series, including cases with predominant accumulation of MM2C prions, may be needed to generalize the implications of the present study. However, the conclusion of the present study is supported by several lines of evidence as follows. First, sCJD MM1+2C prions and MM/MV1 prions showed the same transmission properties also in a transmission study using nonhuman primates 29. Second, the absence of successful transmission 22 or extremely low transmissibility 4 of sCJD MM2C prions has also been revealed by transmission studies using other lines of PrP‐humanized knock‐in mouse. Finally, the incidence rate of non‐plaque‐type dCJD (68%) among the total dCJD cases in Japan perfectly matches the sum total of the incidence rates of sCJD MM/MV1 (40%) and MM/MV1+2C (28%) among all sCJD cases in European countries 30, 37 where the causative dura mater was collected from human cadavers. As virtually all Japanese dCJD patients were of the same 129M/M genotype 25, reflecting overwhelming predominance of the 129M/M homozygosity in the general population in Japan 8, the disease phenotypes of the dCJD patients simply depended on the prion strain infected 19. Indeed, the incidence rate of plaque‐type dCJD (32%) among the total dCJD cases in Japan is also close to the total sum of the incidence rates of its causative origins [ie, sCJD VV2 (15%), MV2K (8%) and MV2K+2C (3%)] in European countries 30. Taken together, the present study suggests that not only sCJD MM/MV1 but also MM/MV1+2C can be a causative origin of non‐plaque‐type dCJD.
The reasons for the lack of transmissibility of sCJD MM/MV2C prions remain unknown. As the disease duration of sCJD MM/MV2C patients is the longest among sCJD subgroups 30, the replication of MM/MV2C prions may not proceed efficiently. This seems to be the same situation as that of self‐propagating misfolded proteins that are transmissible between neighboring cells within a patient but are not transmissible between individuals, so‐called “prionoid” 1. These self‐propagating misfolded proteins can be efficiently transmitted to “primed” animals such as overexpressors or disease‐prone mutants 6, 11, 21. Similarly, MM/MV2C prions can also be transmissible to primed animals such as transgenic mice overexpressing human PrP 20, 24, or transgenic mice overexpressing bank vole PrP 36, with a 100% attack rate. The lack of transmissibility of MM/MV2C prions to non‐primed hosts suggests that CJD patients showing neuropathological and biochemical characteristics of MM/MV1+2C, MM/MV2C or MV2K+2C can be diagnosed as “sporadic” CJD even if they have a medical history of known risk factors for iatrogenic CJD such as dural grafting or treatment with growth hormone. An alternative explanation is that the PrP‐humanized knock‐in mice, humans or nonhuman primates 29 were much more susceptible to MM/MV1 prions than to MM/MV2C prions and this prevented the appearance of histopathological or biochemical features related to MM/MV2C prions despite the existence of undetectable levels of MM/MV2C prions in the brain. To test this possibility, serial passages from MM1+2C prion‐inoculated mice to animals that are not permissive to MM1 prions but susceptible to MM2C prions will be needed in the future.
The clinical features of the sCJD MM/MV1+2C patients can differ from those of MM/MV1 patients depending on the extent of the MM/MV2C pathology 5, 28. The disease duration became significantly longer and the cerebellar signs less frequent with increasing MM/MV2C pathology 28. In contrast, the transmissibility of sCJD MM/MV1+2C prions was identical to that of MM/MV1 prions regardless of the abundance of MM/MV2C prions in the present study, as MM/MV2C prions lacked transmissibility. This would be an important finding from the viewpoint of public health. As the current decontamination/disinfection procedures against CJD prions were developed before genotyping and molecular classification become available, the effectiveness of the current procedures against sCJD MM/MV1+2C prions has not been tested. However, the present study suggests that the current procedures, considered to be effective against MM/MV1 prions, are sufficient for the decontamination/disinfection of MM/MV1+2C prions as well.
In conclusion, sCJD MM/MV1+2C prions and MM/MV1 prions showed identical transmission properties. With respect to public health issues, the present study suggests that sCJD MM/MV1+2C prions and MM/MV1 prions can be considered as the same entity, apart from the classification of sCJD patients.
Conflicts of interest
The authors declare that they have no conflict of interest.
Acknowledgments
We thank the members of the Creutzfeldt–Jakob Disease Surveillance Committee in Japan, Creutzfeldt–Jakob disease specialists in prefectures, and Creutzfeldt–Jakob disease patients and families for providing important clinical information. We thank Y. Ishikawa, H. Kudo, M. Yamamoto and A. Yamazaki for their excellent technical assistance, and B. Bell for critical review of the manuscript. This study was supported by Grants‐in‐Aid from the Ministry of Health, Labor and Welfare of Japan (A.K., M.T. and S.M.), Grants‐in‐Aid for Scientific Research from JSPS (A.K., M.T. and T.K.), a grant from MEXT for the Joint Research Program of the Research Center for Zoonosis Control, Hokkaido University (T.K.), and a Grant‐in‐Aid for Scientific Research on Innovative Areas from MEXT (T.K.).
