

Abstract
Inherited heart disease causing electric instability in the heart has been suggested to be a risk factor for sudden unexpected death in epilepsy (SUDEP). The purpose of this study was to reveal the correlation between epilepsy‐related sudden unexpected death (SUD) and inherited heart disease. Twelve epilepsy‐related SUD cases (seven males and five females, aged 11–78 years) were examined. Nine cases fulfilled the criteria of SUDEP, and three cases died by drowning. In addition to examining three major epilepsy‐related genes, we used next‐generation sequencing (NGS) to examine 73 inherited heart disease‐related genes. We detected both known pathogenic variants and rare variants with minor allele frequencies of <0.5%. The pathogenicity of these variants was evaluated and graded by eight in silico predictive algorithms. Six known and six potential rare variants were detected. Among these, three known variants of LDB3, DSC2 and KCNE1 and three potential rare variants of MYH6, DSP and DSG2 were predicted by in silico analysis as possibly highly pathogenic in three of the nine SUDEP cases. Two of three cases with desmosome‐related variants showed mild but possible significant right ventricular dysplasia‐like pathology. A case with LDB3 and MYH6 variants showed hypertrabeculation of the left ventricle and severe fibrosis of the cardiac conduction system. In the three drowning death cases, one case with mild prolonged QT interval had two variants in ANK2. This study shows that inherited heart disease may be a significant risk factor for SUD in some epilepsy cases, even if pathological findings of the heart had not progressed to an advanced stage of the disease. A combination of detailed pathological examination of the heart and gene analysis using NGS may be useful for evaluating arrhythmogenic potential of epilepsy‐related SUD.
Keywords: autopsy, drowning, epilepsy, inherited heart disease, next‐generation sequencing, pathology, sudden death
Introduction
People with epilepsy have an increased risk of premature death compared with the general population and the risk of sudden unexpected death (SUD) is approximately 24 times higher 20. The term “sudden unexpected death in epilepsy” (SUDEP) has been defined as sudden unexpected, witnessed or unwitnessed, non‐traumatic and non‐drowning death in patients with epilepsy, with or without evidence of seizure and excluding documented status epilepticus, where postmortem examination does not reveal an anatomical or toxicological cause of death 40. The mechanism of SUDEP remains undetermined. Although apnea during seizure or cardiac arrhythmogenic events are frequently noted, it has been proposed that multifactorial processes other than respiratory and/or cardiac factors, such as drug, metabolic or environmental factors, may contribute to SUDEP 17, 27. In addition, accidental events, such as trauma and drowning are significant complications of epilepsy 8, 55. The mechanism of such death has been considered to be epilepsy attack‐related but has not been fully explored. Chiba and Nishida et al 13 considered bathing‐related death in the general population and showed that asymptomatic ventricular tachycardia occurred in older people while sitting in hot water; this arrhythmia developed within 5 minutes after immersion. Therefore, we suggest that some epilepsy‐related drowning deaths can occur after an arrhythmogenic event. In the general population, it can be difficult to diagnose arrhythmogenic SUD by routine autopsy examination, because some cases do not show any significant pathological change 62. Recently, arrhythmogenic events due to inherited heart disease have been considered a significant cause of SUD and molecular analyses at autopsy of “negative” or “unexplained” SUD cases, termed “molecular autopsy”, have been performed 3, 26. These examinations, mainly targeting cardiac channel‐related genes, detected several gene variants 24, 61, 62. Many ion channel genes regulating the central control of cardiac function are also expressed in the brain and some epilepsy candidate genes that are correlated with the occurrence of epilepsy and SUDEP encode ion channels 14, 48, 49, 50, 60. Next‐generation DNA sequencing (NGS) is an exciting advance in the technologies available to the life sciences 38 and allows the examination of large numbers of study samples on a massive scale. NGS can be applied to targeted panels containing 20–80 genes for comprehensive testing of inherited arrhythmia or cardiomyopathy 36. Although few molecular autopsy reports for SUD using NGS have been published 4, 14, the procedure may be useful for postmortem examination of epilepsy‐related SUD. Coll et al 14 showed the significance of using NGS for genetic analysis of SUDEP cases. We recently reported that the combination of careful pathological examination and molecular autopsy using NGS is useful for detecting subclinical or early clinical signs of non‐channelopathy inherited heart disease in cases of sudden unexplained death syndrome (SUDS) 26. Here, we attempted detailed pathological examination of the heart and genetic screening using NGS to detect arrhythmogenic potential in the victims of epilepsy‐related SUD.
Methods
Subjects
Our department performed 1241 full autopsies from 2008 to 2014. Seventeen of these autopsy cases had been diagnosed before autopsy with epilepsy by a neurologist or psychologist. Clinical records, including electrocardiograms (ECG) of each victim while alive, were obtained. Other disease condition could cause epilepsy‐like symptoms were excluded in the clinical record. SUDEP cases were classified as definite SUDEP, definite SUDEP plus or possible SUDEP 41. The ethical committee of Toyama University approved this study, which was performed in accordance with the ethical standards established in the 1964 Declaration of Helsinki.
The cause of death in five cases was explained: death due to a house fire, aortic dissection, bronchopneumonia, suicide by jumping from a height and drug intoxication. These cases were excluded from the study. The remaining 12 cases were considered to be epilepsy‐related SUD (seven were male and five were female, aged 11–78, mean age 50.5 ± 22.5 years) (Table 1). All 12 cases were unwitnessed at the time of collapse, and did not receive intensive resuscitation. Also, all cases did not have an individual or family history of status epilepticus. Nine cases (52.9%) (cases 1–9) fulfilled the criteria of SUDEP. The age of subjects ranged from 20 to 78 years, and three were female. Mean age was 52.6 ± 20.0 years. The range of the age of the first visit to hospital owing to epilepsy was 3–49 years. In all nine cases, there was no history of status epilepticus. Six of nine cases were diagnosed as focal seizures, and the other three cases were generalized seizure. Five of the nine SUDEP cases were found dead in bed, and their deaths were likely to have occurred during sleep. One case was found in her home, and the other three cases were found dead outside their homes. The posture at death was prone in six cases, and supine in three cases. Five of the nine cases regularly attended hospital for follow‐up visits and were medicated with anti‐epileptic drugs (cases 2, 4, 6, 7 and 8), and the remaining four cases did not have a history of visiting hospital in the year before death. In addition to epilepsy, cases 2 and 5 had a history of obesity, hypertension and hyperlipidemia, but case 5 was uncooperative in taking medication. Case 7 had a history of hypertension, and was medicated with a β‐blocker. Case 9 had a history of traumatic brain injury following a fall from height about 25 years before death.
Table 1.
Summary of clinical and toxicological data of epilepsy‐related sudden unexpected death cases. Abbreviations: SUDEP, sudden unexpected death in epilepsy; ECG, electrocardiogram; NA, not applicable; ND, not detected; RVH, right ventricular hypertrophy; CM, carbamazepine (Reference range; 4‐12 μg/mL); VPA, sodium valproate (Reference range; 50–100 μg/mL); PHT, phenytoin (Reference range; 10‐20 μg/mL); HT, hypertension; HL, hyperlipidemia; LQT. long QT interval; PB, phenobarbital (Reference range; 10‐ 40 μg/mL); ST, sinus tachycardia. DM, diabetes mellitus.
| Clinical | ||||||||
|---|---|---|---|---|---|---|---|---|
| Case | Age | Sex |
Onset(y.o) |
Scene | posture | ECG | Anti‐epileptic drug (mg/L) | Other past history |
| SUDEP | ||||||||
| 1 | 72 | F | 12 | In home | prone | NA | ND | None |
| 2 | 53 | M | 8 | Out door | prone | RVH | CM; 6, VPA; 70.2, PHT; 15 | Obesity, HT, HL |
| 3 | 26 | M | 13 | In bed | spine | NA | ND | None |
| 4 | 70 | F | 10 | Out door | prone | LQT | PHT; 2, PB; 6.5 | None |
| 5 | 48 | M | 23 | In bed | prone | ST | ND | Obesity, HT, HL |
| 6 | 68 | F | 15 | In bed | prone | LQT | PHT: 10.5 | None |
| 7 | 78 | M | 10 | Out door | prone | Normal | VPA; 62 | HT |
| 8 | 20 | M | 3 | In bed | spine | NA | CM; 2.6 | None |
| 9 | 54 | M | 49 | In bed | spine | ST | ND | Trauma |
| Drowning | ||||||||
| 10 | 11 | F | 3 | In bath | Normal | CM; 1.1, PHT; 7.6 | None | |
| 11 | 69 | F | 56 | In pool | LQT | VPA; 78.2, PB; 20.2 | Stroke, HT, DM | |
| 12 | 37 | M | 7 | In bath | NA | ND | HT | |
The three cases not categorized as SUDEP (Cases 10–12) died by drowning. The ages of these subjects were 11, 69 and 37, respectively. The age of the first visit to hospital owing to epilepsy was 3, 56 and 7 years. All cases showed classical drowning signs at the scene of death. All cases regularly attended follow‐up visits to the hospital and were medicated with anti‐epileptic drugs. Case 11 was found dead in a swimming pool, and cases 10 and 12 were found dead in hot bathwater. Case 12 was diagnosed as generalized seizure, and the other two cases were focal seizure. The epilepsy of case 11 was considered to result from stroke. No individual or family history of status epilepticus was found in all three cases. In addition to epilepsy, cases 11 and 12 had a history of hypertension, and case 11 also had diabetes mellitus. Case 11 was prescribed a Ca‐blocker and anti‐diabetic drugs, and case 12 was prescribed a β‐blocker.
