

Abstract
In multiple sclerosis, cerebellar symptoms are associated with clinical impairment and an increased likelihood of progressive course. Cortical atrophy and synaptic dysfunction play a prominent role in cerebellar pathology and although the dentate nucleus is a predilection site for lesion development, structural synaptic changes in this region remain largely unexplored. Moreover, the mechanisms leading to synaptic dysfunction have not yet been investigated at an ultrastructural level in multiple sclerosis. Here, we report on synaptic changes of dentate nuclei in post‐mortem cerebella of 16 multiple sclerosis patients and eight controls at the histological level as well as an electron microscopy evaluation of afferent synapses of the cerebellar dentate and pontine nuclei of one multiple sclerosis patient and one control. We found a significant reduction of afferent dentate synapses in multiple sclerosis, irrespective of the presence of demyelination, and a close relationship between glial processes and dentate synapses. Ultrastructurally, we show autophagosomes containing degradation products of synaptic vesicles within dendrites, residual bodies within intact‐appearing axons and free postsynaptic densities opposed to astrocytic appendages. Our study demonstrates loss of dentate afferent synapses and provides, for the first time, ultrastructural evidence pointing towards neuron‐autonomous and neuroglia‐mediated mechanisms of synaptic degradation in chronic multiple sclerosis.
Keywords: autophagy, cerebellum, dentate nucleus, multiple sclerosis, synapses
INTRODUCTION
The cerebellum is a predilection site for lesion development in multiple sclerosis (MS) and an important determinant of disability 29. The presence of cerebellar symptoms is associated with a progressive disease course 14, and cerebellar cortical atrophy and diffuse pathology 7, 10 are independent contributors to cognitive and motor impairment 4, 31. These structural changes are further accompanied by functional alterations in cerebellar connectivity, implicating early synaptic dysfunction 5, 20.
So far, few histopathological studies have explored the extent of neuronal, axonal and neurite loss in the main neuronal populations of the cerebellum. In areas of cerebellar cortical demyelination, but not in the non‐demyelinated MS cortex, a reduction in the number of Purkinje cells has consistently been reported 7, 10, 19. Regardless of the reduction in Purkinje cells no differences in synaptic density in the granular cell layer have been observed 7, 10. Despite the preferential localization of demyelinated lesions to the dentate nucleus 12, the main relay station of the cerebellum, the extent of neuronal and synaptic loss in this region has not yet been characterized.
Synaptic loss has been demonstrated in the forebrain 8, 16, 30 and hippocampus 6 of MS patients, and an association between glial processes and synapses which might contribute to synaptic reduction has been described 13. In animal models of chronic neurodegeneration, a close relationship between activated microglia and synapses has also been observed, yet ultrastructural analyses indicate that synaptic degeneration occurs primarily via neuron‐autonomous mechanisms 15, a possibility that has thus far not been explored in MS. Here, we determine the extent of synaptic loss in the dentate nucleus, the main relay center in the cerebellum, in chronic MS and provide for the first time ultrastructural evidence of synaptic loss and dissociation as well as lysosomal degradation of synaptic constituents in one MS patient. Our results provide evidence suggesting that at least two different mechanisms, a glia‐mediated and a neuron‐autonomous process, are associated with synaptic loss in MS.
MATERIALS AND METHODS
Patients and tissue samples
For light microscopy studies, formalin‐fixed and paraffin‐embedded CNS tissue blocks were obtained from the archives of the Institute of Neurology, McGill University, Montreal, Canada and the Institute of Neuropathology, University Medical Center, Göttingen (UMG), Germany, in agreement with the ethics committee of the UMG and the Montreal Neurological Institute. Blocks containing the cerebellar dentate nucleus were sampled from 16 MS patients and eight age‐matched controls without neurological disease or evidence of neuropathology at autopsy. The MS patients were divided in two groups based on the presence (MS‐D, n = 7) or absence (MS‐ND, n = 9) of demyelinated plaques in the dentate nucleus. Clinical information on the MS patients studied can be found in Table 1. All lesions were classified as inactive lesions according to the criteria of Brück and co‐workers 2.
Table 1.
Clinicopathological characteristics of MS patients.
| Case | Age/Sex | Disease course | Disease duration* | Cause of death | Symptoms | Location of demyelinated lesions at autopsy | Pontine, midbrain or thalamic lesions |
Demyelination Dentate Ncl. |
|---|---|---|---|---|---|---|---|---|
| 1 | 57/M | SPMS | 12 | Congestive heart failure; pancreatic adenocarcinoma | Diplopia; bilateral nuclear ophthalmoplegia; ataxia; tremor and right‐sided hemiparesis | Large periventricular lesions | Few perivascular plaques at the junction of thalamus and internal capsule; demyelination of medial longitudinal fasciculus, the lateral aspects of tegmental structures and partial demyelination of pyramidal tracts | Yes |
| 2 | 39/F | SPMS | N/A | Pneumonia | N/A | Multiple bilateral WM lesions | Diffuse pontine myelin loss; no thalamic or midbrain lesions | Yes |
| 3 | 52/F | PPMS | 8 | Urinary tract infection; sepsis; cardiopulmonary arrest | Progressive loss of right‐sided vision; left‐sided ataxia and bilateral paresthesias; subsequent blindness and tetraparesis | Multiple lesions in the brain, brainstem, spinal cord and cerebellum | Extensive myelin loss in thalamus; periaqueductal demyelination and between tegmentum and base of the pons | Yes |
| 4 | 54/M | SPMS | 11 | Metastatic squamous cell carcinoma of the tonsil | Retrobulbar neuritis; left hemiparesis | Multiple bilateral plaques in the brain; lesions in the brain stem and cerebellum | Demyelination of left inferior peduncle and spinothalamic tract; no thalamic or midbrain lesions | Yes |
| 5 | 57/F | SPMS | 9 | N/A | N/A | Multiple periventricular frontal, parietal and parieto‐occipital WM lesions | Small bilateral demyelinated lesions in thalamus; no lesions in midbrain or pons | Yes |
