

Abstract
Rhabdoid phenotype and loss of SMARCB1 expression in a brain tumor are characteristic features of atypical teratoid/rhabdoid tumors (ATRT). Rare non‐rhabdoid brain tumors showing cribriform growth pattern and SMARCB1 loss have been designated cribriform neuroepithelial tumor (CRINET). Small case series suggest that CRINETs may have a relatively favorable prognosis. However, the long‐term outcome is unclear and it remains uncertain whether CRINET represents a distinct entity or a variant of ATRT. Therefore, 10 CRINETs were clinically and molecularly characterized and compared with 10 ATRTs of each of three recently described molecular subgroups (i.e. ATRT‐TYR, ATRT‐SHH and ATRT‐MYC) using Illumina Infinium HumanMethylation450 arrays, FISH, MLPA, and sequencing. Furthermore, outcome was compared to a larger cohort of 27 children with ATRT‐TYR. Median age of the 6 boys and 4 girls harboring a CRINET was 20 months. On histopathological examination, all CRINETs demonstrated a cribriform growth pattern and distinct tyrosinase staining. On unsupervised cluster analysis of methylation data, all CRINETs examined exclusively clustered within the ATRT‐TYR molecular subgroup. As ATRT‐TYR, CRINETs mainly showed large heterozygous 22q deletions (9/10) and SMARCB1 mutations of the other allele. In two patients, SMARCB1 mutations were also present in the germline. Estimated mean overall survival in patients with CRINETs was 125 months (95% confidence interval 100–151 months) as compared to only 53 (33–74) months in patients with ATRTs of the ATRT‐TYR subgroup (Log‐Rank P < 0.05). In conclusion, CRINET represents a SMARCB1‐deficient non‐rhabdoid tumor, which shares molecular similarities with the ATRT‐TYR subgroup but has distinct histopathological features and favorable long‐term outcome.
Keywords: atypical teratoid/rhabdoid tumor, copy number alterations, DNA methylation profiling, SMARCB1/INI1, tyrosinase
Introduction
Rhabdoid phenotype and loss of SMARCB1 (also known as hSNF5/INI1) protein expression are characteristic features of atypical teratoid/rhabdoid tumors (ATRT). Apart from genetic alterations affecting the SMARCB1 region on chromosome 22q, ATRT show stable genomic profiles without further recurrent chromosomal alterations 9. On an epigenetic level, however, ATRT has recently been shown to be a heterogeneous disease comprised of three different molecular subgroups (i.e. ATRT‐TYR, ATRT‐SHH and ATRT‐MYC), which are characterized by distinct methylome profiles, enhancer landscapes and subgroup‐specific regulatory networks 13. The same holds true for histopathologic features encountered in ATRT, which are remarkably diverse. In addition to rhabdoid tumor cells, areas with primitive neuroectodermal, mesenchymal and epitheloid features are commonly encountered 3, 7. For rare non‐rhabdoid brain tumors showing a cribriform growth pattern and loss of SMARCB1 expression the term cribriform neuroepithelial tumor (CRINET) has been coined 8. Small case series and individual case reports suggest that CRINETs may have a relatively favorable prognosis 2, 4, 8, 11. As yet, however, little is known on long‐term outcome of CRINET and there is uncertainty whether CRINET represents a distinct entity or a variant of ATRT. We thus aimed to further characterize the clinical and molecular features of CRINET as compared with ATRT. Here we show that CRINET is a tumor with distinct histopathologic features, molecular similarities with the ATRT‐TYR subgroup and favorable long‐term outcome.
