

Abstract
The causes of most neurodegenerative diseases are attributed to multiple genetic and environmental factors interacting with one another. Above all, inflammation in the nervous system has been implicated in many neurodegenerative diseases. Still, the roles of neuroinflammation in disease mechanisms and the triggers of inflammatory responses in disease‐inflicted brain tissues seem to remain unclear. This review will examine previous studies that had been done from genetic, pathological and epidemiological perspectives. These studies assess the involvement of neuroinflammation in synucleinopathies, a group of neurodegenerative diseases that are characterized by deposition of α‐synuclein aggregates such as Parkinson's disease, dementia with Lewy bodies and multiple system atrophy. The review will also discuss the role of α‐synuclein aggregates in triggering inflammatory responses from glial cells. It is expected that a precise assessment of the roles and mechanisms of neuroinflammation in neurodegenerative diseases will pave the way for the development of disease‐modifying drugs.
Keywords: synuclein, Parkinson's disease, inflammation, microglia, eurodegeneration
Introduction
Neurodegenerative diseases are characterized by the deposition of specific proteins. A group of such diseases exhibit an aggregation of α‐synuclein, a neuronal protein whose function may involve the regulation of synaptic transmission 38. This group of diseases, collectively referred to as synucleinopathies, includes, Parkinson's disease (PD), dementia Lewy bodies (DLB), multiple system atrophy (MSA) and more than half the cases of Alzheimer's disease (AD).
Human genetics corroborated the importance of α‐synuclein in neurodegenerative diseases, particularly in PD. Thus far, five missense mutations and gene multiplication mutations have been linked to early onset, familial PD 2. More importantly, genome‐wide association studies identified SNCA, the gene for α‐synuclein, as the factor most strongly associated with sporadic PD, in all the cohorts examined in the studies 46, 48.
α‐synuclein aggregates accumulate in specific intracellular structures that characterize the diseases. In PD and DLB, α‐synuclein aggregates are deposited in the intraneuronal structures referred to as Lewy bodies (LBs) and Lewy neurites (LNs). In MSA, glial cytoplasmic inclusions in oligodendrocytes are the major pathological features of α‐synuclein aggregates 24. Moreover, LBs also occur in AD, although their roles in pathogenesis have been rarely investigated.
The role of α‐synuclein in neuronal dysfunction and neurodegeneration has been the major focus of the research. In addition to direct effects of α‐synuclein on neurons, recent evidence suggests its importance in glial involvement of the pathogenesis in synucleinopathies. One of the glial functions that are considered relevant to pathogenesis is inflammatory response. Neuroinflammation is one of the common pathological characteristics of major neurodegenerative diseases such as AD and PD 26. Previously, neuroinflammation was simply regarded as a response to neurodegeneration in these diseases. However, recent studies reveal that neuroinflammation could be the trigger or the key player in the onset of diseases by creating a pathogenic microenvironment. For PD, specifically, post‐mortem studies show that neuroinflammatory processes are large aspects of the disease. This chapter will review the recent advancement of findings on the role of α‐synuclein in the inflammatory glial activation.
Role of Neuroinflammation in Synucleinopathies
Immune cell activation
Brains from PD patients show extensive microglial activation and infiltration of blood‐derived mononuclear phagocytes and lymphocytes 51. The activation of microglial cells, the cells that act as the first line defense of the central nervous system, is detected by their expression of MHC class II molecules in the parkinsonian nigra. Microglia are activated in response to neurological injuries. They can be activated as a response to a direct stimulation by environmental or endogenous toxins, thereby undergoing a reactive microgliosis process. Although microglia possess neurotrophic and neuroprotective functions, it has also become clear that microglia exert neurotoxicity and develop neurodegenerative diseases as they become activated by pathogenic stimulations 22. These changes are identified by morphological and immunophenotypic changes. Once microglial cells are activated, they produce toxic oxygen‐derived and nitrogen‐derived products, as well as various cytokines, to contribute to neurodegeneration.
In addition to the activated microglia, cytokines increase in amount in PD 27. The induction of cytokines activates the inflammatory processes in the brain microenvironments. The neurotoxicity of inflammatory cytokines could be caused by a receptor binding on affected neurons or by glial‐cell activation that amplifies the production of inflammatory factors.
