

Abstract
Aquaporin‐4 (AQP4) and glutamate transporter‐1 (GLT‐1) represent the major water and glutamate astrocyte buffering gateways in the brain. Utilizing perilesional ischemic human cortices, we have performed here for the first time an integrative assessment on both AQP4 and GLT‐1, and on their proximity to blood vessels and neurons. Counting the relative number of AQP4±/GLT‐1±/glial fibrillary acidic protein± cells showed that double‐positive variants were overall most frequent, and their number tended to decrease from organized and recent perilesional cortices to controls. AQP4/GLT‐1 colocalization showed higher coefficients for the perilesional cortices compared with controls, suggesting an increased water/glutamate‐buffering capability. Distance frequency analysis of AQP4/GLT‐1 in relationship to neurons showed that both markers were concentrated at 20–40 μm around the perikarya; with AQP4 being more abundant in close proximity, these differences were not being driven by changes in neuropil density alone. Our study suggests a dual, simultaneous astrocytic function depending on the relative distance to neurons and vessels, with increased water and glutamate‐buffering capability in the mid perineuronal space, and an increased water‐buffering capability in the immediate perineuronal space, even higher than around vessels. Thus, adding specific AQP4/GLT‐1 modulator agents selectively depending on the acute/chronic phase of stroke might increase the efficacy of existing treatments.
Keywords: aquaporin 4, astrocytes, blood vessels, glutamate transporter 1, ischemic stroke, neurons
Introduction
Ischemic strokes result from lack of blood flow to the brain and represent up to 70% of all stroke patients 10, 37. Classical treatment options during the acute phase of an ischemic event are centered on maintaining or regaining vascular permeability, preventing brain edema, and avoiding hypotension and secondary complications. In stroke, it is well established that cerebral edema can be divided, depending on its physiopathology, into cytotoxic (or cellular) edema and vasogenic edema 17. Cytotoxic edema occurs rapidly in anoxic–ischemic conditions that lead to energy depletion and failure of the membrane adenosine triphosphate‐dependent Na+/K+ pump to preserve the ion gradients of the intercellular space, and as a result, osmosis leads to a shift of water content toward the intracellular compartment 9. Vasogenic edema, on the other hand, is the most common type of brain edema and is classically described as the water influx toward the neuropil following the failure of the ischemic blood–brain barrier (BBB) to maintain an adequate filter 17. The only currently accepted nonsurgical approach in the treatment of brain edema is parenteral administration of hypertonic solutions, which rapidly reduces brain volume and lowers intracranial pressure by shifting water from brain to blood 34. This is generally effective only after the first 2 days from the onset of the disease, as it only address the vasogenic edema 2.
The aquaporins (AQPs) are a family of water channel proteins first identified in red blood cells, which seem to be the key players in mediating water equilibrium around cellular membranes, and among which AQP4 is the most abundant in the nervous tissue 4, 7, 43. Water balance in the brain is thus regulated mainly by AQP4 channels, which are concentrated especially in perivascular astrocytic end feet 27, 28. Although it is well known now that AQP4 expression levels are modulated by an ischemic event, little is known about the precise dynamics and the factors that drive these variations 25, 30, 31. After an ischemic stroke produced by arterial occlusion, as a model of mainly cytotoxic edema, mice lacking AQP4 show significantly decreased cerebral edema and improved neurological outcomes compared with control animals 25, 30, 32. On the contrary, in a freeze injury model of vasogenic brain edema, AQP4‐deficient mice had increased brain swelling and worse neurological outcomes 31. Some results are even more controversial, with other data revealing a heterogeneous expression pattern of AQP4, namely, a reduced expression in brain injuries with impaired BBB and vasogenic edema, and nonsignificant changes in cytotoxic edema associated with an intact BBB 16. Despite these contradictory data, little has been done to characterize AQP4 expression changes at the cellular level on human stroke brain tissue, or toward a morphometric analysis of its disposition around blood vessels.
Aside from edema and its direct life‐threatening complications, energy failure in the ischemic brain tissue leads to ion gradient loss on the cellular membranes 9. This is followed by excessive release of glutamate, which together with an impaired glutamate reuptake promotes neuronal excitotoxicity and free radical‐induced neuronal death 8, 20. Glutamate transporter 1 (GLT‐1, or excitatory amino acid transporter 2) protein is the main gate of glutamate reuptake in the central nervous system, driving glutamate accumulation in the astrocytes 41. GLT‐1‐deficient mice show increased glutamate toxicity in prone conditions, and overexpression of GLT‐1 significantly reduces glutamate overflow and cell death improving the clinical recovery in animal models of ischemic stroke 12, 42. Moreover, it has been demonstrated that lack of AQP4 downregulates astrocytic expression of GLT‐1, and in fact AQP4 and GLT‐1 seem to exist in astrocytic membranes as a functional complex 14, 48, 49. Again, despite these facts, an in‐depth morphometric characterization of GLT‐1 relationship with AQP4 and the neurons on human stroke pathology is also lacking.
Altogether, the aim of our study was to perform for the first time on human stroke pathology a close correlative investigation of AQP4 and GLT‐1 expression patterns, as well as to approach their relationships with the neuronal and vascular compartments during cerebral ischemic event.
Materials and Methods
Patients and tissue processing
Formalin‐fixed paraffin‐embedded archived brain tissue blocks were selected from 12 ischemic stroke patients and four age controls (Table 1). A written informed consent was obtained for each patient from their relatives, accepting tissue prelevation for research purposes. All the patients included in this study presented to the Clinic of Neurology (UMF Craiova) and were followed utilizing classical investigation and therapeutic protocols specific for ischemic stroke. Patients presenting within the first 5 days after the onset received antiplatelet or anticoagulant therapy and cerebral depletives, whereas patients with chronic lesions received only antiplatelet/anticoagulant therapy. Moreover, for both categories, associated comorbidities received specific therapeutic options. Because of the occurrence of secondary lesions, edema and brain herniation, superimposed respiratory or cardiovascular pathology, eventual death occurred in these patients and necropsy was performed. Overall, the average ages (±standard deviation, SD) of the patients included in the study were of 74.25 ± 8.80 years for the control group, and 68.75 ± 6.57 for the ischemic stroke group.
Table 1.
Description of the patients included in this study
| Group | Age | Gender | Survival (days)* | Pathology |
|---|---|---|---|---|
| R1 | 56 | M | 5 | Bilateral hippocampal softening (acute temporal lesion) |
| R2 | 67 | M | 20 | Cortical ischemic event (both occipital lobes, right hippocampus, brain stem) |
| R3 | 68 | F | 15 | Acute cortical ischemic event of the frontotemporal lobes, left hemisphere |
| R4 | 77 | F | 2 | Acute cortical ischemic event with hemorrhagic transformation (all lobes of the right hemisphere). The right dentate cerebellar nucleus is also involved |
| O1 | 59 | F | 45 | Old cortical and subcortical ischemic events (left frontal and left striatal body) |
| O2 | 66 | M | 63 | Large old right temporoparietal ischemic stroke |
| O3 | 68 | M | 39 | Old cortical and subcortical ischemic lesion (with cavitation and visible gliotic scar). It involves the cortex and the white matter of the temporal and parietal right lobes |
| O4 | 68 | F | 48 | Old cystic cavitation in the white matter beneath the left temporal lobe |
| O5 | 71 | M | 31 | Old cortical and subcortical ischemic event of the right temporal lobe |
| O6 | 73 | M | 38 | Old cortical and subcortical ischemic event of the parietal lobe, left hemisphere |
| O7 | 74 | F | 55 | Right old temporal cortical and subcortical ischemic event. Right hippocampus is involved |
| O8 | 78 | F | 49 | Old ischemic lesion of the right hippocampus and occipital lobe |
| C1 | 68 | M | – | Lung tumor pathology |
| C2 | 69 | F | – | Digestive tumor pathology |
| C3 | 85 | M | – | Bilateral bronchopneumonia, tuberculosis |
| C4 | 75 | M | – | Digestive tumor pathology |
*Survival time is considered after the first stroke.
C = control patient; O = organizing old ischemic event; R = recent stroke.
Clinical, imaging, macroscopic and microscopic criteria were utilized for identifying recent and old infarcts. (i) Aside from focal neurological deficits/language impairment, computed tomography imaging provided supporting data beginning with 24 h after the onset: sulcal effacement with loss of gray–white matter distinction, gray matter hypodensities, edema formation and shifting of the infarcted tissue in the acute and subacute phase; marked hypodensity in the area supplied by an artery; and lack of mass effect for chronic lesions. (ii) At necropsy, macroscopic neuropathological evaluation confirmed focal brain matter softening and discoloration in recent lesions, with yellowish gliotic areas and cavitations in organized lesions. (iii) Microscopically, a classical array of neuronal eosinophilia, shrunken perikaryons and kariopicnosis/karyolysis/karyorrhexis, liquefactive necrosis, reactive gliosis, neuronal loss, cortical vacuolation and sometimes petechial hemorrhages characterized recent ischemic lesions, while cystic cavitation with dense glial scar formation completed the view of organizing lesions. These features allowed the pathological stratification of the patients with (i) recent lesions (clinically acute and subacute, in the first month of evolution) (four patients) and (ii) chronic, organized lesions (clinically, after the first month of evolution) (eight patients). Age‐matched controls showed mild cortical neuronal loss, perivascular or subleptomeningeal accumulation of corpora amylacea and some degree of vascular hyalinization.