| 6 | 62/M | N/A | N/A | Myocardial infarction; pneumonia | N/A | Frontal and periventricular WM lesions; involvement of cerebellum, spinal cord, hippocampus and brainstem | Multiple lesions in thalamus, pons and midbrain | Yes |
| 7 | 29/M | Acute MS | N/A | Malignant brain edema after car accident | Tetraparesis; respiratory insufficiency | Multiple plaques in frontal and temporal WM | Bilateral thalamic lesions; inflammatory perivascular infiltrates in the cerebellar peduncles and pons | Yes |
| 8 | 69/M | SPMS | 7 | Lung fibrosis; granulomatous bronchopneumonia | Left facial pain; progressive right‐sided sensorimotor hemiparesis | Multiple lesions in both cerebral hemispheres and brainstem | Demyelination of the inferior portion of the left internal capsule; lesions in the left corticospinal and corticobulbar tracts; no pontine lesions | No |
| 9 | 49/F | Acute MS | <1 | Pneumonia | Burning dysesthesia in the right auricular and occipital region and the left arm; weakness of the left leg and rapid progression to tetraparesis | Large confluent lesions in spinal cord and medulla oblongata | No demyelinating lesions in thalamus, midbrain or pons | No |
| 10 | 57/M | PPMS | 23 | Sudden death | Progressive paraparesis; autonomic symptoms; nystagmus; ataxia | Two demyelinating lesions in thalamus and pons | Right cerebral peduncle and inferior internal capsule; left basis pontis | No |
| 11 | 60/M | PPMS | 26 | Cardiac arrest | Slowly progressive right‐sided spastic hemiparesis | Mostly periventricular lesions; involvement of the left internal capsule | No demyelinating lesions in thalamus, midbrain or pons | No |
| 12 | 51/M | PPMS | 8 | Pulmonary embolism after cervical laminectomy | Progressive sensorimotor spastic tetraparesis and autonomic dysfunction | Scattered brain lesions; severe spinal cord involvement | Demyelination around inferior colliculus and posterior portion of the right brachium conjunctivum; no thalamic lesions | No |
| 13 | 59/F | RRMS | 5 | Metastatic breast cancer | Optic neuritis and bilateral optic nerve atrophy; paresthesias and reduced proprioception in both feet | Long standing lesions of both optic nerves | No demyelinating lesions in thalamus, midbrain or pons | No |
| 14 | 66/F | PPMS | 30 | Sudden death through cardiac arrest | Severe tetraparesis and dysarthria | Small frontal WM lesions; extensive spinal cord involvement | Slight myelin loss in the midbrain; no pontine or thalamic lesions | No |
| 15 | N/A | N/A | N/A | N/A | N/A | N/A | N/A | No |
| 16 | 36/M | PPMS | 3 | Pneumonia; pulmonary embolism | Progressive sensorimotor tetraparesis | Multiple spinal cord and bilateral WM brain lesions | Bilateral cerebral peduncles and left medial lemniscus; periventricular lesions; left cerebellar peduncle | No |
M: Male. F: Female; N/A: Not available. MS: Multiple sclerosis. PPMS: Primary progressive multiple sclerosis. RRMS: Relapsing remitting multiple sclerosis. SPMS: Secondary progressive multiple sclerosis. WM: White matter.
*Disease duration in years.
Suitably fixed and embedded tissue for electron microscopy was available from two previously published cases: One 39 year‐old woman with a 14‐year history of MS and an initially relapsing‐remitting disease course which developed to a progressive course three years before death, and a 55 year‐old man with no history of neurological disease 17.
Histological techniques, image acquisition and processing
For light microscopy studies, paraffin‐embedded tissue sections with a thickness of 2‐3 µm were deparaffinized, pretreated and stained with haematoxylin‐eosin (HE), Luxol Fast Blue/periodic‐acid Schiff (LFB/PAS) or immunohistochemically as described previously 26. Antibody specifications can be found in Supporting Information Table S1.
Color microphotographs of tissue sections were acquired with a XM10 camera (Olympus, Germany) mounted on a BX51 light microscope (Olympus, Germany) using a 100x oil‐immersion objective and the cellSense Dimension 1.7.1. software (Olympus, Germany). An inverted brightfield microscope (BZ‐9000E, KEYENCE Deutschland, Germany) and the BZ‐II Analyzer software (KEYENCE Deutschland, Germany) were used for magnifications of 100x or 200x. Individual images were mosaic‐merged with the BZ‐II Viewer software v2.2 (KEYENCE Deutschland, Germany). For confocal microscopy images, fluorescence signals were collected with an LSM 510 Meta confocal microscope (Carl Zeiss, Germany) and 40x or 63x oil immersion objectives using the Zen software (Carl Zeiss, Germany).
Post‐acquisition image processing was done using the image analysis software FIJI 24. The number of axosomatic synapses was determined by counting synaptophysin‐positive structures at the soma membrane of 5 adjacent neurons of the dentate nucleus in demyelinated areas (n = 9), in MS patients without demyelination of the dentate gyrus (n = 7) and in healthy controls (n = 8). Neurons were morphologically defined as large cells containing abundant synaptophysin‐negative cytoplasm, and a pale, spherical nucleus in hematoxylin staining. When present, one or more clearly identifiable nucleoli served as additional criterion. Only neurons located in the center of the grey matter band of the dentate nucleus were included. The number of dentate neurons was determined by manually counting neuronal somata in the entire length of the dentate nucleus. The length of the dentate nucleus was digitally traced and measured and the results are reported as cell counts per mm length. The area of individual neuronal somata was determined manually. At least 23 neurons per patient were analyzed.
The Purkinje cell density was determined by manually counting the number of neuronal somata in the cerebellar cortex located superior to the dentate nucleus, corresponding to the anterior quadrangular lobule. The number of cells per field of view in ten fields was quantified (magnification: 400×). The presence of cerebellar cortical demyelination was determined by immunohistochemistry using antibodies against myelin basic protein (MBP) and proteolipid protein (PLP), and the size of cortical demyelinated lesions was qualitatively defined as small, large or confluent. Granule and Purkinje cell layers and the cerebellar cortex located superior to the dentate nucleus, used for Purkinje cell quantification, were assessed separately.
