

Abstract
Accumulation, aggregation and deposition of Aβ peptides are pathological hallmarks in the brains of individuals affected by Alzheimer's disease (AD) or by cerebral β‐amyloid angiopathy (Aβ‐CAA). While Aβ is a peptide of yet largely unknown function, it is constantly produced in the human brain where it normally remains in a soluble state. However, Aβ peptides are aggregation prone by their intrinsic ability to adopt alternative conformations rich in β‐sheet structure that aggregate into oligomeric as well as fibrillar formations. This transition from soluble to aggregated state has been hypothesized to initiate the pathological cascade and is therefore subject to intensive research. Mounting evidence suggests prion‐like templated misfolding as the biochemical phenomenon responsible for promoting progressive Aβ aggregation. Here, we review studies in vitro and in vivo that suggest that cerebral Aβ aggregation may indeed progress via prion‐like templated misfolding. The implications of these findings are discussed with respect to understanding initiation and progression of the disease and to developing therapeutics.
Keywords: Aβ, Alzheimer's disease, cerebral β‐amyloidosis, cerebral beta‐amyloid angiopathy, prion‐like aggregation, seeding, templated misfolding
Introduction
More than 100 years ago, neuropathologist, Alois Alzheimer, discovered peculiar changes in the brains of individuals who had died from dementia: amyloid plaques, neurofibrillary tangles and, to varying extent, vascular amyloidosis 3, 31, 53, 58. Today, we know that extracellular amyloid plaques and vascular amyloid are proteinaceous deposits mainly composed of Aβ peptides 51. In contrast, neurofibrillary tangles are intracellular aggregates of hyperphosphorylated tau proteins 50. Normally retaining a soluble state, Aβ peptides generated through proteolytic cleavage from the amyloid precursor protein (APP), as well as tau proteins, are integrated into cellular physiology 33, 50. However, in Alzheimer's disease (AD), Aβ peptides and tau proteins adopt alternative, misfolded conformations rich in β‐sheet structure that renders them aggregation prone and promotes association into oligomeric as well as fibrillar structures. The abundance of these pathological deposits increases with disease severity and compromises synaptic and neuronal function. In advanced stages of AD, vast areas of the brain are affected 13, particularly in the so‐called default mode network 78, and significant synaptic and neuronal loss causes progressive dementia 79.
The amyloid cascade hypothesis of AD proposes that Aβ misfolding and aggregation are the central and earliest events in the disease and trigger a sequence of pathological changes that result in tau hyperphosphorylation and aggregation, brain inflammation and ultimately synapse as well as neuronal loss, and dementia 35. Conclusive evidence is derived largely from rare forms of familial AD (<3%) caused by mutations in APP or in the catalytic subunits of the γ‐secretase complex, that is, presenilin 1 (PS1) and presenilin 2 (PS2), responsible for releasing Aβ peptides from APP 33. These mutations lead to subtle changes in APP metabolism with increased production of Aβ peptides and/or to generation of more aggregation prone variants 7, 12, 16, 19, 20, 52, 76. Similarly, an increase in aggregation prone Aβ peptides caused by increased production and/or reduced degradation and clearance is hypothesized to initiate the more common sporadic cases of AD. Aβ aggregates, especially small units termed oligomers, have been shown to be neurotoxic and to alter synaptic function 57, although by themselves they may not be sufficient to lead to dementia, correlating only insufficiently with cognitive decline 14, 47, 54, 59, 91. Current state of knowledge suggests that Aβ toxicity in AD is at least partially mediated via tau hyperphosphorylation and aggregation that promote neuronal dysfunction 38, 72, 88. This part of the mini‐symposium focuses on the mechanisms and consequences of Aβ aggregation. Initiation and spreading of tau aggregation and concomitant pathological changes are reviewed in Clavaguera et al (this issue).
AD is not the only disorder with signature Aβ aggregation. In cerebral β‐amyloid angiopathy (Aβ‐CAA), a sporadic or rare hereditary pathological condition in the elderly, characteristic Aβ deposits are found almost exclusively in the walls of small‐ to medium‐sized blood vessels in the leptomeninges and in the cortex 9. These vascular Aβ deposits can weaken vessel walls and compromise vascular function. Abundant vascular Aβ deposits can cause intracerebral microbleeds or hemorrhages as well as neurodegeneration and cognitive impairment. Pathological accumulation of Aβ peptides has also been described in sporadic inclusion body myositis (sIBM), an inflammatory and degenerative muscle disease 5, 60, 87; however, the contribution of Aβ aggregation to sIBM is controversial 32.
While it seems evident that the transition from soluble Aβ peptides to misfolded and aggregated versions plays an important role in these diseases, many questions still remain open and urgently await answers in order to develop efficacious treatments. These unanswered questions include: How are misfolding and aggregation of normally soluble Aβ peptides initiated? With disease progression, Aβ aggregation affects vast areas and neuronal networks of the central nervous system (CNS)—what is the relationship of these aggregates? Do they form independently at the different sites? Or are they related?
Interestingly, new insights arise from similarities between Aβ aggregation and protein aggregation observed in prion disorders, similarities with regard to a biochemical phenomenon termed templated misfolding.
Nucleation‐Dependent Polymerization, Seeding and Prion‐Like Templated Misfolding
It has been proposed that Aβ aggregation as well as the aggregation of other amyloidogenic proteins follows a nucleation‐dependent polymerization process similar to crystallization 36, 40. Such polymerization reactions consist of two phases: the initial nucleation phase (“lag phase”) and the growth phase (Figure 1A). During the nucleation phase, the native monomer undergoes a series of kinetically unfavorable conformational changes and misfolded or oligomeric species can be formed. These are unstable because the intra‐ and intermolecular interactions do not outweigh the entropic costs of misfolding and aggregation. However, under certain conditions an ordered, putatively multimeric nucleus can be formed. Once the nucleus is present, more cognate proteins can be incorporated relatively rapidly into the aggregate (“growth phase”). In this model, aggregation is concentration‐ and time‐dependent, and the rate‐limiting step of fibrillization is the formation of the nucleus. Moreover, fragmentation of fibrils generates new seeds and boosts the polymerization cascade 25. Seeding, which is the addition of a preformed nucleus, circumvents the lag phase and initiates immediate aggregate formation (Figure 1B). Support for nucleation‐dependent polymerization of Aβ peptides is derived from in vitro aggregation of synthetic or recombinant Aβ peptides in solution 48, 61, 64, 77. With regard to AD, this model takes into account that Aβ fibrillization in the human brain is normally a slow process: initial amyloid plaque formation may take decades in individuals who develop AD and may never even occur in histologically significant amounts at very old age 14. However, once Aβ aggregation has been initiated, the misfolding and deposition seems to inevitably progress.