Epidemiology
Nonsteroidal anti‐inflammatory drugs, or NSAIDs, are commonly used analgesics, specifically used to treat inflammatory responses. Studies have found that NSAIDs lower the risk of neurodegenerative diseases, such as AD and PD (reviewed in 27). Although certain types of NSAIDs like aspirin displayed controversial effects, ibuprofen seemed to show more consistent preventive effects on the risk of PD 4, 5 Eventually, a prospective study showed the protective effects only with ibuprofen, and not with aspirin, acetoaminophen or other NSAIDs 18.
Animal studies
Many experiments in regards to inflammatory responses in neurodegenerative diseases have been tested in rodent models. Some evidence of LPS‐mediated neurotoxicity reveals that inflammatory responses from non‐neuronal cells are ample enough to cause dopaminergic neuron loss. Systemic administration of LPS led to inflammatory responses in CNS of rats and mice, some with dopaminergic neurodegeneration 44 and others without signs of neurodegeneration 34. Low‐dose intraperitoneal administration of LPS for prolonged periods elicited persistent neuroinflammation in both wild‐type and parkin−/− mice 17. Inflammatory and oxidative stress responses to the LPS administration did not differ significantly between the two genotypes. However, only parkin−/− mice displayed subtle fine‐motor deficits and selective loss of dopaminergic neurons in the substantia nigra 17. This study suggests that loss of parkin function increases the vulnerability of nigral dopaminergic neurons to inflammation‐related degeneration.
Studies with minocycline
Among many anti‐inflammatory drugs, which have been tested on animal models and preclinical studies, minocycline is an antibiotic that possesses anti‐inflammatory and antiapoptotic effects as well as neuroprotective effect during neurodegeneration in the animal models of several neurodegenerative diseases. In mice, minocycline attenuated 1‐methyl‐4‐phenyl‐1,2,3,6‐tetrahydropyridine (MPTP)‐induced dopaminergic neuronal death, nitotyrosine formation and microglial activation 54. This drug also exhibited beneficial effects in rodent models of AD 8, 45, ALS 56, 57 and HD 6, 53. However, studies indicated that minocycline might actually worsen neurodegeneration if the wrong dose and a route are used to treat rodents or non‐human primates 11, 12, and the results of clinical trials for PD and MSA with minocycline turned out to be either unsuccessful or uncertain 13, 31.
Microglia Activation in Synucleinopathies
Microglial cells mainly engage in the immunity of the CNS; the establishment of innate immune system in the CNS is attributed to their prenatal invasion 50. The origin of microglia has been revealed to be the yolk sac macrophages during the early development of brain, and they are known to remain until adulthood 21. In terms of their normal phenotypes, microglia can display several different characteristics: resting, activated but nonphagocytic, activated phagocytic 22. Resting microglia branch out extensively, contact neuronal synapses and potentially undergo transformation in response to external stimuli such as injuries or neuronal death 52. Although their corresponding roles in diseases have not yet been revealed, morphological characteristics as well as transforming features demonstrate that microglia are similar to macrophages 37. Particularly in AD, the mixed phenotypes—neuroprotective and neuroinflammatory—of microglia have befuddled researchers in regards to understanding the exact disease mechanism and coming up with therapeutic measures 19.
In synucleinopathies, microglia are distinguishable from astrocytes in that they do not accumulate cytoplasmic α‐synuclein but rather degrade the internalized α‐synuclein rapidly and give rise to adequate morphological changes 42. Experimentally, however, microglia can be manipulated to slow down the degradation 42, which is likely to occur in aged and disease‐inflicted brains. Another pathology observable in PD patients is the presence of internalized, nondegraded neuromelamin granules in the cell bodies of microglia 43. As these granules as well as α‐synuclein accumulate more and more in the extracellular space as neuronal loss progress, failure to clear them may occur as a result of some abnormality in microglia 24.