For the percentage of area, the DAB signal of GFAP, Iba1 or KiM1P images of the grey matter band of the dentate nucleus was segmented using the color deconvolution plugin 23 and quantified using the particle analyzer function. Post acquisition processing of confocal images was done with the Imaris software (Imaris x64 7.4.0, Bitplane, USA).
Electron microscopy
Electron microscopy studies were performed on ultrathin sections of CNS tissue. Samples were taken from the dentate nucleus and paramedian parts of the pontine nuclei. Epon blocks of the MS patient were selected to contain both demyelinated and myelinated regions. All images were captured with a transmission electron microscope (EM 10C, Zeiss) using a Mega View III digital camera (Olympus) and Analysis software (Soft Imaging System, Münster, Germany).
Statistical analyses
Data were analyzed using Graph Pad Prism (GraphPad Software, La Jolla, CA, USA) or R software 18. Student's t‐test, one‐way ANOVA with post‐hoc Tukey/Dunnett or Kruskal‐Wallis (KS) with Dunn's multiple comparisons tests were used where appropriate. Data represent mean ± standard error of the mean (SEM) unless otherwise stated. Asterisks correspond to P‐values of < 0.05 (*), <0.01 (**) and <0.001 (***).
RESULTS
Dentate nucleus neurons and synapses are reduced in multiple sclerosis
We determined the number and density of synaptic dentate afferents by comparing the number of axosomatic synaptic boutons in dentate neurons from MS patients with (MS‐D, n = 7) or without (MS‐ND, n = 9) demyelination of the dentate nucleus and in age‐matched controls (n = 8) as described in the methods section (Figure 1A). The overall mean number of synaptophysin‐positive axosomatic structures in the dentate nucleus of MS patients (11.1 ± 0.3, n = 16) was significantly reduced as compared to controls (19.5 ± 0.5, n = 8) (t 22 = 4.255, P = 0.0003). A significant difference in mean synapse number per neuron was seen for the three groups (F(2,21) = 8.6, P = 0.0018), and the mean synapse number was significantly reduced in both demyelinated (D: 11.9 ± 2.0, n = 7) and non‐demyelinated (ND: 11.0 ± 2.4, n = 9) dentate nuclei of MS patients compared to controls (19.1 ± 1.9, n = 8) (P < 0.01, Tukey post hoc test). However, the decrease was not more pronounced in the demyelinated lesions (P > 0.05, Tukey post hoc test), indicating that local demyelination does not directly influence synapse reduction in the dentate nucleus. Also, the number of dentate neurons differed among groups, with a significant reduction in MS cases with demyelination compared to controls (F(2,22) = 4.630, P = 0.021; P < 0.05, Bonferroni's multiple comparison test). Morphologically, dentate neurons in demyelinated areas appeared shrunken and hyperchromatic (Figure 1B), and although an increased frequency of neurons with smaller area were found in MS‐D, these differences failed to reach statistical significance (control: 496.8 ± 76.9 µm, n = 8; MS: 414.4 ± 38.6 µm, n = 16; t 22=1.076, P = 0.29) (Supporting information Figure S2). Analogous to synapse number, mean synaptic density in MS was lower than in controls (F(2,21) = 5.604, P = 0.01) irrespective of focal cerebellar demyelination (synapses/µm2 neuron: 0.03 ± 0.004, n = 9 (ND); 0.032 ± 0.004, n = 7 (D); P > 0.05, Tukey post hoc test) (Figure 1B).
Figure 1.
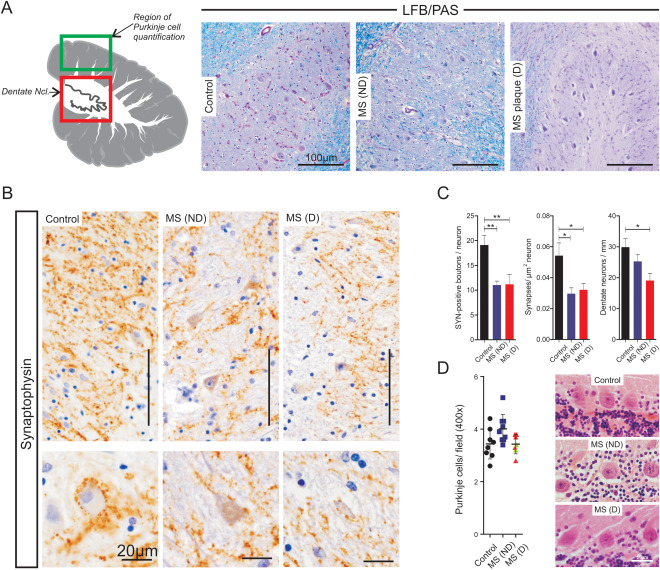
Reduction of axosomatic synaptic terminals in the dentate nucleus of multiple sclerosis patients. A. Schematic representation of the cerebellum (left) showing the regions analyzed (red square: dentate nucleus, green square: Purkinje cell layer in the anterior quadrangular lobe). Representative microphotographs of Luxol Fast Blue/periodic acid‐Schiff (LFB/PAS) stained dentate nuclei of control (left panel) and multiple sclerosis patients with (MS‐D, right panel) or without (MS‐ND, middle panel) demyelination. B. Synaptophysin immunohistochemistry (IHC) reveals an evenly distributed staining of the neuropil in control subjects (left panel), MS‐ND (middle panel) and MS‐D (right panel). Individual synaptic terminals can be seen surrounding neuronal somata (bottom inset). C. Quantification of the total number (left panel) and density (middle panel) of axosomatic synapses showing a significant decrease in multiple sclerosis (ND: blue; D: red) compared to controls (black). Significant reduction in the number of dentate neurons in MS (D) as compared to controls (right panel) D. Representative microphotographs (right panel) and quantification (left panel) of Purkinje cells revealed no significant differences between control subjects and multiple sclerosis irrespective of demyelination. Bars represent mean values ± standard error of the mean. Points represent averages of single cases. Points in green represent cases with cortical demyelination in the quantification area. Asterisks indicate P < 0.05 (*) and P < 0.001 (**). Unless explicitly stated otherwise, scale bars represent 100 µm.