Figure 1.
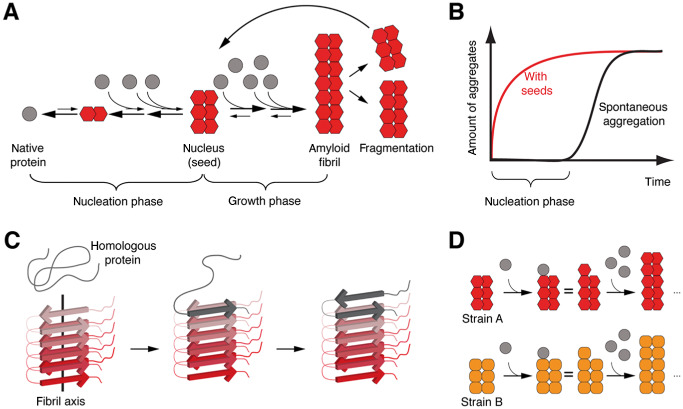
Nucleation‐dependent polymerization, seeding and templated misfolding. A. Under certain circumstances, some proteins can adopt an alternative, misfolded conformation that allows for multimerization. The initial conformational changes are kinetically unfavorable and may not occur under physiological conditions. However, once a nucleus (“seed,” putatively multimeric) is formed, the properties of the nucleus allow for relatively rapid incorporation of homologous proteins into amyloid fibrils. Fragmentation of fibrils generates more nuclei resulting in a vicious cycle of progressive protein misfolding and aggregation. B. Schematic representation of protein aggregation kinetics. Spontaneous aggregation is delayed because of the thermodynamically unfavorable nucleus formation. Addition of preformed seeds leads to immediate aggregate formation and circumvents the nucleation phase. C. Structure analysis of amyloid fibrils reveals typical sets of in‐register β‐sheets arranged parallel to the fibril axis. This structure has been suggested to serve as a template for the congruent incorporation of homologous, native or partially unfolded proteins that orient their peptide chains accordingly, hence termed templated misfolding. D. Some proteins can adopt diverse misfolded conformations that can be associated with different pathogenic properties. These conformational differences are referred to as “strains” and can be propagated by templated misfolding.
Interestingly, structure analysis of fibrils formed by amyloidogenic proteins or fragments thereof provides a plausible and attractive explanation for the efficient incorporation of homologous molecules into the preformed nucleus. Amyloid fibrils were found to typically consist of sets of β‐sheets arranged parallel to the fibril axis, with β‐sheets either parallel or antiparallel and usually in‐register 24. The side chains within and in between the sheets are tightly interdigitated and form a dry steric zipper devoid of water in the interface. Such structures allow for compact stacking of molecules and are stabilized by intermolecular interaction. Homologous molecules can be incorporated accordingly and this leads to progressive growth of the amyloid fibril (Figure 1C). For Aβ fibrils, such structures were proposed from analyses of synthetic or recombinant Aβ peptides aggregated in vitro by several groups 8, 18, 48, 63, 77, 84, 85. Although detailed structural information of Aβ fibrils in brain tissue is not available yet, it is conceivable that similar structures are formed in vivo 44, 61. Such fibrils can grow from one or both ends, depending on the polarity of the fibril. The growth rate depends (i) on the availability of soluble, homologous molecules for incorporation; (ii) on the efficiency of incorporation; and (iii) on the rate of fragmentation that generates more molecular interfaces for interaction of nucleus and homologous molecules. So, essentially proteins in their abnormal multimeric conformation seem to directly interact with native or partially unfolded homologous proteins and template them into the abnormal, misfolded form. Therefore, this phenomenon has also been termed “templated misfolding.” In case a given protein can adopt two or more differently organized aggregate formations that are reliably sustained by templated misfolding, these distinct aggregates are known as different “strains” of aggregates 2, 24 (Figure 1D). Different strains of aggregates can have different pathological properties and may differ in their growth and fragmentation rates 2.
The principle of templated misfolding was first proposed and is best characterized for the misfolding cascade of prion proteins 17, 66 (Kretzschmar and Tatzelt, this issue). Only recently similar mechanisms of misfolding are discussed for other proteins associated with neurodegenerative diseases, for example, Aβ, tau (Clavaguera et al, this issue), α‐synuclein (George et al this issue) as well as several others 65, 67. With respect to proteins for which this principle of misfolding is still under debate, it is often referred to as “prion‐like” misfolding. However, an important difference exists between the prion diseases and other disorders with putative “prion‐like” protein misfolding: only prion diseases have been shown to be “infectious” in a sense that the disease‐causing agent can be acquired from the environment and “infect” a new organism. There is no evidence that any of the other disorders with putative “prion‐like” protein aggregation are “infectious” or “transmissible” in a similar sense. This is likely because of the absence of an “infectious cycle” 1. AD is a disorder that only occurs in humans and the signature protein aggregates are confined to the individual's brain 21, 83. Moreover, clinical characteristics of AD do not suggest an infectious nature 1. Even in patients treated with cadaver‐derived human growth hormone, no evidence for iatrogenic transmissibility of AD or other protein aggregation disorders was found, except prion diseases 37. However, within the brain, templated misfolding of Aβ peptides may explain disease progression.
Evidence for Prion‐Like Templated Misfolding of Aβ
In the human brain, Aβ peptides are constantly generated from APP by proteolytic cleavage by β‐secretase (BACE‐1) generating the N‐terminus and by the γ‐secretase complex generating the C‐terminus 33. γ‐Secretase cleavage generates Aβ peptides of various lengths ranging from 37 to 43 residues 68. The 40‐residue peptide Aβ (1–40) is the most abundant species in normal brain, followed by Aβ (1–42) species 94. In general, longer Aβ peptides are more aggregation prone than shorter ones 15, 68.