Chromogenic immunohistochemistry
Single immunohistochemistry was performed for all the antibodies enumerated in Table 2. Briefly, after antigen retrieval, sections were first incubated for 30 minutes in a 1% hydrogen peroxide solution. The sections were next blocked for 1 h in 3% skim milk (Bio‐Rad, München, Germany), then they were incubated with the primary antibodies for 18 h at 4°C, and the next day the signal was amplified for 30 minutes utilizing a species‐specific human‐adsorbed peroxidase polymer‐based system (Nikirei‐Histofine, Tokyo, Japan). The signal was detected with 3,3′‐diaminobenzidine (Dako, Glostrup, Denmark), and the slides were coverslipped in DPX (Sigma‐Aldrich, Hamburg, Germany) after a hematoxylin staining. Each experiment included normal brain tissue as positive controls for the existence of known structures (ie, astrocytes, neuronal silhouettes and blood vessels, respectively) (Supporting Information Figure S1A), whereas negative controls were obtained by omitting the primary antibodies (Supplementary Figure S1B).
Table 2.
The antibodies utilized in this study
| Name | Clonality | Staining pattern | Immunization target | Dilution | Source |
|---|---|---|---|---|---|
| Anti‐AchR | Goat, polyclonal | Neuronal membrane | A synthetic peptide KRPGEDKVRPACQHKQ from an internal region of human nicotinic acetylcholine receptor alpha 7 | 1:100 | NBP1‐52375, Novus Biologicals, Cambridge, UK |
| Anti‐aquaporin 4 | Rabbit, polyclonal | Aquaporin 4 on astrocyte membrane | Amino acids 244–323, C‐terminal, of human aquaporin 4 | 1:1000 | sc‐20812, Santa Cruz, Heidelberg, Germany |
| Anti‐collagen type IV | Mouse, IgG1k, clone CIV22 | Basement membranes | Purified pepsin fragments of type IV collagen isolated from human kidney | 1:100 | M0785, Dako, Glostrup, Denmark |
| Anti‐collagen type IV | Rabbit, polyclonal | Basement membranes | Collagen type IV from human and bovine placenta | 1:5000, with tyramide | NB120‐6586, Novus Biologicals |
| Anti‐CD31 | Mouse, IgG1k, clone JC70A | Vascular endothelium | Cell membrane preparation from the spleen of a patient with hairy cell leukemia | 1:100 | M0823, Dako |
| Anti‐GFAP | Mouse, IgG1k, clone GA5 | Astrocyte cytoskeleton | Glial fibrillary acidic protein from porcine spinal cord | 1:500 | ABIN125137, Antibodies online, Aachen, Germany |
| Anti‐GFAP | Rabbit, polyclonal | Astrocyte cytoskeleton | GFAP isolated from cow spinal cord | 1:20 000 | Z0334, Dako |
| Anti‐GLT‐1 (EAAT2) | Mouse, IgG2a, clone 1H8 | GLT‐1 molecule on astrocyte membrane | Recombinant protein corresponding to the C‐terminal domain of human glutamate transporter 1 protein | 1:10 | LS‐C87838, Lifespan Biosciences, Seattle, WA, USA |
Antigen retrieval utilized: microwaving in 0.1 M Citrate pH = 6.
AchR = acetylcholine receptor; EAAT = excitatory amino acid transporter; GFAP = glial fibrillary acidic protein; GLT‐1 = glutamate transporter‐1; IgG = immunoglobulin G.
Fluorescent immunohistochemistry
After confirmation on single immunohistochemistry, double‐fluorescent immunodetections were performed in order to study the relationship between AQP4 and the neurons (nicotinic acetylcholine receptor alpha 7; AchR), astrocytes [mouse anti‐human glial fibrillary acidic protein (GFAP), GLT‐1], endothelia and their basement membranes (CD31, mouse anti‐human collagen IV) (Table 2). To assess the relationship of AQP4 to another membrane astrocyte marker, or with the endothelium and the basement membranes, a series of sections from all cases were double stained for GFAP (rabbit polyclonal) and GLT‐1, and another series was triple stained for AQP4, collagen IV (rabbit anti‐human) and vascular endothelium (CD31). Moreover, GLT‐1 was also studied in combination with collagen IV (rabbit anti‐human) and AchR.
For AQP4‐based double stainings, as well as for GFAP (rabbit polyclonal)/GLT‐1 staining, after antigen retrieval, sections were blocked for 1 h in 3% skim milk, and incubated overnight with the pair of primary antibodies. Next day, the sections were incubated with a mixture of goat anti‐rabbit Alexa Fluor 594, and goat anti‐mouse Alexa Fluor 488‐labeled secondary antibodies (Invitrogen, Life Technologies GmbH, Darmstadt, Germany; 1:300, 1 h at room temperature).
For AQP4/AchR immunostaining, AchR was visualized first in order to prevent any cross‐reactivity between the detection systems. Thus, the sections were incubated overnight with the AchR antibody, next day the signal was amplified with an anti‐goat biotinylated secondary antibody (1:100, Dako) and detected with a streptavidin Alexa Fluor 488 (1:300, 1 h at room temperature, Invitrogen). Then the procedure was repeated for the AQP4 primary antibody in conjunction with the goat anti‐rabbit Alexa Fluor 594 secondary antibody. GLT‐1 and collagen IV (rabbit anti‐human) were simultaneously detected with species‐specific Alexa Fluor antibodies (anti‐mouse Alexa Fluor 594/anti‐rabbit Alexa Fluor 488), while for GLT‐1/AchR double labeling, the neuronal marker was amplified utilizing a biotinylated secondary antibody and a streptavidin Alexa Fluor 488 as described above.
For the triple stainings (AQP4/collagen IV/CD31), the slides were first incubated with the rabbit anti‐human collagen IV, amplified with a human‐adsorbed anti‐rabbit peroxidase tagged polymer (Nikirei‐Histofine) for 30 minutes, and visualized with Alexa Fluor 488‐labeled tyramide (Invitrogen), with a precipitation time of 1 minute. After tyramide precipitation, the anti‐collagen IV primary antibody was removed from the tissue by incubating the slides in a 25‐mM glycine‐HCl, 1% sodium dodecyl sulfate, pH 2 buffer for 60 minutes at 60°C, as previously described by us 33. This antibody elution method has also been tested for the present experimental setup and indicated a complete removal of the primary anti‐collagen IV antibody after direct detection with another ant‐rabbit fluorescently labeled antibody (Supporting Information Figure S1C–F). The next two primary antibodies were added on the sections (AQP4—rabbit and CD31—mouse), followed by a mixture of goat anti‐rabbit Alexa Fluor 555 antibody (Invitrogen, 1:400) and goat anti‐mouse biotinylated antibody (Dako, 1:300) for 30 minutes at room temperature. A streptavidin Alexa Fluor 594 (Invitrogen, 1:300, 1 h at room temperature) finalized the immunodetection.
In all cases, the slides were counterstained with 4′,6‐diamidino‐2‐phenylindole (DAPI) (Invitrogen) for 15 minutes, incubated for 20 s in a 0.3% Sudan Black (Sigma‐Aldrich), alcoholic solution to reduce lipofuscin autofluorescence, washed in distillate water and coverslipped with a fluorescence anti‐fading mounting medium (Dako) 45.
Image analysis and quantification
Light microscopy and fluorescent images were grabbed utilizing a Nikon Eclipse 90i motorized microscope (Apidrag, Bucharest, Romania) equipped with a 5‐megapixel Nikon DS‐Fi1 CCD color camera (Apidrag), and a 1.4‐megapixel high‐quantum efficiency monochrome Rolera‐XR cooled CCD camera (QImaging, Surrey, British Columbia, Canada), together with the Image‐Pro Plus AMS 7 image analysis software (Media Cybernetics, Bethesda, MD, USA). Because of the abrupt variations and clear‐cut laminar expression of AQP4 and GLT‐1 in the glial scar tissue (see the Results section), all the semiquantitative studies performed further on involved exclusively the immediately perilesional cortices.
Pictures for image analysis have been collected randomly from the areas of interest (closest perilesional cortices and corresponding cortices from control patients), counting between five and nine microscopic fields for each region and experimental setup. Images were obtained by sequential scanning of each channel with specific pairs of highly selective custom‐made filters in order to eliminate the cross‐talk of the fluorophores and to ensure a reliable quantification for DAPI, Alexa 488, Alexa 555 and Alexa 594 spectra (Chroma Technology Corp., Bellows Falls, VT, USA). All images were originally stored in Image‐Pro Plus's proprietary format, then they were subjected to a blind deconvolution algorithm based on a multi‐pass adaptive point spread function subtraction of diffracted light (AutoDeblur, Image‐Pro Plus; Media Cybernetics).