The mean number of Purkinje cells was similar in both the control (3.44 ± 0.2, n = 8) and MS (ND: 4.0 ± 0.18, n = 9; D: 3.4 ± 0.14, n = 7) cohort, irrespective of demyelination in the dentate nucleus (H(2)= 5.6, P = 0.0617), suggesting that Purkinje cell loss is not the major factor contributing to synaptic loss in the dentate nucleus in MS.
Since synapse reduction has been associated with glial activation in the cerebral cortex and hippocampus in MS 13, 16, we evaluated the extent of microglial activation and astrocyte reactivity in the dentate nucleus in our cohort. In both MS and controls we observed reactive astrocytes and activated microglia among the neurons of the dentate nucleus (Figure 2A,B). Morphologically, in the cases with demyelination the reactive gliosis seemed more pronounced (Figure 2A, left panel), yet the mean percentage of total glial fibrillary acidic protein (GFAP)‐positive area did not reveal significant differences between the groups (H(2)= 5.5, P = 0.06407) (Figure 2B). Furthermore, the percentage of area of the dentate nucleus occupied by macrophages/activated microglia (KiM1P, Iba‐1) did not differ between controls and MS, regardless of the myelination status (KiM1P: H(2):4.56, P = 0.1; Iba‐1: H(2)=2.6, P = 0.27) (Figure 2B).
Figure 2.
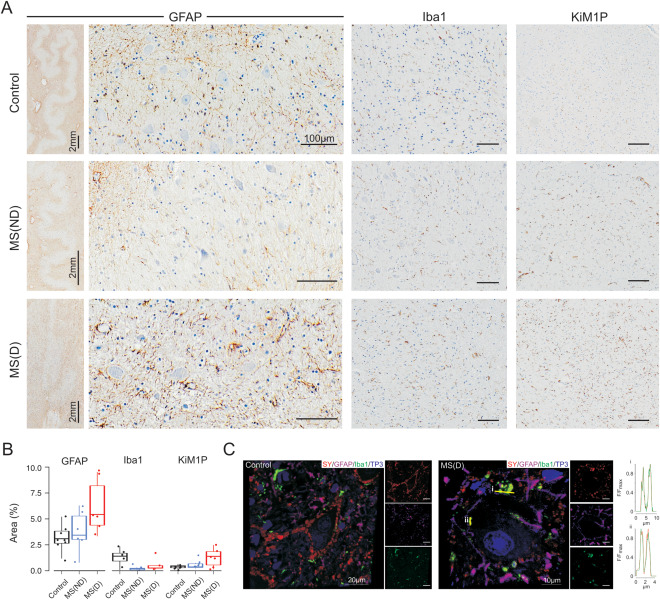
Reactive astrocytes and microglia in the dentate nucleus. A. GFAP‐IHC of the dentate nucleus of control (upper row, left panel), multiple sclerosis without demyelination (MS‐ND, middle row, left panel) and multiple sclerosis with demyelination (MS‐D, bottom row, left panel) showing the presence of reactive astrocytes around the dentate nucleus neurons. Note the higher density of reactive gliosis in the tissue surrounding the neuron band (left inset). Iba‐1‐ (middle panel) and KiM1P‐IHC (right panel) showing a discrete macrophage/microglial activation (KiM1P) in control (top row) and MS (middle and bottom rows) and no significant differences in the relative area occupied by microglia (Iba1). B. Box plots and individual data points of the mean percentage of area occupied by GFAP (left panel), Iba1 (middle panel) and KiM1P (right panel) showing no significant differences between the control (black), MS‐ND (blue) and MS‐D (red) cohorts. C. Confocal microscopy images of dentate nucleus neurons immunofluorescently labeled with synaptophysin (SY, red), GFAP (purple), Iba1 (green) and ToPro‐3 as a nuclear staining (TP3, blue) in control (left) and MS‐D (right). Note the reduced number of synapses in MS and the colocalization between SY and Iba1 expressed as a normalized fluorescence (F) line intensity profile of two selected regions (i and ii). GFAP = Glial fibrillary acidic protein. IHC = Immunohistochemistry. Unless otherwise stated, scale bars = 100 µm.
We further explored the relationship between glial processes and synapses by means of confocal microscopy. As observed in light microscopy evaluations, the dentate nucleus of MS patients had a decreased synaptic density. Both MS and controls revealed a close relationship between glial processes (GFAP/Iba‐1) and synaptophysin‐positive signals. Synaptophysin colocalized predominantly with Iba‐1 rather than GFAP (Figure 2C), and most of the Iba‐1‐positive signals in MS were present at synaptophysin‐positive structures, while control samples also revealed abundant Iba‐1‐positive processes not directly associated with synapses (Figure 2C).
Synaptic stripping and lysosomal degradation in the cerebellar dentate and pontine nuclei of multiple sclerosis patients
Ultrastructurally, the axosomatic synapses on individual neurons appeared reduced in areas of demyelination in the dentate nucleus (Figure 3A). Occasionally, axonal boutons containing synaptic vesicles and mitochondria remained in contact with the soma membrane, but were surrounded by widened intercellular clefts (Figure 3A,B). Many presynaptic‐like profiles were separated from the soma membrane by glial processes, which predominantly showed astrocytic characteristics (Figure 3B,C). Also, free postsynaptic densities appeared opposed to astrocytic appendages (Figure 3C,D) while lamellar extensions and fibrous astrocytes enwrapped somata and large dendritic profiles of the surrounding neuropil (Figure 3D,E).
Figure 3.
