

Abstract
Glioblastoma may develop rapidly without evidence for precursor lesions (primary glioblastomas), or progress from diffuse or anaplastic astrocytomas (secondary glioblastomas). Despite having distinct genetic profiles, these glioblastoma subtypes have similar histological features. We hypothesized that the highly malignant phenotype of glioblastoma may be attributable to genetic alterations that are common to both glioblastoma subtypes. In the present study, we first searched for commonly (>35%) amplified genes in glioblastomas with IDH1 mutation (a hallmark of secondary glioblastoma) and those without IDH1 mutation (typical for primary glioblastoma) in data from The Cancer Genome Atlas (TCGA). A total of 25 genes were identified, of which 21 were located at 7q31‐34. We then screened 264 gliomas (70 glioblastomas, 112 diffuse astrocytomas, 82 oligodendrogliomas) for gain of the MET at 7q31.2 with quantitative polymerase chain reaction (PCR). MET gain was detected in primary glioblastomas (47%) and secondary glioblastomas (44%), suggesting that this genetic alteration plays a role in the pathogenesis of both glioblastoma subtypes. MET gain was also common in diffuse astrocytomas (38%), but less frequent in oligodendrogliomas (16%). MET gain in diffuse astrocytomas was associated with shorter survival (median, 43.0 vs. 70.7 months; P = 0.004), suggesting that MET gain is a useful prognostic marker for diffuse astrocytomas.
Keywords: diffuse astrocytoma, MET, primary glioblastoma, secondary glioblastoma
Introduction
Gliomas account for up to 70% of primary brain tumors in adults, with glioblastoma (WHO grade IV) being the most common and malignant histologic type 25. The 5‐year survival of patients with glioblastoma is <3% 25, despite multimodal therapy with surgery, radiotherapy and chemotherapy. Most glioblastomas are considered to arise with a short clinical history, and in the absence of less malignant precursor lesions (primary glioblastoma). Other glioblastomas (secondary glioblastomas) develop slowly from diffuse astrocytoma (WHO grade II) or anaplastic astrocytoma (WHO grade III). Primary and secondary glioblastomas carry distinct genetic alterations, with IDH1 mutations being the most reliable genetic marker to distinguish between these glioblastoma subtypes 12, 51. On the other hand, the histological features of primary and secondary glioblastomas are similar, suggesting that they may share genetic alterations that play important roles in their development.
Diffuse astrocytomas tend to progress to secondary glioblastomas, but time to progression varies considerably among patients. In one study, time to progression of diffuse astrocytoma to glioblastoma ranged from 7 to 133 months 48, while in another study, the interval before malignant transformation of low‐grade astrocytomas ranged from 39 to 119 months 45. Little is known about the molecular mechanisms underlying the rapid progression of diffuse astrocytomas. We hypothesized that this may be at least partly attributable to the presence of genetic alterations that are essential for the glioblastoma phenotype, and that such genetic alterations may be common to both primary and secondary glioblastomas.
The objective of this study was to identify molecular markers that are prognostic for unfavorable outcome of patients with diffuse astrocytoma. We first analyzed The Cancer Genome Atlas (TCGA) dataset and found frequent gain at 7q31 in both glioblastomas with IDH1 mutation (typical of secondary glioblastoma) and those without IDH1 mutation (typical of primary glioblastoma). We then decided to focus on the MET gene at 7q31.2, which is known to be involved in the pathogenesis of a variety of neoplasms 7, 50, and screened for MET gain in 112 diffuse astrocytomas, 82 oligodendrogliomas (WHO grade II), 34 primary glioblastomas and 36 secondary glioblastomas.
Materials and methods
Analysis of TCGA data
We used the TCGA data portal (http://cancergenome.nih.gov/), which contains copy number data for 372 glioblastomas (of these, eight glioblastomas had IDH1 mutations). A log2 ratio of >0.5 was considered as copy‐number gain. Furthermore, TCGA data were analyzed using cBio Cancer Genomics Portal (http://www.cbioportal.org/public‐portal/index.do), provided by the Memorial Sloan‐Kettering Cancer Center. For comparison of the results obtained, the Rembrandt database (http://rembrandt.nci.gov) was used.