In vitro synthetic or recombinant Aβ peptides readily form aggregates of different kinds: low to high n oligomers have been described as well as protofibrils, annular aggregates and mature fibrillar structures 90. Aggregation in vitro is concentration‐dependent and the precise conformation depends on the Aβ species studied (eg, Aβ40 or Aβ42) as well as on agitation, temperature, pH and peptide concentration. A detailed comparison of the structures of Aβ oligomers and fibrils is beyond the scope of this review and has been described elsewhere 24, 26, 86. Interestingly, Aβ aggregation in vitro can be initiated by the addition of preformed Aβ aggregates (“seeds”) 36, 40, 74, 93. This circumvents the lag phase and supports the concept of nucleation‐dependent aggregation. When different polymorphs of Aβ fibrils were used for in vitro seeding, the structural properties of the seeds were reliably propagated to the seeded aggregates indicating templated misfolding 64.
However, most evidence for prion‐like templated misfolding of Aβ comes from inoculation studies in transgenic (tg) rodents. The full spectrum of AD is a human‐specific disorder and only partially similar pathological changes including cerebral Aβ deposition were found in other mammals. Although the reasons for this are not fully understood, it is likely a combination of differences in APP and Aβ sequence as well as shorter lifespan of other mammals in comparison to the aging human society. Therefore, numerous tg mice and a few tg rats overexpressing human APP and generating human Aβ peptides were made and are often used to study cerebral β‐amyloidosis 4, 41, 70. Most of these tg rodents recapitulate age‐related onset of Aβ aggregation in the brain in the form of parenchymal plaques and vascular β‐amyloid very similar to the lesions characteristic for AD. The age‐of‐onset varies between the different tg lines and depends on the levels of APP overexpression and related Aβ levels. Insertion of APP mutations associated with familial AD (FAD) or additional overexpression of human presenilin variants with AD‐causing mutations promotes earlier Aβ deposition.
Several laboratories have studied “seeded” induction of cerebral Aβ aggregation in tg rodents 43, 55, 56, 92. The experimental paradigm typically uses dilute homogenates from brains of AD patients or of aged APP tg mice with abundant cerebral β‐amyloidosis. Minute amounts of these homogenates are injected into the brains of young, predepositing APP tg rodents and the development of cerebral β‐amyloidosis is monitored over time.
The essential findings are (see also Figure 2) as follows: Brain extracts from AD patients or from aged APP tg mice containing in vivo generated Aβ aggregates are capable of inducing premature cerebral β‐amyloidosis in young APP tg rodents 22, 34, 43, 45, 55, 56, 73, 80, 92. Interestingly, extracts derived from aged APP tg mice with abundant cerebral β‐amyloidosis were similarly effective as extracts from AD cases ruling out that factors exclusive to the human brain are essential 55. In contrast, control extracts from healthy individuals or aged wild‐type mice without Aβ aggregates fail to induce cerebral β‐amyloidosis 22, 45, 55. Likewise, induction of cerebral β‐amyloidosis is prevented when Aβ peptides are immunodepleted from the extract or when extracts are treated with formic acid resolving any tertiary protein structure. This indicates that misfolded Aβ peptides are essential components of the “seeding extract” 55. In addition, seeding is significantly reduced when Aβ peptides are neutralized by anti‐Aβ antibodies 55. Induced cerebral β‐amyloidosis was found to be time‐dependent and occurs in histologically detectable amounts with a delay of several months 22, 34, 45, 55, 92. Induction of cerebral β‐amyloidosis was also found to depend on the concentration of seeds applied 55. This supports the concept that more seeds, that is, more interfaces for templated misfolding can more efficiently recruit soluble Aβ peptides for templated misfolding.
Figure 2.
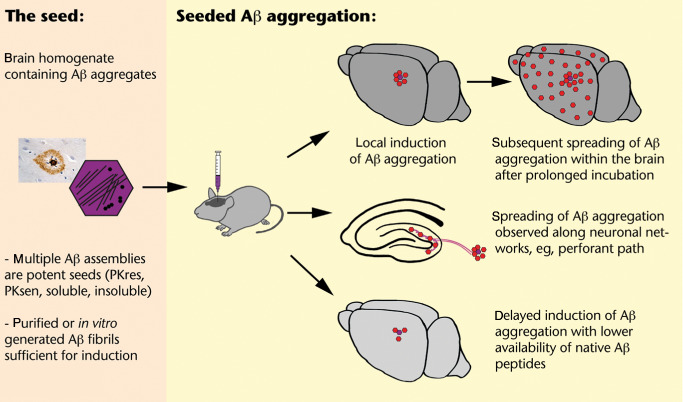
Schematic overview on induction and spreading of Aβ deposits in brains of young APP transgenic mice following intracerebral seeding with brain homogenates containing aggregated Aβ peptides 22 , 23 , 34 , 45 , 55 , 80 . [PKres = proteinase K resistant; PKsen = proteinase K sensitive under standard conditions (50 μg PK/mL homogenate, 30 min, 37°C)].
The lag time until induced Aβ deposits are immunohistochemically detectable is not only dependent on the amount of seeds applied but also on the level of APP overexpression in the tg line used. Lower APP overexpression and thus lower availability of soluble Aβ peptides for misfolding goes along with an extended lag phase 34, 55, 56. In the brains of non‐tg mice, no induced Aβ deposition was found with the same kind and amount of extracts and same incubation time 43, 55. Murine Aβ sequence differs from human Aβ sequence by three amino acids and can possibly not easily accommodate the misfolded amyloid state of human Aβ peptides. This again would indicate specificity of templated misfolding. However, it should not be overlooked that, all other rodent lines inoculated generate higher amounts of Aβ peptides because of tg overexpression. Therefore, it is conceivable that lowest availability of soluble Aβ peptides in non‐tg mice is not sufficient for seeded induction of histopathologically detectable Aβ aggregation within the lifespan of these mice although templated misfolding may occur at low levels.
Interestingly, multiple Aβ assemblies seem capable of seeding Aβ aggregation, that is, Aβ aggregates resistant or sensitive to standard proteinase K treatment, soluble or insoluble aggregates when differentiated by 100 000 × g ultracentrifugation, and may thus represent a continuum of seeding‐capable Aβ aggregates 45. Soluble seeds seemed to exert high activity, probably because of the presence of many molecular interfaces for templated misfolding 45. This interpretation is also supported by the observation that fragmentation increases the seeding capacity of a given extract 45.