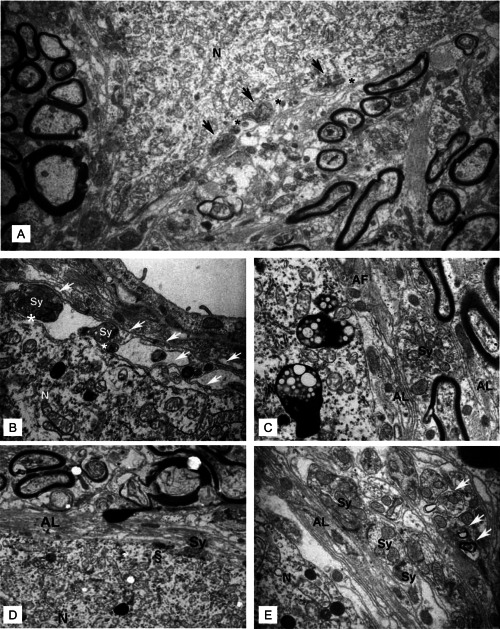
A. Representative electron microscopic image showing a neuronal cell body (N) with only few axosomatic synaptic contacts (arrows). Increased intercellular spaces lie adjacent to the synaptic boutons (asterisks). Magnification: 9000×. B. Thin glial processes (arrows) enwrap synaptic boutons (Sy) on the soma. Widened intercellular spaces are seen between pre‐ and postsynaptic surface membranes (asterisks). C. Astrocytic lamellae (AL) and fibrous processes (AF) separate synaptic boutons (Sy) from the soma membrane. (D and E) Stacks of astrocytic lamellae (AL) insulate the largely synapse‐free soma from the surrounding neuropil. Characteristic synaptic structures persist in apposition to glial processes. Synapse without presynaptic vesicles (Sy), vacant postsynaptic density (§) without presynaptic element (D). Note that in contrast to the few axosomatic synaptic features (D), synapses in (E) appear considerably better preserved in the surrounding neuropil. In part, synapses of the neuropil undergo lysosomal degradation (arrows). Unless otherwise stated, images were obtained at a magnification of 40,000×.
The remaining synaptic vesicles also exhibited ultrastructural alterations. Synaptic vesicles appeared aggregated, closely abutted and dislocated from the active zone. At the periphery of the vesicle pool multi‐vesicular fusion (focal vacuolization of presynaptic elements) could be observed (Figure 4A,B).
Figure 4.
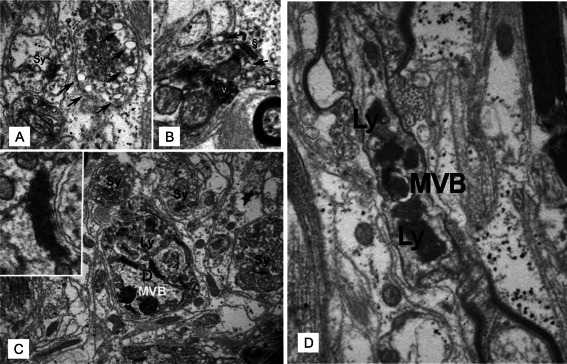
Ultrastructural changes and lysosomal degradation of synapses in the dentate nucleus in multiple sclerosis. A. Presynaptic bouton with accumulation (v) and selective fusion of synaptic vesicles (arrows) at the periphery of the vesicle pool. The bouton appears without any postsynaptic density, thus not forming synaptic contact. Note the glycogen pools in processes of surrounding astrocytes. An adjacent morphologically normal synapse (Sy) is also depicted. B. Synaptic contact with presynaptic vesicles (v) are focally accumulated and dislocated from the active zone (§). Vesicles are heterogeneous and show fusion (arrows) at the periphery of the vesicle pool. Magnification: 40,000×. C. Remodelling process in a dendrite. Multivesicular bodies (MVB) and secondary lysosomes (Ly) with recognizable structural elements such as condensed lamellae of endoplasmic reticulum (inset) appear in a dendrite (D), which is surrounded by intact synapses (Sy). Magnification: 40,000x; inset: 80,000×. D. Secondary lysosomes (Ly) and a multivesicular body (MVB) transported in an intact axon at the node of Ranvier. Magnification: 15,000×.
Further, autophagosomes (cytolysosomes) loaded with degradation products of synaptic vesicle aggregates were recognized in dendrites (Figure 4C), suggesting that lysosomal degradation is also involved in synaptic remodeling of MS patients. Interestingly, residual bodies were present in myelinated axons which otherwise appeared intact (Figure 4D), indicating that, in addition to local storage of degradation products in neurons, axons with intact intracellular transport systems may remove residues of synaptic regression from demyelinated plaques and non‐demyelinated zones.
In the paramedian pontine nuclei, another major relay station of motor information, synaptic vesicles were also characterized by aggregation (Supporting information Figure S1A–C), dislocation from the active zones and peripheral multi‐vesicular fusion (Supporting information Figure S1D), and cytolysosomes and residual bodies containing remnants of presynaptic elements appeared to be transported in dendrites (Supporting information Figure S1G,H).
In contrast to the dentate nucleus, we did not observe glial processes separating synaptic boutons from the cell soma. Nevertheless, processes of filamentous astrocytes embracing synaptic structures (Supporting information Figure S1E,F) and free postsynaptic densities opposed to astrocytic processes (Supporting information Figure S1F) were detected.
DISCUSSION
In this report, we show a reduction in the number and density of axosomatic synapses in the cerebellar dentate nucleus of MS patients and provide, for the first time, ultrastructural evidence suggesting at least two distinct mechanisms of synaptic pathology in MS, namely a glia‐mediated and a neuron‐autonomous process. Also, atrophy and reduction of dentate neurons was observed. Our findings underscore the importance of synaptic and neuronal reduction in cerebellar pathophysiology and support synaptic loss as a widespread phenomenon in MS, as shown here for the chronic disease stage. Most importantly, we bring forward a neuron‐autonomous mechanism previously unreported in the synaptic pathology of MS.
Synaptic loss may occur via: 1 Degeneration of afferent neurons, transection of their axons or functional silencing of afferent input (de‐afferentiation); 2 glia‐mediated synaptic removal (eg synaptic stripping), 3 retrograde trans‐synaptic degeneration or 4 neuron‐autonomous mechanisms such as autophagy or lysosomal degradation of synaptic components 15, 28.