ECGs of six SUDEP cases and two drowning cases were available. The QT interval was measured in II and V5 or V6, and was corrected by Bazzet's formula (QTc‐QT/RR0.5). The definition of long QT interval was >450 ms in men and >460 ms in women, in accordance with ANA/ACCF/HRS recommendations 53. A mild prolonged QT interval was found in cases 4, 6 and 11, and their QTcs were 0.470, 0.469 and 0.475, respectively.
Ethanol was not detected in the blood of any case. Anti‐epileptic drugs were detected in five SUDEP and two drowning cases. The blood content of all prescribed anti‐epileptic drugs was examined and related to generally used guidelines for the therapeutic and toxic levels of drugs 51. In cases 4, 8 and 10, the blood content of drugs was under therapeutic levels. In the other cases, the content was within the therapeutic level of each drug.
Pathological examination of the brain and heart
Pathological examination of the brain and heart was performed according to previously reported methods 25, 43.
Brain
All brains were fixed in 20% buffered formalin for at least 2 weeks prior to sampling. Specimens of frontal, parietal, temporal and occipital neocortex, amygdala, hippocampus, basal ganglia, hypothalamus, thalamus, midbrain, pons, medulla, cerebellar cortex and dentate nucleus were sampled. All sections were cut and stained with Luxol fast blue hematoxylin eosin. Gallyas‐Braak and Holzer stainings were also performed 43.
Heart
The heart was excised and dissected free from the great vessels. All epicardial coronary arteries were cut transversely at 5 mm intervals and decalcified as required. The right and left ventricles were cut at 1 cm intervals parallel to the levels of the papillary muscle from the apex. Sections at the level of the papillary muscle and the apex of heart muscle were examined in detail histologically. Blocks containing the sinoatrial node and atrioventricular conduction system were excised. These blocks were processed and embedded in paraffin, and 3 μm‐thick sections (at 30 μm intervals) were obtained from each block. Histological sections were stained with hematoxylin and eosin or Elastica‐Masson stains 25. Evaluation and diagnosis of heart disease were conducted according to standardized protocols 6, 24.
Molecular testing
Genomic DNA was extracted directly from whole blood using a QIAamp DNA Mini Kit (Qiagen Science, Germantown, MD, USA). We designed a custom AmpliSeq panel of PCR primers (Life Technologies, Carlsbad, CA, USA) using Ion AmpliSeq designer software (www.ampliseq.com) to target all exons of 73 genes associated with cardiac disorders, including cardiomyopathies and channelopathies (Table 2). This custom panel, which consisted of two separate PCR primer pools and produced a total of 1870 amplicons, was used to generate target amplicon libraries. Genomic DNA samples were each PCR‐amplified using the custom‐designed panel and Ion AmpliSeq Library Kit v2.0 (Life Technologies) in accordance with the manufacturer's instructions. Various samples were distinguished using an Ion Xpress Barcode Adapters Kit (Life Technologies) and then pooled in equimolar concentrations. Emulsion PCR and Ion Sphere Particle (ISP) enrichment were performed with an Ion PGM Template OT2 200 Kit (Life Technologies) in accordance with the manufacturer's instructions. ISPs were loaded on an Ion 316 Chip v2 and sequenced with an Ion PGM Sequencing 200 Kit (Life Technologies). All variants derived from PGM sequencing were prioritized and then confirmed by Sanger sequencing to validate the NGS results. Additionally, we examined three genes (SCN1A, SCN2A and KCNA1) known to correlate with SUDEP because of expression in both brain and heart by Sanger sequencing 37, 63. For Sanger sequencing, the nucleotide sequences of the amplified fragments were analyzed by direct sequencing in both directions using a BigDye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems, Foster City, CA, USA) and an ABI 3130xl automated sequencer (Applied Biosystems).
Table 2.
List of the 73 genes analyzed that are associated with inherited cardiac disease.
| ABCC9, ACTC1, ACTN2 AKAP9, ANK2, BAG3, BMPR1A, CACNA1C, CACNB2, CALR3, CAPN3, CAV3, COL4A1, DES, DMD, DSC2, DSG2, DSP, ELN, EMD, GAA, GATA4, GLA, GPD1L, HCN4, JUP, KCNE1, KCNE2, KCNE3, KCNH2, KCNJ2, KCNQ1, KRAS, LAMP2, LDB3, LMNA, MYBPC3, MYH11, MYH6, MYH7, MYL2, MYL3, MYLK, MYOZ2, NKX2‐5, NRAS, PKP2, PLN, PRKAG2, PTPN11, RAF1, RPS7, RYR2, SCN1B, SCN3B, SCN4B, SCN5A, SGCD, SLC25A4, SMAD3, SNTA1, SOS1, STARD3, TAZ, TBX5, TGFBR1, TGFBR2, TMEM43, TNNC1, TNNI3, TNNT2, TPM1, VCL |
Data analysis
Torrent Suite software and Ion Reporter Software 5.0 (Life Technologies) were used to perform primary to tertiary analyses, including optimized signal processing, base calling, sequence alignment, and variant analysis. We then obtained functional and/or segregation data on previously reported variants from the Human Gene Mutation Database (BIOBASE, Wolfenbüttel, Germany) and ClinVar (http://www.ncbi.nlm.nih.gov/clinvar). We identified known disease‐causing variants (known variants) when there was at least one study that evaluated the variant as pathogenic in both databases. Then, the allelic frequency of all detected variants was determined across the East Asian (EAS) population database of 4327 individuals in the Exome Aggregation Consortium (ExAC). The known variants were divided into common known variants with a minor allele frequency (MAF) of ≥0.5% and other rare known variants 31, 46.
In the next step, all variants with a MAF of ≥0.5% among the ExAC (EAS) population were filtered out. Variants for which the MAF had not yet been determined were also defined as rare variants.
We then assessed all the detected variants with eight in silico predictive algorithms [Functional Analysis through Hidden Markov Models Ver.2.3 (FATHMM), MutationAssessor, SIFT Sequence (SIFT), Align GVGD, MutationTaster, PolyPhen‐2, Protein Variation Effect Analyzer (PROVEAN), and Combined Annotation‐Dependent Depletion (CADD)] to evaluate their pathogenicity. The URL for each algorithm and the conditions used to evaluate pathogenicity are listed in Table 3. Subsequently, we assessed the possible pathogenicity of each variant by the algorithm rating of “pathogenicity”: 0–2, low; 3–5, intermediate; and 6–8, high. Finally, with regard to rare variants, only variants with intermediate and high levels of possible pathogenicity were classified as “potential rare variants.”
Table 3.
In silico algorithms used to predict variant pathogenicity. Abbreviations: FATHMM, Functional Analysis through Hidden Markov Models; SIFT, SIFT Sequence; PROVEAN, Protein Variation Effect Analyzer; CADD, Combined Annotation‐Dependent Depletion.
| Name | Website | Indication of pathogenicity |
|---|---|---|
| FATHMM | http://fathmm.biocompute.org.uk | Damaging |
| MutationAssessor | http://mutationassessor.org | Medium, high |
| SIFT | http://sift.jcvi.org | Damaging |
| Aligh GVGD | http://agvgd.iarc.fr/index.php | ≥C15 |
| Mutation taster | http://www.mutationtaster.org | Disease causing |
| PolyPhen‐2 | http://genetics.bwh.harvard.edu/pph2 | Probably damaging, possibly damaging |
| PROVEAN | http://provean.jcvi.org/index.php | Deleterious |
| CADD | http://cadd.gs.washington.edu | Score ≥ 10 |
Analysis of rare variants using controls
Differences in proportions of known and potential rare variants vs. controls from the ExAC (EAS) were assessed using Fisher's exact test with P < 0.05 considered statistically significant. Genotype data were used in a case–control analysis of known and candidate potential variants identified in this study. Potential pathogenicity of the variants was evaluated based on allele frequency, as recommended by recent guidelines for interpreting sequence variants 54.