PD is characterized by extensive microglial activation in the brain regions, such as the striatum and the substantia nigra 30, 43. Compared to MSA, however, PD is associated with relatively limited signs of neuronal inflammation 20, 32. In PD, activated microglia have been identified in the SN in the vicinity of degenerating dopaminergic neurons, as supported by PET imaging from both human studies and animal models 9, 29. Alternatively, experimental suppression of such microglial activation turned out to reduce degeneration of dopaminergic neurons, which indicates that microglial immune responses may account for some aspects of neurodegeneration 54. Enzymes and cytokines expressed by the activated phagocytic microglia are in fact involved in inflammatory processes. Also, the brain autopsies performed on PD patients and animal models showed increased expression of such inflammatory factors 7, 28. Although the aforementioned roles of microglia have been identified, they are simultaneously being questioned by more recent reviews that suggest microglial dysfunction and degeneration in PD 22. For example, it has been postulated that chronic microglial activation could lead to microglial over‐reaction that could cause microglial degeneration.
Activation of Microglia by Extracellular α‐Synuclein
One of the critical questions regarding the mechanism of microglia activation in PD and other synucleinopathies is what triggers microglia to turn on the pro‐inflammatory responses. Here, we provide evidence supporting the role of neuron‐released α‐synuclein in microglia activation.
Secretion of neuronal α‐synuclein
Elucidation of the normal biology of α‐synuclein is critical to thorough understanding of synucleinopathies. Although the protein was once considered solely an intracellular component, further studies have shown the presence of α‐synuclein inside vesicles and the subsequent secretion via exocytosis 41. This section will examine different mechanisms of exocytosis, introduce pathways of α‐synuclein secretion from neuronal cells, and illustrate favorable conditions in which the secretion is promoted.
Unconventional exocytosis is a general term that refers to the exocytosis pathways that proceed through ER‐Golgi‐independent pathways. A small proportion of cellular α‐synuclein is secreted from neuronal cells through unconventional exocytosis as their normal life cycle 41. The precise mechanism of secretion remains unknown. A few groups of researchers have reported exosome‐associated exocytosis, which is one type of unconventional exocytosis derived from the fusion of the plasma membrane and multivesicular bodies 1, 15. However, significance of such association with exosomes is yet to be determined. When it comes to α‐synuclein, exosome‐associated exocytosis represents only a small fraction of the entire extracellular α‐synuclein. The exosome mechanism was later challenged by another group as the genetic ablation of VPS4, a gene involved in exosome formation, led to an increased amount of α‐synclein secretion 25. Nevertheless, we cannot rule out the possibility that the exosome‐associated α‐synuclein, though accounting for only a little proportion of the total secreted α‐synuclein, may play a critical function in the pathogenesis of synucleinopathies 10.
As an alternative mechanism of secretion, Ejlerskov et al. 14 suggested that exophagy might play a role in α‐synuclein secretion. Exophagy refers to a type of unconventional exocytosis mediated by the fusion of the plasma membrane and autophagosome (or amphisome). Similar to the exosome pathway, exophagy could also give rise to the release of small vesicles to the extracellular space. Therefore, it is not certain whether the vesicle‐associated α‐synuclein in the culture medium should be attributed to the exosome or the exophagy pathway. Exophagy may not be the sole mechanism of α‐synuclein exocytosis, as the inhibition of the early step of autophagy increased α‐synuclein secretion 40.
While most studies on the secretion of α‐synuclein have focused on the unconventional exocytosis, a recent study suggests the role of the ER‐Golgi‐dependent classical pathway on activation of the cultured enteric neurons. 23.
α‐Synuclein in its nature is not secreted in the same amount or in the same rate at all times. Instead, it has the tendency to be secreted more actively under certain conditions. One of such conditions is a situation in which protein‐misfolding stress is induced. This has been demonstrated by the treatment of inhibitors that hinder proper maintenance of cellular proteostasis, which resulted in an increased vesicle translocation and secretion of α‐synuclein 33. In addition to different inhibitors, there are other specific molecules such as dopamine and 4‐hydroxy‐2‐nonenal, which can promote chemical modifications and oligomerization of α‐synuclein and its secretion into extracellular space 3, 39. Moreover, physical stresses like application of heat shock have been reported to increase α‐synuclein secretion 15.