Two‐signal colocalization studies further utilized these deconvolved images and were based on a fix thresholding for signal selection in their respective channels (thresholds approved initially by D.P. and C.M. after visualizing image examples, and kept constant afterward for the whole analysis). Together with the fact that exposure times were kept constant during image acquisition for each immunolabeling, these ensured the consistency of the analysis. Colocalization results were reported as Pearson's correlation coefficients of the intensity distribution between the two channels 51.
In order to study the randomness of the proximity of non‐colocalizing AQP4/GLT‐1 and the perikaryons (immunolabeled for AchR) or blood vessels (immunolabeled for collagen IV), we further investigated the frequency distribution of AQP4/GLT‐1 signal pixels function of their distances to the closest neighboring neuronal or vascular silhouettes, based on an algorithm developed by Media Cybernetics in response to our request (Figure 1). For this study, AchR/collagen IV signals (Alexa 488) were kept as the green channel, whereas AQP4 (Alexa 555)/GLT‐1 (Alexa 594) signal was pseudocolored as red in the respective RGB series of images. The analysis algorithm itself started with creating and saving a histogram‐based profile of the AQP4/GLT‐1‐positive pixels (on the correspondent red channel), so that all the acquired images would be processed based on this unique profile. After this segmentation, the outlines of the AQP4/GLT‐1 signal were saved. Neurons/vessels were also selected based on a histogram profile on their specific channel (green), and a mask was created after this selection. This mask was inversed and a non‐integer distance map filter algorithm was applied generating a distance map image where each pixel's intensity was in fact the distance measured to the nearest black pixel. On this distance map image, we overlaid the AQP4/GLT‐1 outlines previously saved, and we measured their mean intensities—expressed now as the mean distance to the edge of the nearest neuronal/vascular outline. This whole algorithm, together with an initial step of image calibration (the analysis being performed on 40× images, 42 455.48 μm2), was recorded as a macro command in Image–Pro Plus and was executed on each saved RGB image, exporting in an Excel spreadsheet the mean distances as microns. In order to observe the neuropil density changes during ischemia, the above distance–frequency algorithm was also applied to study the interrelation between neurons and blood vessels labeled for AchR and collagen IV.
Figure 1.

Image processing for distance frequency analysis. For aquaporin 4 (AQP4)–neuronal membrane distance frequency analysis, images captured from slides double stained for AQP4 (red) and nicotinic acetylcholine receptor (AchR) (green) (A) were first processed for extracting and saving the outlines of the AQP4‐positive pixels based on a constant histogram‐based profile on the red channel (B). Neuron masks were also created with the aid of another histogram‐based profile in the green channel, respectively (C), and a three‐dimensional distance map filter algorithm was applied to generate a distance map image (D) where each pixel's intensity represents its distance to the nearest black pixel from the mask itself. The outlines of the AQP4 signal were then overlaid on the distance map image (E), and their mean intensities (representing now the mean distances to the edge of the nearest neuronal outline) were saved for each image. An identical algorithm was utilized for AQP4–vascular basement membranes distance frequency analysis based on the AQP4 (red) and collagen IV (Col. IV) (green) immunofluorescence signals (F–J). Scale bars represent 50 μm.
In addition, AQP4, GLT‐1 and GFAP single‐ or double‐positive astrocytes were directly counted on randomly chosen 10 cortical areas (20×, 169 819.58 μm2) for each considered case, on AQP4/GFAP, AQP4/GLT‐1 and GLT‐1/GFAP double‐labeled slides. A cell was counted only when its nucleus could be clearly identified on DAPI staining and was deemed positive/negative independently for the cytoskeletal GFAP and respectively one of the two membrane markers. As individual cells could not be followed up in areas with diffuse gliosis or diffuse AQP4 and GLT‐1 staining, these regions were rejected for this analysis (Figure 2A,B). All images utilized in this algorithm were also deconvolved (as described above).
Figure 2.
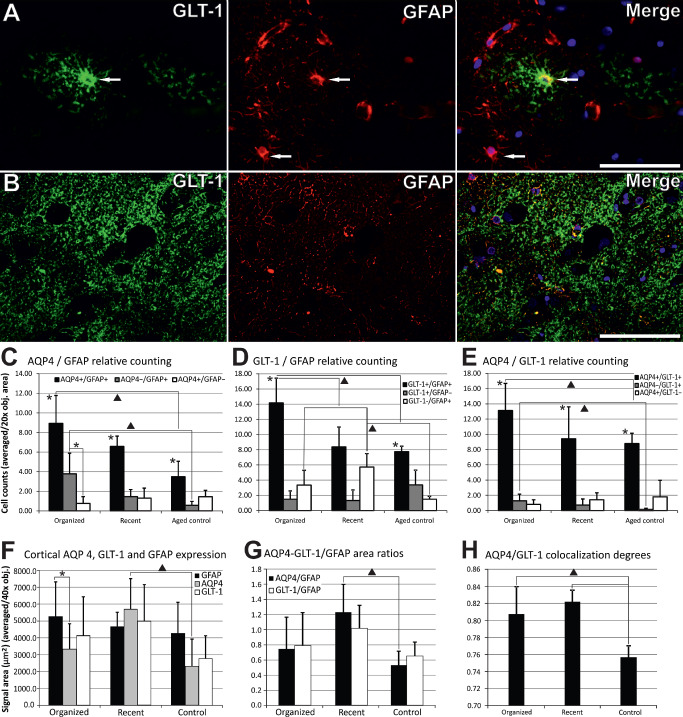
Semiquantitative analysis of aquaporin 4 ( AQP 4), glial fibrillary acidic protein ( GFAP ) and glutamate transporter 1 protein ( GLT ‐1). For direct double/single‐labeled cell counting (AQP4, GFAP and GLT‐1), only clear‐cut cells with visible nuclei were counted (arrows in A for example), while rejecting areas with a diffuse‐like signal (B). Counting showed that double AQP4+/GFAP+, GLT‐1+/GFAP+ and AQP4+/GLT‐1+ cells' population decreased from organized to recent lesional and control cortices (C–E). AQP4–/GFAP+ and AQP4–/GLT‐1+ populations followed the same global trend (C, E), while GLT‐1–/GFAP+ cells were most numerous in recent lesions (D). Semiquantitative expression area analysis shows that AQP4 has a higher expression for recent perilesional cortices compared with control cortices, and has a significantly lower expression than GFAP only for organized perilesional areas (F). AQP4/GFAP area ratios were higher for recent perilesional cortices compared again with control areas, while GLT‐1/GFAP area ratios followed the global trend of AQP4/GFAP (G). AQP4/GLT‐1 correlation coefficients had the lowest values in control cortices (H). * represents statistical significance on Student's t‐test; ▲ represents statistical significance on one‐way analysis of variance with post hoc comparisons using the Tukey's test. Error bars represent standard deviation.
Statistical analysis
All the data were represented graphically and further analyzed utilizing Microsoft Excel 2007 and SPSS 10.0 (SPSS, Inc., Chicago, IL, USA).
For the AQP4/GLT‐1 neuron/vessel distance maps, data were represented graphically as the mean frequency of AQP4‐positive pixels (normalized for a 40× objective area) from the nearest neuron/vessel 11. In order to compare these nonlinear frequency distribution curves, we have performed a one‐way analysis of variance (ANOVA) taking into account all values recorded for each curve from all component cases. Specific paired significances were further evaluated through post hoc comparisons using Tukey's honestly significant difference (HSD) test. Furthermore, we have also analyzed data coming from different distance intervals in order to best characterize any statistical differences between these distribution curves.
The diameters of the vessels included in these proximity studies ranged between 2.90 and 44.66 μm, with an average of 6.85 μm, a median of 6.17 μm and a standard deviation of 3.71 μm (Figure 3F).
Figure 3.