Results
Summary of pathological findings
Summary of pathologic and genetic analysis results are listed in Table 4. Five of the nine SUDEP cases showed structural disorder in the brain associated with epilepsy. Hippocampal sclerosis was found in four cases (Figure 1A,B), and an old cerebral contusion was present in case 9 (Figure 1C). Among the three drowning cases, case 10 showed hippocampal sclerosis and Case 11 showed an old cerebral infarction of the left putamen (Figure 1D).
Table 4.
Summary of pathological and genetic data of epilepsy‐related sudden unexpected death cases. Abbreviations: BW, brain weight; HW, heart weight; LQT, long QT interval; SUDEP, sudden unexpected death in epilepsy; ND, not detected; F, focal epilepsy; HS, hippocampal sclerosis; FaRV, fatty replacement of right ventricle; G, generalized epilepsy; del, deletion; H‐Tb, hypertrabeculation; f‐CS, fibrosis of conduction system; ASD, atrial septal defect; RVH, right ventricular hypertrophy; LVH, left ventricular hypertrophy; OCI, old cerebral infarction. Genes in bold‐type indicate high possible pathogenicity.
| Neuropathology | Cardiac pathology | Genetic | |||||||
|---|---|---|---|---|---|---|---|---|---|
| BW | Findings | HW | Findings | Known | Classification | ||||
| Case | (g) | (g) | LQT | Common | Rare | Potential |
(seizure/cause) |
||
| SUDEP | |||||||||
| 1 | 1180 | HS | 300 | ND | F/HS | ||||
| 2 | 1208 | HS | 410 | FaRV | SCN5A, DSC2 (del)* | F/HS | |||
| 3 | 1429 | ND | 250 | H‐Tb, f‐CS | LDB3 | MYH6 | G/unknown | ||
| 4 | 1260 | ND | 288 | ASD, RVH | + | F/unknown | |||
| 5 | 1592 | ND | 525 | LVH, FaRV | DSC2 (del)* | DSP | G/unknown | ||
| 6 | 1148 | HS | 325 | FaRV | + | KCNE1 | DSC2 | DSG2 | F/HS |
| 7 | 1272 | HS | 439 | LVH | SCN5A | MYBPC3 | F/HS | ||
| 8 | 1576 | ND | 344 | ND | G/unknown | ||||
| 9 | 1506 | Old injury | 287 | ND | DMD | F/trauma | |||
| Drowning | |||||||||
| 10 | 1216 | HS | 176 | ND | F/HS | ||||
| 11 | 1329 | OCI | 419 | LVH | + | ANK2x2 | F/stroke | ||
| 12 | 1600 | ND | 425 | LVH | SCN5A | G/unknown | |||
*The pathogenicity of DSC2 (del) could not be estimated.
Figure 1.
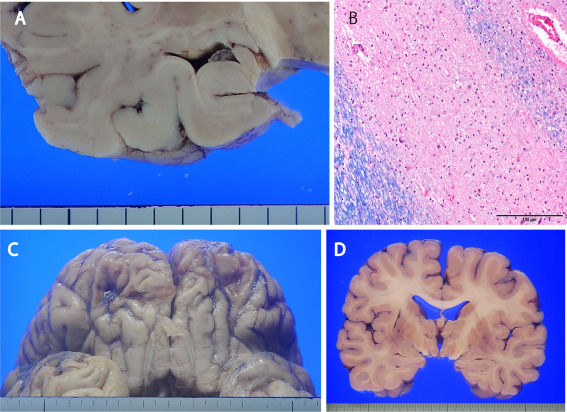
Neuropathology of the epilepsy‐related sudden unexpected death cases. A. Atrophy of the left hippocampus was found in case 1. B. Marked neuronal loss in left‐side CA1 in case 1 (Luxol fast blue/hematoxylin eosin). C. Old contusion injury of the base of the frontal lobe (case 9). D. Old infarction of the left putamen (case 11). Scale bar = 100 µm (B).
Gross examination of the heart showed cases 2, 5 and 6 to have fatty replacement of the right ventricle (Figure 2A). Case 3 showed both mild hypertrabeculation of the left ventricle and dilatation of the right ventricle (Figure 2B). Case 4 showed an atrioseptal defect of the secundum type, and the diameter of the defect was about 1 cm. Mild right ventricular hypertrophy and fatty replacement was also seen (Figure 2C,D). Microscopically, ischemic necrosis of myocytes and significant atherosclerosis of the coronary artery with narrowing >50% was not found in any case. Fibrofatty replacement of the right ventricle was found in cases 2, 5 and 6 (Figure 2E), and interstitial fibrosis of left ventricle was found in case 3 (Figure 2F). A few small inflammatory foci were found in the right ventricle of cases 5 and 6 (Figure 2G,H). Mild hypertrophy of myocytes without myocardial disarray was found in the left ventricle of cases 5, 7 and two drowning cases (cases 11 and 12). Severe interstitial fibrosis of the sinoatrial node and atrioventricular conduction system were found in the heart of case 3, compared with an age matched control case, other SUDEP and drowning cases (Figure 3A,B).
Figure 2.
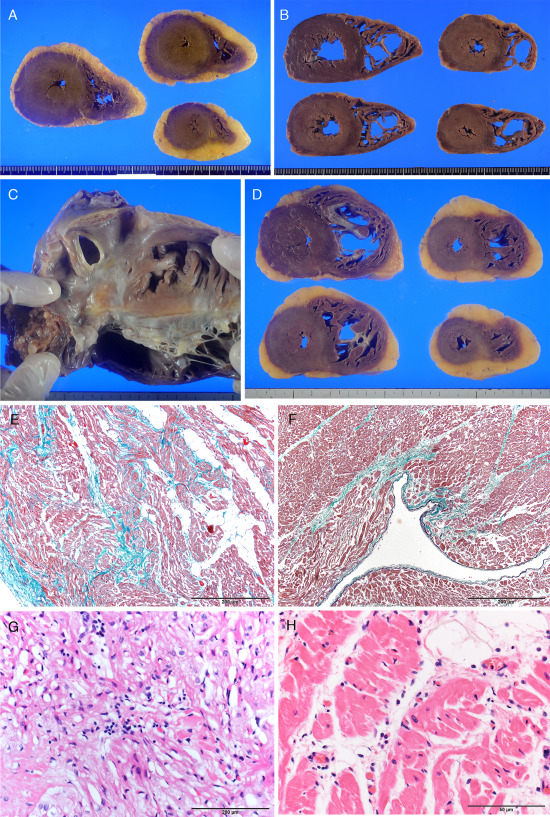
Heart pathology of the epilepsy‐related sudden unexpected cases. A. Fatty replacement of both ventricles in case 6. B. Hypertrabeculation of the left ventricle and dilatation of the right ventricular cavity in case 3. C. Atrioseptal defect in case 4. D. Mild right ventricular hypertrophy and fatty replacement in case 4. E. Fibrofatty replacement of the right ventricle of case 2 (Elastica‐Masson). F. Interstitial fibrosis of the left ventricle in case 3. G,H: Small inflammatory focus in the right ventricle of case 5 (G) and 6 (H) (Hematoxylin eosin). Scale bar = 200 µm (E), 100 µm (F), 50 µm (G,H).
Figure 3.
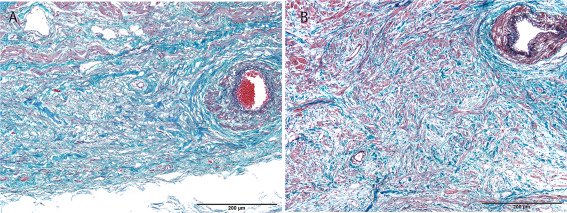
Sinoatrial node of case 3(A) and the control subject (B). Decreased numbers of conduction fibers in the node of case 3 compared with the control case (Elastica‐Masson), Scale bar = 200 µm.
Classification of the nine SUDEP cases, defined cases 1,8 and 9 without preexisting clinical conditions and pathological findings as Definite SUDEP, while the other six cases with minor but possible significant lesion in the heart were defined as Definite SUDEP plus.
NGS
The gene analysis results are shown in Tables 4, 5, 6, 7. The in silico analysis results for all variants detected are shown in Table 7.
Table 5.