Interestingly, induced deposits are first apparent locally at the site of injection 22, 34, 55. With prolonged incubation times, induced Aβ aggregates increase locally but also appear in neuroanatomically connected brain regions: inoculations into the entorhinal cortex induced Aβ deposits in the outer molecular layer of the dentate gyrus, that is, the terminal zone of projections of the perforant path which connects the entorhinal cortex to the hippocampus 22, 42 and vice versa 89. It was observed that local application of Aβ seeds can induce widespread Aβ deposition throughout cortex and hippocampus in APP tg mice that would otherwise not have any histochemically detectable Aβ deposition at this age 34. This suggests that Aβ aggregates may actively or passively traffic to neighboring and interconnected brain areas. Remarkably, peripheral application of Aβ seeds can also induce cerebral β‐amyloidosis in young APP tg mice 23. However, it is unclear how this finding would relate to the human situation.
Recently, Aβ fibrils purified from β‐amyloid‐laden mouse brains as well as fibrils generated in vitro from synthetic Aβ were shown to be capable of inducing cerebral β‐amyloidosis in young APP tg mice and are discussed as the final proof that misfolded Aβ by itself promotes prion‐like templated misfolding and induces associated pathology 80.
Perhaps the most convincing studies suggesting prion‐like templated misfolding of Aβ peptides are observations of reliable propagation of conformational strains. It is well known that Aβ deposits in the AD brain are heterogeneous in histopathological appearance both within and among brain regions and patients. Amyloid plaques are diffuse or compact, smaller or larger in appearance and this is also reflected in the biochemical composition 49, 82, 83. When different extracts were used for the inoculation studies, derived from either APPPS1 tg mice with small and compact, more punctate plaques 69 or from APP23 tg mice with larger and more filamentous and diffuse plaques 81, the characteristic plaque morphology of the donor was replicated in the host (Figure 3) 55. This indicates that the type of Aβ aggregates present in the seeding extract has a major influence in shaping the induced deposits. This could be conveyed by prion‐like templated misfolding. At present, it is not well known if polymorphic Aβ “strains” are also polyfunctional and are linked to differences in toxicity or the clinical spectrum of AD.
Figure 3.
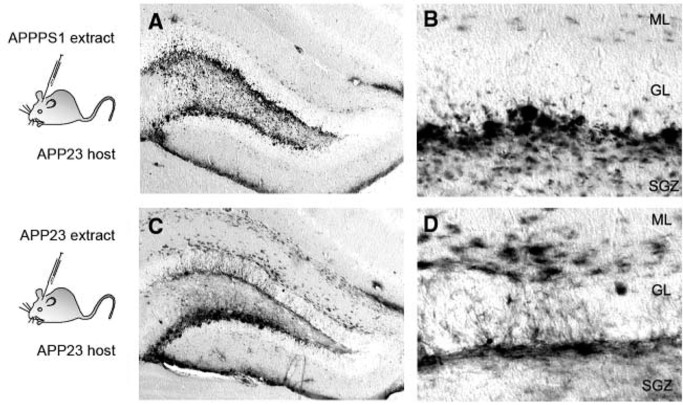
Seeding in young APP23 transgenic hosts induced different morphotypes of Aβ deposits when different extracts were used for intracerebral inoculations. Note the more punctate appearance of induced deposits with APPPS1 extract, especially in the subgranular cell layer (SGZ) (A, B) in contrast to more filamentous and diffuse Aβ deposits induced with APP23 extract (C, D). The morphotypes of the induced Aβ deposits resemble the Aβ deposits found in the brains of the donors of the extracts [from Meyer‐Luehmann et al 55. Reprinted with permission from AAAS]. ML = molecular layer; GL = granular cell layer.
Taken together, all these studies in vitro and in vivo support the concept of prion‐like templated misfolding of Aβ peptides as defined earlier. Although tg rodents predisposed to developing cerebral β‐amyloidosis were used, the observation of induced Aβ deposits in tg lines without Aβ deposits within the normal lifespan suggests true seeded conversion of normally soluble Aβ peptides and not simply acceleration of an ongoing process 56, 73. However, other possible explanations should not be neglected and will largely have to take into account that inoculation of Aβ aggregates may cause secondary effects that indirectly promote Aβ aggregation, especially in environments with unnaturally high concentrations of Aβ peptides. Regarding the observed spreading of Aβ deposits within neuronal networks, indirect effects of compromised neuronal functions that may affect APP processing or Aβ levels are conceivable. Moreover, these experiments in tg rodents focused on Aβ aggregation, and therefore do not allow a conclusion if induction of cerebral Aβ deposition would provoke the whole spectrum of clinical AD. However, it was repeatedly observed that the induced deposits resemble typical AD‐like Aβ deposits and can be accompanied by dystrophic neurites with typical hyperphosphorylation of tau proteins, as well as micro‐ and astrogliosis 22, 23, 34, 45, 55, 92. While induced deposits may be largely diffuse initially, a more compact and congophilic appearance was noted with longer incubation times, indicating maturation toward more pathological lesions 45. In addition, region‐specific differences in induced deposits may point to the importance of additional cellular factors shaping Aβ aggregation, for example, induced deposits in the striatum of APP23 tg mice were largely diffuse within the observation time whereas cortical or hippocampal inoculation resulted in more compact deposits 22. Last but not least, minute amounts of Aβ aggregates may be sufficient to trigger progressive tau aggregation and pathology 11, 29.
Naturally, no such inoculation studies can be undertaken in humans. Attempts in nonhuman primates were initially inconclusive 30, but subsequent work has shown that intracerebral inoculation of crude homogenates from AD brain into marmosets can induce β‐amyloid deposition after incubation times of at least 5–6 years 6, 71. However, because of the high prevalence of AD and Aβ‐CAA and the suggestive spreading within brain networks 13, 78, it will be important to further probe this concept for understanding the human disorders and for developing therapeutics.
Implications of Prion‐Like Templated Misfolding of Aβ Peptides
These findings have multiple implications with regard to future research and to the development of therapeutics for the treatment and the prevention of Aβ‐misfolding disorders. Disease‐specific Aβ aggregation in the brains of individuals in prodromal AD stages has been detected many years to decades before clinical symptoms manifest 39, 62. Aβ seeds if present in body fluids (eg, CSF) could serve as an early and disease‐specific biomarker, complementing currently used measurements of total CSF Aβ and tau levels 10. However, it is at present unknown if any kind of Aβ seed exists outside the brain, and if so, how efficient its capability to transmit the misfolded state via templated misfolding would be. Evidently, if such seeds would exist, accidental transmission between individuals must be avoided, particularly as the seeds may be chemically stable and resistant to some common inactivation procedures.