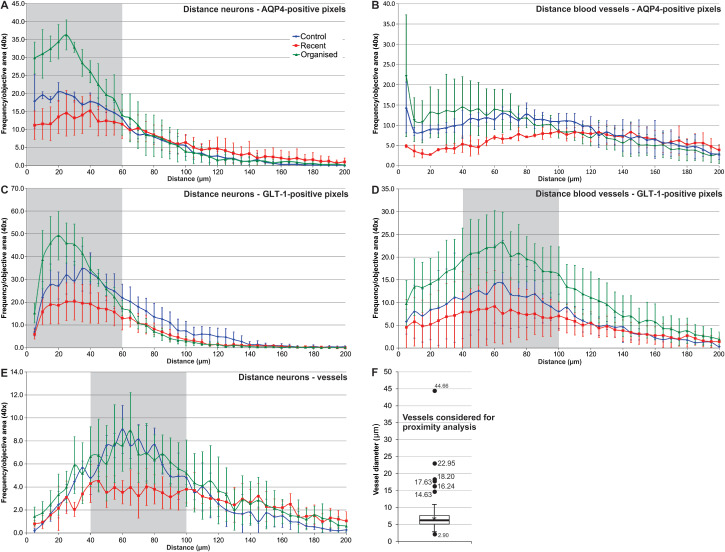
Aquaporin 4 ( AQP 4)/glutamate transporter 1 ( GLT ‐1) distance frequency distribution in relationship with neurons and blood vessels. AQP4‐positive pixels aggregated around neuronal membranes (stained for nicotinic acetylcholine receptor, AchR), with a peak at around 20–40 μm distance from the AchR signals, for all instances studied (A); organized perilesional cortical areas had a denser AQP4 distribution, especially in the 0–60 μm perineuronal interval compared with recent perilesional and control cortices. AQP4‐positive pixels were grouped close around blood vessels' basement membranes (<10 μm), and if considering only the data for the first 100 μm around the blood vessels, the perilesional recent cortices showed significant lower AQP4 pixel frequencies compared with the other two categories (B). GLT‐1‐positive pixels also aggregated around AchR signals, mostly at around 20–40 μm, but with lower density in the immediate neuronal vicinity, compared with AQP4 (C). GLT‐1 distribution around blood vessels showed a peak for all studied instances at 40–100 μm around the vessels, organized lesions having higher densities compared with recent and organized lesions; compared with AQP4, there was no frequency peak in the immediate perivascular compartment (D). AchR‐positive pixels' aggregation around vessels (collagen IV) revealed a peak at a distance of 40–100 μm, with no difference between organized lesions and controls; recent lesions showing a lower plateau in this interval, probably because of edema (E). Also, the amplitude of the AchR collagen IV mapping was decreased compared with the other four instances studied. The diameters of the vessels included in these proximity studies ranged between 2.90 and 44.66 μm, with an average of 6.85 μm (*), a median of 6.17 μm (horizontal bar), a standard deviation of 3.71 μm (error bars) and a Q1–Q3 interquartile interval of 2.34 μm (box plot) (F). Points indicate the average values; error bars represent standard deviation (A–E). Gray‐shaded areas represent intervals for which frequency distributions could statistically differentiate between all three pathological instances considered (one‐way analysis of variance with Tukey's post hoc comparisons); error bars represent standard deviation (A–E).
For the semiquantitative assessment of AQP4, GLT‐1 and GFAP‐positive astrocytes, the signal areas were represented as averages for a 40× objective area, and the expression of the markers was compared between the three histological states (control tissue, recent and organized lesions) utilizing a one‐way ANOVA with the Tukey's correction as post hoc analysis.
For AQP4/GFAP colocalization study, correlation coefficients were first averaged for each image and each patient, data for different pathologies being averaged and compared utilizing again a one‐way ANOVA with Tukey's post hoc analysis. Direct AQP4, GLT‐1 and GFAP single‐ or double‐labeled cell counts were averaged (as number of cells/20× objective area) for each case and pathology, then a one‐way ANOVA with Tukey's post hoc analysis was performed to assess their differences. In all cases, P < 0.05 was used to indicate statistical significance.
Results
General characterization of AQP4 and GLT‐1 expression patterns
Single immunohistochemistry for AQP4 revealed a dense‐diffuse expression pattern throughout the isocortex in aged control tissue, while almost no signal could be detected in the white matter (Figure 4A). The signal looked more diffuse in the middle cortical layers, and more compact in the lower and upper cortical layers, showing a strong line at the level of the pia. Higher magnification objectives revealed mainly a granular vesicular pattern surrounding an astrocyte‐like nucleus (Figure 5A) or, rarely, a more diffuse pattern surrounding obvious neuronal silhouettes (Figure 5D). In surrounding perilesional isocortices, AQP4 showed a relative sparing of the middle cortical layers, with a dense‐diffuse distribution in the lower and upper cortical layers, and with a diffuse‐like involvement of the white matter (Figure 4B). Areas of acutely hypoxic eosinophilic neurons showed no obvious differences from control cortical regions, having a more conserved AQP4 bilaminar expression. Close to old ischemic liquefaction cores, a mixed diffuse granular pattern could be noticed especially in the immediate vicinity of the cavitations (Figure 4D), the immunolabeling clearly decreasing in area and intensity with increasing distance from the cavitation cores (Figures 4D and 6A). Pia and perivascular glia limitans had a strong reactivity in all cases, with regional differences that did not allow for further stratifications between aged controls, recent or old ischemic events.
Figure 4.

Immunoexpression patterns of aquaporin 4 ( AQP 4) and glutamate transporter 1 protein ( GLT ‐1), low power view. AQP4 shows a dense‐diffuse expression pattern in all cortical layers with sparing of the white matter in the cortices of aged control patients (A), whereas the expression is restricted mostly to the upper and lower cortical layers together with a diffuse pattern in the white matter, for perilesional cortices (B). GLT‐1 exhibits the same dense‐diffuse expression, but restricted even more to the upper and lower cortical layers, and with no obvious difference between age controls (data not shown) and perilesional cortices (C). Near an old cavitation site, AQP4 is present in the inner wall of the glial scar, the immunostaining variably diminishing with the increasing distance from the cavity (D). Scale bars represent 200 μm.
Figure 5.
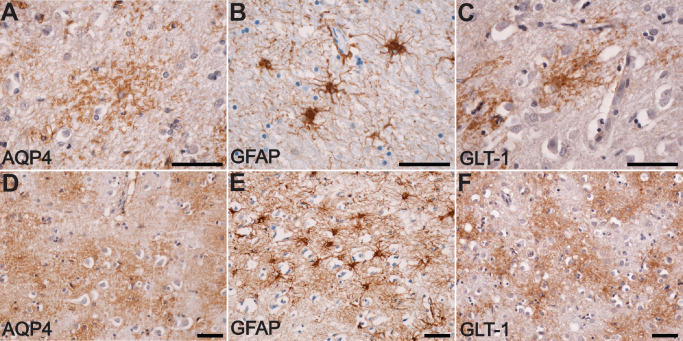
Immunoexpression patterns of aquaporin 4 ( AQP 4) and glutamate transporter 1 protein ( GLT ‐1), high power view. AQP4 shows mainly a granular–vesicular expression pattern (A) that does not resemble the cytoskeleton of the astrocytes stained for glial fibrillary acidic protein (GFAP) (B), but looks more similar to the expression pattern of GLT‐1 (C). Less frequently, a cloudy and diffuse‐like pattern for AQP4 expression could be noted (D), and this again was not similar to anti‐GFAP staining (E), but resembled more to the anti‐GLT‐1 immunostaining (F). Scale bars represent 50 μm.
Figure 6.
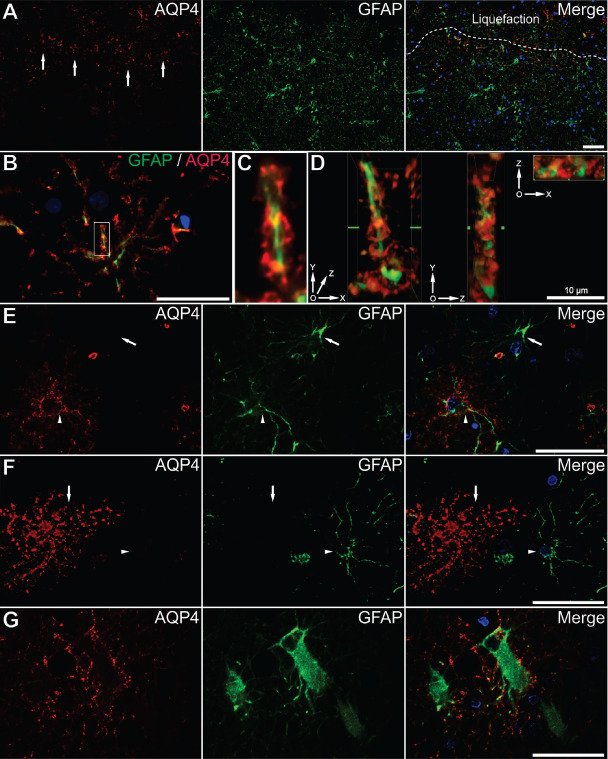
Characterization of aquaporin 4 ( AQP 4) in the astrocytic compartment. Immediately around the cavitation cores, AQP4 (red) is present especially in the inner rim of glial cells [stained for glial fibrillary acidic protein (GFAP) green] (arrows in series A). Dotted line in series A delimitates the cavitation core of the infarct. AQP4 signal surrounds the arms of an astrocyte's cytoskeleton (B). An inset from image B is enlarged in image C, and after z‐scanning and deconvolution, three‐dimensional projections show that AQP4 does not colocalize with GFAP, but rather surrounds the tortuosities of this astrocytic process on its entire length (D). No vessel was present around this cell supporting the idea that AQP4 is not only expressed in astrocytic end feet. Not all GFAP‐positive astrocytes were also positive for AQP4, the arrow depicts a GFAP+/AQP4− astrocyte and the arrow head illustrates a GFAP+/AQP4+ astrocyte (E). Not all AQP4+ astrocytes were GFAP+, the arrow illustrating a GFAP−/AQP4+ astrocyte, and the arrow head a GFAP+/AQP4− astrocyte (F). Perilesional gemistocytes also express AQP4, but restricted mostly to their processes (G). Scale bars represent 50 μm unless denominated otherwise.