References
- 1. Aguzzi A (2009) Cell biology: beyond the prion principle. Nature 459:924–925. [DOI] [PubMed] [Google Scholar]
- 2. Asano M, Mohri S, Ironside JW, Ito M, Tamaoki N, Kitamoto T (2006) vCJD prion acquires altered virulence through trans‐species infection. Biochem Biophys Res Commun 342:293–299. [DOI] [PubMed] [Google Scholar]
- 3. Bartz JC, Kramer ML, Sheehan MH, Hutter JAL, Ayers JI, Bessen RA, Kincaid AE (2007) Prion interference is due to a reduction in strain‐specific PrPSc levels. J Virol 81:689–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bishop MT, Will RG, Manson JC (2010) Defining sporadic Creutzfeldt‐Jakob disease strains and their transmission properties. Proc Natl Acad Sci U S A 107:12005–12010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cali I, Castellani R, Alshekhlee A, Cohen Y, Blevins J, Yuan J et al (2009) Co‐existence of scrapie prion protein types 1 and 2 in sporadic Creutzfeldt‐Jakob disease: its effect on the phenotype and prion‐type characteristics. Brain 132:2643–2658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Clavaguera F, Bolmont T, Crowther RA, Abramowski D, Frank S, Probst A et al (2009) Transmission and spreading of tauopathy in transgenic mouse brain. Nat Cell Biol 11:909–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Dickinson AG, Fraser H, Meikle VM, Outram GW (1972) Competition between different scrapie agents in mice. Nat New Biol 237:244–245. [DOI] [PubMed] [Google Scholar]
- 8. Doh‐ura K, Kitamoto T, Sakaki Y, Tateishi J (1991) CJD discrepancy. Nature 353:801–802. [DOI] [PubMed] [Google Scholar]
- 9. Grathwohl KUD, Horiuchi M, Ishiguro N, Shinagawa M (1996) Improvement of PrPSc‐detection in mouse spleen early at the preclinical stage of scrapie with collagenase‐completed tissue homogenization and Sarkosyl‐NaCl extraction of PrPSc . Arch Virol 141:1863–1874. [DOI] [PubMed] [Google Scholar]
- 10. Hizume M, Kobayashi A, Teruya K, Ohashi H, Ironside JW, Mohri S, Kitamoto T (2009) Human prion protein (PrP) 219K is converted to PrPSc but shows heterozygous inhibition in variant Creutzfeldt‐Jakob disease infection. J Biol Chem 284:3603–3609. [DOI] [PubMed] [Google Scholar]
- 11. Kane MD, Lipinski WJ, Callahan MJ, Bian F, Durham RA, Schwarz RD et al (2000) Evidence for seeding of beta‐amyloid by intracerebral infusion of Alzheimer brain extracts in beta‐amyloid precursor protein‐transgenic mice. J Neurosci 20:3606–3611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kitamoto T, Muramoto T, Hilbich C, Beyreuther K, Tateishi J (1991) N‐terminal sequence of prion protein is also integrated into kuru plaques in patients with Gerstmann‐Sträussler syndrome. Brain Res 545:319–321. [DOI] [PubMed] [Google Scholar]
- 13. Kitamoto T, Shin R‐W, Doh‐ura K, Tomokane N, Miyazono M, Muramoto T, Tateishi J (1992) Abnormal isoform of prion proteins accumulates in the synaptic structures of the central nervous system in patients with Creutzfeldt‐Jakob disease. Am J Pathol 140:1285–1294. [PMC free article] [PubMed] [Google Scholar]
- 14. Kitamoto T, Ohta M, Doh‐ura K, Hitoshi S, Terao Y, Tateishi J (1993) Novel missense variants of prion protein in Creutzfeldt‐Jakob disease or Gerstmann‐Sträussler syndrome. Biochem Biophys Res Commun 191:709–714. [DOI] [PubMed] [Google Scholar]
- 15. Kitamoto T, Mohri S, Ironside JW, Miyoshi I, Tanaka T, Kitamoto N et al (2002) Follicular dendritic cell of the knock‐in mouse provides a new bioassay for human prions. Biochem Biophys Res Commun 294:280–286. [DOI] [PubMed] [Google Scholar]
- 16. Kobayashi A, Asano M, Mohri S, Kitamoto T (2007) Cross‐sequence transmission of sporadic Creutzfeldt‐Jakob disease creates a new prion strain. J Biol Chem 282:30022–30028. [DOI] [PubMed] [Google Scholar]
- 17. Kobayashi A, Sakuma N, Matsuura Y, Mohri S, Aguzzi A, Kitamoto T (2010) Experimental verification of a traceback phenomenon in prion infection. J Virol 84:3230–3238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kobayashi A, Iwasaki Y, Otsuka H, Yamada M, Yoshida M, Matsuura Y (2013) Deciphering the pathogenesis of sporadic Creutzfeldt‐Jakob disease with codon 129 M/V and Type 2 abnormal prion protein. Acta Neuropathol Commun 1:74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kobayashi A, Matsuura Y, Mohri S, Kitamoto T (2014) Distinct origins of dura mater graft‐associated Creutzfeldt‐Jakob disease: past and future problems. Acta Neuropathol Commun 2:32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Korth