Our current understanding of the kinetics of Aβ aggregation and the indications for prion‐like templated misfolding highlights several possibilities for therapeutic intervention: First, by keeping Aβ levels below the critical (local) concentration for aggregation. This could be achieved by inhibition or modulation of the enzymes releasing Aβ from its precursor 28, 95, by increasing Aβ degradation 75 or by promoting its clearance. Second, by removing existing aggregates. Some enzymes have been shown to be capable of degrading or dissociating small Aβ aggregates 75. In addition, immunotherapy was shown to be efficient in removing Aβ deposits 46. Third, by capping templated misfolding or by promoting the formation of less toxic aggregates. In principle, the cascade of progressive templated misfolding would be stopped if the molecular interfaces for templated misfolding are not available 24. Similarly, if differentially toxic Aβ aggregate versions exist, a shift toward less toxic and more biologically inert forms could be beneficial. Fourth, preventing the spreading of Aβ aggregation to neighboring or interconnected tissue may limit the detrimental effects and concomitant progressive cognitive deficits. Fifth, and maybe most importantly, compounds that preserve neuronal and synaptic integrity and function could keep dementia at bay. The prediction would be that all disease‐modifying strategies are most effective as prevention or in the early stages of the disease because severe pathology and massive neuron loss in late stages are likely irreversible. Such early or preventive therapeutics may have to be taken for many years or decades, and therefore, side effects should be avoided and well‐tolerated yet effective medication would be desirable. This is especially challenging as we yet lack a good understanding of the physiological functions of APP, its diverse fragments including Aβ and the enzymes involved in APP processing. In the end, likely not one but several of the possible therapeutic approaches outlined above may be needed for preventing cognitive decline. There is a silver lining for causative or preventive AD treatment as several compounds targeting Aβ‐generating enzymes, anti‐Aβ immunotherapy and others are already being tested in clinical trials and some are under consideration for preventive trials 27. While Aβ aggregation may be the most relevant trigger for Aβ‐CAA, it may only be the starting point for AD. Changes in tau phosphorylation and eventually tau aggregation may be the effectors leading to AD neurodegeneration 38, 72, 88. As tau aggregation likewise seems to propagate by prion‐like templated misfolding (Clavaguera et al, this issue), it will be important for any Aβ‐targeting AD therapeutics to stop Aβ aggregation before tau aggregation proceeds beyond the point of no return.
Conclusions
Prion‐like templated misfolding of Aβ peptides provides a plausible framework to explain the progressive accumulation and spreading of Aβ aggregation within the AD brain. This concept has far‐reaching implications for possible therapeutic interventions. In particular, the need for early intervention before a fatal cascade of Aβ misfolding leads to neuropathological damage beyond repair. However, more work is needed to fully understand the characteristics of this process and its contribution to AD and other Aβ‐misfolding disorders. At present, prion‐like templated misfolding is discussed as a driving force in several neurodegenerative disorders. This could unify experimental and translational approaches to these increasingly prevalent and devastating disorders.
Funding
The work in our laboratory was supported by grants from the Competence Network on Degenerative Dementias (BMBF‐01G10705) and the Alzheimer Forschung Initiative e.V. (AFI).
Conflict of Interest
The author declares that she has no conflicting interest related to this article.
Acknowledgments
The author would like to thank Mathias Jucker, Lary Walker, Matthias Staufenbiel and all members of the laboratory for the support and helpful discussion.
References
- 1. Aguzzi A (2009) Cell biology: beyond the prion principle. Nature 459:924–925. [DOI] [PubMed] [Google Scholar]
- 2. Aguzzi A, Heikenwalder M, Polymenidou M (2007) Insights into prion strains and neurotoxicity. Nat Rev Mol Cell Biol 8:552–561. [DOI] [PubMed] [Google Scholar]
- 3. Alzheimer A (1907) Über eine eigenartige Erkrankung der Hirnrinde. Allg Zeitschr Psychiatr 64:146–148. [Google Scholar]
- 4. Ashe KH, Zahs KR (2010) Probing the biology of Alzheimer's disease in mice. Neuron 66:631–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Askanas V, Engel WK (2006) Inclusion‐body myositis: a myodegenerative conformational disorder associated with Abeta, protein misfolding, and proteasome inhibition. Neurology 66(Suppl. 1):S39–S48. [DOI] [PubMed] [Google Scholar]
- 6. Baker HF, Ridley RM, Duchen LW, Crow TJ, Bruton CJ (1994) Induction of beta (A4)‐amyloid in primates by injection of Alzheimer's disease brain homogenate. Comparison with transmission of spongiform encephalopathy. Mol Neurobiol 8:25–39. [DOI] [PubMed] [Google Scholar]
- 7. Bentahir M, Nyabi O, Verhamme J, Tolia A, Horre K, Wiltfang J et al (2006) Presenilin clinical mutations can affect gamma‐secretase activity by different mechanisms. J Neurochem 96:732–742. [DOI] [PubMed] [Google Scholar]
- 8. Benzinger TL, Gregory DM, Burkoth TS, Miller‐Auer H, Lynn DG, Botto RE, Meredith SC (1998) Propagating structure of Alzheimer's beta‐amyloid(10‐35) is parallel beta‐sheet with residues in exact register. Proc Natl Acad Sci U S A 95:13407–13412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Biffi A, Greenberg SM (2011) Cerebral amyloid angiopathy: a systematic review. J Clin Neurol 7:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Blennow K, Zetterberg H, Fagan AM (2012) Fluid biomarkers in Alzheimer disease. Cold Spring Harb Perspect Med 2:a006221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bolmont T, Clavaguera F, Meyer‐Luehmann M, Herzig MC, Radde R, Staufenbiel M et al (2007) Induction of tau pathology by intracerebral infusion of amyloid‐beta‐containing brain extract and by amyloid‐beta deposition in APPxTau transgenic mice. Am J Pathol 171(6):2012–2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Borchelt DR, Thinakaran G, Eckman CB, Lee MK, Davenport F, Ratovitsky T et al (1996) Familial Alzheimer's disease‐linked presenilin 1 variants elevate Abeta1‐42/1‐40 ratio in vitro and in vivo . Neuron 17:1005–1013. [DOI] [PubMed] [Google Scholar]