Although both are glial markers, the staining pattern of AQP4 did not show too much resemblance with that of GFAP (Figure 5B,E). This was in fact anticipated, as AQP4 is a membrane‐bound protein, while GFAP is a cytoskeleton intermediate filament. We have thus further utilized an antibody raised against the GLT‐1 astrocyte glutamate transporter protein as a selective astrocyte membrane marker. Assessing the expression patterns of GLT‐1, we noticed that it was generally expressed in the upper and lower cortical layers, with almost no expression in the pia, middle cortical layers and the white matter (Figure 4C). There were no obvious differences between age controls and perilesional stroke areas. Around the ischemic old cavities, GLT‐1 was present at distance from the glial scar, as compared with the immediately perilesional expression of AQP4 (Figure 7A). On a closer view, the immunolabeling for GLT‐1 resembled much more the granular and diffuse‐like patterns described for AQP4 (Figure 5C,F) compared with GFAP.
Figure 7.
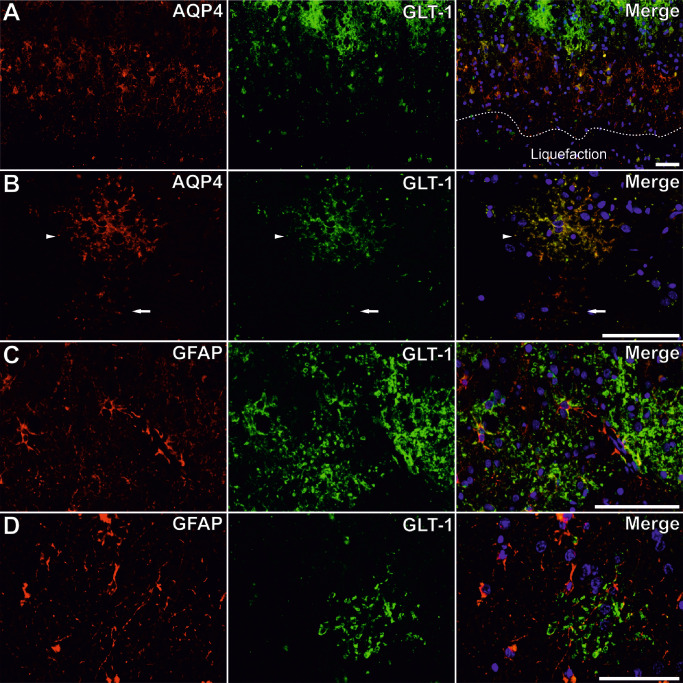
Glutamate transporter 1 protein ( GLT ‐1) and aquaporin 4 ( AQP 4) co‐expression in different types of astrocytes. Although AQP4 (red) is mostly expressed in the vicinity of the scar, GLT‐1 (green) is not present in close proximity of the scar (A). Dotted line in series A delimitates the cavitation core of the infarct. Not all AQP4+ astrocytes are GLT‐1+ (arrow in series B), and they do colocalize on the entire surface of other astrocytes (arrowhead in series B). GLT‐1 does not predominate around blood vessels, where glia limitans (GFAP, red) is present (C), and another example of such a GFAP+/GLT‐1+ astrocyte without a blood vessels in its surroundings (D). Scale bars represent 50 μm.
Distribution of AQP4 in the astrocytic compartment
We next looked at AQP4/GFAP double immunolabelings for characterization of these cells. As already described, at the very edge of the glial scar surrounding old cavitation lesions, the inner layer of astrocytes composing the glial scar expressed AQP4 more than those in the outer layers (Figures 4D and 6A). Three‐dimensional reconstructions showed that AQP4 surrounded the GFAP+ cytoskeleton not only at the end feet, but even in astrocyte processes close to the cell body and on the cell body membrane itself (Figure 6B–D). Although in the majority of the cases there was a strong co‐expression of AQP4 and GFAP in the cytoskeleton and the membrane of the same cell, we have identified rare AQP4−/GFAP+ and AQP4+/GFAP− cells (Figure 6E,F), and this was confirmed later on by directly counting AQP4±/GFAP± cells (Figure 2C). In some reactive astrocytes, AQP4 showed an evident loss of reactivity on the body membranes, retaining its expression only on some of the cells' processes (Figure 6G).
In the cortices, directly counted AQP4+/GFAP+ cells dominated the AQP4±/GFAP± cells' population (Student's t‐test in each group, P < 0.05), and their number decreased from organized and recent perilesional cortices toward controls (8.92 ± 2.85, 6.58 ± 1.05 and 3.49 ± 1.57 cells) (Figure 2C). A one‐way ANOVA found a significant global difference for the number of double‐positive cells for the three conditions [F(2, 25) = 29.394, P < 0.001], and post hoc comparisons using the Tukey's HSD test indicated a difference only between organized areas and control cortices (P < 0.001). Also for AQP4−/GFAP+ cells, their number differed globally [F(2, 24) = 4.66, P < 0.05], with significant differences only between organized areas (3.78 ± 2.10) and control cortices (0.57 ± 0.40) (P < 0.05). No difference could be observed for AQP4+/GFAP− cells. AQP4−/GFAP+ cells were significantly higher in number than AQP4+/GFAP− cells only for organized lesions (3.78 ± 2.10 vs. 0.76 ± 0.70 cells) (Student's t‐test, P < 0.001).
As revealed by deconvolution and three‐dimensional reconstructions, AQP4 was expressed around GFAP+ cytoskeleton (Figure 6B–D). We have next examined semiquantitatively the signal areas extracted from the individual GFAP and AQP4/GLT‐1 channels captured in correspondent double‐fluorescence images for aged control brain tissue, recent ischemic and old perilesional cortices (Figure 2F,G). GFAP expression increased from aged control cortices to recent and organized perilesional cortices, but because of the variations, especially in control and old brain tissues, these differences could not be deemed significant (Figure 2F). AQP4 expression was higher in recent lesions compared with control tissue (one‐way ANOVA with post hoc comparisons, P < 0.05), while the difference did not attain statistical significance for organized areas. For the old perilesional and control tissues, AQP4 expression was lower compared with GFAP levels, and this attained significance only for old perilesional areas (Student's t‐test, P < 0.05). Although the differences were nonsignificant (due again to area‐to‐area variations), this ratio was inversed for incipient ischemic perilesional cortices. AQP4/GFAP area ratios showed global differences between the three groups on a one‐way ANOVA testing [F(2,30) = 14.472, P < 0.001], but with significant between‐pair differences only between recent perilesional and control cortices (post hoc comparisons, P < 0.001) (Figure 2G).
Colocalization of AQP4 and GLT‐1
After characterization of GLT‐1 protein as showing very similar expression patterns to those of AQP4, and thus a membrane‐like localization (Figures 5, 6B and 7B), we have turned to a closer characterization of their relationship. In what it regarded evident differences, it was clear that in the glial scar surrounding old liquefaction cavities, GLT‐1 was mostly expressed at some distance from the wall of the cavity, whereas AQP4 was present especially in the inner part of the glial scar (Figure 7A).
GLT‐1 expression areas had the tendency to be higher than AQP4 on corresponding areas from organized and control cortices, but this difference was not found significant (Student's t‐test, P > 0.05) (Figure 2F). A one‐way ANOVA test also found no global difference for the GLT‐1 expression areas between the three categories (one‐way ANOVA test, P > 0.05). Moreover, GLT‐1/GFAP area ratios showed no differences between the three studied groups (one‐way ANOVA test, P > 0.05), or compared with AQP4/GFAP corresponding indexes (Student's t‐test, P > 0.05) (Figure 2G). On perilesional cortices, directly counted GLT‐1+/GFAP+ cells dominated the GLT‐1±/GFAP± cells' population (Student's t‐test, P ≤ 0.05; except for recent areas where there was no significant difference with the GLT‐1−/GFAP+ cells), and their number decreased from organized and recent perilesional cortices toward controls (14.08 ± 3.73, 8.30 ± 2.27 and 7.75 ± 0.71 cells) [one‐way ANOVA, F(2, 23) = 60.188, P < 0.001; with post hoc comparisons significant for organized‐recent (P < 0.05) and organized‐control area pairs (P < 0.001)] (Figure 2D). Most of the inert astrocytes (in respect with their GLT‐1 expression, namely, GLT‐1−/GFAP+ cells) were found in recent lesions [one‐way ANOVA, F(2, 23) = 4.89, P < 0.01; with post hoc comparisons significant for recent‐organized (P < 0.05) and recent‐control area pairs (P < 0.01)] (Figure 2D). The double AQP4/GLT‐1‐positive cells also gradually decreased in number from organized (13.13 ± 3.55 cells) to recent (9.43 ± 4.19 cells) and control cortices (8.79 ± 1.36 cells) [one‐way ANOVA, F(2, 25) = 71.467, P < 0.01; with post hoc comparisons significant only for organized‐control area pairs (P < 0.01)], and the AQP4−/GLT‐1+ cells followed the same trend (1.27 ± 0.88, 0.71 ± 0.81 and 0.13 ± 0.18 cells) [one‐way ANOVA, F(2, 25) = 7.112, P < 0.05 with post hoc comparisons significant only for organized‐control area pairs (P < 0.05)] (Figure 2E). No significant difference could be found for AQP4+/GLT‐1‐ cells' number variations. Overall, AQP4+/GLT‐1+ cells were the most abundant type of AQP4±/GLT‐1± cells in each of the three pathological instances (Student's t‐test, P < 0.001). Observing GFAP/GLT‐1 double staining, it was clear that most perivascular end feet did not co‐express GLT‐1 (Figure 7C), as well as many other astrocytes' branches, but on the other hand, the vast majority of GLT‐1+ cells were also GFAP+ (Figures 2D and 7D).