Materials and Methods
Samples and patients
Formalin‐fixed paraffin‐embedded (FFPE) samples of 10 CRINETs were collected from the archives of the Institute of Neuropathology Münster and by contacting institutions, which had previously published CRINET cases 2, 11, 19. Our tumor bank received local ethical committee approval (Ethics committee of the University Hospital Münster) and parents had given informed consent for scientific use of the archival samples. Follow‐up information for all patients was obtained by contacting treating physicians. For clustering analyses, available clinical and molecular data of 10 ATRTs of each of three recently described molecular subgroups (i.e. ATRT‐TYR, ATRT‐SHH and ATRT‐MYC) 13 were evaluated (for characteristics see Supporting Information Table S1). Furthermore, outcome of patients with CRINET was compared to a larger cohort of 27 patients with ATRT‐TYR (including eight of the ATRT‐TYR cases used for clustering analyses), for which information on overall survival was available. Protein expression of SMARCB1 and tyrosinase was examined using immunohistochemistry 13, 14. Fluorescence in situ hybridization (FISH) analyses of the SMARCB1 region and SMARCB1 sequencing were performed as described previously 17 and Multiplex ligation‐dependent probe amplification (MLPA) was carried out using the SALSA MLPA P258 (SMARCB1) kit (MRC‐Holland, Amsterdam, the Netherlands) according to the manufacturer's protocol.
DNA methylation array processing
For DNA methylation profiling of CRINETs, we used Illumina Infinium HumanMethylation450 Bead Chip arrays according to the manufacturer's instructions and protocols at the German Cancer Research Center (DKFZ) Genomics and Proteomics Core Facility. DNA methylation data were generated from FFPE tissue samples using 250 ng of DNA as input material. All DNA methylation analyses were performed in R version 3.2.0 (R Development Core Team, 2015). The following criteria were applied to filter the data: removal of probes targeting sex chromosomes (n = 11 551), removal of probes containing a single nucleotide polymorphism (dbSNP132 Common) within five base pairs of and including the targeted CpG‐site (n = 24 536), and probes not uniquely mapping to the human reference genome (hg19) allowing for one mismatch (n = 9993). In total, 438 370 probes were kept for analysis. Unsupervised hierarchical clustering of the samples was performed using the 5000 most variably methylated probes across the dataset and the 1‐Pearson correlation coefficient as the distance measure. Data were compared to 450k methylation data generated in 30 ATRTs of each of three recently described molecular subgroups (i.e. ATRT‐TYR, ATRT‐SHH and ATRT‐MYC) 13 deposited in GEO (accession number GSE70460). The cluster dendrogram was formed by using average linkage as agglomeration method. To re‐order probes for the heatmap visualization, probes were clustered by agglomerative hierarchical clustering using 1‐centered Pearson correlation as distance measure and average linkage as agglomeration method. Copy‐number variation (CNV) analysis from 450k methylation array data was performed using the conumee Bioconductor package version 1.0.0 (http://bioconductor.org/packages/release/bioc/html/conumee.html). Scoring of chromosomal gains and losses was performed by manual inspection of each profile.
Statistical analysis
Continuous and categorical variables were compared using Mann–Whitney‐U‐Test and Chi square test, respectively. Survival analysis was performed using Kaplan‐Meier estimation for survival curves and the Log‐rank test using IBM SPSS 23 software (release 23.0). Overall survival time was defined as the time from the date of diagnosis to the date of death. For all analyses, P < 0.05 was considered to be significant.
Results
The median age of the 6 boys and 4 girls harboring a CRINET was 20 months (range 10–129 months). CRINETs were either located supratentorially [midline in the vicinity of the third ventricle (4/10), near the lateral ventricles (2/10)], or infratentorially in the posterior fossa (4/10 cases; see Table 1 for detailed patient characteristics). Based on neuroradiological findings and clinical features, the possibility of a choroid plexus tumor had been considered initially in the majority of patients (6/10). On histopathological examination, all CRINETs were characterized by the presence of cribriform strands and ribbons. In more compact areas, small lumina were also present, but rhabdoid tumor cells showing eosinophilic cytoplasms and eccentric nuclei with prominent nucleoli were absent (Figure 1A). All CRINETs showed loss of tumoral SMARCB1 protein expression (Figure 1B). The median Ki67/MIB1 proliferation index was 29% (range: 15%–35%). Furthermore, 10/10 CRINETs exhibited staining for tyrosinase (Figure 1C), which was also observed in 9/10 ATRTs of the ATRT‐TYR subgroup, but only in 1/10 tumors of the ATRT‐SHH and ATRT‐MYC subgroup, respectively, (Chi‐square: 29.2, df:3; P < 0.00001). Tyrosinase staining in CRINETs was cytoplasmic and heterogeneous as observed in ATRTs of the ATRT‐TYR subgroup.