Tumor samples
A total of 264 gliomas were obtained from the Department of Neuropathology, University Hospital Zurich; Switzerland, the Department of Neuropathology, University Frankfurt, Germany; the Departments of Neuropathology and Neurosurgery, University Hospital Essen, Germany; the Department of Pathology, Gunma University, Japan; the Institute of Neuropathology and Department of Neurosurgery, University Hospital Munster, Germany; Institute of Neuroscience, Bordeaux, France; and the Department of Neurosurgery, University Hospital Bern, Switzerland. Histologically, these tumors were classified as diffuse astrocytoma WHO grade II (112 cases; mean age, 39 years), oligodendroglioma WHO grade II (82 cases; mean age, 43 years), primary glioblastoma (34 cases; mean age, 59 years) and secondary glioblastoma (36 cases; mean age, 41 years). The diagnosis of secondary glioblastoma was made on the basis of clinical information. All primary glioblastomas lacked IDH1 mutations, whereas 27 out of 36 (75%) secondary glioblastomas were found to have IDH1 mutations. Median survival of patients with primary glioblastomas, secondary glioblastomas, diffuse astrocytomas or oligodendrogliomas was 8.0 months, 7.8 months, 70.7 months or 112.5 months, respectively. Mean survival of patients with primary and secondary glioblastoma was 9.7 ± 6.2 months and 13.7 ± 14.9 months, respectively (P = 0.145), and that of patients with glioblastoma without and with IDH1 mutations was 9.2 ± 5.8 months and 15.9 ± 16.2 months, respectively (P = 0.015). Genetic alterations in these tumors have been reported previously 17, 34. Survival analyses were carried out for patients who were followed up for at least 80 months after the date of surgery. The study was approved by the International Agency for Research on Cancer Ethics Committee.
Quantitative polymerase chain reaction (PCR)
DNA was extracted from formalin‐fixed and paraffin‐embedded histological sections as previously reported 34. Quantitative PCR was carried out using an iCycler (Bio‐Rad, Hercules, CA, USA) in a 20 μL reaction mixture composed of 10 μL 2× iQ SYBR Green Supermix, 2 μL H2O, 1.6 μL template DNA (approx. 20 ng/μL) and primers. MET primers were designed for exon 20/21. The primer sequences were 5′‐TCC TAC AAC CCG AAT ACT GC‐3′ (sense) and 5′‐GGT GCC AGC ATT TTA GCA TT‐3′ (antisense; product, 155 bp). As a reference, beta‐globin (at 11p15.5) was used as previously reported 24. Primer sequences for beta‐globin were 5′‐GTG CAC CTG ACT CCT GAG GAG A‐3′ (sense) and 5′‐CCT TGA TAC CAA CCT GCC CAG‐3′ (antisense; product, 102 bp). The primers were designed using Primer3 online software (http://frodo.wi.mi.edu/primer3/input.htm). DNA was first denatured at 95°C for 12 minutes, followed by 40 cycles of denaturation at 95°C for 20 s, annealing at 55°C for 20 s and extension at 72°C for 45 s. The final extension step was at 72°C for 2 minutes. Each plate contained measurements for the target and the reference genes. Each reaction was carried out in triplicate. We calculated the PCR cycle umber (Ct) value, δ‐Ct [Ct (MET) − Ct (beta‐globin)] and δδ‐Ct [δ‐Ct (tumor) − δ‐Ct (normal)] values, and the relative copy number (2‐δδ‐Ct) as previously reported 31. For normalization, DNA was extracted from paraffin‐embedded sections of 10–11 samples of normal tissue. A tolerance interval (TI) with a confidence interval (CI) of 95% was determined from the standard deviation of normal DNA, as reported previously 31. Gain was considered as a copy number > 2.699 (Figure 1).
Figure 1.
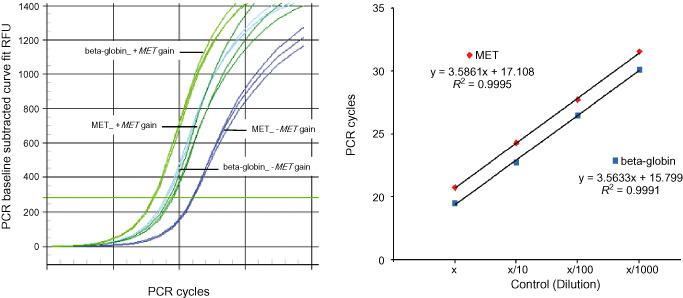
Quantitative PCR showing MET gain in low‐grade diffuse gliomas. The left‐hand figure shows the original quantitative PCR graph for low‐grade diffuse gliomas with (+MET gain) and without (−MET gain) MET gain. Relative CT values for MET and beta‐globin at different concentrations of normal control DNA are shown. The slopes of the curves are similar, suggesting equal efficiencies of the two PCR reactions (right).