Following the colocalization study, most AQP4+ astrocytes were also positive for GLT‐1 (Figures 2H and 7B). Control cases showed the lowest colocalization degree (0.756 ± 0.013), lower than the colocalization levels for organized (0.807 ± 0.032) and recent perilesional tissues (0.821 ± 0.013) [one‐way ANOVA, F(2, 16) = 3.149, P < 0.01; with post hoc comparisons significant for organized‐control (P < 0.05) and recent‐control area pairs (P < 0.001)] (Figure 2H). Although there was a slight tendency for the recent perilesional glial cells to have the highest AQP4/GLT‐1 colocalization, this difference was not significant because of the inter‐case heterogeneity.
Aquaporin 4 distribution in relationship with the neuronal compartment
Regardless of its glial cell localization, we noticed that AQP4 expression patterns tended to concentrate around neuronal silhouettes labeled for AchR, in both control and perilesional cortices (Figure 8A,B). Given the observed aggregation of AQP4 around neurons, we next evaluated its distance frequency distribution relative to the neuronal silhouettes (Figure 3A). There was a higher frequency of AQP4 staining around the neurons for all studied cases, with a peak for the 20–40 μm distance interval, the stained pixels' frequency decreasing with further increasing distance. Old ischemic cortical regions showed the highest frequency of AQP4 signal in the area of 0–60 μm around the neuronal membranes, and there were significant differences between all three conditions in this perineuronal distance interval [one‐way ANOVA, F(2, 131) = 84.681, P < 0.001, with post hoc comparisons using the Tukey's test (P ≤ 0.01)]. For the 0–70 μm interval around the neurons, only recent perilesional areas had a significantly different distribution compared with the other two conditions, and no differences could be observed if we considered the data beyond 100 μm (one‐way ANOVA with post hoc comparisons using the Tukey's test).
Figure 8.
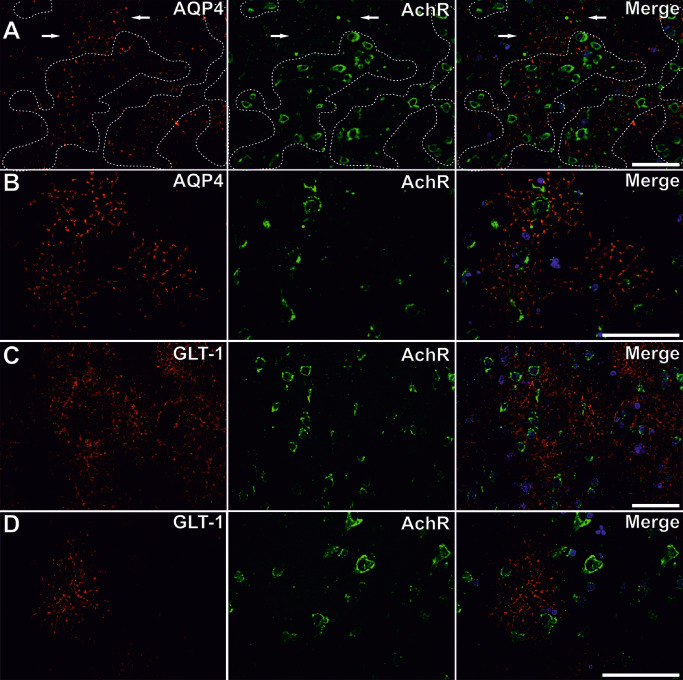
Aquaporin 4 ( AQP 4) expression in relationship with the neuronal compartment. For both control (A) and perilesional cortices (B), AQP4 (red) aggregates around neuronal silhouettes [membranes stained for the nicotinic acetylcholine receptor (AchR) green]. Dotted line in series A encircles neurons showing that only little AQP4‐positive areas are not situated in the vicinity of neuronal membranes (arrows). GLT‐1 (red) is also expressed preferentially around neurons (AchR, green) (C, D). Scale bars represent 50 μm.
GLT‐1 distribution in relationship with the neuronal compartment
In analogy to AQP4, we have next sought to characterize GLT‐1‐positive pixels' distribution around the perikaryons (Figures 3C and 8C,D). The distance frequency distribution mapping for GLT‐1 and AchR revealed again an increase in pixels' frequencies in the 20–40 μm perineuronal areas, but compared with AQP4, GLT‐1 pixels showed a drop in frequency in the immediate neuronal proximity. Considering the data in the 0–60 μm distance interval around the neurons, there were significant differences between all three conditions [one‐way ANOVA, F(2, 165) = 37.871, P < 0.001, with post hoc comparisons using the Tukey's test (P < 0.001)]. Moreover, a paired comparison showed that GLT‐1 recorded higher frequency values compared with AQP4 pixels in the same perineuronal distance intervals (Student's t‐test, P < 0.05).
AQP4 distribution in the perivascular compartment
Triple immunofluorescence for AQP4, collagen IV and CD31 confirmed that AQP4 was strongly expressed in the perivascular glia limitans, but also on the complete membrane surface of some astrocytes that apparently did not show any contact with an endothelium or vascular basement membrane (Figure 9A). When analyzing vessels and perivascular regions, in both control and ischemic perilesional sites, we noticed AQP4 to be present with no clear‐cut preference around vessels with intact or discontinuous endothelium/basement membrane (Figure 9B).
Figure 9.
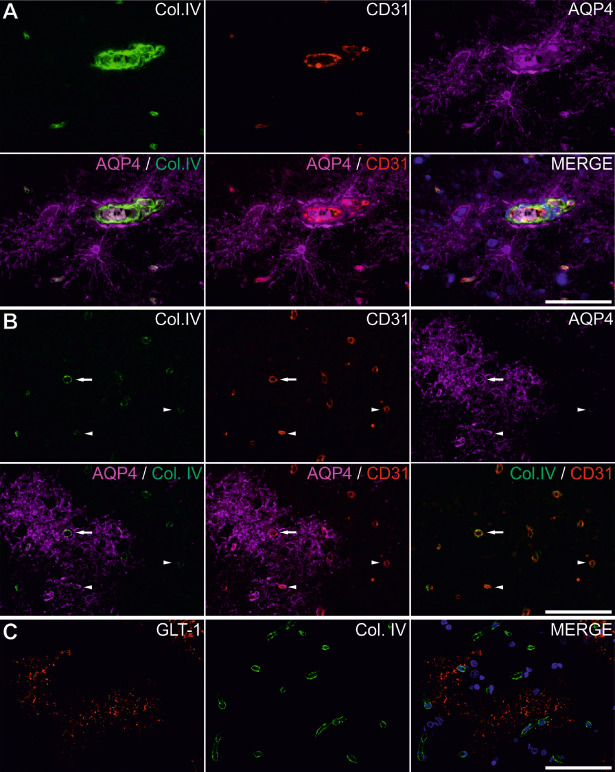
Aquaporin 4 (AQP4) and glutamate transporter 1 (GLT‐1) expression patterns in the perivascular space. Triple immunofluorescence for AQP4, collagen IV (Col. IV) and CD31 on control tissue shows AQP4 strong immunolabeling of astrocytic end feet but also bodies and complete astrocytic extensions. The strongest signal comes around a medium‐sized blood vessel with thickened and bisected basement membrane (series A). In perilesional areas, basement membrane rough integrity does not predict the association with the more diffuse surrounding AQP4 expression (B). Arrows represent a vessel with apparent integral basement membrane but heavily surrounded by AQP4, whereas arrowheads show vessels with much fainter and apparently discontinuous basement membranes, but variable associated with surrounding AQP4 (series B). On double immunohistochemistry for GLT‐1 and collagen IV, GLT‐1 does not seem to have any predilection for the perivascular space (C). Scale bars represent 50 μm.
Next, we wanted to assess any putative variations for the distributions of AQP4 water channels around the blood vessels, by analyzing the frequency distribution of AQP4 signal pixels around the collagen IV silhouettes (Figure 3B). This distribution showed first that for control and organized areas, modest peak frequencies were recorded either in the close proximity of the blood vessels (under 10 μm away) or in the range of 60–120 μm from the basement membranes. Recent perilesional areas showed an actual drop in frequency around the blood vessels compared with the other two conditions, and statistical analysis of the data considered for the 0–100 μm distance interval revealed that only recent perilesional areas could be found significantly different from the other two conditions {one‐way ANOVA [F(2, 279) = 24.547, P < 0.001, with post hoc comparisons using the Tukey's test (P < 0.001)]}.
GLT‐1 distribution in the perivascular compartment and control of the imaging algorithm
We next turned to the GLT‐1 distribution in relationship with the vascular compartment, on GLT‐1–collagen IV double immunolabelings (Figures 3D and 9C). After plotting the GLT‐1 distance frequency distribution around vessels, organized perilesional cortices showed the highest values compared with both other instances for the whole 0–200 μm perivascular interval [one‐way ANOVA (2, 599) = 69.261, P < 0.001, with post hoc comparisons using Tukey's test (P < 0.001)]. However, a statistically significant difference between all three pathological states was found only for the 40–100 μm perivascular interval [one‐way ANOVA (2, 194) = 82.126, P < 0.001, with post hoc comparisons using the Tukey's test (P ≤ 0.05)].