The conditions identified to increase the secretion of α‐synuclein so far led to the increase of misfolded proteins, either by interfering with the protein quality control system or by increasing chemical modifications of the protein. These results suggest that the secretion mechanism is sensitive to protein conformation, selecting misfolded and/or aggregated proteins for vesicular packaging and secretion. However, as we discussed in this section, the details of mechanism remain largely unknown.
Activation of microglia by neuron‐derived α‐synuclein
α‐synuclein‐mediated inflammatory responses have been demonstrated in rat primary microglia cultures, human microglia cultures and monocytic cell line THP‐1, when treated with exogeneous α‐synuclein. These studies show that α‐synuclein activates microglia to produce extra‐cellular superoxide, increases microglial intracellular ROS concentrations (iROS) and induces morphological changes in microglia. α‐synuclein is also shown to be phagocytized by microglia, and the production of microglial ROS in response to α‐synuclein is inhibited by cytochalasin D, implying that phagocytosis is a critical component of the mechanism of α‐synuclein‐induced microglial activation. In the culture of dopaminergic neurons, α‐synuclein fails to show dopaminergic neurotoxicity at low concentrations, while the same concentration of α‐synuclein is strongly neurotoxic when dopaminergic neurons are cultured with glia. Therefore, in addition to the direct neurotoxicity of α‐synuclein, an indirect mechanism through the inflammatory responses from glial cells could account for α‐synuclein‐induced neurodegeneration.
These observations with the recombinant proteins have recently been validated with neuron‐derived α‐synuclein. α‐synuclein preparations that are secreted from differentiated SH‐SY5Y neuroblastoma cells activate microglia, exhibiting morphological changes, increase proliferation, iROS, and cytokine production and increase motility 36, 47.
For the activation of microglia, Toll‐like receptor 2 (TLR2) acts as the receptor for neuron‐secreted α‐synuclein (Figure 1). TLR2 mediates both the uptake of α‐synuclein and the signaling leading to inflammation. Interaction between α‐synuclein and TLR2 has been shown on the surface of microglia, and these proteins co‐localized in the intracellular compartments after internalization. TLR2 is absolutely required for the activation of microglia; however, TLR2 depletion leads to about 70% reduction in α‐synuclein uptake. These suggest that TLR2 is the only receptor mediating the α‐synuclein‐induced signaling, but there might be other receptor molecules mediating the internalization of this protein. In fact, TLR4 has been suggested to be involved in α‐synuclein‐dependent activation of microglia and astrocytes 16. This study suggested that TLR4 acts as a modulator of proinflammatory responses in glia and the production of reactive oxygen species induced by α‐synuclein. The same group also suggested that TLR4 plays an important role in α‐synuclein a clearance in a mouse model of MSA 49. In addition, scavenger receptors and integrins might serve as receptors for α‐synuclein 35, 55. The precise functions of these cell surface proteins in the inflammatory activation of microglia and in α‐synuclein clearance remain to be determined.
Figure 1.
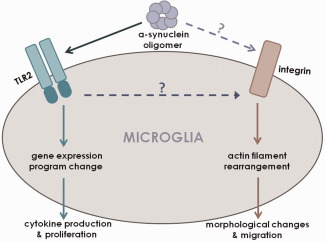
Receptors for oligomeric α‐synuclein in microglia. Oligomeric α‐synuclein that is secreted from neuronal cells activate microglial cells through two receptor systems, TLR2 and β1‐integrin. Activation of TLR2 changes gene expression program and induces the production of inflammatory cytokines, while activation of β1‐integrin leads to actin cytoskeleton rearrangement resulting in morphological changes and increased motility. Whether α‐synuclein activates β1‐integrin through direct interaction or through an indirect mechanism via the TLR2 signaling is unclear.
Each of the cell surface receptors mentioned above may interact with specific forms of α‐synuclein 36. TLR2 is one of the pattern‐recognition receptors, and is recognized by and responds only to certain forms of α‐synuclein. Only certain types of oligomers, not monomers or various types of aggregates including fibrils, could activate TLR2. The oligomers that can activate TLR2 are detected only in the secreted α‐synuclein, but not in the cytosolic fractions of neuronal cells.