Table 1.
Patient characteristics. Clinical and molecular characteristics of 10 patients harboring a CRINET (CR = complete remission; DOD = dead of disease; WT = wildtype).
| # | Age at diagnosis (months) | Sex | Tumor location | Copy number alterations on analysis 450k data | SMARCB1 region FISH | SMARCB1 region MLPA | SMARCB1 sequencing | Germ line status | Adjuvant therapy | Follow‐up (months) | Status | Comments |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 26 | male | Infratentorial (fourth ventricular region) | large heterozygous deletion 22q | heterozygous deletion | heterozygous deletion (delTBX1_SMARCB1) | c.367>T p.Gln123* (Exon 4) | not available | CPT‐SIOP‐2000 (etoposide, cyclophosphamide, and vincristine) | 86 | CR | |
| 2 | 10 | male | Infratentorial (fourth ventricular region) | large heterozygous deletion 22q | not available | heterozygous deletion (delTBX1_SMARCB1) | c.492duplCCTT p.Pro165Leufs*6 (Exon 4) | not available | CPT‐SIOP‐2000 (etoposide, cyclophosphamide, and vincristine), radiotherapy (54G) | 139 | CR | Case 1 reported by Hasselblatt et al 8and Gessi et al 6 |
| 3 | 21 | male | Infratentorial (fourth ventricular region) | large heterozygous deletion 22q | heterozygous deletion | heterozygous deletion + duplEx6 SMARCB1 (reported by Ibrahim et al) | duplEx6 SMARCB1 (reported by Ibrahim et al) | duplEx6 SMARCB1 [reported by Ibrahim et al 11] | COG‐99703 (cisplatin, etoposide, cyclophosphamide and vincristine) followed by three rounds of high‐dose chemotherapy (carboplatin, thiotepa) with stem‐cell rescue. | 68 | CR | Case reported by Ibrahim et al. 11 |
| 4 | 27 | female | Supratentorial (third ventricular region) | large heterozygous deletion 22q | not available | delTBX1_NIPSNAP1 + delEx7‐9 SMARCB1 | WT | not available | HIT‐SKK‐2000 (including methotrexate, cyclophosphamide, and vincristine) | 124 | CR | Case 2 reported by Hasselblatt et al 8 and Gessi et al 6 |
| 5 | 11 | female | Supratentorial (third ventricular region) | large heterozygous deletion 22q | heterozygous deletion | delTBX1_NIPSNAP1 + delEx7 SMARCB1 | WT | not available | no information available | 10 | CR | |
| 6 | 20 | female | Supratentorial (third ventricular region) | large heterozygous deletion 22q | heterozygous deletion | not available | WT | WT | EU‐RHAB protocol,radiotherapy (proton‐radiation) | 78 | CR | |
| 7 | 20 | male | supratentorial | large heterozygous deletion 22q | not available | not available | not available | not available | CPT‐SIOP‐2000 (etoposide, cyclophosphamide, and vincristine) | 36 | CR | |
| 8 | 14 | male | Supratentorial (lateral ventricle) | large heterozygous deletion 22q | heterozygous deletion | heterozygous deletion (delTBX1_NIPSNAP1) | c.986 + 1G>T (Intron 7) | WT [reported by Arnold et al 2] | COG‐99703 (cisplatin, etoposide, cyclophosphamide and vincristine) followed by three rounds of high‐dose chemotherapy (carboplatin, thiotepa) with stem‐cell rescue. | 43 | CR | Case reported by Arnold et al 2 |
| 9 | 14 | male | Supratentorial (lateral ventricle) | absent | not available | no copy number alterations | homozygous c.367>T p.Gln123* (Exon 4) | not available | Patient died from respiratory failure before therapy could be initiated | 1 | DOD | Case reported by Park et al 19 |
| 10 | 129 | female | Supratentorial (third ventricular region) | large heterozygous deletion 22q | heterozygous deletion 22q | heterozygous deletion (delTBX1_NIPSNAP1) | c.1142C>G p.Thr381Arg(Exon 9) | c.1142C>G (Exon 9) | EU‐RHAB protocol,radiotherapy | 16 | CR | Case 3 reported by Gessi et al 12 |
Figure 1.