Statistical analyses
Statistical analyses were carried out using SPSS Statistics 15.0 Software for Windows (SPSS Inc., Chicago, IL, USA). The Fisher's exact test was applied for the evaluation of associations between clinical parameters and genetic alterations. The Cox proportional hazards model was implemented to examine the effect of different combinations of genetic alterations after adjusting for age at diagnosis (<40 years vs. ≥40 years) and sex (female vs. male). Factors with no significant association were eliminated (P ≥ 0.05). The remaining factors in the multivariate analysis (P < 0.05) were assumed to be independent predictors of survival. Kaplan–Meier survival statistics were used to compare survival curves in the different patient groups. The log‐rank test was used to compare the different survival curves. The figures were created using StatView software (SAS Institute Inc., Cary, NC, USA).
Results
Analysis of TCGA data
With the criterion log2 ratio of >0.5, we identified 25 genes that showed gain in >35% cases of glioblastomas with IDH1/2 mutations (8 cases) and those without IDH1/2 mutations (364 cases). Except for CFHR3 (located at 1q31.3), LCE3B (1q21.3), LCE3C (1q21.3), TARP (7p14.1), all the other 21 genes (ANKRD7, ASZ1, C7ORF58, CAPZA2, CFTR, CTTNBP2, ING3, KCND2, NAA38, MET, MGAM, PRSS2, PTPRZ1, ST7, ST7‐AS1, ST7‐AS2, ST7‐OT3, ST7‐OT4, TSPAN12, WNT16, WNT2) were located at 7q31‐34. The cBio Cancer Genomics Portal showed that 68% of glioblastomas showed gain of all 21 genes, and 71%–74% of glioblastomas showed gain in at least 1 of these 21 genes.
The MET gene at 7q31.2 was selected for further analysis, as it is a well‐characterized oncogene implicated in the pathogenesis and progression of a variety of tumors 2, 36. In the cBio Cancer Genomics Portal, MET gain was present in 72% of glioblastomas, and was associated with significantly shorter patient survival (P = 0.035). The Rembrandt database also showed MET gain in 133 of 181 (74%) glioblastomas, and in 58 of 98 (59%) WHO grade II/III astrocytomas.
Frequency of MET gain in glioma
We screened for MET gain in 112 diffuse astrocytomas, 82 oligodendrogliomas, 34 primary glioblastomas and 36 secondary glioblastomas with quantitative PCR. MET gain was detected at similar frequencies in primary (16/34; 47%) and secondary (16/36; 44%) glioblastomas, and in glioblastomas without and with IDH1 mutations (20/43; 46.5% vs. 12/27, 44.4%). MET gain was significantly more frequent in glioblastoma patients aged >40 years than in younger patients [29/54 (54%) vs. 3/16 (19%); P = 0.021]. In the majority of cases, the level of MET gain was low (2.7–5.0 copies), except for four secondary glioblastomas and one primary glioblastoma, which showed 11–21 MET copies.
MET gain was also common in diffuse astrocytomas (43/112; 38%), but less frequent in oligodendrogliomas (13/82; 16%; P = 0.001; Table 1). Gain of more than five copies of MET was found in two diffuse astrocytomas, but in none of the oligodendrogliomas. MET gain was significantly more frequent in low‐grade gliomas with TP53 mutations than in those without TP53 mutation (41% vs. 20%; P = 0.002). There was no significant difference in the frequency of MET gain in younger (<40 years) and older (>40 years) patients with diffuse astrocytoma (Fisher's exact test; P = 0.41).
Table 1.
Frequency of MET gain in glioma
| Tumor type | Tumors with MET gain (%) |
|---|---|
| Diffuse astrocytoma (WHO grade II) | 43/112 (38)** |
| Oligodendroglioma (WHO grade II) | 13/82 (16) |
| Primary glioblastoma (WHO grade IV) | 16/34 (47) |
| Secondary glioblastoma (WHO grade IV) | 16/36 (43) |
**Significantly more frequent than in oligodendrogliomas (P = 0.001).
Timing of MET gain during astrocytoma progression
Pairs of tumor tissues of diffuse astrocytoma and the subsequently developing secondary glioblastoma were available for 14 patients. In one patient, MET gain was present both in the first biopsy (diffuse astrocytoma) and the second biopsy (secondary glioblastoma), whereas in five patients, MET gain was observed only in the secondary glioblastoma.