Known variants in the epilepsy‐related sudden unexpected death cases. Abbreviations: MAF, minor allele frequency; ExAC, Exome Aggregation Consortium database; AFR, African/African American; AMR, Latino; EAS, East Asian; FIN, Finnish; NFE, Non‐Finnish European; SAS, South Asian; OTH, Other; Path, possible pathogenicity; Ref, references; LQTS, long QT syndrome; BrS, Brugada syndrome; SSS, sick sinus syndrome; Int, intermediate; DCM, dilated cardiomyopathy; LVNC, left ventricular non‐compaction; ARVC, arrythmogenic right ventricular cardiomyopathy; HCM, hypertrophic cardiomyopathy; ND, not described. Bold shows a MAF of the variants in EAS cohort in ExAC.
| Gene | Case | Transcript | Protein | dbSNP | Disease | MAF | Path | Ref |
|---|---|---|---|---|---|---|---|---|
| ExAC | ||||||||
| SCN5A | 2,7,12 | NM_198056.2 | p.Arg1193Gln | rs41261344 | LQTS, BrS, SSS | ALL:A=0.62% − AFR:0% − AMR:0.069% − EAS:7.09% − SAS:0.049% − NFE:0.13% − FIN:0.13% − OTH:0.32% | Int | (29.65) |
| DSC2 | 2,4 | NM_024422.3 | p.Gly790del | rs377272752 | ARVC | ALL:0.14% − AFR:0% − AMR:0% − EAS:1.77% − SAS:0.067% − NFE:0% − FIN:0.015% − OTH:0.22% | ND | 42 |
| LDB3 | 3 | NM_007078.2 | p.Asp673Asn | rs45514002 | DCM, LVNC | ALL:A=0.017% − AFR:0.0096% − AMR:0% − EAS:0.16% − SAS:0% − NFE:0.0090% − FIN:0% − OTH:0% | High | (2.67) |
| DSC2 | 6 | NM_024422.3 | p.Thr275Met | ND | ARVC | ALL:T=0.0025% − AFR:0% − AMR:0% − EAS:0% − SAS:0.012% − NFE:0.0015% − FIN:0% − OTH:0% | High | 21 |
| KCNE1 | 6 | NM_001127670.2 | p.Asp85Asn | rs1805128 | LQTS | ALL:A=0.92% − AFR:0.18% − AMR:0.19% − EAS:0.56% − SAS:0.14% − NFE:1.32% − FIN:1.65% − OTH:0.77% | High | 66 |
| MYBPC3 | 7 | NM_000256.3 | p.Thr1046Met | rs371061770 | HCM | ALL:T=0.0058% − AFR:0% − AMR:0% − EAS:0.058% − SAS:0% − NFE:0.0030% − FIN:0% − OTH:0% | Low | 1 |
Table 6.
Potential rare variants in the epilepsy‐related sudden unexpected death cases. Abbreviations: MAF, minor allele frequency; ExAC, Exome Aggregation Consortium database; AFR, African/African American; AMR, Latino; EAS, East Asian; FIN, Finnish; NFE, Non‐Finnish European; SAS, South Asian; OTH, Other; Path, possible pathogenicity; LQTS, long QT syndrome; DCM, dilated cardiomyopathy; ARVC, arrhythmogenic right ventricular cardiomyopathy; SVAS, supravalvular aortic stenosis; HCM, hypertrophic cardiomyopathy; ND, not described. Bold shows a MAF of the variants in EAS cohort in ExAC.
| Gene | Case | Transcript | Protein | dbSNP | Disease | MAF | Path | |
|---|---|---|---|---|---|---|---|---|
| ExAC | ||||||||
| MYH6 | 3 | NM_002471.3 | p.Ala822Thr | rs138419275 | DCM, HCM | ALL:A = 0.015% ‐ AFR:0% − AMR:0% − EAS:0.21% − SAS:0% − NFE:0% − FIN:0% − OTH:0% | High | |
| DSP | 5 | NM.004415.2 | p.Leu2628Pro | rs147484870 | ARVC | ALL:C = 0.013% − AFR:0% − AMR:0% − EAS:0.19% − SAS:0% − NFE:0% − FIN:0% − OTH:0% | High | |
| DSG2 | 6 | NM_001943.3 | p.Pro927Leu | rs146402368 | ARVC | ALL:T = 0.027% − AFR:0% − AMR:0% − EAS:0.37% − SAS:0% − NFE:0% − FIN:0% − OTH:0% | High | |
| DMD | 9 | NM_004006.2 | p.Arg395Gly | rs148511512 | DCM | ALL:A = 0.018% − AFR:0% − AMR:0% − EAS:0.24% − SAS:0% − NFE:0% − FIN:0% − OTH:0% | Int | |
| ANK2 | 11 | NM_001148.4 | p.Ser105Thr | ND | LQTS | − | Int | |
| ANK2 | 11 | NM_001148.4 | p.Glu1934Val | ND | LQTS | ALL:T = 0.00082% − AFR:0% − AMR:0.0086% − EAS:0% − SAS:0% − NFE:0% − FIN:0% − OTH:0% | Int |
Table 7.
Results of in silico analysis. Bold shows pathogenic condition in each in silico algorithm. Abbreviations: Path, Possible Pathogenicity; FATHMM, Functional Analysis through Hidden Markov Models; SIFT, SIFT Sequence; PROVEAN, Protein Variation Effect Analyzer; CADD, Combined annotation dependent depletion.
| Gene | Path | Case | In silico algorithm | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| FATHEM |
Mutation‐Assessor |
SIFT | Align GVGD | MutationTaster | Polyphen‐2 | PROVEAN | CADD | |||
| Known variants | ||||||||||
|
LDB3 p.Asp673Asn |
High | 3 | Damaging | Low | Damaging | C15 | Disease causing | Probably damaging | Deleterious | 34 |
|
DSC2 p.Thr275Met |
High | 6 | Tolerated | Medium | Damaging | C65 | Disease causing | Probably damaging | Deleterious | 23.8 |
|
KCNE1 p.Asp85Asn |
High | 6 | Damaging | Medium | Damaging | C15 | Polymorphism | Possibly damaging | Deleterious | 21.8 |
|
SCN5A p.Arg1193Gln |
Int | 2,7,12 | Damaging | Low | Tolerated | C0 | Polymorphism | Possibly damaging | Neutral | 12.32 |
|
MYBPC3 p.Thr1046Met |
Low | 7 | Tolerated | Medium | Tolerated | C0 | Polymorphism | Benign | Neutral | 18.56 |
|
DSC2 p.Gly790del |
ND | 2,5 | NE | NE | NE | NE | NE | NE | Deleterious | 12.91 |
| Potential rare variants | ||||||||||
|
MYH6 p.Ala822Thr |
High | 3 | Tolerated | Medium | Damaging | C55 | Disease causing | Possibly damaging | Deleterious | 20.3 |
|
DSP p.Leu2628Pro |
High | 5 | Tolerated | Medium | Damaging | C0 | Disease causing | Probably damaging | Deleterious | 26.6 |
|
DSG2 p.Pro927Leu |
High | 6 | Tolerated | Medium | Damaging | C25 | Disease causing | Probably damaging | Deleterious | 23.8 |
|
DMD p.Arg395Gly |
Int | 9 | Tolerated | Low | Tolerated | C0 | Disease causing | Possibly damaging | Neutral | 19.27 |
|
ANK2 p.Ser105Thr |
Int | 11 | Tolerated | Neutral | Damaging | C0 | Disease causing | Probably damaging | Deleterious | 29.3 |
|
ANK2 p.Glu1934Val |
Int | 11 | Tolerated | Low | Damaging | C0 | Polymorphism | Possibly damaging | Neutral | 24.8 |
|
PKP2 p.Gln220Arg |
Low | 1 | Tolerated | Neutral | Tolerated | C0 | Polymorphism | Benign | Neutral | 6.161 |
|
DSP p.Lys1581Glu |
Low | 7,12 | Tolerated | Low | Damaging | C0 | Polymorphism | Benign | Neutral | 21.8 |
|
DSG2 p.Val1040Ile |
Low | 8 | Tolerated | Low | Tolerated | C0 | Polymorphism | Benign | Neutral | 9.253 |
Six known variants were found in five of the nine SUDEP cases and in one of the three drowning cases (Table 4, 5) 1, 2, 21, 29, 42, 65, 66, 67. Among known variants, the MAFs of SCN5A_p.Arg1193Gln, DSC2_p.Gly790del and KCNE1_p.Asp85Asn were 7.09%, 1.77% and 0.56%, respectively, and those of the other three known variants were under 0.5%. Two in silico tools (PROVEAN and CADD) evaluated DSC2_p.Gly790del as pathogenic. The other six algorithms could not evaluate deletion variants. Therefore, the evaluation of pathogenicity of this variant by in silico analysis was reserved. In silico analysis of the other variants showed that the possibility of pathogenicity was high for LDB3 in case 3 and DSC2 and KCNE1 in case 6, intermediate for SCN5A, and low for MYBPC3.
Nine variants were identified as rare variants, and three of the nine were filtered out because in silico analysis indicated low possible pathogenicity (Table 7). Among the SUDEP cases, four potential rare variants were found, and two ANK2 variants were present in one death by drowning case (case 11). Among the six variants, two ANK2 variants were channelopathy‐related, and four (MYH6, DSP, DSG2 and DMD) were cardiomyopathy‐related. The possible pathogenicity was intermediate for three variants (in DMD and two in ANK2), and high for three (in DSP, MYH6 and DSG). Cases 2, 3, 5, 6, 7 and 11 had multiple variants (Table 4), and cases 3 and 6 had two or three variants with high possible pathogenicity. The sequence data of all variants with high possible pathogenicity are shown in Supporting Information Figure S1.