Independent of TLR2, neuron‐secreted α‐synuclein increases the motility of microglia through β1‐integrin (Figure 1) 47. However, the integrin pathway is not required for the cytokine production in response to α‐synuclein. Therefore, it appears that separate receptor/signaling systems are working together to trigger full spectrum of microglial activation in exposure to neuron‐secreted α‐synuclein. The fact that separate signaling systems are responsible for different aspects of microglia activation opens up the possibility that microglia can be manipulated in such a way that beneficial clearance function is activated and detrimental inflammation is blocked.
Conclusions
Neuroinflammation is a major component of pathological changes linked to many neurodegenerative diseases. Whether inflammation plays an important role in initiation and progression of the diseases is still unclear. However, mounting evidence suggests that the following might be the case: inflammatory responses of microglia might contribute to the pathogenesis of PD and other synucleinopathies as shown by recent studies in synucleinopathy models. In order for such inflammatory responses of microglia to occur, they can be activated by oligomeric forms of neuron‐secreted α‐synuclein through specific receptor/signaling systems. Within those systems, at least two—TLR2 and β1‐integrin—pathways are involved in microglia activation in which the former is responsible for microglia proliferation and cytokine production, while the latter increases microglial motility (Figure 1). In conclusion, as the significance of microglia in the onset and progression of synucleinopathies have been highlighted over time, manipulation of microglia as means of inhibiting deleterious inflammatory responses while enhancing clearance function in the diseases may bring about positive disease‐modifying effects.
Acknowledgements
This study was supported by a grant from Seoul National University Hospital (2016), by the National Research Foundation (NRF) grant funded by the Korean Government (MEST) (No. 2015R1A2A10052540, 2015R1A2A1505366), and by the Korea Health Technology R&D Project, Ministry of Health & Welfare, Republic of Korea (HI14C0093).
References
- 1. Alvarez‐Erviti L, Seow Y, Schapira AH, Gardiner C, Sargent IL, Wood MJ, Cooper JM (2011) Lysosomal dysfunction increases exosome‐mediated alpha‐synuclein release and transmission. Neurobiol Dis 42:360–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Appel‐Cresswell S, Vilarino‐Guell C, Encarnacion M, Sherman H, Yu I, Shah B et al (2013) Alpha‐synuclein p.H50Q, a novel pathogenic mutation for Parkinson's disease. Mov Disord 28:811–813. [DOI] [PubMed] [Google Scholar]
- 3. Bae EJ, Ho DH, Park E, Jung JW, Cho K, Hong JH et al (2013) Lipid peroxidation product 4‐hydroxy‐2‐nonenal promotes seeding‐capable oligomer formation and cell‐to‐cell transfer of alpha‐synuclein. Antioxid Redox Signal 18:770–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chen H, Jacobs E, Schwarzschild MA, McCullough ML, Calle EE, Thun MJ, Ascherio A (2005) Nonsteroidal antiinflammatory drug use and the risk for Parkinson's disease. Ann Neurol 58:963–967. [DOI] [PubMed] [Google Scholar]
- 5. Chen H, Zhang SM, Hernan MA, Schwarzschild MA, Willett WC, Colditz GA et al (2003) Nonsteroidal anti‐inflammatory drugs and the risk of Parkinson disease. Arch Neurol 60:1059–1064. [DOI] [PubMed] [Google Scholar]
- 6. Chen M, Ona VO, Li M, Ferrante RJ, Fink KB, Zhu S et al (2000) Minocycline inhibits caspase‐1 and caspase‐3 expression and delays mortality in a transgenic mouse model of Huntington disease. Nat Med 6:797–801. [DOI] [PubMed] [Google Scholar]