MET gain and clinical outcome
The median overall survival of patients with diffuse astrocytoma showing MET gain (10 cases) was significantly shorter [log‐rank test; 43.0 months (95% CI, 42.5–43.5)] than that of patients without MET gain (53 cases) [70.7 months (95% CI, 55.0–86.4); P = 0.004] (Figure 1). In multivariate analysis after adjusting for patient age and sex, MET gain remained a significant prognostic factor for poorer survival among patients with diffuse astrocytoma [Cox‐Regression; HR 2.96 (95% CI, 1.43–6.15); P = 0.004]. MET gain did not affect the survival of patients with all glioblastomas combined (P = 0.39), primary glioblastomas (P = 0.89), secondary glioblastomas (P = 0.29) (Figure 2), glioblastomas with IDH1 mutations (P = 0.26) or those without IDH1 mutations (P = 0.9)
Figure 2.
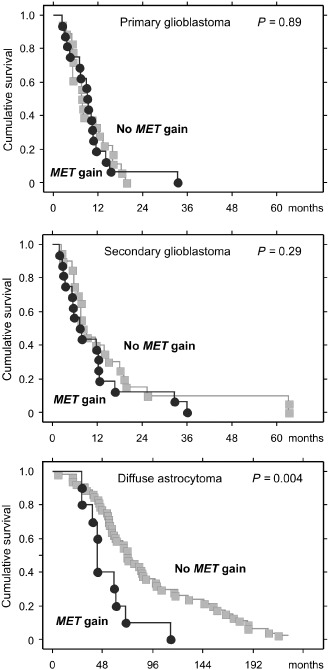
Cumulative survival of patients with diffuse astrocytoma is significantly shorter for tumors with MET gain than for tumors without MET gain (median overall survival, 43.0 months vs. 70.7 months; P = 0.004). There was no significant difference in patient survival for glioblastomas with or without MET gain.
Discussion
To identify genes that are commonly amplified in both primary and secondary glioblastomas, we first analyzed TCGA data, using IDH1 mutations as a molecular marker of secondary glioblastomas 32, 33. We found 25 genes that showed gene copy‐number gain in more than one‐third of glioblastomas with or without IDH1 mutations. Interestingly, most of these genes (21/25; 84%) were located at 7q31‐34, where gain has been observed frequently in a variety of human neoplasms, including gastric cancer (46%) 29 and fibrosarcoma (44%) 40. Gain at 7q was reported in 38%–79% of glioblastomas 4, 20 and in 21%–50% of low‐grade gliomas 13, 42. Gain at 7q was associated with a shorter survival of patients with high‐grade astrocytomas 20, 49.
We chose the MET gene at 7q31.2 for detailed analysis because this gene has been demonstrated to play important roles in pathogenesis and progression in a variety of human neoplasms 4, 7, 50. The MET gene encodes the receptor tyrosine kinase MET, comprising an α‐chain linked by a disulfide bridge to a β‐chain. The α‐chain is located extracellularly and carries the binding site for the substrate, whereas the β‐chain traverses the membrane and includes the cytoplasmic kinase domain and the carboxy‐terminal, which is essential for downstream signaling 36. The MET receptor is the only known target of hepatocyte growth factor (HGF) 2. Binding of HGF leads to MET dimerization and the subsequent autophophorylation of tyrosine residues at the carboxy‐terminal binding site. This is followed by the activation of downstream signaling cascades 9, including PI3K/Akt, Ras/MAPK and STAT pathways 5. MET signaling is therefore involved in a variety of cellular functions, such as cell proliferation, survival, apoptosis, invasion and angiogenesis 2. Dysregulation of the MET pathway may also play important roles during the epithelial–mesenchymal transition, tumor invasion, progression and metastasis 9.
Abnormal MET signaling may be caused by gene amplification, overexpression, missense mutations, translocations, and ligand‐dependent autocrine or paracrine mechanisms 44. MET missense mutations have been detected in a small fraction of papillary renal cell carcinomas (13%) 41, lung cancer (13%) 19 and mesothelioma (9%) 15, but rarely in gliomas (<2%) 1. MET overexpression has been observed in a variety of human neoplasms, including ovarian cancer (30%–67%) 26, 50, colorectal cancer (78%) 26, chordomas (77%) 47. Immunohistochemistry has revealed MET overexpression in glioblastomas (34%–88%) 11, 30 and low‐grade astrocytomas (21%) 30. In glioblastomas, MET expression was higher in recurrent tumors than in primary tumors, and glioblastoma patients with MET overexpression had a significantly shorter progression‐free survival time (6.1 months vs. 11.5 months) 23. However, little is known about MET gain in glioma.