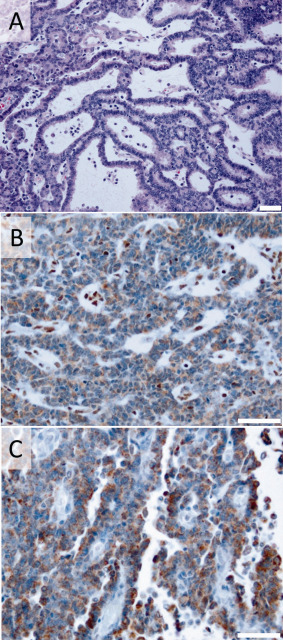
Histopathology of CRINET. Hematoxylin and eosin staining (A), immunohistochemistry for SMARCB1 (B) and tyrosinase (C) in a representative CRINET case. Note loss of nuclear SMARCB1 staining in the tumor cells, which is retained in the nuclei of non‐neoplastic cells (internal positive control) as well as distinct staining of tumor cells for tyrosinase. Scale bars denote 50 µm.
On unsupervised cluster analysis of methylation profiles using the 5000 most differentially methylated CpG sites across all samples, all eight CRINETs for which sufficient DNA was available for examination, exclusively clustered within the ATRT‐TYR molecular subgroup (Figure 2A). Copy‐number profiles as derived from intensity measures of the methylation probes indicated 22q losses affecting the SMARCB1 region as the only recurrent alteration in CRINETs with a pattern very similar to that seen in ATRT‐TYR tumors (Figure 2B).
Figure 2.
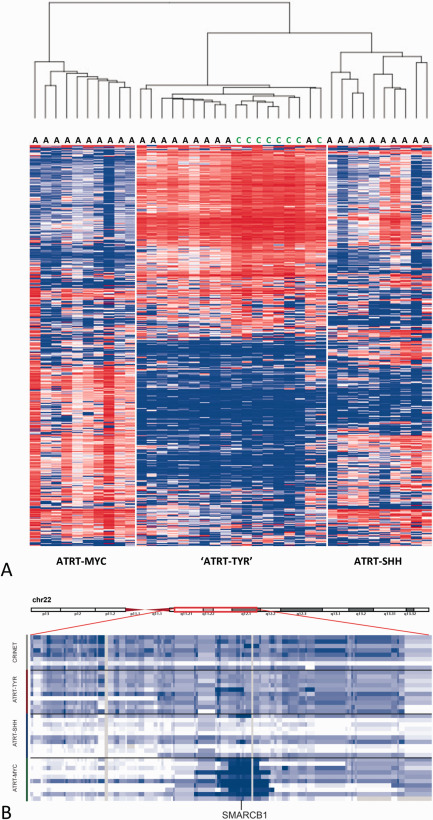
Molecular profiling of CRINET vs. ATRT. Heatmap showing unsupervised clustering of methylation profiles of 38 samples using the 5000 most variable probes (A) as well as copy number alterations affecting chromosome 22q (B) of 8 CRINETs and ATRTs of the molecular subgroups TYR, SHH and MYC (n = 10 each). Note that all CRINETs cluster within the ATRT‐TYR subgroup and (like ATRT‐TYR) show large heterozygous deletions of 22q affecting the SMARCB1 locus.
FISH and/or MLPA analyses confirmed the presence of large heterozygous 22q deletions affecting the SMARCB1 region in 9/10 CRINET cases. The only CRINET without a large heterozygous 22q deletion showed loss of heterozygosity for a truncating SMARCB1 mutation affecting exon 4 (c.367C > T p.Gln123*). In the remaining CRINET cases, a truncating SMARCB1 mutation affecting exon 4 (c.367C > T p.Gln123*), an exon 4 mutation resulting in a frameshift (c.492duplCCTT p.Pro165Leufs*6) and a mutation affecting intron 7 (c.986 + 1G > T) were identified as a “second hit,” while two cases showed additional small SMARCB1 deletions on MLPA (delEx7, delEx7‐Ex9, Figure 3). Furthermore, an exon 9 missense mutation (c.1142C > G) expected to result in disturbed splicing and an exon 6 duplication were encountered in two CRINETs, both mutations also being present in the germline of the respective patients. In the boy harboring a germline exon 6 duplication, the mutation was inherited from the apparently healthy mother and grandmother. The grandmotheŕs brother had died due to a brain tumor. The grandmotheŕs first cousin and her 3 children all carry the same mutation. The two daughters both had SMARCB1‐negative pediatric brain tumors initially interpreted as “ependymoma with monosomy 22” 22. One survived, the other one passed away as a teenager. The son never developed a tumor. The surviving daughter has 3 children, two of whom carry the mutation. Her oldest son presented with a CRINET in 2013 [reported by Dunham 4 but not included in the present series and is in complete remission.