Lastly, in order to evaluate the significance of our findings over the general neuropil density changes during stroke in the perilesional cortices, we have analyzed the distance–frequency distributions for vessels around neurons in our three pathological instances based on collagen IV/AchR double stainings (Figure 3E). The resulting graph revealed a relative small amplitude AchR peak at around 40–100 μm around vessels for control and organized perilesional cortices, and that for no distance interval these two instances could be differentiated. However, recent perilesional cortices showed significantly smaller density values compared with other two instances only for the 40–100 μm perineuronal interval [one‐way ANOVA (2, 181) = 38.431, P < 0.001, with post hoc comparison using Tukey's test (P < 0.001)].
Discussion
Although therapies targeting brain edema have a long‐standing history in the treatment of stroke, they are mostly aimed at the end result of the physiopathological chain, namely, the volumetric brain matter increase 1, 34, 39, or at managing its symptoms, whereas the exact mechanisms leading to edema development are still incompletely understood 9. One of the main key players in buffering water transfer across BBB is AQP4, located especially on the perivascular and subleptomeningeal astrocytic end feet 27, 28. Although a large number of studies have addressed this issue, the complete physiological and physiopathological picture of AQP4 function is far from complete 25, 30, 31, 32. On the other hand, energy depletion in the astrocytic compartment following ischemia/anoxia also interferes with the re‐uptake of glutamate primarily because of the loss of function of the astrocytic membrane‐associated GLT‐1 glycoprotein, resulting in a further increase of the abnormal accumulation of synaptic glutamate and the consecutive activation of glutamate‐induced toxicity cascade 22, 42. GLT‐1 transporter is responsible for the bulk of glutamate reuptake in the central nervous system 41, and GLT‐1‐deficient mice show increased glutamate toxicity in prone conditions 42. Recent data suggest that AQP4 and the GLT‐1 glycoprotein exist in astrocytic membranes as a functional complex, as neuromyelitis optica‐immunoglobulin G autoantibodies directed against AQP4 lead to a concomitant loss of GLT‐1 13, 14. Given their functional differences, however, little is known about their effective distribution in relationship with the perivascular and perineuronal glial sectors.
In the present study, we have evaluated for the first time the expression of AQP4 and GLT‐1 at the tissue level in the perilesional cortices of recent and old ischemic infarctions compared with aged control human patients. Paralleling other studies on both animal models and human stroke patients, we have shown a general AQP4 increase in the perilesional vicinity, as well as in the perilesional cortices and their correspondent subpial and subependymal regions 3, 30, 35. Furthermore, we have found that while a dense‐diffuse expression pattern was present throughout the cortex in aged controls, perilesional cortices showed a more clear segregation of the signal for the upper and lower cortical layers, together with a diffuse pattern in the white matter. This corresponds well to the fact that water diffusion is maximum in the sub‐pial cortical layers, in the deep layers near the white matter and along the white matter tracts themselves 23, 46. Also, we showed that in both perilesional cortices and control patients, AQP4 expression was not restricted to perivascular and submeningeal astrocytic end feet, but was present on the membranes of their entire cell bodies and extensions, suggesting that water buffering toward and from the blood vessels might not be its only function 44. These associate well with the biphasic AQP4 expression found after transient focal cerebral ischemia in mice, in which after an initial expression peak localized at the astrocytic end feet, the second wave of expression shifted its localization to the entire membrane of the astrocyte 35.
In order to see the relative distribution of AQP4 and GLT‐1 in the astrocytic compartment, we next counted directly the number of AQP4±/GLT‐1±/GFAP± cells on double‐labeled slides. AQP4+/GFAP+, GLT‐1+/GFAP+ and AQP4+/GLT‐1+ represented the most abundant types of astrocyte, and these densities increased gradually (although not significantly for all cases) from control to perilesional recent and organized cortices, showing that most of this reaction consists in astrocyte hyperplasia/astrocytosis and thus is not functionally inert. As in stroke both astrocytosis and hypertrophy/astrogliosis are encountered, and as we could not show a direct correlation between AQP4 and GFAP expression levels, it would be difficult to speculate here to what extent astrogliosis occurs compared with astrocytosis in this sector.
GLT‐1 expression pattern overlapped well on the AQP4 expression, and direct cell counting confirmed that most of the AQP4‐positive astrocytes were also positive for GLT‐1. Moreover, the correlation coefficients and the number of double‐positive cells were higher in lesional cortices compared with controls, a trend which was also shared by AQP4− cells (either GFAP+ or GLT‐1+), but not by GLT‐1− cells (either GFAP+ or AQP4+). Although this approach had the major caveat that it utilized only images where individual cells could be reliably counted (and rejecting diffuse‐like signal patterns), it still underlined subtle differences in the cortical distribution of the two markers even in the same cell for the three histological instances considered. Together with the fact that we and others have shown AQP4 expression extending on the astrocyte membranes other than the end feet themselves 35, and that practically no other class of glial cells other than astrocytes express AQP4 40, these data suggest a more intense water‐buffering activity for these astrocytes during the acute ischemic event compared with those found in age controls. This is supported by imaging data showing that edema occurs most frequently in the initial phases of an ischemic stroke, especially if we do not take into account secondary hemorrhagic transformations 26. In contrast to perilesional cortices, AQP4 was expressed as a rim in the immediate vicinity of the liquefactive cavitations, an analog expression pattern around the infarct cores being also documented by other studies 38, 40. It seems not only that AQP4 mediates water buffering itself in a tissue area that lost its BBB bordering, but also that it facilitates water influx across the advancing edges of migrating astroglia, as a reduced migration of reactive astrocytes toward the site of injury, and overall an impaired glial scar formation was observed in a cortical stab injury model on AQP4‐null mice 38.
Examined more closely, the astrocytes composing the perivascular glia limitans or the inner wall of the glial scar surrounding the liquefaction cores (deemed positive for AQP4) were mostly negative for GLT‐1, suggesting that these “interfacing” set of astrocytes have indeed more water‐buffering function rather than a glutamate‐buffering role. However, in the perilesional cortices, at distance from the vascular discontinuity areas, this clear stratification was lost. This finding suggests that at distance from the BBB opening, AQP4‐positive cells shift their phenotype to buffer also the glutamate levels by overexpressing the GLT‐1 transporter, thus presenting AQP4‐positive cells as a heterogeneous class of astrocytes depending on the functional requirements of the surrounding neuropil. Although no other data are available at the moment regarding the dynamics of GLT‐1 in post‐stroke conditions for human pathology, it could be that despite the fact that AQP4+/GLT‐1+ cells increase in number and signal colocalization in acute stroke, the incapacity of GLT‐1 to buffer nonphysiologically large amounts of glutamate, as well as with the overexpression of neuronal glutamate receptors, may still incline the balance toward neurotoxicity 50.
To assess the spatial relationship between AQP4/GLT‐1 and neurons, we performed a distance–frequency distribution study for AQP4 and GLT‐1 together with the neuronal membranes. This analysis showed a shift of AQP4‐positive pixels toward the vicinity of neuronal compartment in chronic perilesional cortices compared with control and recent perilesional cortices. It was interesting to see that for all instances, AQP4 pixels did not peak immediately in contact with the neurons, but around 20–40 μm from the closest neuronal membranes.
For GLT‐1, the change resembled AQP4's dynamics, but with higher amplitude for the 20–40 μm interval and a drastic drop for the <20 μm close perineuronal domain. Together with the increasing colocalization of GLT‐1 and AQP4 in these areas, this supports the idea of an increased glutamate–water buffering domain that develops as a response to ischemic conditions, a duality that is not maintained in the very close vicinity of the neurons themselves. Glutamate has also been suggested to escape from the synaptic clefts, generating an extrasynaptic glutamate pool, or the so‐called glutamate spillover phenomenon 5, 6, 19, 29. As our data suggest, it seems that this perineuronal glutamate‐diffusing compartment becomes more abundant in pathological conditions, but further analysis on nonaged or non‐injured cellular setups are needed to confirm/infirm the presence of a closer immediately perineuronal glutamate uptake compartment.
Next, we looked at the AQP4/GLT‐1 distribution around blood vessels. AQP4 was strongly expressed in the perivascular astrocytic end feet, and its presence/absence was not dependent on the integrity or on the discontinuity of the endothelium or basal lamina, for both control and ischemic perilesional sites, as shown by our triple labeling for AQP4, collagen IV and CD31. Recent data utilizing an embolic model of focal cerebral ischemia in Wistar rats to mimic the ischemia‐induced BBB breakdown revealed that at 5 and 25 h after ischemia induction, the morphology of endothelial tight junction complexes showed no ultrastructural changes in regions with massive albumin leakage into the neuropil 18. Thus, it becomes clear that such differentiation in AQP4 expression in the vascular vicinity can be made only if these subtle changes closely following BBB dysfunction would be available for study, details that were beyond the possibility of classical fluorescence microscopy. We have also performed a distance–frequency distribution analysis for AQP4 together with the vascular basement membranes. As expected, we obtained for all instances an AQP4 peak expression in contact with the basement membranes, but it was of interest to note that in the range of 0–100 μm around blood vessels, recent perilesional cortices showed a significant drop in AQP4 pixel distribution compared with control and old perilesional cortices, suggesting that early ischemic events impair the perivascular distribution of AQP4 channels. It is known that in response to injury, astrocytes retract their end feet from blood vessels, leading to an increased BBB permeability, and these data fit well with our finding 15, 24, 36. Although described as tightly associated with AQP4, the expression pattern of GLT‐1 around vessels did not show a common distribution with that of AQP4, probably reflecting that the massive water‐buffering capabilities needed around hindered BBB do not justify its presence in this sector.