We show here that MET gain is common in both primary and secondary glioblastomas (47% vs. 44%), indicating that abnormal MET signaling is involved in the pathogenesis of both glioblastoma subtypes. This was consistent with the TCGA data, in which 41% (151/372) of glioblastomas showed low‐level amplification determined by the criterion of a log2 ratio > 0.5. It is of interest to note that MET gain was more frequent in patients aged ≥40 years (29/54; 54%) than younger patients with glioblastoma (3/16; 19%; P = 0.021). This is consistent with previous findings that 7q gain was associated with older age in patients with astrocytic tumors 4, 20.
Diffuse astrocytomas tend to progress to a more malignant histologic type, that is, anaplastic astrocytomas and eventually secondary glioblastomas 13. However, time until progression varies significantly between patients 18, 38, 48. Several clinical prognostic factors for poorer survival of patients with diffuse astrocytoma have been reported, including old age at diagnosis 35, 43, incomplete resection 37, 46 and predominant presence of gemistocytes 35. The genetic hallmark of diffuse astrocytomas is co‐presence of IDH1 mutations and TP53 mutations 17. Several studies have shown that IDH1 mutations are a significant prognostic marker for favorable outcome of patients with diffuse astrocytoma 6, 12, 39. In contrast, the prognostic value of TP53 mutations has been controversial. In a study of astrocytomas and oligoastrocytomas (n = 159), cumulative progression‐free survival was significantly shorter for patients with tumors with TP53 mutation, but there was no impact on overall survival 38. Other studies have shown that TP53 mutations were marginally associated 14 or not associated 35 with poorer patient outcome. In the present study, we show that MET gain was also common (38%) in diffuse astrocytomas, and importantly, the presence of MET gain was significantly associated with poor clinical outcome.
MET gain and MET overexpression are associated with higher tumor grades of ovarian clear cell adenocarcinomas 50. In gastric cancer, MET expression correlates with advanced tumor stage with liver metastasis 3. MET overexpression was significantly associated with rapid tumor recurrence in patients with non‐small cell lung carcinoma (NSCLC) 8. In astrocytomas, MET expression levels increased with higher WHO grades 2, 28, 30. In the present study, biopsies with diffuse astrocytoma and secondary glioblastoma were available for 14 patients. In one patient, MET gain was found in both the biopsies with diffuse astrocytoma and secondary glioblastoma, whereas in five cases, MET gain was observed only in secondary glioblastoma. Thus, MET gain is associated with astrocytoma progression, and is usually a late event; but if present in diffuse astrocytomas, this predicts poorer clinical outcome of patients.
The level of MET gain detected in the present study was relatively low (three to five copies) in the majority of gliomas. However, low‐level gain may also have biologically significant effects. MET gain (four to five copies), detected by quantitative PCR and fluorescence in situ hybridization (FISH) in 10%–21% of gastric cancer, was significantly associated with a poorer prognosis 10, 22. In patients with NSCLC with an increased copy‐number (more than five copies) of MET detected by FISH, survival was significantly shorter than in patients without MET gain 7.
The targeting of receptor tyrosine kinases is a promising therapeutic strategy for glioblastomas. MET inhibitors are emerging drugs for the treatment of different cancers and are currently undergoing phase I, II and III clinical trials in multiple types of tumors 44. MET monoclonal antibody inhibits MET phosphorylation, cell proliferation, migration and apoptosis in U87 glioblastoma cells 27. As MET activation is involved in the development of resistance to epidermal growth factor receptor (EGFR) inhibitors in glioblastoma 16, and inhibition of the MET pathway can overcome resistance to EGFR pathway inhibition in human EGFR‐hyperactivated glioblastoma xenografts 21, combined treatment with EGFR inhibitors and MET inhibitors may be a promising additional treatment strategy. The present study provides evidence that both primary and secondary glioblastomas, as well as a fraction of diffuse astrocytomas with MET gain, may benefit from such treatment.
Acknowledgments
Daniela Pierscianek was supported by the Fritz‐Thyssen Foundation.