Figure 3.
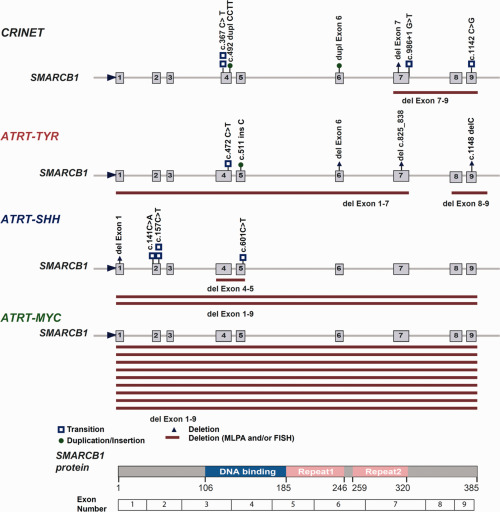
Spectrum of SMARCB1 mutations in CRINET vs. ATRT. SMARCB1 mutations encountered on FISH, MLPA and sequencing in CRINETs as compared to ATRTs of the molecular subgroups TYR, SHH and MYC (n = 10 each). Note similar distribution of mutations across the SMARCB1 gene in CRINET and ATRT‐TYR. Plotted are only hits affecting the second SMARCB1 allele. For more details, see Table 1 and Supporting Information Table S1.
Importantly, except for one child, who died one month postoperatively from respiratory failure, to our knowledge all patients with CRINET of the present series are alive and well. On Kaplan Meyer analysis of survival data, mean overall survival was 125 months (95% confidence interval 100‐151 months) and thus longer as compared to the patients of the ATRT groups used for genetic profiling and methylation clustering [ATRT‐TYR: 37 (18–56) months, ATRT‐SHH:16 (8–25) months, ATRT‐MYC: 13 (5–22) months, Log‐Rank P < 0.05]. Given the molecular similarities of CRINET and ATRT‐TYR, we next examined outcome of patients with CRINETs as compared to a larger cohort of 27 patients harboring ATRTs of the ATRT‐TYR subgroup. Here, significantly longer overall survival as compared to ATRT‐TYR patients [53 (33–74) months] could be confirmed (Log‐Rank P < 0.05, Figure 4).
Figure 4.
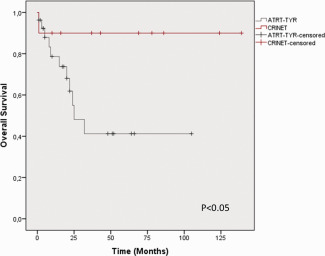
Prognosis of CRINET vs. ATRT‐TYR. On Kaplan Meier analysis of survival data, the 10 patients with CRINET show longer overall survival as compared to a cohort of 27 patients harboring ATRT‐TYR, P < 0.05 Log rank test).
Discussion
The key finding of our study is that genetic and epigenetic profiles of CRINET are highly similar to those of ATRT‐TYR, representing one of the three recently described molecular subgroups of ATRT 13. On gene expression profiling, ATRT‐TYR is characterized by overexpression of melanosomal markers, such as tyrosinase and MITF. Another characteristic feature of this subgroup is the overexpression of genes involved in ciliogenesis 13. The observation that protein expression of tyrosinase was not only present in ATRT‐TYR, but also in CRINET, suggests similarities between CRINET and ATRT‐TYR also at gene expression level. Another common feature of CRINET and ATRT‐TYR is the presence of large heterozygous 22q deletions affecting the SMARCB1 region, which are relatively rare in ATRT of the SHH and MYC subgroups 13. This finding could well point to a role of gene dosage of other genes on 22q in the biology of CRINET and ATRT‐TYR.