In our study we did not observe differences in Purkinje cell density between patients with or without demyelination in the dentate nucleus and controls. Also, we only observed cortical demyelination in the area of quantification in two cases (Supporting Information Table S2). In this respect our results are in line with previous reports showing similar Purkinje cell counts in the non‐demyelinated cerebellar cortex in MS and controls 10, 19. Although in our cohort, the two cases with cortical demyelination in the area of quantification did not reveal a markedly reduced Purkinje cell density, a reduction in Purkinje cell numbers within areas of cortical cerebellar demyelination has previously been reported 10, 19, thus suggesting that the degeneration of afferent input may contribute to cerebellar synaptic loss in MS.
Similarly, we could observe that neuronal loss in the dentate nucleus was more pronounced in cases with local demyelination. Given the frequent presence of lesions in midbrain, thalamus and pons in the MS cohort, it cannot be excluded that besides local influences, neuronal loss could be partly explained by retrograde trans‐synaptic degeneration. However, it is unclear whether this degenerative phenomenon also contributes to the loss of synaptic afferents observed in the dentate nucleus.
In contrast, we observed that synaptic reduction in the dentate nucleus occurred independently of local demyelination. These results are in line with other autopsy studies in which a demyelination‐independent synaptic pathology was observed in the insular, frontotemporal and occipital lobes suggesting a primary synaptic pathology in the normal appearing grey matter in MS 8.
Although contributing influences such as a reduced afferent input by axonal transport dysfunction or axon transection and retrograde degeneration of efferent axons are likely to be important contributors to synaptic loss 8, neuroinflammation also seems to play a prominent role in synaptic pathology 3. It has been proposed that part of the irreversible dendritic pathology may be mediated by inflammatory cytokines from infiltrating T cells and activated microglia 3, 9, 13, 33.
Microglia engulfment of complement‐tagged synapses in the hippocampus and a close apposition of microglia to cortical neurites have been reported in MS 13, 16, suggesting an association between microglia and synaptic reduction. Consistent with these studies we show a colocalization of predominantly microglial process and synapses and provide ultrastructural evidence that synaptic boutons are separated from the soma by microglial and astrocytic processes. However, we did not find microglial digestion of synaptic elements, supporting the view that the close association of synapses and microglia might be a reaction to ‐ and not the cause of ‐ synaptic degeneration 13, 15. Alternatively, the apposition of microglial appendages to cerebellar dentate afferents could be a response to changes in synaptic activity 32 due in part to aberrant expression of sodium channels in Purkinje cells 21.
In our study, no significant differences in Iba‐1 and KiM1P immunohistochemistry were observed, suggesting that the level of microglia activation is not associated with evident morphological changes Also, whether the activation state of the microglia influences synaptic reduction in MS and neuroinflammation is still a matter of debate. For instance, it has been shown that microglia engulfment of synapses in MS is triggered by the activation of the C1q‐C3 axis of the complement system without activation of the terminal effector C5b9 complex, suggesting a non‐inflammatory process and only slight microglia activation 13. Similarly, in the experimental autoimmune encephalomyelitis (EAE) model the peripheral elevation of TNF‐α resulted in an increased turnover of cortical dendritic spines and axonal boutons that was independent of microglial activation 33. However, stereotaxic injection of heat‐killed Bacillus Calmette–Guerin in the rat cortex induced extensive microglial activation which resulted in about 45% synaptic displacement by microglia 27. These observations suggest that microglia might participate in synaptic loss through different initiating and effector mechanisms.
In the light microscopic evaluation, we observed morphological differences of the astrocytes in demyelinated lesions, as expected for the formation of a glial scar in chronic MS plaques. However, when the GFAP‐positive area was quantified in demyelinated lesions a trend towards an increased area was observed which failed to reach statistical significance. In view of the obvious morphological differences and the clear trend to an increased GFAP‐positive area in demyelinated lesions, the lack of statistical significance is likely caused by the effects of limited sampling and probably does not reflect a lack of differences in the level of astrocytic activation.
Furthermore, at the ultrastructural level, we observed free postsynaptic densities apposed to astrocytic appendages. To our knowledge, a role for astrocytes in synaptic degeneration has not been described in human neuroinflammation. In murine models of axotomy, interdigitations between synapses and astroglia 11 and enlargement of astrocytic processes 32 have been been proposed to play a role in synaptic reorganization, protection and neuronal survival 11, 32, yet further evidence is required for a direct mechanistic extrapolation to the human disease.
Ultrastructurally, we identified lysosomal degradation of synaptic components in two relay stations of motor information, namely the cerebellar dentate and the pontine nuclei. The presence of autophagosomes points towards autophagy, a lysosomal degradation pathway for cytoplasmic components, as a potential neuron‐autonomous mechanism contributing to synaptic reduction in MS. Synaptic autophagy is commonly found in CNS pathology and has been described in models of neurodegeneration 1, ischemic injury 22 and axotomy 28. Furthermore, lysosomal degradation of synaptic components is transiently increased in the motor cortex after facial nerve transection, coinciding with cortical synaptic reorganization 11. The difficulties associated with the acquisition of suitable CNS material for electron microscopy preclude the undertaking of large scale ultrastructural studies in MS. Although our study is the first to provide ultrastructural data suggesting autophagy as a mechanism underlying synaptic pathology in chronic MS, the evidence presented here derives from the study of a single MS case. Therefore the implications of this pathway in synaptic degeneration and restructuring in human neuroinflammation warrant further investigation.
All in all and considering the limitations of the limited sample size our data support the idea that, in MS, synaptic pathology is multifactorial and does not only depend on anterograde and retrograde axonal dysfunction and loss but also on functional synaptic changes that lead to an increased surveillance of synapses by astrocytes and microglia, with or without active synaptic removal, eg, stripping. Simultaneously, the changes in activity could induce synaptic autophagy 25, which might further be promoted by the neurodegenerative process of the afferent neurons. The reduction of synapses independent of local demyelination highlights their exquisite vulnerability to the disease process in MS.