References
- 1. Cancer Genome Atlas Research Network (2008) Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 455:1061–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Abounader R, Laterra J (2005) Scatter factor/hepatocyte growth factor in brain tumor growth and angiogenesis. Neuro Oncol 7:436–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Amemiya H, Kono K, Itakura J, Tang RF, Takahashi A, An FQ et al (2002) c‐Met expression in gastric cancer with liver metastasis. Oncology 63:286–296. [DOI] [PubMed] [Google Scholar]
- 4. Arslantas A, Artan S, Oner U, Muslumanoglu MH, Ozdemir M, Durmaz R et al (2007) Genomic alterations in low‐grade, anaplastic astrocytomas and glioblastomas. Pathol Oncol Res 13:39–46. [DOI] [PubMed] [Google Scholar]
- 5. Birchmeier C, Birchmeier W, Gherardi E, Vande Woude GF (2003) Met, metastasis, motility and more. Nat Rev Mol Cell Biol 4:915–925. [DOI] [PubMed] [Google Scholar]
- 6. Bourne TD, Schiff D (2010) Update on molecular findings, management and outcome in low‐grade gliomas. Nat Rev Neurol 6:695–701. [DOI] [PubMed] [Google Scholar]
- 7. Cappuzzo F, Marchetti A, Skokan M, Rossi E, Gajapathy S, Felicioni L et al (2009) Increased MET gene copy number negatively affects survival of surgically resected non‐small‐cell lung cancer patients. J Clin Oncol 27:1667–1674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cheng TL, Chang MY, Huang SY, Sheu CC, Kao EL, Cheng YJ, Chong IW (2005) Overexpression of circulating c‐met messenger RNA is significantly correlated with nodal stage and early recurrence in non‐small cell lung cancer. Chest 128:1453–1460. [DOI] [PubMed] [Google Scholar]
- 9. Gherardi E, Birchmeier W, Birchmeier C, Vande WG (2012) Targeting MET in cancer: rationale and progress. Nat Rev Cancer 12:89–103. [DOI] [PubMed] [Google Scholar]
- 10. Graziano F, Galluccio N, Lorenzini P, Ruzzo A, Canestrari E, D'Emidio S et al (2011) Genetic activation of the MET pathway and prognosis of patients with high‐risk, radically resected gastric cancer. J Clin Oncol 29:4789–4795. [DOI] [PubMed] [Google Scholar]
- 11. Hirose Y, Kojima M, Sagoh M, Murakami H, Yoshida K, Shimazaki K, Kawase T (1998) Immunohistochemical examination of c‐Met protein expression in astrocytic tumors. Acta Neuropathol 95:345–351. [DOI] [PubMed] [Google Scholar]
- 12. Houillier C, Wang X, Kaloshi G, Mokhtari K, Guillevin R, Laffaire J et al (2010) IDH1 or IDH2 mutations predict longer survival and response to temozolomide in low‐grade gliomas. Neurology 75:1560–1566. [DOI] [PubMed] [Google Scholar]
- 13. Idbaih A, Carvalho SR, Criniere E, Marie Y, Carpentier C, Boisselier B et al (2008) Genomic changes in progression of low‐grade gliomas. J Neurooncol 90:133–140. [DOI] [PubMed] [Google Scholar]
- 14. Ishii N, Tada M, Hamou MF, Janzer RC, Meagher‐Villemure K, Wiestler OD et al (1999) Cells with TP53 mutations in low grade astrocytic tumors evolve clonally to malignancy and are an unfavorable prognostic factor. Oncogene 18:5870–5878. [DOI] [PubMed] [Google Scholar]
- 15. Jagadeeswaran R, Ma PC, Seiwert TY, Jagadeeswaran S, Zumba O, Nallasura V et al (2006) Functional analysis of c‐Met/hepatocyte growth factor pathway in malignant pleural mesothelioma. Cancer Res 66:352–361. [DOI] [PubMed] [Google Scholar]
- 16. Jun HJ, Acquaviva J, Chi D, Lessard J, Zhu H, Woolfenden S et al (2011) Acquired MET expression confers resistance to EGFR inhibition in a mouse model of glioblastoma multiforme. Oncogene, in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kim YH, Nobusawa S, Mittelbronn M, Paulus W, Brokinkel B, Keyvani K et al (2010) Molecular classification of low‐grade diffuse gliomas. Am J Pathol 177:2708–2714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kraus JA, Bolln C, Wolf HK, Neumann J, Kindermann D, Fimmers R et al (1994) TP53 alterations and clinical outcome in low grade astrocytomas. Genes Chromosomes Cancer 10:143–149. [DOI] [PubMed] [Google Scholar]