Importantly, despite the highly similar (epi)genetic profiles of CRINETs and ATRT‐TYR tumors, patients harboring CRINETs experience relatively favorable long‐term outcomes and do much better than patients with ATRT‐TYR tumors. This situation is reminiscent of desmoplastic medulloblastomas, which are also genetically and epigenetically indistinguishable from classic medulloblastomas of the SHH subtype, but still show a difference in outcome 15, 16 and provides another example that information obtained on histopathologic analysis and molecular profiling is not redundant, but rather complementary.
Taking into account the limitations of this retrospective series, which combines outcome data of patients treated at various institutions across several countries, our data suggest that CRINET responds well to chemotherapy protocols commonly employed for the treatment of malignant brain tumors and ATRT. What makes the difference between the relatively favorable biological behavior of CRINET and the dismal outcome of ATRT‐TYR? The absence of recurrent genomic alterations apart from 22q losses in CRINET (also a characteristic feature of ATRT‐TYR) 6, 9, 12, 21 argues against a role of losses or gains of other chromosomal regions in the biology of CRINET. Even though the possibility of point mutations of genes putatively modifying the detrimental effects of SMARCB1‐deficiency cannot be excluded, it is tempting to speculate that the type of SMARCB1 mutation may determine tumor phenotype and prognosis.
The distribution of mutations across the SMARCB1 gene encountered in CRINET seems to be quite similar to that of ATRT‐TYR, but distinct from those seen in ATRT‐SHH or ATRT‐MYC. Some mutations encountered in CRINET such as c.367C > T have also been described in ATRT 5, but histopathological features and molecular subgroup information of these published cases are unknown. Interestingly, the somatic mutation affecting intron 7 in the CRINET of patient #8 (c.986 + 1G > T) has been previously described in a family with rhabdoid tumor predisposition syndrome and been shown to result in the exclusion of exon 7 on RNA level 5. A similar mutation in the donor splice site of exon 7 has been reported in a family with pediatric posterior fossa brain tumors diagnosed as ATRT or choroid plexus carcinoma 24. Germline duplications affecting exon 6 of the SMARCB1 gene have also been reported in a family with rhabdoid tumor predisposition syndrome and schwannomatosis 23. The family of the CRINET patient harboring a SMARCB1 germline exon 6 duplication, however, rather shows similarities to a previously described Dutch family 1. In this family, two patients survived for an unusually long time. Of note, both tumors had been initially reported as “anaplastic ependymomas” and showed growth in strands and ribbons, i.e. histopathological features reminiscent of CRINET 18 and a SMARCB1 mutation affecting a splice site (c.500 + 1G > A) could be demonstrated in the patients and their unaffected fathers 1. In schwannomatosis patients, it has been suggested that synthesis of an altered SMARCB1 protein (either by translation re‐initiation or encoded by missense, splice‐site mutations or in‐frame deletions) may prevent the development of malignant rhabdoid tumors 10. Neither exon 1 mutations nor the mosaic SMARCB1 staining pattern typical for schwannomatosis‐associated schwannomas 20 were encountered in CRINET patients. Nevertheless, as the BAF47 antibody is directed against a relatively C‐terminal epitope (amino acids 257‐359), the possibility that a truncated SMARCB1 protein with some residual function could be responsible for the less aggressive biological behavior of CRINET cannot be entirely excluded and warrants attention in future studies. Furthermore, the potential prognostic role of molecular subgrouping in ATRT will need to be determined in the carefully characterized patient cohorts of large international registries.
In conclusion, CRINET represents a SMARCB1‐deficient non‐rhabdoid tumor, which shares molecular similarities with the ATRT‐TYR subgroup but has distinct histopathological features and favorable long‐term outcome.
Supporting information
Additional Supporting Information may be found in the online version of this article at the publisher's web‐site:
Table S1. Characteristics of the AT/RT control group. Characteristics of 30 patients harboring AT/RT that were employed for as controls for clustering analyses and molecular profiling (WT = wildtype).