CONFLICT OF INTEREST
The authors do not report any conflict of interest.
Supporting information
Additional Supporting Information may be found in the online version of this article at the publisher's web‐site:
Figure S1. Degeneration of synapses in the demyelinated pons. (A) An accumulation of synaptic vesicles (v) in the boutons is accompanied by multivesicular bodies (MVB) at the nerve endings and often lacks postsynaptic densities (B). (C and D) Other presynaptic endings exhibit heterogeneous vesicles that are focally accumulated (v) and selectively fused (arrows) at the periphery of the vesicle pool. The entire vesicle aggregate is dislocated from the active zone (§). Magnification (A to D): 40,000x. (E) Vacant postsynaptic densities (§) are accompanied by processes of filamentous astrocytes (AF) (60,000x). (F) Filamentous astroglial process (AF) covering a vacant dendritic postsynaptic density (§). (G) Lysosomal degradation (black arrow) of the synaptic vesicles. On the right a vacant postsynaptic density (§) is situated on a dendrite. Magnification (F and G): 40,000x. (H) Synaptic bouton showing dense aggregation of vesicles. The middle part of the dendrite undergoes the formation of an autophagosome (15,000x).
Figure S2. Area of dentate neurons. Combined dot‐ and boxplot showing no statistically significant differences among groups: Control, multiple sclerosis (MS) with (D) or without (ND) demyelination of the dentate nucleus. The area of single dentate neurons in µm2 is depicted by individual points. Each color corresponds to one patient. Box plots illustrate the median and 25th and 75th percentiles.
Table S1. Primary Antibodies.
Table S2. Cerebellar cortical demyelination in MS cases.
ACKNOWLEDGMENTS
This work was supported by grants from the Deutsche Forschungsgemeinschaft to W.B. and C.S. (SFB‐TRR 43 “The brain as a target of inflammatory processes”), the Forschungsförderprogramm of the Faculty of Medicine, Georg‐August‐University, Göttingen (A.B.F.), and the Gemeinnützige Hertie Foundation (W.B., C.S.). J.P.A was supported by grants from the MS Society of Canada. We recognize the excellent technical support from Heidi Brodmerkel, Katja Schulz and Brigitte Maruschak. We thank Cynthia Bunker for language editing.
REFERENCES
- 1. Adalbert R, Nogradi A, Babetto E, Janeckova L, Walker SA, Kerschensteiner M et al (2009) Severely dystrophic axons at amyloid plaques remain continuous and connected to viable cell bodies. Brain 132:402–416. [DOI] [PubMed] [Google Scholar]
- 2. Brück W, Schmied M, Suchanek G, Brück Y, Breitschopf H, Poser S et al (1994) Oligodendrocytes in the early course of multiple sclerosis. Ann Neurol 35:65–73. [DOI] [PubMed] [Google Scholar]
- 3. Centonze D, Muzio L, Rossi S, Furlan R, Bernardi G, Martino G (2010) The link between inflammation, synaptic transmission and neurodegeneration in multiple sclerosis. Cell Death Differ 17:1083–1091. [DOI] [PubMed] [Google Scholar]
- 4. Deppe M, Tabelow K, Krämer J, Tenberge J‐G, Schiffler P, Bittner S et al, (2016) Evidence for early, non‐lesional cerebellar damage in patients with multiple sclerosis: DTI measures correlate with disability, atrophy, and disease duration. Mult Scler 22:73–84. [DOI] [PubMed] [Google Scholar]
- 5. Dogonowski A‐M, Andersen KW, Madsen KH, Sørensen PS, Paulson OB, Blinkenberg M, Siebner HR (2014) Multiple sclerosis impairs regional functional connectivity in the cerebellum. NeuroImage 4:130–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Dutta R, Chang A, Doud MK, Kidd GJ, Ribaudo MV, Young EA et al (2011) Demyelination causes synaptic alterations in hippocampi from multiple sclerosis patients. Ann Neurol 69:445–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Howell OW, Schulz‐Trieglaff EK, Carassiti D, Gentleman SM, Nicholas R, Roncaroli F, Reynolds R (2015) Extensive grey matter pathology in the cerebellum in multiple sclerosis is linked to inflammation in the subarachnoid space. Neuropath Appl Neuro 41:798–813. [DOI] [PubMed] [Google Scholar]
- 8. Jürgens T, Jafari M, Kreutzfeldt M, Bahn E, Brück W, Kerschensteiner M, Merkler D (2016) Reconstruction of single cortical projection neurons reveals primary spine loss in multiple sclerosis. Brain 139:39–46. [DOI] [PubMed] [Google Scholar]
- 9. Kreutzfeldt M, Bergthaler A, Fernandez M, Brück W, Steinbach K, Vorm M et al, (2013) Neuroprotective intervention by interferon‐γ blockade prevents CD8+ T cell–mediated dendrite and synapse loss. J Exp Med 210:2087–2103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kutzelnigg A, Faber‐Rod JC, Bauer J, Lucchinetti CF, Sorensen PS, Laursen H et al, (2007) Widespread demyelination in the cerebellar cortex in multiple sclerosis. Brain Pathol 17:38–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Laskawi R, Landgrebe L, Wolff JR (1996) Electron microscopical evidence of synaptic reorganization in the contralateral motor cortex of adult rats following facial nerve lesion. ORL J Oto‐Rhino‐Lary 58:266–270. [DOI] [PubMed] [Google Scholar]
- 12. Lumsden CE (1970) The neuropathology of multiple sclerosis. In: Handbook of Clinical Neurology, Pierre J. Vinken , George W. Bruyn (eds), Chapter 8, pp. 217–309. North‐Holland Publishing Company: Amsterdam. [Google Scholar]
- 13. Michailidou I, Willems JGP, Kooi E‐J, van Eden C, Gold SM, Geurts JJG et al (2015) Complement C1q‐C3–associated synaptic changes in multiple sclerosis hippocampus. Ann Neurol 77:1007–1026. [DOI] [PubMed] [Google Scholar]