- 19. Krishnaswamy S, Kanteti R, Duke‐Cohan JS, Loganathan S, Liu W, Ma PC et al (2009) Ethnic differences and functional analysis of MET mutations in lung cancer. Clin Cancer Res 15:5714–5723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kunwar S, Mohapatra G, Bollen A, Lamborn KR, Prados M, Feuerstein BG (2001) Genetic subgroups of anaplastic astrocytomas correlate with patient age and survival. Cancer Res 61:7683–7688. [PubMed] [Google Scholar]
- 21. Lal B, Goodwin CR, Sang Y, Foss CA, Cornet K, Muzamil S et al (2009) EGFRvIII and c‐Met pathway inhibitors synergize against PTEN‐null/EGFRvIII+ glioblastoma xenografts. Mol Cancer Ther 8:1751–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lee J, Seo JW, Jun HJ, Ki CS, Park SH, Park YS et al (2011) Impact of MET amplification on gastric cancer: possible roles as a novel prognostic marker and a potential therapeutic target. Oncol Rep 25:1517–1524. [DOI] [PubMed] [Google Scholar]
- 23. Liu W, Fu Y, Xu S, Ding F, Zhao G, Zhang K et al (2011) c‐Met expression is associated with time to recurrence in patients with glioblastoma multiforme. J Clin Neurosci 18:119–121. [DOI] [PubMed] [Google Scholar]
- 24. Lo YM, Tein MS, Lau TK, Haines CJ, Leung TN, Poon PM et al (1998) Quantitative analysis of fetal DNA in maternal plasma and serum: implications for noninvasive prenatal diagnosis. Am J Hum Genet 62:768–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Louis DN, Ohgaki H, Wiestler OD, Cavenee WK (eds) (2007) WHO Classification of Tumours of the Central Nervous System. IARC: Lyon. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ma PC, Tretiakova MS, MacKinnon AC, Ramnath N, Johnson C, Dietrich S et al (2008) Expression and mutational analysis of MET in human solid cancers. Genes Chromosomes Cancer 47:1025–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Martens T, Schmidt NO, Eckerich C, Fillbrandt R, Merchant M, Schwall R et al (2006) A novel one‐armed anti‐c‐Met antibody inhibits glioblastoma growth in vivo. Clin Cancer Res 12:6144–6152. [DOI] [PubMed] [Google Scholar]
- 28. Moriyama T, Kataoka H, Koono M, Wakisaka S (1999) Expression of hepatocyte growth factor/scatter factor and its receptor c‐Met in brain tumors: evidence for a role in progression of astrocytic tumors (Review). Int J Mol Med 3:531–536. [DOI] [PubMed] [Google Scholar]
- 29. Morohara K, Nakao K, Tajima Y, Nishino N, Yamazaki K, Kaetsu T et al (2005) Analysis by comparative genomic hybridization of gastric cancer with peritoneal dissemination and/or positive peritoneal cytology. Cancer Genet Cytogenet 161:57–62. [DOI] [PubMed] [Google Scholar]
- 30. Nabeshima K, Shimao Y, Sato S, Kataoka H, Moriyama T, Kawano H et al (1997) Expression of c‐Met correlates with grade of malignancy in human astrocytic tumours: an immunohistochemical study. Histopathology 31:436–443. [DOI] [PubMed] [Google Scholar]
- 31. Nigro JM, Takahashi MA, Ginzinger DG, Law M, Passe S, Jenkins RB, Aldape K (2001) Detection of 1p and 19q loss in oligodendroglioma by quantitative microsatellite analysis, a real‐time quantitative polymerase chain reaction assay. Am J Pathol 158:1253–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Nobusawa S, Watanabe T, Kleihues P, Ohgaki H (2009) IDH1 mutations as molecular signature and predictive factor of secondary glioblastomas. Clin Cancer Res 15:6002–6007. [DOI] [PubMed] [Google Scholar]
- 33. Ohgaki H, Kleihues P (2011) Genetic profile of astrocytic and oligodendroglial gliomas. Brain Tumor Pathol 28:177–183. [DOI] [PubMed] [Google Scholar]
- 34. Ohgaki H, Dessen P, Jourde B, Horstmann S, Nishikawa T, Di Patre PL et al (2004) Genetic pathways to glioblastoma: a population‐based study. Cancer Res 64:6892–6899. [DOI] [PubMed] [Google Scholar]