- 13. Braak H, Braak E (1991) Neuropathological stageing of Alzheimer‐related changes. Acta Neuropathol 82:239–259. [DOI] [PubMed] [Google Scholar]
- 14. Braak H, Braak E (1997) Frequency of stages of Alzheimer‐related lesions in different age categories. Neurobiol Aging 18:351–357. [DOI] [PubMed] [Google Scholar]
- 15. Burdick D, Soreghan B, Kwon M, Kosmoski J, Knauer M, Henschen A et al (1992) Assembly and aggregation properties of synthetic Alzheimer's A4/beta amyloid peptide analogs. J Biol Chem 267:546–554. [PubMed] [Google Scholar]
- 16. Citron M, Westaway D, Xia W, Carlson G, Diehl T, Levesque G et al (1997) Mutant presenilins of Alzheimer's disease increase production of 42‐residue amyloid beta‐protein in both transfected cells and transgenic mice. Nat Med 3:67–72. [DOI] [PubMed] [Google Scholar]
- 17. Colby DW, Prusiner SB (2011) Prions. Cold Spring Harb Perspect Biol 3:a006833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Colletier JP, Laganowsky A, Landau M, Zhao M, Soriaga AB, Goldschmidt L et al (2011) Molecular basis for amyloid‐beta polymorphism. Proc Natl Acad Sci U S A 108:16938–16943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. De Jonghe C, Esselens C, Kumar‐Singh S, Craessaerts K, Serneels S, Checler F et al (2001) Pathogenic APP mutations near the gamma‐secretase cleavage site differentially affect Abeta secretion and APP C‐terminal fragment stability. Hum Mol Gen 10:1665–1671. [DOI] [PubMed] [Google Scholar]
- 20. Duff K, Eckman C, Zehr C, Yu X, Prada CM, Perez‐tur J et al (1996) Increased amyloid‐beta42 (43) in brains of mice expressing mutant presenilin 1. Nature 383:710–713. [DOI] [PubMed] [Google Scholar]
- 21. Duyckaerts C, Delatour B, Potier MC (2009) Classification and basic pathology of Alzheimer disease. Acta Neuropathol 118:5–36. [DOI] [PubMed] [Google Scholar]
- 22. Eisele YS, Bolmont T, Heikenwalder M, Langer F, Jacobson LH, Yan ZX et al (2009) Induction of cerebral beta‐amyloidosis: intracerebral versus systemic Abeta inoculation. Proc Natl Acad Sci U S A 106:12926–12931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Eisele YS, Obermuller U, Heilbronner G, Baumann F, Kaeser SA, Wolburg H et al (2010) Peripherally applied Abeta‐containing inoculates induce cerebral beta‐amyloidosis. Science 330:980–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Eisenberg D, Jucker M (2012) The amyloid state of proteins in human diseases. Cell 148:1188–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Falsig J, Nilsson KP, Knowles TP, Aguzzi A (2008) Chemical and biophysical insights into the propagation of prion strains. HFSP J 2:332–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Fandrich M (2012) Oligomeric intermediates in amyloid formation: structure determination and mechanisms of toxicity. J Mol Biol 421:427–440. [DOI] [PubMed] [Google Scholar]
- 27. Gerald Z, Ockert W (2012) Alzheimer's disease market: hope deferred. Nat Rev Drug Discov 12:19–20. [DOI] [PubMed] [Google Scholar]
- 28. Ghosh AK, Brindisi M, Tang J (2012) Developing β‐secretase inhibitors for treatment of Alzheimer's disease. J Neurochem 120(Suppl. 1):71–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gotz J, van Chen F, Dorpe J, Nitsch RM (2001) Formation of neurofibrillary tangles in P301l tau transgenic mice induced by Abeta 42 fibrils. Science 293:1491–1495. [DOI] [PubMed] [Google Scholar]
- 30. Goudsmit J, Morrow CH, Asher DM, Yanagihara RT, Masters CL, Gibbs CJ Jr, Gajdusek DC (1980) Evidence for and against the transmissibility of Alzheimer disease. Neurology 30:945–950. [DOI] [PubMed] [Google Scholar]
- 31. Graeber MB, Mehraein P (1999) Reanalysis of the first case of Alzheimer's disease. Eur Arch Psychiatry Clin Neurosci 249(Suppl. 3):10–13. [DOI] [PubMed] [Google Scholar]
- 32. Greenberg SA (2009) Inclusion body myositis: review of recent literature. Curr Neurol Neurosci Rep 9:83–89. [DOI] [PubMed] [Google Scholar]
- 33. Haass C, Kaether C, Thinakaran G, Sisodia S (2012) Trafficking and proteolytic processing of APP. Cold Spring Harb Perspect Med 2:a006270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hamaguchi T, Eisele YS, Varvel NH, Lamb BT, Walker LC, Jucker M (2012) The presence of Aβ seeds, and not age per se, is critical to the initiation of Aβ deposition in the brain. Acta Neuropathol 123:31–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hardy J, Selkoe DJ (2002) The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science 297:353–356. [DOI] [PubMed] [Google Scholar]
- 36. Harper JD, Lansbury PT Jr (1997) Models of amyloid seeding in Alzheimer's disease and scrapie: mechanistic truths and physiological consequences of the time‐dependent solubility of amyloid proteins. Annu Rev Biochem 66:385–407. [DOI] [PubMed] [Google Scholar]
- 37. Irwin DJ, Abrams JY, Schonberger LB, Leschek EW, Mills JL, Lee VM, Trojanowski JQ (2013) Evaluation of potential infectivity of Alzheimer and Parkinson disease proteins in recipients of cadaver‐derived human growth hormone. JAMA Neurol 4:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ittner LM, Ke YD, Delerue F, Bi M, van Gladbach A, Eersel J et al (2010) Dendritic function of tau mediates amyloid‐beta toxicity in Alzheimer's disease mouse models. Cell 142:387–397. [DOI] [PubMed] [Google Scholar]