- 14. Novotna M, Paz Soldan MM, Abou Zeid N, Kale N, Tutuncu M, Crusan DJ et al (2015) Poor early relapse recovery affects onset of progressive disease course in multiple sclerosis. Neurology 85:722–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Perry VH, O'Connor V (2010) The role of microglia in synaptic stripping and synaptic degeneration: A revised perspective. ASN Neuro 2:e00047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Peterson JW, Bö L, Mörk S, Chang A, Trapp BD (2001) Transected neurites, apoptotic neurons, and reduced inflammation in cortical multiple sclerosis lesions. Ann Neurol 50:389–400. [DOI] [PubMed] [Google Scholar]
- 17. Prineas JW, Connell F (1979) Remyelination in multiple sclerosis. Ann Neurol 5:22–31. [DOI] [PubMed] [Google Scholar]
- 18. R Core Team (2015) R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing: Vienna, Austria. [Google Scholar]
- 19. Redondo J, Kemp K, Hares K, Rice C, Scolding N, Wilkins A (2015) Purkinje cell pathology and loss in multiple sclerosis cerebellum. Brain Pathol 25:692–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Romascano D, Meskaldji D‐E, Bonnier G, Simioni S, Rotzinger D, Lin Y‐C et al (2015) Multicontrast connectometry: A new tool to assess cerebellum alterations in early relapsing‐remitting multiple sclerosis. Hum Brain Mapp 36:1609–1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Roostaei T, Sadaghiani S, Park MT, Mashhadi R, Nazeri A, Noshad S et al (2016) Channelopathy‐related SCN10A gene variants predict cerebellar dysfunction in multiple sclerosis. Neurology 86:410–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ruan YW, Han XJ, Shi ZS, Lei ZG, Xu ZC (2012) Remodeling of synapses in the CA1 area of the hippocampus after transient global ischemia. Neuroscience 218:268–277. [DOI] [PubMed] [Google Scholar]
- 23. Ruifrok AC, Johnston DA (2001) Quantification of histochemical staining by color deconvolution. Anal Quant Cytol Histol 23:291–299. [PubMed] [Google Scholar]
- 24. Schindelin J, Arganda‐Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T et al (2012) Fiji: an open‐source platform for biological‐image analysis. Nat Meth 9:676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Shehata M, Matsumura H, Okubo‐Suzuki R, Ohkawa N, Inokuchi K (2012) Neuronal stimulation induces autophagy in hippocampal neurons that is involved in AMPA receptor degradation after chemical long‐term depression. J Neurosci 32:10413–10422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Stadelmann C, Kerschensteiner M, Misgeld T, Brück W, Hohlfeld R, Lassmann H (2002) BDNF and gp145trkB in multiple sclerosis brain lesions: neuroprotective interactions between immune and neuronal cells?. Brain 125:75–85. [DOI] [PubMed] [Google Scholar]
- 27. Trapp BD, Wujek JR, Criste GA, Jalabi W, Yin X, Kidd GJ et al, (2007) Evidence for synaptic stripping by cortical microglia. Glia 55:360–368. [DOI] [PubMed] [Google Scholar]
- 28. Viscomi M, Molinari M (2014) Remote neurodegeneration: Multiple actors for one play. Mol Neurobiol 50:368–389. [DOI] [PubMed] [Google Scholar]
- 29. Waxman SG, Kantarci O (2016) The cerebellar channelopathy of multiple sclerosis. Neurology 86:406–407. [DOI] [PubMed] [Google Scholar]
- 30. Wegner C, Esiri M, Chance S, Palace J, Matthews P (2006) Neocortical neuronal, synaptic, and glial loss in multiple sclerosis. Neurology 67:960–967. [DOI] [PubMed] [Google Scholar]
- 31. Weier K, Penner IK, Magon S, Amann M, Naegelin Y, Andelova M et al, (2014) Cerebellar abnormalities contribute to disability including cognitive impairment in multiple sclerosis. PLoS One 9:e86916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Yamada J, Nakanishi H, Jinno S (2011) Differential involvement of perineuronal astrocytes and microglia in synaptic stripping after hypoglossal axotomy. Neuroscience 182:1–10. [DOI] [PubMed] [Google Scholar]
- 33. Yang G, Parkhurst CN, Hayes S, Gan W‐B (2013) Peripheral elevation of TNF‐α leads to early synaptic abnormalities in the mouse somatosensory cortex in experimental autoimmune encephalomyelitis. Proc Natl Acad Sci USA 110:10306–10311 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting Information may be found in the online version of this article at the publisher's web‐site:
Figure S1. Degeneration of synapses in the demyelinated pons. (A) An accumulation of synaptic vesicles (v) in the boutons is accompanied by multivesicular bodies (MVB) at the nerve endings and often lacks postsynaptic densities (B). (C and D) Other presynaptic endings exhibit heterogeneous vesicles that are focally accumulated (v) and selectively fused (arrows) at the periphery of the vesicle pool. The entire vesicle aggregate is dislocated from the active zone (§). Magnification (A to D): 40,000x. (E) Vacant postsynaptic densities (§) are accompanied by processes of filamentous astrocytes (AF) (60,000x). (F) Filamentous astroglial process (AF) covering a vacant dendritic postsynaptic density (§). (G) Lysosomal degradation (black arrow) of the synaptic vesicles. On the right a vacant postsynaptic density (§) is situated on a dendrite. Magnification (F and G): 40,000x. (H) Synaptic bouton showing dense aggregation of vesicles. The middle part of the dendrite undergoes the formation of an autophagosome (15,000x).
Figure S2. Area of dentate neurons. Combined dot‐ and boxplot showing no statistically significant differences among groups: Control, multiple sclerosis (MS) with (D) or without (ND) demyelination of the dentate nucleus. The area of single dentate neurons in µm2 is depicted by individual points. Each color corresponds to one patient. Box plots illustrate the median and 25th and 75th percentiles.
Table S1. Primary Antibodies.
Table S2. Cerebellar cortical demyelination in MS cases.