- 35. Okamoto Y, Di Patre PL, Burkhard C, Horstmann S, Jourde B, Fahey M et al (2004) Population‐based study on incidence, survival rates, and genetic alterations of low‐grade astrocytomas and oligodendrogliomas. Acta Neuropathol 108:49–56. [DOI] [PubMed] [Google Scholar]
- 36. Organ SL, Tsao MS (2011) An overview of the c‐MET signaling pathway. Ther Adv Med Oncol 3:S7–S19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Peraud A, Ansari H, Bise K, Reulen HJ (1998) Clinical outcome of supratentorial astrocytoma WHO grade II. Acta Neurochir (Wien) 140:1213–1222. [DOI] [PubMed] [Google Scholar]
- 38. Peraud A, Kreth FW, Wiestler OD, Kleihues P, Reulen HJ (2002) Prognostic impact of TP53 mutations and P53 protein overexpression in supratentorial WHO Grade II astrocytomas and oligoastrocytomas. Clin Cancer Res 8:1117–1124. [PubMed] [Google Scholar]
- 39. Sanson M, Marie Y, Paris S, Idbaih A, Laffaire J, Ducray F et al (2009) Isocitrate dehydrogenase 1 codon 132 mutation is an important prognostic biomarker in gliomas. J Clin Oncol 27:4150–4154. [DOI] [PubMed] [Google Scholar]
- 40. Schmidt H, Taubert H, Wurl P, Kappler M, Lange H, Bartel F et al (2002) Gains of 12q are the most frequent genomic imbalances in adult fibrosarcoma and are correlated with a poor outcome. Genes Chromosomes Cancer 34:69–77. [DOI] [PubMed] [Google Scholar]
- 41. Schmidt L, Junker K, Nakaigawa N, Kinjerski T, Weirich G, Miller M et al (1999) Novel mutations of the MET proto‐oncogene in papillary renal carcinomas. Oncogene 18:2343–2350. [DOI] [PubMed] [Google Scholar]
- 42. Schrock E, Blume C, Meffert MC, du MS, Bersch W, Kiessling M et al (1996) Recurrent gain of chromosome arm 7q in low‐grade astrocytic tumors studied by comparative genomic hybridization. Genes Chromosomes Cancer 15:199–205. [DOI] [PubMed] [Google Scholar]
- 43. Shafqat S, Hedley Whyte ET, Henson JW (1999) Age‐dependent rate of anaplastic transformation in low‐grade astrocytoma. Neurology 52:867–869. [DOI] [PubMed] [Google Scholar]
- 44. Sharma N, Adjei AA (2011) In the clinic: ongoing clinical trials evaluating c‐MET‐inhibiting drugs. Ther Adv Med Oncol 3:S37–S50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Thon N, Eigenbrod S, Kreth S, Lutz J, Tonn JC, Kretzschmar H et al (2012) IDH1 mutations in grade II astrocytomas are associated with unfavorable progression‐free survival and prolonged postrecurrence survival. Cancer 118:452–460. [DOI] [PubMed] [Google Scholar]
- 46. van Veelen ML, Avezaat CJ, Kros JM, van Putten W, Vecht C (1998) Supratentorial low grade astrocytoma: prognostic factors, dedifferentiation, and the issue of early versus late surgery. J Neurol Neurosurg Psychiatry 64:581–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Walter BA, Begnami M, Valera VA, Santi M, Rushing EJ, Quezado M (2011) Gain of chromosome 7 by chromogenic in situ hybridization (CISH) in chordomas is correlated to c‐MET expression. J Neurooncol 101:199–206. [DOI] [PubMed] [Google Scholar]
- 48. Watanabe K, Sato K, Biernat W, Tachibana O, von Ammon K, Ogata N et al (1997) Incidence and timing of p53 mutations during astrocytoma progression in patients with multiple biopsies. Clin Cancer Res 3:523–530. [PubMed] [Google Scholar]
- 49. Wiltshire RN, Herndon JE, Lloyd A, Friedman HS, Bigner DD, Bigner SH, McLendon RE (2004) Comparative genomic hybridization analysis of astrocytomas: prognostic and diagnostic implications. J Mol Diagn 6:166–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Yamamoto S, Tsuda H, Miyai K, Takano M, Tamai S, Matsubara O (2012) Accumulative copy number increase of MET drives tumor development and histological progression in a subset of ovarian clear‐cell adenocarcinomas. Mod Pathol 25:122–130. [DOI] [PubMed] [Google Scholar]
- 51. Yan H, Parsons DW, Jin G, McLendon R, Rasheed BA, Yuan W et al (2009) IDH1 and IDH2 mutations in gliomas. N Engl J Med 360:765–773. [DOI] [PMC free article] [PubMed] [Google Scholar]