The prevalence of the 12 detected variants in the epilepsy‐related SUD cases and the ExAC EAS (East Asian subpopulation) was compared: ANK2_p.Ser105Thr was not found in ExAC, and the prevalence of three known variants (LDB3_p.Asp673Asn, DSC2_ p.Thr275Met and MYBPC3_p.Thr1046Met) and of three potential rare variants (DSP_p.Leu2628Pro, DMD_p.Arg395Gly and ANK2_p.Glu1934Val) was significantly higher in epilepsy‐related SUD cases than control cases (Table 8).
Table 8.
Assessment of the frequency of the identified candidate variants in control population data. Abbreviations: Alt; Alternative; HGVD, Human Genetic Variation Database; ExAC (EAS), East Asian (EAS) population database in the Exome Aggregation Consortium (ExAC) Database. Bold text indicates a significant difference between cases and controls from the ExAC (EAS).
| Identified variants | Cases n = 12 | ExAC (EAS) n = 4327 | Fisher's exact test p‐value | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Alt Number | Hemizygote Number | Allele Number | Homozygote Number | Alt Number | Hemizygote Number | Allele Number | Homozygote Number | ||
| Known variants | |||||||||
| LDB3 p.Asp673Asn | 1 | — | 24 | 0 | 14 | — | 8654 | 0 | 0.0407 |
| DSC2 p.Thr275Met | 1 | — | 24 | 0 | 0 | — | 8638 | 0 | 0.0028 |
| KCNE1 p.Asp85Asn | 1 | — | 24 | 0 | 48 | — | 8636 | 0 | 0.1275 |
| SCN5A p.Arg1193Gln | 3 | — | 24 | 0 | 461 | — | 6498 | 17 | 0.2415 |
| MYBPC3 p.Thr1046Met | 1 | — | 24 | 0 | 5 | — | 8584 | 0 | 0.0166 |
| DSC2 p.Gly790del | 2 | — | 24 | 0 | 153 | — | 8642 | 1 | 0.0679 |
| Potential rare variants | |||||||||
| MYH6 p.Ala822Thr | 1 | — | 24 | 0 | 18 | — | 8648 | 0 | 0.0513 |
| DSP p.Leu2628Pro | 1 | — | 24 | 0 | 16 | — | 8634 | 0 | 0.0461 |
| DSG2 p.Pro927Leu | 1 | — | 24 | 0 | 32 | — | 8624 | 0 | 0.0878 |
| DMD p.Arg395Gly | 1 | 1 | 17 | 0 | 16 | 5 | 6632 | 0 | 0.0426 |
| ANK2 p.Ser105Thr | 1 | — | 24 | 0 | — | — | — | — | |
| ANK2 p.Glu1934Val | 1 | — | 24 | 0 | 0 | — | 8608 | 0 | 0.0028 |
The known or potential rare variants that fulfill the criteria of the present study were not found in SCN1A, SCN2A and KCNA1. Final assessment of the classification of seizures and possible cause of epilepsy by reference to the recent report of the International League Against Epilepsy is summarized in Table 4 10.
Discussion
Diagnosis and classification of SUDEP
The definition and classification of SUDEP by Nashef et al 41 is considered to provide reliable ascertainment of incidence, monitoring of trends and comparison between studies. Among candidate SUDEP cases, some had preexisting physiological conditions and/or pathological conditions that may have caused sudden death, or were not clearly determined as the cause of death. Therefore, they additionally proposed to differentiate such cases into a new category “Definite SUDEP plus” to avoid differentiating such cases into “Possible SUDEP”; these cases tended to be excluded from some research studies of epilepsy‐related sudden death. Nashef et al 41 noted that this new category provides the opportunity to investigate the preexisting condition of undetermined significance. Although the evaluation of preexisting physiological conditions or pathological lesions must be subjective to some extent, in addition adequate or satisfactory anatomical, histopathological and toxicological examination, our study aims to show the significance of detailed genetic examination targeting inherited heart disease.
A large cohort study showed that about 40% of epilepsy cases fulfill the criteria for SUDEP. Many autopsied cases in our forensic autopsy unit have had an unusual death or the cause of death is undetermined before autopsy. The relatively high incidence of SUDEP in our study may be caused by differences in the autopsied population compared with other studies.
Advantages and limitations of gene analysis by NGS for epilepsy‐related SUD
Tu et al 64 examined three long QT syndrome‐related genes in 68 SUDEP cases and revealed 6 (13%) non‐synonymous (amino acid changing) variants in KCNH2 (n = 2) and SCN5A (n = 4). The recent study by Bagnall et al 4 showed more cases than expected had clinically relevant channelopathy‐related gene variants for cardiac arrhythmia. Four of 61 SUDEP cases had variants in common genes, and nine cases had variants with possible pathogenic potential. The detected variants in our study were not only channelopathy‐related genes, but also cardiomyopathy‐related gene variants with high possible pathogenicity in both SUDEP and epilepsy‐related drowning cases. This is a notable result of the present study. Death by drowning is excluded from the recent SUDEP criteria; however, it is not known whether the mechanism of epilepsy‐related drowning is the same as that of SUDEP. The pathogenesis in postictal drowning death cases might be identical to that of SUDEP, at least in some cases.
Recent studies show NGS to be useful for considering the mechanism of SUD or severe arrhythmogenic events 3, 26; however, population database and/or in silico predictive tools are frequently needed to evaluate the pathogenicity of candidate variants without confirmation of a positive relationship between clinical presentation and genetic variation. Further, Le Scouarnec 34 and Kapplinger 33 recently reported that many of the same variants were identified in Brugada syndrome patients and controls, indicating that identification of a variant does not demonstrate the presence of disease. Additionally, they commented that previous publication of a given known variants as a pathogenic variant does not guarantee its pathogenicity.
The recent consensus guidelines for the interpretation of sequence variants recommend that several in silico analyses should be deployed to evaluate the pathogenicity of arrhythmia‐related gene variants, because most algorithms for missense variant prediction are 65%–80% accurate when examining known disease variants 54. Following these guidelines, we used eight in silico tools to generate improved risk stratification of the detected variants. According to the guidelines 54, a variant that was absent in a population database and six variants with a significantly higher prevalence compared with qualifying controls were evaluated as moderate and strong possible pathogenic variants, respectively.
On the other hand, the prevalence of known or highly possible pathogenic variants was not always significantly different between SUDEP or drowning cases and qualifying controls. This may be caused by the limited number of the SUDEP or drowning cases examined in the present study. Therefore, additional cases may be essential to determine the statistical significance of the prevalence of each variant.
NGS, in silico predictive algorithms, and analyzing the prevalence of candidate variants do not yet represent a complete genetic analysis. In addition, Behr et al 9 reported that sudden arrhythmogenic death syndrome autopsy cases may have family members with overt cardiomyopathy. Alternatively, the abnormality may have been undetectable at autopsy because of an inadequate autopsy or the unavoidable limitations of autopsy sectioning. As in our previous report 26, the present study showed that careful pathological examination with reference to clinical information, in parallel with genetic analysis, can be highly beneficial for detecting the phenotype‐genotype correlation of SUDEP cases, and that such combined examination may lead to more accurate postmortem diagnosis of SUDEP cases with inherited heart diseases. In the present study, to prevent over‐estimation, we did not positively conclude that both known variants with high MAF and potential rare variants with intermediate possible pathogenicity have significant potential for inducing SUDEP, when phenotype‐genotype correlation was not evident. However, solely from the results of this study, we should not conclude that these variants are “benign.”
Evaluation of gene variants in SUDEP cases
Three SUDEP cases had five variants (in LDB, MYH6, DSP, DSC2 and DSG2) that had low MAF and that in silico analysis predicted to have high possible pathogenicity. Three of the five were desmosome‐related gene variants (in DSP, DSC2, DSG2) that are responsible of arrhythmogenic right ventricular cardiomyopathy (ARVC). These ARVC‐related pathogenic variants may eventually lead to myocyte necrosis, and subsequent induction of an inflammatory response to necrosis and fibrofatty replacement, which are histological hallmarks of ARVC 11. Although the pathological changes in cases 2, 5 and 6 had not progressed to an advanced form of ARVC, fatty infiltration with minimal fibrosis and inflammatory foci found in cases 5 and 6 are consistent with DSP in case 5, and DSC2 and DSG2 in case 6 26, 35. Kapplinger et al 32 demonstrated that, especially for desmosome‐related genes, the background noise of innocent variants is large. However, a serious arrhythmogenic event or sudden death may occur in a patient with ARVC genetic variants before morphological changes in the heart are apparent 7, 26. Furthermore, some investigators propose that Brugada syndrome shares features with ARVC, thus opening the possibility that they represent two poles of the same disease spectrum, ultimately leading to increased risk of sudden death 15. Brugada syndrome patients show minor right ventricle structural abnormalities 12, and desmosomal mutation carriers can experience ventricular fibrillation and SUD without overt structural disease 16. Possible high risk genetic variants with the mild but uncommon right ventricular pathology seen in cases 5 and 6 may indicate considerable risk for an arrhythmogenic event.