- 7. Choi DK, Pennathur S, Perier C, Tieu K, Teismann P, Wu DC et al (2005) Ablation of the inflammatory enzyme myeloperoxidase mitigates features of Parkinson's disease in mice. J Neurosci 25:6594–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Choi Y, Kim HS, Shin KY, Kim EM, Kim M, Kim HS et al (2007) Minocycline attenuates neuronal cell death and improves cognitive impairment in Alzheimer's disease models. Neuropsychopharmacology 32:2393–404. [DOI] [PubMed] [Google Scholar]
- 9. Croisier E, Moran LB, Dexter DT, Pearce RK, Graeber MB (2005) Microglial inflammation in the parkinsonian substantia nigra: relationship to alpha‐synuclein deposition. J Neuroinflammation 2:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Danzer KM, Kranich LR, Ruf WP, Cagsal‐Getkin O, Winslow AR, Zhu L et al (2012) Exosomal cell‐to‐cell transmission of alpha synuclein oligomers. Mol Neurodegener 7:42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Diguet E, Fernagut PO, Wei X, Du Y, Rouland R, Gross C et al (2004) Deleterious effects of minocycline in animal models of Parkinson's disease and Huntington's disease. Eur J Neurosci 19:3266–3276. [DOI] [PubMed] [Google Scholar]
- 12. Diguet E, Gross CE, Tison F, Bezard E (2004) Rise and fall of minocycline in neuroprotection: need to promote publication of negative results. Exp Neurol 189:1–4. [DOI] [PubMed] [Google Scholar]
- 13. Dodel R, Spottke A, Gerhard A, Reuss A, Reinecker S, Schimke N et al (2010) Minocycline 1‐year therapy in multiple‐system‐atrophy: effect on clinical symptoms and [(11)C] (R)‐PK11195 PET (MEMSA‐trial). Mov Disord 25:97–107. [DOI] [PubMed] [Google Scholar]
- 14. Ejlerskov P, Rasmussen I, Nielsen TT, Bergstrom AL, Tohyama Y, Jensen PH, Vilhardt F (2013) Tubulin polymerization‐promoting protein (TPPP/p25alpha) promotes unconventional secretion of alpha‐synuclein through exophagy by impairing autophagosome‐lysosome fusion. J Biol Chem 288:17313–17335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Emmanouilidou E, Melachroinou K, Roumeliotis T, Garbis SD, Ntzouni M, Margaritis LH et al (2010) Cell‐produced alpha‐synuclein is secreted in a calcium‐dependent manner by exosomes and impacts neuronal survival. J Neurosci 30:6838–6851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fellner L, Irschick R, Schanda K, Reindl M, Klimaschewski L, Poewe W et al (2013) Toll‐like receptor 4 is required for alpha‐synuclein dependent activation of microglia and astroglia. Glia 61:349–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Frank‐Cannon TC, Tran T, Ruhn KA, Martinez TN, Hong J, Marvin M et al (2008) Parkin deficiency increases vulnerability to inflammation‐related nigral degeneration. J Neurosci 28:10825–10834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gao XA, Chen HL, Schwarzschild MA, Ascherio A (2011) Use of ibuprofen and risk of Parkinson disease. Neurology 76:863–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gentleman SM (2013) Review: microglia in protein aggregation disorders: friend or foe? Neuropathol Appl Neurobiol 39:45–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gerhard A, Banati RB, Goerres GB, Cagnin A, Myers R, Gunn RN et al (2003) [11C](R)‐PK11195 PET imaging of microglial activation in multiple system atrophy. Neurology 61:686–689. [DOI] [PubMed] [Google Scholar]
- 21. Ginhoux F, Lim S, Hoeffel G, Low D, Huber T (2013) Origin and differentiation of microglia. Front Cell Neurosci 7:45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Graeber MB, Streit WJ (2010) Microglia: biology and pathology. Acta Neuropathol 119:89–105. [DOI] [PubMed] [Google Scholar]
- 23. Grathwohl SA, Steiner JA, Britschgi M, Brundin P (2013) Mind the gut: secretion of alpha‐synuclein by enteric neurons. J Neurochem 125:487–490. [DOI] [PubMed] [Google Scholar]