Acknowledgments
Susanne Peetz‐Dienhart and Yvonne Crede provided expert technical assistance. The authors also acknowledge the excellent technical support of the Genomics and Proteomics core facility of the German Cancer Research Center (DKFZ) and the technical staff of the molecular cytogenetic laboratory of the Institute of Human Genetics in Kiel. This study was supported by Deutsche Krebshilfe (DKH110267) and IZKF Münster (Ha3/019/15). R. Schneppenheim and F. Oyen are supported by Fördergemeinschaft Kinderkrebszentrum Hamburg e.V., R. Siebert and S. Bens are supported by KinderKrebsInitiative Buchholz/Holm‐Seppensen. The authors thank the DKFZ‐Heidelberg Center for Personalized Oncology (DKFZ‐HIPO) for technical support and funding through HIPO project number H049.
References
- 1. Ammerlaan AC, Ararou A, Houben MP, Baas F, Tijssen CC, Teepen JL et al (2008) Long‐term survival and transmission of INI1‐mutation via nonpenetrant males in a family with rhabdoid tumour predisposition syndrome. Br J Cancer 98:474–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Arnold MA, Stallings‐Archer K, Marlin E, Grondin R, Olshefski R, Biegel JA, Pierson CR (2013) Cribriform neuroepithelial tumor arising in the lateral ventricle. Pediatr Dev Pathol 16:301–307. [DOI] [PubMed] [Google Scholar]
- 3. Biegel JA, Rorke LB, Packer RJ, Emanuel BS (1990) Monosomy 22 in rhabdoid or atypical tumors of the brain. J Neurosurg 73:710–714. [DOI] [PubMed] [Google Scholar]
- 4. Dunham C (2015) Uncommon pediatric tumors of the posterior fossa: pathologic and molecular features. Childs Nerv Syst 31:1729–1737. [DOI] [PubMed] [Google Scholar]
- 5. Eaton KW, Tooke LS, Wainwright LM, Judkins AR, Biegel JA (2011) Spectrum of SMARCB1/INI1 mutations in familial and sporadic rhabdoid tumors. Pediatr Blood Cancer 56:7–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gessi M, Japp AS, Dreschmann V, Zur Muhlen A, Goschzik T, Dorner E, Pietsch T (2015) High‐resolution genomic analysis of cribriform neuroepithelial tumors of the central nervous system. J Neuropathol Exp Neurol 74:970–974. [DOI] [PubMed] [Google Scholar]
- 7. Haberler C, Laggner U, Slavc I, Czech T, Ambros IM, Ambros PF et al (2006) Immunohistochemical analysis of INI1 protein in malignant pediatric CNS tumors: lack of INI1 in atypical teratoid/rhabdoid tumors and in a fraction of primitive neuroectodermal tumors without rhabdoid phenotype. Am J Surg Pathol 30:1462–1468. [DOI] [PubMed] [Google Scholar]
- 8. Hasselblatt M, Oyen F, Gesk S, Kordes U, Wrede B, Bergmann M et al (2009) Cribriform neuroepithelial tumor (CRINET): a nonrhabdoid ventricular tumor with INI1 loss and relatively favorable prognosis. J Neuropathol Exp Neurol 68:1249–1255. [DOI] [PubMed] [Google Scholar]
- 9. Hasselblatt M, Isken S, Linge A, Eikmeier K, Jeibmann A, Oyen F et al (2013) High‐resolution genomic analysis suggests the absence of recurrent genomic alterations other than SMARCB1 aberrations in atypical teratoid/rhabdoid tumors. Genes Chromosomes Cancer 52:185–190. [DOI] [PubMed] [Google Scholar]
- 10. Hulsebos TJ, Kenter S, Verhagen WI, Baas F, Flucke U, Wesseling P (2014) Premature termination of SMARCB1 translation may be followed by reinitiation in schwannomatosis‐associated schwannomas, but results in absence of SMARCB1 expression in rhabdoid tumors. Acta Neuropathol 128:439–448. [DOI] [PubMed] [Google Scholar]
- 11. Ibrahim GM, Huang A, Halliday W, Dirks PB, Malkin D, Baskin B et al (2011) Cribriform neuroepithelial tumour: novel clinicopathological, ultrastructural and cytogenetic findings. Acta Neuropathol 122:511–514. [DOI] [PubMed] [Google Scholar]