- 39. Jack CR Jr, Knopman DS, Jagust WJ, Shaw LM, Aisen PS, Weiner MW et al (2010) Hypothetical model of dynamic biomarkers of the Alzheimer's pathological cascade. Lancet Neurol 9:119–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Jarrett JT, Lansbury PT Jr (1993) Seeding “one‐dimensional crystallization” of amyloid: a pathogenic mechanism in Alzheimer's disease and scrapie? Cell 73:1055–1058. [DOI] [PubMed] [Google Scholar]
- 41. Jucker M (2010) The benefits and limitations of animal models for translational research in neurodegenerative diseases. Nat Med 16:1210–1214. [DOI] [PubMed] [Google Scholar]
- 42. Jucker M, Walker LC (2011) Pathogenic protein seeding in Alzheimer disease and other neurodegenerative disorders. Ann Neurol 70:532–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kane MD, Lipinski WJ, Callahan MJ, Bian F, Durham RA, Schwarz RD et al (2000) Evidence for seeding of beta‐amyloid by intracerebral infusion of Alzheimer brain extracts in beta‐amyloid precursor protein‐transgenic mice. J Neurosci 20:3606–3611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kuo YM, Kokjohn TA, Beach TG, Sue LI, Brune D, Lopez JC et al (2001) Comparative analysis of amyloid‐beta chemical structure and amyloid plaque morphology of transgenic mouse and Alzheimer's disease brains. J Biol Chem 276:12991–12998. [DOI] [PubMed] [Google Scholar]
- 45. Langer F, Eisele YS, Fritschi SK, Staufenbiel M, Walker LC, Jucker M (2011) Soluble Aβ seeds are potent inducers of cerebral β‐amyloid deposition. J Neurosci 31:14488–14495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Liu YH, Giunta B, Zhou HD, Tan J, Wang YJ (2012) Immunotherapy for Alzheimer disease: the challenge of adverse effects. Nat Rev Neurol 8:465–469. [DOI] [PubMed] [Google Scholar]
- 47. Lue LF, Kuo YM, Roher AE, Brachova L, Shen Y, Sue L et al (1999) Soluble amyloid beta peptide concentration as a predictor of synaptic change in Alzheimer's disease. Am J Pathol 15(3):853–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Luhrs T, Ritter C, Adrian M, Riek‐Loher D, Bohrmann B, Dobeli H et al (2005) 3D structure of Alzheimer's amyloid‐beta(1‐42) fibrils. Proc Natl Acad Sci U S A 102:17342–17347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Maarouf CL, Daugs ID, Spina S, Vidal R, Kokjohn TA, Patton RL et al (2008) Histopathological and molecular heterogeneity among individuals with dementia associated with Presenilin mutations. Mol Neurodegener 3:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Mandelkow EM, Mandelkow E (2012) Biochemistry and cell biology of tau protein in neurofibrillary degeneration. Cold Spring Harb Perspect Med 2:a006247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Masters CL, Selkoe DJ (2012) Biochemistry of amyloid β‐protein and amyloid deposits in Alzheimer disease. Cold Spring Harb Perspect Med 2:a006262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Mastrangelo P, Mathews PM, Chishti MA, Schmidt SD, Gu Y, Yang J et al (2005) Dissociated phenotypes in presenilin transgenic mice define functionally distinct gamma‐secretases. Proc Natl Acad Sci U S A 102:8972–8977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Maurer K, Volk S, Gerbaldo H (1997) Auguste D and Alzheimer's disease. Lancet 349(9064):1546–1549. [DOI] [PubMed] [Google Scholar]
- 54. McLean CA, Cherny RA, Fraser FW, Fuller SJ, Smith MJ, Beyreuther K et al (1999) Soluble pool of Abeta amyloid as a determinant of severity of neurodegeneration in Alzheimer's disease. Ann Neurol 46(6):860–866. [DOI] [PubMed] [Google Scholar]
- 55. Meyer‐Luehmann M, Coomaraswamy J, Bolmont T, Kaeser S, Schaefer C, Kilger E et al (2006) Exogenous induction of cerebral beta‐amyloidogenesis is governed by agent and host. Science 313:1781–1784. [DOI] [PubMed] [Google Scholar]
- 56. Morales R, Duran‐Aniotz C, Castilla J, Estrada LD, Soto C (2012) De novo induction of amyloid‐β deposition in vivo . Mol Psychiatry 17:1347–1353. [DOI] [PubMed] [Google Scholar]
- 57. Mucke L, Selkoe DJ (2012) Neurotoxicity of amyloid β‐protein: synaptic and network dysfunction. Cold Spring Harb Perspect Med 2:a006338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Muller U, Winter P, Graeber MB (2013) A presenilin 1 mutation in the first case of Alzheimer's disease. Lancet Neurol 12(2):129–130. [DOI] [PubMed] [Google Scholar]
- 59. Naslund J, Haroutunian V, Mohs R, Davis KL, Davies P, Greengard P, Buxbaum JD (2000) Correlation between elevated levels of amyloid beta‐peptide in the brain and cognitive decline. JAMA 283:1571–1577. [DOI] [PubMed] [Google Scholar]
- 60. Nogalska A, D'Agostino C, Engel WK, Klein WL, Askanas V (2010) Novel demonstration of amyloid‐β oligomers in sporadic inclusion‐body myositis muscle fibers. Acta Neuropathol 120:661–666. [DOI] [PubMed] [Google Scholar]
- 61. Paravastu AK, Qahwash I, Leapman RD, Meredith SC, Tycko R (2009) Seeded growth of β‐amyloid fibrils from Alzheimer's brain‐derived fibrils produces a distinct fibril structure. Proc Natl Acad Sci U S A 106:7443–7448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Perrin RJ, Fagan AM, Holtzman DM (2009) Multimodal techniques for diagnosis and prognosis of Alzheimer's disease. Nature 461:916–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Petkova AT, Ishii Y, Balbach JJ, Antzutkin ON, Leapman RD, Delaglio F, Tycko R (2002) A structural model for Alzheimer's beta‐amyloid fibrils based on experimental constraints from solid state NMR. Proc Natl Acad Sci U S A 99:16742–16747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Petkova AT, Leapman RD, Guo Z, Yau WM, Mattson MP, Tycko R (2005) Self‐propagating, molecular‐level polymorphism in Alzheimer's beta‐amyloid fibrils. Science 307:262–265. [DOI] [PubMed] [Google Scholar]
- 65. Polymenidou M, Cleveland DW (2012) Prion‐like spread of protein aggregates in neurodegeneration. J Exp Med 209:889–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Prusiner SB (1982) Novel proteinaceous infectious particles cause scrapie. Science 216:136–144. [DOI] [PubMed] [Google Scholar]