- 24. Halliday GM, Stevens CH (2011) Glia: initiators and progressors of pathology in Parkinson's disease. Mov Disord 26:6–17. [DOI] [PubMed] [Google Scholar]
- 25. Hasegawa T, Konno M, Baba T, Sugeno N, Kikuchi A, Kobayashi M et al (2011) The AAA‐ATPase VPS4 regulates extracellular secretion and lysosomal targeting of alpha‐synuclein. PloS One 6:e29460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Heneka MT, Kummer MP, Latz E (2014) Innate immune activation in neurodegenerative disease. Nat Rev Immunol 14:463–477. [DOI] [PubMed] [Google Scholar]
- 27. Hirsch EC, Hunot S (2009) Neuroinflammation in Parkinson's disease: a target for neuroprotection? Lancet Neurol 8:382–397. [DOI] [PubMed] [Google Scholar]
- 28. Hunot S, Boissiere F, Faucheux B, Brugg B, Mouatt‐Prigent A, Agid Y, Hirsch EC (1996) Nitric oxide synthase and neuronal vulnerability in Parkinson's disease. Neuroscience 72:355–363. [DOI] [PubMed] [Google Scholar]
- 29. Hurley SD, O'Banion MK, Song DD, Arana FS, Olschowka JA, Haber SN (2003) Microglial response is poorly correlated with neurodegeneration following chronic, low‐dose MPTP administration in monkeys. Exp Neurol 184:659–668. [DOI] [PubMed] [Google Scholar]
- 30. Imamura K, Hishikawa N, Sawada M, Nagatsu T, Yoshida M, Hashizume Y (2003) Distribution of major histocompatibility complex class II‐positive microglia and cytokine profile of Parkinson's disease brains. Acta Neuropathol 106:518–526. [DOI] [PubMed] [Google Scholar]
- 31. Investigators NN‐P (2006) A randomized, double‐blind, futility clinical trial of creatine and minocycline in early Parkinson disease. Neurology 66:664–671. [DOI] [PubMed] [Google Scholar]
- 32. Ishizawa K, Komori T, Sasaki S, Arai N, Mizutani T, Hirose T (2004) Microglial activation parallels system degeneration in multiple system atrophy. J Neuropathol Exp Neurol 63:43–52. [DOI] [PubMed] [Google Scholar]
- 33. Jang A, Lee H‐J, Suk J‐E, Jung J‐W, Kim K‐P, Lee S‐J (2010) Non‐classical exocytosis of α‐synuclein is sensitive to folding states and promoted under stress conditions. J Neurochem 113:1263–1274. [DOI] [PubMed] [Google Scholar]
- 34. Jeong H‐K, Jou I, Joe E‐H (2010) Systemic LPS administration induces brain inflammation but not dopaminergic neuronal death in the substantia nigra. Exp Mol Med 42:823–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kim C, Cho ED, Kim HK, You S, Lee HJ, Hwang D, Lee SJ (2014) β1‐integrin‐dependent migration of microglia in response to neuron‐released alpha‐synuclein. Exp Mol Med 46:e91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kim C, Ho DH, Suk JE, You S, Michael S, Kang J et al (2013) Neuron‐released oligomeric alpha‐synuclein is an endogenous agonist of TLR2 for paracrine activation of microglia. Nat Commun 4:1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kreutzberg GW (1996) Microglia: a sensor for pathological events in the CNS. Trends Neurosci 19:312–328. [DOI] [PubMed] [Google Scholar]
- 38. Lashuel HA, Overk CR, Oueslati A, Masliah E (2013) The many faces of alpha‐synuclein: from structure and toxicity to therapeutic target. Nat Rev Neurosci 14:38–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lee HJ, Baek SM, Ho DH, Suk JE, Cho ED, Lee SJ (2011) Dopamine promotes formation and secretion of non‐fibrillar alpha‐synuclein oligomers. Exp Mol Med 43:216–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lee HJ, Cho ED, Lee KW, Kim JH, Cho SG, Lee SJ (2013) Autophagic failure promotes the exocytosis and intercellular transfer of alpha‐synuclein. Exp Mol Med 45:e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lee HJ, Patel S, Lee SJ (2005) Intravesicular localization and exocytosis of alpha‐synuclein and its aggregates. J Neurosci 25:6016–6024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lee HJ, Suk JE, Bae EJ, Lee SJ (2008) Clearance and deposition of extracellular alpha‐synuclein aggregates in microglia. Biochem Biophys Res Commun 372:423–428. [DOI] [PubMed] [Google Scholar]