The heart of case 3 with MYH6 and LDB3 variants showed hypertrabeculation of the left ventricle, suggesting mild left ventricular non‐compaction, and severe interstitial fibrosis of the sinoatrial node. LDB3 is a disease‐causing gene of dilated cardiomyopathy and left ventricular non‐compaction 67. On the other hand, MYH6 is a possible disease‐causing gene of sick sinus syndrome 28. Few studies have reported pathological analysis of the heart in SUDEP; however, Opeskin et al 47 proposed that microscopic conduction system abnormalities may contribute to the occurrence of SUDEP. Both hypertrabeculation suggestive mild left ventricular non‐compaction and conduction system abnormality may have increased the potential of SUD in case 3.
The interpretation of pathogenicity of the three known but not rare variants found in the present SUDEP cases may be difficult to conclude. Mutation in KCNQ1 or KCNE1 subunits can cause congenital channelopathy leading to deafness, cardiac arrhythmia and epilepsy 5. KCNE1_p.Asp85Asn, found in case 6 has been shown in several studies to be a common variant or associated with an acquired prolonged QT interval 30, 52; however, two recent clinical and physiological studies showed the variant is functional and may be clinically important 44, 45. The variant may cause prolonged QT interval in case 6, and may have the potential to cause SUD because in silico analysis predicted high possible pathogenicity. Epilepsy attack can prolong the QT interval directly, especially if hypercapnia, hypoxia or catecholamine release occur. Therefore, genetic prolonged QT interval may be associated with SUDEP because of increased risk of torsade de pointes 58. In a similar fashion, although some studies conclude that SCN5A_p.Arg1193Gln is not pathogenic owing to high incidence 22, 31, it may be difficult to conclude that the variant is “benign” because it was demonstrated to cause loss of function by electrophysiological analysis 29. The pathogenicity of these single variants of KCNE1 or SCN5A may not be high, and these variants may not be a major causative factor for SUDEP. We assume that these variants might synergistically increase the risk of arrhythmogenic events in combination with other risk factors in epilepsy patients. An in‐frame deletion variant of DSC2 has already been evaluated as pathogenic by a family study 42, despite its high MAF and a single amino‐acid deletion. Further case studies might be useful to reexamine the significance of these “common” known variants. Furthermore, as shown in Table 5, it is interesting that SCN5A_p.Arg1193Gln and DSC2_p.Gly790del had a higher incidence in the east Asian population compared with other populations.
The significance of multiple variants with possible pathogenicity by for sudden death or a serious arrhythmogenic event, as seen in Cases 2, 3, 5, 6, 7 and 11 has not yet been explored. We assume that dual pathogenic mutations may increase the risk of SUDEP; however, further case studies are needed.
Evaluation of gene variants in drowning cases
Although the number of cases was limited, cases 11 and 12 showed possible significant findings. Case 11 showed two mutations in ANK2. These variants may have caused prolonged QT when case 11 was alive, and may have contributed to their SUD. This case possibly experienced arrhythmogenic events that caused their drowning. Analysis of genes involved in inherited heart disease may be useful for considering the pathogenesis of both SUDEP and epilepsy‐related drowning cases.
Other factors relevant to epilepsy‐related SUD
Although overdose of administered drugs was not evident in the present study, we should note that a few case studies showed some anti‐epileptic drugs, such as phenytoin or carbamazepine, might induce QT prolongation or other arrhythmia 19, 59. Autopsies of epilepsy‐related SUD should carefully evaluate the effect of anti‐epileptic drugs and all drugs that have a possible effect on the cardiovascular system. On the other hand, six SUDEP cases and one drowning case did not take adequate doses of prescribed drugs; therefore, low levels of anti‐epileptic drug may be a risk factor in epilepsy‐related death, as shown in a previous study 39. In case 4, Atrioseptal defect with mild right ventricular hypertrophy were incidental findings and both cardiac pathology and poor intake of anti‐epileptic drugs may have contributed to SUD.
The present study could not find an obvious correlation between epilepsy‐related SUD and factors other than cardiac factors, such as neuropathology, scene of death, type of seizure or estimated cause of epilepsy. Many incidences of arrhythmogenic SUD in the general population may require both a permissive myocardial substrate and an inciting trigger for its occurrence 24, 25. In addition to exercise, sleeping, effect of administered drugs as discussed, emotional stress such as extreme anger or sadness, seizure and related respiratory dysfunction may be strong inciting factors for a serious arrhythmogenic event 18, 37, 60. Seizure attack can also induce arrhythmia in humans 68 and in an animal model 57. The Mortemus study 56 identified that respiratory dysfunction frequently occurs preceding SUDEP. Beside channelopathies, other inherited heart diseases might contribute to death triggered by respiratory failure due to an epilepsy attack. The present study may show that multi‐faceted examination, including genetic analysis of inherited heart disease‐related genes, may be useful for the diagnosis and risk evaluation of epilepsy‐related SUD. In the classification of SUDEP cases, genetic analysis has not yet been extensively applied, but this may prove useful in the future.
The complex genetics of SUDEP parallels that of sudden cardiac death 23. However, molecular autopsy for sudden cardiac death or SUDEP will contribute to the early detection of relevant disease and may help to identify risk for arrhythmogenic and/or epileptic events. Careful counseling of family members by appropriate medical personnel, including neurologists, cardiologists, medical geneticists, paramedics and pathologists will contribute to the prevention of SUDEP or sudden cardiac death of family members 14.
Conclusions
The present study used NGS to identify highly probable pathogenic desmosome‐, dilated cardiomyopathy‐ and conduction disease‐related gene variants associated with mild but highly probable pathology in three SUDEP cases. A death by drowning case with a double ANK2 variant is also significant. Careful evaluation of the pathogenicity of detected gene variants is essential, and the combination of referring to clinical information, detail pathological examination and adequate molecular analysis of inherited heart disease‐related genes by NGS can become a valid approach for correctly diagnosing epilepsy‐related SUD, even in the absence of the clinical appearance or typical gross pathology suggesting advanced inherited heart disease.
Conflict of Interest
The authors declare no conflicts of interest.
Supporting information
Additional Supporting Information may be found in the online version of this article at the publisher's website:
Figure S1. Variants with high possible pathogenicity in epilepsy‐related sudden unexpected death cases. A: In‐frame deletion variant of DSC2 in cases 2 and 5. B, C: MYH6 (B) and LDB3 (C) variants in case 3. D: DSP variant in case 5. E‐G: KCNE1 (E), DSC2 (F) and DSG2 (G) variants in case 6.
Acknowledgments
The authors thank Ms. Syuko Matsumori, Ms. Tamae Sasakura, Mr. Noboru Onozuka, and Mr. Osamu Yamamoto for their technical assistance. This work was supported in part by a KAKENHI grant from JSPS, Japan, to Y.H. (15k08867) and Presidential Discretionary Funds, University of Toyama 2014, to N.N.