- 12. Jackson EM, Sievert AJ, Gai X, Hakonarson H, Judkins AR, Tooke L et al (2009) Genomic analysis using high‐density single nucleotide polymorphism‐based oligonucleotide arrays and multiplex ligation‐dependent probe amplification provides a comprehensive analysis of INI1/SMARCB1 in malignant rhabdoid tumors. Clin Cancer Res 15:1923–1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Johann P, Erkek S, Zapatka M, Kerl K, Buchhalter I, Hovestadt V et al (2016) Atypical teratoid/rhabdoid tumor (ATRT) comprises three epigenetic subgroups with distinct enhancer landscapes. Cancer Cell 29:379–393. [DOI] [PubMed] [Google Scholar]
- 14. Judkins AR, Mauger J, Ht A, Rorke LB, Biegel JA (2004) Immunohistochemical analysis of hSNF5/INI1 in pediatric CNS neoplasms. Am J Surg Pathol 28:644–650. [DOI] [PubMed] [Google Scholar]
- 15. Kool M, Jones DT, Jager N, Northcott PA, Pugh TJ, Hovestadt V et al (2014) Genome sequencing of SHH medulloblastoma predicts genotype‐related response to smoothened inhibition. Cancer Cell 25:393–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kool M, Korshunov A, Remke M, Jones DT, Schlanstein M, Northcott PA et al (2012) Molecular subgroups of medulloblastoma: an international meta‐analysis of transcriptome, genetic aberrations, and clinical data of WNT, SHH, Group 3, and Group 4 medulloblastomas. Acta Neuropathol 123:473–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kordes U, Gesk S, Frühwald MC, Graf N, Leuschner I, Hasselblatt M et al (2010) Clinical and molecular features in patients with atypical teratoid rhabdoid tumor or malignant rhabdoid tumor. Genes Chromosomes Cancer 49:176–181. [DOI] [PubMed] [Google Scholar]
- 18. Nijssen PC, Deprez RH, Tijssen CC, Hagemeijer A, Arnoldus EP, Teepen JL et al (1994) Familial anaplastic ependymoma: evidence of loss of chromosome 22 in tumour cells. J Neurol Neurosurg Psychiatry 57:1245–1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Park JY, Kim E, Kim DW, Chang HW, Kim SP (2012) Cribriform neuroepithelial tumor in the third ventricle: a case report and literature review. Neuropathology 32:570–576. [DOI] [PubMed] [Google Scholar]
- 20. Patil S, Perry A, Maccollin M, Dong S, Betensky RA, Yeh TH et al (2008) Immunohistochemical analysis supports a role for INI1/SMARCB1 in hereditary forms of schwannomas, but not in solitary, sporadic schwannomas. Brain Pathol 18:517–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ruland V, Hartung S, Kordes U, Wolff JE, Paulus W, Hasselblatt M (2014) Choroid plexus carcinomas are characterized by complex chromosomal alterations related to patient age and prognosis. Genes Chromosomes Cancer 53:373–380. [DOI] [PubMed] [Google Scholar]
- 22. Savard ML, Gilchrist DM (1989) Ependymomas in two sisters and a maternal male cousin with mosaicism with monosomy 22 in tumour. Pediatr Neurosci 15:80–84. [DOI] [PubMed] [Google Scholar]
- 23. Swensen JJ, Keyser J, Coffin CM, Biegel JA, Viskochil DH, Williams MS (2009) Familial occurrence of schwannomas and malignant rhabdoid tumour associated with a duplication in SMARCB1. J Med Genet 46:68–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Taylor MD, Gokgoz N, Andrulis IL, Mainprize TG, Drake JM, Rutka JT (2000) Familial posterior fossa brain tumors of infancy secondary to germline mutation of the hSNF5 gene. Am J Hum Genet 66:1403–1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting Information may be found in the online version of this article at the publisher's web‐site:
Table S1. Characteristics of the AT/RT control group. Characteristics of 30 patients harboring AT/RT that were employed for as controls for clustering analyses and molecular profiling (WT = wildtype).