- 67. Prusiner SB (2012) Cell biology. A unifying role for prions in neurodegenerative diseases. Science 336:1511–1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Qi‐Takahara Y, Morishima‐Kawashima M, Tanimura Y, Dolios G, Hirotani N, Horikoshi Y et al (2005) Longer forms of amyloid beta protein: implications for the mechanism of intramembrane cleavage by gamma‐secretase. J Neurosci 25:436–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Radde R, Bolmont T, Kaeser SA, Coomaraswamy J, Lindau D, Stoltze L et al (2006) Abeta42‐driven cerebral amyloidosis in transgenic mice reveals early and robust pathology. EMBO Rep 7:940–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Radde R, Duma C, Goedert M, Jucker M (2008) The value of incomplete mouse models of Alzheimer's disease. Eur J Nucl Med Mol Imaging 35(Suppl. 1):S70–S74. [DOI] [PubMed] [Google Scholar]
- 71. Ridley RM, Baker HF, Windle CP, Cummings RM (2006) Very long term studies of the seeding of beta‐amyloidosis in primates. J Neural Transm 113:1243–1251. [DOI] [PubMed] [Google Scholar]
- 72. Roberson ED, Scearce‐Levie K, Palop JJ, Yan F, Cheng IH, Wu T et al (2007) Reducing endogenous tau ameliorates amyloid beta‐induced deficits in an Alzheimer's disease mouse model. Science 316:750–754. [DOI] [PubMed] [Google Scholar]
- 73. Rosen RF, Fritz JJ, Dooyema J, Cintron AF, Hamaguchi T, Lah JJ et al (2012) Exogenous seeding of cerebral β‐amyloid deposition in βAPP‐transgenic rats. J Neurochem 120:660–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Roychaudhuri R, Yang M, Hoshi MM, Teplow DB (2009) Amyloid beta‐protein assembly and Alzheimer disease. J Biol Chem 284:4749–4753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Saido T, Leissring MA (2012) Proteolytic degradation of amyloid β‐protein. Cold Spring Harb Perspect Med 2:a006379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Scheuner D, Eckman C, Jensen M, Song X, Citron M, Suzuki N et al (1996) Secreted amyloid beta‐protein similar to that in the senile plaques of Alzheimer's disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer's disease. Nat Med 2:864–870. [DOI] [PubMed] [Google Scholar]
- 77. Schmidt M, Sachse C, Richter W, Xu C, Fandrich M, Grigorieff N (2009) Comparison of Alzheimer Abeta(1‐40) and Abeta(1‐42) amyloid fibrils reveals similar protofilament structures. Proc Natl Acad Sci U S A 106:19813–19818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Seeley WW, Crawford RK, Zhou J, Miller BL, Greicius MD (2009) Neurodegenerative diseases target large‐scale human brain networks. Neuron 62:42–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Serrano‐Pozo A, Frosch MP, Masliah E, Hyman BT (2011) Neuropathological alterations in Alzheimer disease. Cold Spring Harb Perspect Med 1:a006189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Stohr J, Watts JC, Mensinger ZL, Oehler A, Grillo SK, DeArmond SJ et al (2012) Purified and synthetic Alzheimer's amyloid beta (Aβ) prions. Proc Natl Acad Sci U S A 109:11025–11030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Sturchler‐Pierrat C, Abramowski D, Duke M, Wiederhold KH, Mistl C, Rothacher S et al (1997) Two amyloid precursor protein transgenic mouse models with Alzheimer disease‐like pathology. Proc Natl Acad Sci U S A 94:13287–13292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Tekirian TL, Saido TC, Markesbery WR, Russell MJ, Wekstein DR, Patel E, Geddes JW (1998) N‐terminal heterogeneity of parenchymal and cerebrovascular Abeta deposits. J Neuropathol Exp Neurol 57:76–94. [DOI] [PubMed] [Google Scholar]
- 83. Thal DR, Capetillo‐Zarate E, Del Tredici K, Braak H (2006) The development of amyloid beta protein deposits in the aged brain. Sci Aging Knowledge Environ 2006:re1. [DOI] [PubMed] [Google Scholar]
- 84. Torok M, Milton S, Kayed R, Wu P, McIntire T, Glabe CG, Langen R (2002) Structural and dynamic features of Alzheimer's Abeta peptide in amyloid fibrils studied by site‐directed spin labeling. J Biol Chem 277:40810–40815. [DOI] [PubMed] [Google Scholar]
- 85. Tycko R (2011) Solid‐state NMR studies of amyloid fibril structure. Ann Rev Phys Chem 62:279–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Tycko R, Wickner RB (2013) Molecular structures of amyloid and prion fibrils: consensus versus controversy. Acc Chem Res. doi: 10.1021/ar300282r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Vattemi G, Nogalska A, King Engel W, D'Agostino C, Checler F, Askanas V (2009) Amyloid‐beta42 is preferentially accumulated in muscle fibers of patients with sporadic inclusion‐body myositis. Acta Neuropathol 117:569–574. [DOI] [PubMed] [Google Scholar]
- 88. Vossel KA, Zhang K, Brodbeck J, Daub AC, Sharma P, Finkbeiner S et al (2010) Tau reduction prevents Abeta‐induced defects in axonal transport. Science 330:198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Walker LC, Callahan MJ, Bian F, Durham RA, Roher AE, Lipinski WJ (2002) Exogenous induction of cerebral beta‐amyloidosis in betaAPP‐transgenic mice. Peptides 23:1241–1247. [DOI] [PubMed] [Google Scholar]
- 90. Walsh DM, Selkoe DJ (2007) A beta oligomers—a decade of discovery. J Neurochem 101:1172–1184. [DOI] [PubMed] [Google Scholar]
- 91. Wang J, Dickson DW, Trojanowski JQ, Lee VM (1999) The levels of soluble versus insoluble brain Abeta distinguish Alzheimer's disease from normal and pathological aging. Exp Neurol 158(2):328–337. [DOI] [PubMed] [Google Scholar]
- 92. Watts JC, Giles K, Grillo SK, Lemus A, DeArmond SJ, Prusiner SB (2011) Bioluminescence imaging of Abeta deposition in bigenic mouse models of Alzheimer's disease. Proc Natl Acad Sci U S A 108:2528–2533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Wetzel R (2006) Kinetics and thermodynamics of amyloid fibril assembly. Acc Chem Res 39:671–679. [DOI] [PubMed] [Google Scholar]
- 94. Wolfe MS (2007) When loss is gain: reduced presenilin proteolytic function leads to increased Abeta42/Abeta40. Talking point on the role of presenilin mutations in Alzheimer disease. EMBO Rep 8:136–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Wolfe MS (2012) γ‐Secretase as a target for Alzheimer's disease. Adv Pharmacol 64:127–153. [DOI] [PubMed] [Google Scholar]