References
- 1. Amendola LM, Dorschner MO, Robertson PD, Salama JS, Hart R, Shirts BH et al (2015) Actionable exomic incidental findings in 6503 participants: challenges of variant classification. Genome Res 25:305–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Arimura T, Hayashi T, Terada H, Lee SY, Zhou Q, Takahashi M et al (2004) A Cypher/ZASP mutation associated with dilated cardiomyopathy alters the binding affinity to protein kinase C. J Biol Chem 279:6746–6752. [DOI] [PubMed] [Google Scholar]
- 3. Bagnall RD, Das KJ, Duflou J, Semisarian C (2014) Exome analysis‐based molecular autopsy in cases of sudden unexplained death in the young. Heart Rhythm 11:655–662. [DOI] [PubMed] [Google Scholar]
- 4. Bagnall RD, Crompton DE, Petrovski S, Lam L, Cutmore C, Garry SI et al (2016) Exome‐based analysis of cardiac arrhythmia, respiratory control and epilepsy genes in sudden unexpected death in epilepsy. Ann Neurol 79:522–534. [DOI] [PubMed] [Google Scholar]
- 5. Barro‐Soria R, Rebolledo S, Liin SI, Perez ME, Sampson KJ, Kass RS, Larsson HP (2014) KCNE1 divides the voltage sensor movement in KCNQ1/KCNE1 channels into two steps. Nat Commun 5:3750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Basso C, Burke M, Fornes P, Gallagher PJ, de Gouveia RH, Sheppard M et al (2008) Guidelines for autopsy investigation of sudden cardiac death. Virchows Arch 452:11–18. [DOI] [PubMed] [Google Scholar]
- 7. Basso C, Corrado D, Marcus FI, Nava A, Thiene G (2009) Arrhythmogenic right ventricular cardiomyopathy. Lancet 373:1289–1300. [DOI] [PubMed] [Google Scholar]
- 8. Begni E (2009) Accident and injuries in patients with epilepsy. Expert Rev Neurother 9:291–298. [DOI] [PubMed] [Google Scholar]
- 9. Behr ER, Dalageorgou C, Christiansen M, Syrris P, Hughes S, Esteban MTT et al (2008) Sudden arrhythmogenic death syndrome: familial evaluation identifies inheritable heart disease in the majority of families. Eur Heart J 29:1670–1680. [DOI] [PubMed] [Google Scholar]
- 10. Berg AT, Berkovic SF, Brodie MJ, Buchhalter J, Cross JH, van Emde Boas W et al (2010) Revised terminology and concepts for organization of seizures and epilepsies: report of the ILAE commission on classification and terminology, 2005‐2009. Epilepsia 51:676–685. [DOI] [PubMed] [Google Scholar]
- 11. Burke AP, Farb A, Tashko G, Virmani R (1998) Arrhythmogenic right ventricular cardiomyopathy and fatty replacement of the right ventricular myocardium: are they different diseases? Circulation 28:1571–1580. [DOI] [PubMed] [Google Scholar]
- 12. Catalano O, Antonaci S, Moro G, Mussida M, Frascaroli M, Baldi M et al (2009) Magnetic resonance investigations in brugada syndrome reveal unexpectedly high rate of structural abnormalities. Eur Heart J 30:2241–2248. [DOI] [PubMed] [Google Scholar]
- 13. Chiba T, Yamauchi M, Nishida N, Kaneko T, Yoshizaki K, Yoshioka N (2005) Risk factors of sudden death in the Japanese hot bath in the senior population. Forensic Sci Int 149:151–158. [DOI] [PubMed] [Google Scholar]
- 14. Coll M, Allegue C, Partemi S, Mates J, Del Olmo B, Campuzano O et al (2016) Genetic investigation of sudden unexpected death in epilepsy cohort by panel target resequencing. Int J Legal Med 130:331–339. [DOI] [PubMed] [Google Scholar]
- 15. Corrado D, Basso C, Buja G, Nava A, Rossi L, Thiene G (2001) Right bundle branch block, right precordial ST‐segment elevation, and sudden death in young people. Circulation 103:710–717. [DOI] [PubMed] [Google Scholar]
- 16. Delmar M, McKenna WJ (2010) The cardiac desmosome and arrhythmogenic cardiomyopathies: from gene to disease. Circ Res 107:700–714. [DOI] [PubMed] [Google Scholar]
- 17. Devinsky O (2011) Sudden, unexpected death in epilepsy. N Engl J Med 365:1801–1811. [DOI] [PubMed] [Google Scholar]
- 18. Espinosa PS, Lee JW, Tedrow UB, Bromfield EB, Dworetzky BA (2009) Sudden unexpected near death in epilepsy: malignant arrhythmia from a partial seizure. Neurology 72:1702–1703. [DOI] [PubMed] [Google Scholar]
- 19. Feldman AE, Gibal BE (2013) QTc prolongation by antiepileptic drugs and the risk of torsade de pointes in patients with epilepsy. Epilepsia Behav 26:421–426. [DOI] [PubMed] [Google Scholar]
- 20. Ficker DM, So EL, Shen WK, Annegers JF, O'Brien PC, Cascino GD, Belau PG (1998) Population‐based study of the incidence of sudden unexplained death in epilepsy. Neurology 51:1270–1274. [DOI] [PubMed] [Google Scholar]
- 21. Gehmlich K, Syrris P, Peskett E, Evans A, Ehler E, Asimaki A et al (2011) Mechanistic insights into arrhythmogenic right ventricular cardiomyopathy caused by desmocollin‐2 mutations. Cardiovasc Res 90:77–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ghouse J, Have CT, Weeke P, Bille Nielsen J, Ahlberg G, Balslev‐Harder M et al (2015) Rare genetic variants previously associated with congenital forms of long QT syndrome have little or no effect on the QT interval. Eur Heart J 36:2523–2529. [DOI] [PubMed] [Google Scholar]
- 23. Goldman AM, Behr ER, Semsarian C, Bagnall RD, Sisodiya S, Cooper PN (2016) Sudden enexpected death in epilepsy genetics: molecular diagnosis and prevention. Epilepsia 57:17–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hata Y, Mori H, Tanaka A, Fujita Y, Shimomura T, Tabata T et al (2014) Identification and characterization of a novel genetic mutation with prolonged QT syndrome in an unexplained postoperative death. Int J Legal Med 128:105–115. [DOI] [PubMed] [Google Scholar]
- 25. Hata Y, Kinoshita K, Kudo K, Ikeda N, Nishida N (2015) Anomalous origin of the right coronary artery from the left coronary sinus with intramural course‐comparison between sudden death and non‐sudden death cases. Cardiovasc Pathol 24:154–159. [DOI] [PubMed] [Google Scholar]
- 26. Hata Y, Kinoshita K, Mizumaki K, Yamaguchi Y, Hirono K, Ichida F et al (2016) Postmortem genetic analysis of sudden unexplained death syndrome under 50 years of age: a next‐generation sequencing study. Heart Rhythm (in press) doi: 10.1016/j.hrthm.2016.03.038. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 27. Hirsch LJ, Hauser WA (2004) Can sudden unexplained death in epilepsy be prevented? Lancet 364:2157–2158. [DOI] [PubMed] [Google Scholar]
- 28. Holm H, Gudbjartsson DF, Sulem P, Masson G, Helgadottir HT, Zanon C et al (2011) A rare variant in MYH6 is associated with high risk of sick sinus syndrome. Nat Genet 43:316–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Huang H, Zhao J, Barrane FZ, Champagne J, Chahine M (2006) Nav1.5/R1193Q polymorphism is associated with both long QT and Brugada syndromes. Can J Cardiol 22:309–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kapplinger JD, Tester DJ, Salisbury BA, Carr JL, Harris‐Kerr C, Pollevick GD et al (2009) Spectrum and prevalence of mutations from the first 2,500 consecutive unrelated patients referred for the FAMILION long QT syndrome genetic test. Heart Rhythm 6:1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kapplinger JD, Tester DJ, Alders M, Benito B, Berthet M, Brugada J et al (2010) An international compendium of mutations in the SCN5A‐encoded cardiac sodium channel in patients referred for Brugada syndrome genetic testing. Heart Rhythm 7:33–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kapplinger JD, Landstrom AP, Salisbury BA, Callis TE, Pollevick GD, Tester DJ et al (2011) Distinguishing arrhythmogenic right ventricular cardiomyopathy/dysplasia‐associated mutations from background genetic noise. J Am Coll Cardiol 57:2317–2327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kapplinger JD, Giudicessi JR, Ye D, Tester DJ, Callis TE, Valdivia CR et al (2015) Enhanced Classification of Brugada Syndrome‐Associated and Long‐QT Syndrome‐Associated Genetic Variants in the SCN5A‐Encoded Nav1.5 Cardiac Sodium Channel. Circ Cardiovasc Genet 8:582–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Le Scouarnec S, Karakachoff M, Gourraud JB, Lindenbaum P, Bonnaud S, Portero V et al (2015) Testing the burden of rare variation in arrhythmia‐susceptibility genes provides new insights into molecular diagnosis for Brugada syndrome. Hum Mol Genet 24:2757–2763. [DOI] [PubMed] [Google Scholar]
- 35. Lopez‐Ayala JM, Pastor‐Quirante F, Gonzalez‐Carrillo J, Lopez‐Cuenca D, Sanchez‐Munoz JJ, Oliva‐Sandoval MJ, Gimeno JR (2015) Genetic myocarditis in arrhythmogenic right ventricular dysplasia. Heart Rhythm 12:766–773. [DOI] [PubMed] [Google Scholar]