C, Kaneko K, Groth D, Heye N, Telling G, Mastrianni J et al (2003) Abbreviated incubation times for human prions in mice expressing a chimeric mouse‐human prion protein transgene. Proc Natl Acad Sci U S A 100:4784–4789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Luk KC, Kehm VM, Zhang B, O'Brien P, Trojanowski JQ, Lee VM (2012) Intracerebral inoculation of pathological α‐synuclein initiates a rapidly progressive neurodegenerative α‐synucleinopathy in mice. J Exp Med 209:975–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Moda F, Suardi S, Di Fede G, Indaco A, Limido L, Vimercati C et al (2012) MM2‐thalamic Creutzfeldt‐Jakob disease: neuropathological, biochemical and transmission studies identify a distinctive prion strain. Brain Pathol 22:662–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Noguchi‐Shinohara M, Hamaguchi T, Kitamoto T, Sato T, Nakamura Y, Mizusawa H, Yamada M (2007) Clinical features and diagnosis of dura mater graft associated Creutzfeldt‐Jakob disease. Neurology 69:360–367. [DOI] [PubMed] [Google Scholar]
- 24. Notari S, Xiao X, Espinosa JC, Cohen Y, Qing L, Aguilar‐Calvo P et al (2014) Transmission characteristics of variably protease‐sensitive prionopathy. Emerg Infect Dis 20:2006–2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Nozaki I, Hamaguchi T, Sanjo N, Noguchi‐Shinohara M, Sakai K, Nakamura Y et al (2010) Prospective 10‐year surveillance of human prion diseases in Japan. Brain 133:3043–3057. [DOI] [PubMed] [Google Scholar]
- 26. Parchi P, Giese A, Capellari S, Brown P, Schulz‐Schaeffer W, Windl O et al (1999) Classification of sporadic Creutzfeldt‐Jakob disease based on molecular and phenotypic analysis of 300 subjects. Ann Neurol 46:224–233. [PubMed] [Google Scholar]
- 27. Parchi P, Zou W, Wang W, Brown P, Capellari S, Ghetti B et al (2000) Genetic influence on the structural variations of the abnormal prion protein. Proc Natl Acad Sci U S A 97:10168–10172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Parchi P, Strammiello R, Notari S, Giese A, Langeveld JP, Ladogana A (2009) Incidence and spectrum of sporadic Creutzfeldt‐Jakob disease variants with mixed phenotype and co‐occurrence of PrPSc types: an updated classification. Acta Neuropathol 118:659–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Parchi P, Cescatti M, Notari S, Schulz‐Schaeffer WJ, Capellari S, Giese A et al (2010) Agent strain variation in human prion disease: insights from a molecular and pathological review of the National Institutes of Health series of experimentally transmitted disease. Brain 133:3030–3042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Parchi P, Strammiello R, Giese A, Kretzschmar H (2011) Phenotypic variability of sporadic human prion disease and its molecular basis: past, present, and future. Acta Neuropathol 121:91–112. [DOI] [PubMed] [Google Scholar]
- 31. Prusiner SB, Scott MR, DeArmond SJ, Cohen FE (1998) Prion protein biology. Cell 93:337–348. [DOI] [PubMed] [Google Scholar]
- 32. Puoti G, Giaccone G, Rossi G, Canciani B, Bugiani O, Tagliavini F (1999) Sporadic Creutzfeldt‐Jakob disease: co‐occurrence of different types of PrPSc in the same brain. Neurology 53:2173–2176. [DOI] [PubMed] [Google Scholar]
- 33. Schoch G, Seeger H, Bogousslavsky J, Tolnay M, Janzer RC, Aguzzi A, Glatzel M (2006) Analysis of prion strains by PrPSc profiling in sporadic Creutzfeldt‐Jakob disease. PLoS ONE 3:e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Taguchi Y, Mohri S, Ironside JW, Muramoto T, Kitamoto T (2003) Humanized knock‐in mice expressing chimeric prion protein showed varied susceptibility to different human prions. Am J Pathol 163:2585–2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Uro‐Coste E, Cassard H, Simon S, Lugan S, Bilheude JM, Perret‐Liaudet A et al (2008) Beyond PrPres type 1 / type 2 dichotomy in Creutzfeldt‐Jakob disease. PLoS Pathog 4:e1000029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Watts JC, Giles K, Patel S, Oehler A, DeArmond SJ, Prusiner SB (2014) Evidence that bank vole PrP is a universal acceptor for prions. PLoS Pathog 10:e1003990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Yamada M, Noguchi‐Shinohara M, Hamaguchi T, Nozaki I, Kitamoto T, Sato T (2009) Dura mater graft‐associated Creutzfeldt‐Jakob disease in Japan: clinicopathological and molecular characterization of the two distinct subtypes. Neuropathology 29:609–618. [DOI] [PubMed] [Google Scholar]