- 43. McGeer PL, Itagaki S, Boyes BE, McGeer EG (1988) Reactive microglia are positive for HLA‐DR in the substantia nigra of Parkinson's and Alzheimer's disease brains. Neurology 38:1285–1291. [DOI] [PubMed] [Google Scholar]
- 44. Morrison BE, Marcondes MC, Nomura DK, Sanchez‐Alavez M, Sanchez‐Gonzalez A, Saar I et al (2012) Cutting edge: IL‐13Ralpha1 expression in dopaminergic neurons contributes to their oxidative stress‐mediated loss following chronic peripheral treatment with lipopolysaccharide. J Immunol (Baltimore, Md: 1950) 189:5498–5502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Noble W, Garwood C, Stephenson J, Kinsey AM, Hanger DP, Anderton BH (2009) Minocycline reduces the development of abnormal tau species in models of Alzheimer's disease. FASEB J 23:739–750. [DOI] [PubMed] [Google Scholar]
- 46. Satake W, Nakabayashi Y, Mizuta I, Hirota Y, Ito C, Kubo M et al (2009) Genome‐wide association study identifies common variants at four loci as genetic risk factors for Parkinson's disease. Nat Genet 41:1303–1307. [DOI] [PubMed] [Google Scholar]
- 47. Selkoe D, Dettmer U, Luth E, Kim N, Newman A, Bartels T (2014) Defining the native state of alpha‐synuclein. Neurodegener Dis 13:114–117. [DOI] [PubMed] [Google Scholar]
- 48. Simon‐Sanchez J, Schulte C, Bras JM, Sharma M, Gibbs JR, Berg D et al (2009) Genome‐wide association study reveals genetic risk underlying Parkinson's disease. Nat Genet 41:1308–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Stefanova N, Fellner L, Reindl M, Masliah E, Poewe W, Wenning GK (2011) Toll‐like receptor 4 promotes alpha‐synuclein clearance and survival of nigral dopaminergic neurons. Am J Pathol 179:954–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Streit WJ, Kincaid‐Colton CA (1995) The brain's immune system. Sci Am 273:54–5, 8–61. [DOI] [PubMed] [Google Scholar]
- 51. Vilhardt F (2005) Microglia: phagocyte and glia cell. Int J Biochem Cell Biol 37:17–21. [DOI] [PubMed] [Google Scholar]
- 52. Wake H, Moorhouse AJ, Jinno S, Kohsaka S, Nabekura J (2009) Resting microglia directly monitor the functional state of synapses in vivo and determine the fate of ischemic terminals. J Neurosci 29:3974–3980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Wang X, Zhu S, Drozda M, Zhang W, Stavrovskaya IG, Cattaneo E et al (2003) Minocycline inhibits caspase‐independent and ‐dependent mitochondrial cell death pathways in models of Huntington's disease. Proc Natl Acad Sci USA 100:10483–10487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Wu DC, Jackson‐Lewis V, Vila M, Tieu K, Teismann P, Vadseth C et al (2002) Blockade of microglial activation is neuroprotective in the 1‐methyl‐4‐phenyl‐1,2,3,6‐tetrahydropyridine mouse model of Parkinson disease. J Neurosci 22:1763–1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Zhang W, Dallas S, Zhang D, Guo JP, Pang H, Wilson B et al (2007) Microglial PHOX and Mac‐1 are essential to the enhanced dopaminergic neurodegeneration elicited by A30P and A53T mutant alpha‐synuclein. Glia 55:1178–1188. [DOI] [PubMed] [Google Scholar]
- 56. Zhang W, Narayanan M, Friedlander RM (2003) Additive neuroprotective effects of minocycline with creatine in a mouse model of ALS. Ann Neurol 53:267–270. [DOI] [PubMed] [Google Scholar]
- 57. Zhu S, Stavrovskaya IG, Drozda M, Kim BY, Ona V, Li M et al (2002) Minocycline inhibits cytochrome c release and delays progression of amyotrophic lateral sclerosis in mice. Nature 417:74–78. [DOI] [PubMed] [Google Scholar]