- 36. Lubitz SA, Ellinor PY (2015) Next generation sequencing for the diagnosis of cardiac arrhythmia syndrome. Heart Rhythm 12:1062–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Massey CA, Sowers LP, Dlouhy BJ, Richerson GB (2014) SUDEP mechanisms: the pathway to prevention. Nat Rev Neurol 10:271–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Metzker ML (2010) Sequencing technologies‐the next generation. Nat Rev Genet 11:31–46. [DOI] [PubMed] [Google Scholar]
- 39. Monté CP, Arends JB, Tan IY, Aldenkamp AP, Limburg M, de Krom MC (2007) Sudden unexpected death in epilepsy patients: risk factors. A systematic review. Seizure 16:1–7. [DOI] [PubMed] [Google Scholar]
- 40. Nashef L (1997) Sudden unexpected death in epilepsy: terminology and definitions. Epilepsia 38:S6–S8. [DOI] [PubMed] [Google Scholar]
- 41. Nashef L, So EL, Ryvlin P, Tomson T (2012) Unifying the definition of sudden unexpected death in epilepsy. Epilepsia 53:227–233. [DOI] [PubMed] [Google Scholar]
- 42. Ng D, Johnston JJ, Teer JK, Singh LN, Peller LC, Wynter JS et al (2013) Interpreting secondary cardiac disease variants in an exome cohort. Circ Cardiovasc Genet 6:337–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Nishida N, Yoshida K, Hata Y, Arai Y, Kinoshita K (2015) Pathological features of preclinical or early clinical stages of corticobasal degeneration: a comparison with advanced cases. Neuropathol Appl Neurobiol 41:893–905. [DOI] [PubMed] [Google Scholar]
- 44. Nishio Y, Makiyama T, Itoh H, Sakaguchi T, Ohno S, Gong YZ et al (2009) D85N, a KCNE1 polymorphism, is a disease‐causing gene variant in long QT syndrome. J Am Coll Cardiol 54:812–819. [DOI] [PubMed] [Google Scholar]
- 45. Nof E, Barajas‐Martinez H, Eldar M, Urrutia J, Caceres G, Rosenfeld G et al (2011) LQT5 masquerading as LQT2: a dominant negative effect of KCNE1‐D85N rare polymorphism on KCNH2 current. Europace 13:1478–1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Nunn LM, Lopes LR, Syrris P, Murphy C, Plagnol V, Firman E et al (2015) Diagnostic yield of molecular autopsy in patients with sudden arrhythmogenic death syndrome using targeted exome sequencing. Europace (in press) doi: 10.1093/europace/euv285. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Openski K, Thomas A, Berkovic SF (2000) Does cardia counduction pathology contribure to sudden unexpected death in epilepsy? Epilepsy Res 40:17–24. [DOI] [PubMed] [Google Scholar]
- 48. Parisi P, Oliva A, Coll Vidal M, Partemi S, Campuzano O, Iglesias A et al (2013) Coexistence of epilepsy and Brugada syndrome in a family with SCN5A mutation. Epilepsy Res 105:415–418. [DOI] [PubMed] [Google Scholar]
- 49. Partemi S, Cestèle S, Pezzella M, Campuzano O, Paravidino R, Pascali VL et al (2013) Loss‐of‐function KCNH2 mutation in a family with long QT syndrome, epilepsy, and sudden death. Epilepsia 54:e112–e116. [DOI] [PubMed] [Google Scholar]
- 50. Partemi S, Vidal MC, Striano P, Campuzano O, Allegue C, Pezzella M et al (2015) Genetic and forensic implications in epilepsy and cardiac arrhythmias: a case series. Int J Legal Med 129:495–504. [DOI] [PubMed] [Google Scholar]
- 51. Patsalos PN, Berry DJ, Bourgeois BF, Cloyd JC, Glauser TA, Johannessen SI et al (2008) Antiepileptic drugs–best practice guidelines for therapeutic drug monitoring: a position paper by the subcommission on therapeutic drug monitoring, ILAE Commission on Therapeutic Strategies. Epilepsia 49:1239–1276. [DOI] [PubMed] [Google Scholar]
- 52. Paulussen AD, Gilissen RA, Armstrong M, Doevendans PA, Verhasselt P, Smeets HJ et al (2004) Genetic variations of KCNQ1, KCNH2, SCN5A, KCNE1, and KCNE2 in drug‐induced long QT syndrome patients. J Mol Med 82:182–188. [DOI] [PubMed] [Google Scholar]
- 53. Rautaharju PM, Surawicz B, Gettes LS, Bailey JJ, Childers R, Deal BJ et al American Heart Association Electrocardiography and Arrhythmias Committee, Council on Clinical Cardiology; American College of Cardiology Foundation; Heart Rhythm Society (2009) AHA/ACCF/HRS recommendations for the standardization and interpretation of the electrocardiogram: part IV: the ST segment, T and U waves, and the QT interval: a scientific statement from the American Heart Association Electrocardiography and Arrhythmias Committee, Council on Clinical Cardiology; the American College of Cardiology Foundation; and the Heart Rhythm Society. Endorsed by the International Society for Computerized Electrocardiology. J Am Coll Cardiol 53:982–991. [DOI] [PubMed] [Google Scholar]
- 54. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier‐Foster J et al ACMG Laboratory Quality Assurance Committee. (2015) Standards and guidelines for the interpretation of sequence variants.: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the association for molecular autopsy. Genet Med 17:405–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Ryan CA, Dowling G (1993) Drowning deaths in people with epilepsy. Can Med Assoc J 148:781–784. [PMC free article] [PubMed] [Google Scholar]
- 56. Ryvlin P, Nashef L, Lhatoo SD, Bateman LM, Bird J, Bleasel A et al (2013) Incidence and mechanisms of cardiorespiratory arrests in epilepsy monitoring units (MORTEMUS): a retrospective study. Lancet Neurol 12:966–977. [DOI] [PubMed] [Google Scholar]
- 57. Schraeder PL, Lathers CM (1983) Cardiac neural discharge and epileptogenic activity in the cat: an animal model for unexplained death. Life Sci 32:1371–1382. [DOI] [PubMed] [Google Scholar]
- 58. Shorvon S, Tomson T (2011) Sudden unexpected death in epilepsy. Lancet 378:2028–2038. [DOI] [PubMed] [Google Scholar]
- 59. Stöllberger C, Finsterer J (2004) Cardiorespiratory findings in sudden unexplained/unexpected death in epilepsy (SUDEP). Epilepsy Res 59:51–60. [DOI] [PubMed] [Google Scholar]
- 60. Surges R, Sander JW (2012) Sudden unexpected death in epilepsy: mechanisms, prevalence, and prevention. Curr Opin Neurol 25:201–207. [DOI] [PubMed] [Google Scholar]
- 61. Tester DJ, Ackerman MJ (2007) Postmortem long QT syndrome genetic testing for sudden unexpected death in the young. J Am Coll Cardiol 49:240–246. [DOI] [PubMed] [Google Scholar]
- 62. Tester DJ, Medeiros‐Domingo A, Will ML, Haglund CM, Ackerman MJ (2012) Cardiac channel molecular autopsy: insights from 173 consecutive cases of autopsy‐negative sudden unexpected death referred for postmortem genetic testing. Mayo Clin Proc 87:524–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Tiron C, Campuzano O, Pérez‐Serra A, Mademont I, Coll M, Allegue C et al (2015) Further evidence of the association between LQT syndrome and epilepsy in a family with KCNQ1 pathogenic variant. Seizure 25:65–67. [DOI] [PubMed] [Google Scholar]
- 64. Tu S, Bagnall D, Duflpu J, Semsarian C (2011) Post‐mortem review and genetic analysis od sudden unexpected death in epilepsy (SUDEP) cases. Brain Pathol 21:201–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Vatta M, Dumaine R, Varghese G, Richard TA, Shimizu W, Aihara N et al (2002) Genetic and biophysical basis of sudden unexplained death nocturnal death syndrome (SUNDS), a disease allelic to Brugada syndrome. Hum Mol Genet 11:337–345. [DOI] [PubMed] [Google Scholar]
- 66. Westenskow P, Splawski I, Timothy KW, Keating MT, Sanguinetti MC (2004) Compound mutations: a common cause of severe long‐QT syndrome. Circulation 109:1834–1841. [DOI] [PubMed] [Google Scholar]
- 67. Xing Y, Ichida F, Matsuoka T, Isobe T, Ikemoto Y, Higaki T et al (2006) Genetic analysis in patients with left ventricular noncompaction and evidence for genetic heterogeneity. Mol Genet Metab 88:71–77. [DOI] [PubMed] [Google Scholar]
- 68. Zijlmans M, Flanagan D, Gotman J (2002) Heart rate changes and ECG abnormalities during epileptic seizures: prevalence and definition of an objective clinical sign. Epilepsia 43:847–855. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting Information may be found in the online version of this article at the publisher's website:
Figure S1. Variants with high possible pathogenicity in epilepsy‐related sudden unexpected death cases. A: In‐frame deletion variant of DSC2 in cases 2 and 5. B, C: MYH6 (B) and LDB3 (C) variants in case 3. D: DSP variant in case 5. E‐G: KCNE1 (E), DSC2 (F) and DSG2 (G) variants in case 6.