Finally, we were also interested to check somehow that the changes observed in the distance frequency distributions are not only cause by the density changes that occur during the dynamics of stroke. Indeed edema, neuronal loss, angiogenesis and gliosis participate all at different extents in stroke, and even in normal conditions the interstitial space can vary in volume with up to 60% during the night–day rhythm 47. To this extent we have also performed here a distance–frequency distribution analysis between vessels and neurons. Comparing the resulting data with our AQP4/GLT‐1 study (and especially for the distribution around neurons), both the shapes of the distribution curves and the amplitude of the changes strongly suggested that AQP4/GLT‐1 do not just follow inertly the changes in the neuropil's density.
Altogether, our study suggests a dual and simultaneous astrocytic function depending on the relative distance to neurons and blood vessels, with increased water and glutamate‐buffering capability in the 20–40 μm perineuronal space and water‐buffering capability in the immediate perineuronal space, even higher than around perivascular spaces. As examples, P2X7 receptor (P2X7R) agonists and antagonists are now known to modulate AQP4 expression, and β‐lactam antibiotic ceftriaxone is a potent GLT‐1 stimulator 21, 48. It can be thus conceived that beyond modulating a decrease of AQP4 function during the initial phase of cytotoxic edema with later on increased functionality, with a general increase of GLT‐1 glutamate‐buffering capacity, a preferential expression of the two transporters in the close‐ and mid‐perineuronal/perivascular space would maximize their potential usage in future multimodal and personalized treatments for stroke.
Supporting information
Figure S1. Immunohistochemistry controls. While specific and as expected glial‐like, neuronal and vascular patterns were obtained for the individual antibodies utilized in this study (A), skipping the primary antibodies and adding only the detection systems (anti‐mouse and anti‐rabbit polymer‐HRP and, respectively, anti‐goat‐biotinylated + streptavidin HRP), ensured no false‐positive immunostaining (B). For double immunohistochemistry on two primary rabbit antibodies, after detecting collagen IV (rabbit anti‐human) with anti‐rabbit polymer‐HRP and Alexa‐488 tyramide, slides were subjected (C and D) or not (E and F) to a low pH SDS antibody elution step. Adding another anti‐rabbit Alexa 594 antibody shows no labeling for collagen IV when using the elution buffer (C), but reveals strong cross‐reactivity when not utilizing the elution step (E). Moreover, continuing the immunodetection sequence with the anti‐AQP4 antibody (also rabbit anti‐human) and an anti‐rabbit Alexa 594 for labeling shows besides the new glial signal, again, no cross‐reactivity with collagen IV when utilizing the elution method (D), and a strong cross‐reactivity when not using it (F), proving that PBS washing steps during the second immunoprocedure are not enough to wash away the first primary antibody, which is still well detectable. Scale bars represent 50 μm.
Acknowledgments
We thank Mr. David Wiles (European Technical Support Engineer—Media Cybernetics, Marlow, UK) for providing us with helpful information on creating the scripts necessary to perform the frequency distribution analysis. We would also like to express our gratitude to Professor Florin Gorunescu from the Department of Biostatistics of the University of Medicine and Pharmacy of Craiova for expert reviewing of our statistical data. This paper is supported by the Sectorial Operational Programme Human Resources Development, financed from the European Social Fund and by the Romanian Government under the contract number POSDRU/89/1.5/S/64109 to D.P.
References
- 1. Abou‐Chebl A (2013) Management of acute ischemic stroke. Curr Cardiol Rep 15:348. [DOI] [PubMed] [Google Scholar]
- 2. Amaro S, Soy D, Obach V, Cervera A, Planas AM, Chamorro A (2007) A pilot study of dual treatment with recombinant tissue plasminogen activator and uric acid in acute ischemic stroke. Stroke 38:2173–2175. [DOI] [PubMed] [Google Scholar]
- 3. Aoki K, Uchihara T, Tsuchiya K, Nakamura A, Ikeda K, Wakayama Y (2003) Enhanced expression of aquaporin 4 in human brain with infarction. Acta Neuropathol 106:121–124. [DOI] [PubMed] [Google Scholar]
- 4. Badaut J, Lasbennes F, Magistretti PJ, Regli L (2002) Aquaporins in brain: distribution, physiology, and pathophysiology. J Cereb Blood Flow Metab 22:367–378. [DOI] [PubMed] [Google Scholar]
- 5. Barbour B (2001) An evaluation of synapse independence. J Neurosci 21:7969–7984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Barbour B, Hausser M (1997) Intersynaptic diffusion of neurotransmitter. Trends Neurosci 20:377–384. [DOI] [PubMed] [Google Scholar]
- 7. Benga G (1994) Water channels in membranes. Cell Biol Int 18:829–833. [DOI] [PubMed] [Google Scholar]
- 8. Choi DW (1987) Ionic dependence of glutamate neurotoxicity. J Neurosci 7:369–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Donkin JJ, Vink R (2010) Mechanisms of cerebral edema in traumatic brain injury: therapeutic developments. Curr Opin Neurol 23:293–299. [DOI] [PubMed] [Google Scholar]
- 10. Finsterer J (2010) Management of cryptogenic stroke. Acta Neurol Belg 110:135–147. [PubMed] [Google Scholar]
- 11. Fung AS, Jonkman J, Tannock IF (2012) Quantitative immunohistochemistry for evaluating the distribution of Ki67 and other biomarkers in tumor sections and use of the method to study repopulation in xenografts after treatment with paclitaxel. Neoplasia 14:324–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Harvey BK, Airavaara M, Hinzman J, Wires EM, Chiocco MJ, Howard DB et al (2011) Targeted over‐expression of glutamate transporter 1 (GLT‐1) reduces ischemic brain injury in a rat model of stroke. PLoS ONE 6:e22135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hinson SR, Pittock SJ, Lucchinetti CF, Roemer SF, Fryer JP, Kryzer TJ et al (2007) Pathogenic potential of IgG binding to water channel extracellular domain in neuromyelitis optica. Neurology 69:2221–2231. [DOI] [PubMed] [Google Scholar]
- 14. Hinson SR, Roemer SF, Lucchinetti CF, Fryer JP, Kryzer TJ, Chamberlain JL et al (2008) Aquaporin‐4‐binding autoantibodies in patients with neuromyelitis optica impair glutamate transport by down‐regulating EAAT2. J Exp Med 205:2473–2481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ito U, Hakamata Y, Kawakami E, Oyanagi K (2011) Temporary cerebral ischemia results in swollen astrocytic end‐feet that compress microvessels and lead to delayed [corrected] focal cortical infarction. J Cereb Blood Flow Metab 31:328–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ke C, Poon WS, Ng HK, Pang JC, Chan Y (2001) Heterogeneous responses of aquaporin‐4 in oedema formation in a replicated severe traumatic brain injury model in rats. Neurosci Lett 301:21–24. [DOI] [PubMed] [Google Scholar]
- 17. Klatzo I (1987) Pathophysiological aspects of brain edema. Acta Neuropathol 72:236–239. [DOI] [PubMed] [Google Scholar]
- 18. Krueger M, Hartig W, Reichenbach A, Bechmann I, Michalski D (2013) Blood–brain barrier breakdown after embolic stroke in rats occurs without ultrastructural evidence for disrupting tight junctions. PLoS ONE 8:e56419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kullmann DM, Erdemli G, Asztely F (1996) LTP of AMPA and NMDA receptor‐mediated signals: evidence for presynaptic expression and extrasynaptic glutamate spill‐over. Neuron 17:461–474. [DOI] [PubMed] [Google Scholar]
- 20. Lee JM, Zipfel GJ, Choi DW (1999) The changing landscape of ischaemic brain injury mechanisms. Nature 399:A7–A14. [DOI] [PubMed] [Google Scholar]
- 21. Lee M, Lee SJ, Choi HJ, Jung YW, Frokiaer J, Nielsen S et al (2008) Regulation of AQP4 protein expression in rat brain astrocytes: role of P2X7 receptor activation. Brain Res 1195:1–11. [DOI] [PubMed] [Google Scholar]
- 22. Lepore AC, O'Donnell J, Kim AS, Yang EJ, Tuteja A, Haidet‐Phillips A et al (2011) Reduction in expression of the astrocyte glutamate transporter, GLT1, worsens functional and histological outcomes following traumatic spinal cord injury. Glia 59:1996–2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Leuze CW, Anwander A, Bazin PL, Dhital B, Stuber C, Reimann K et al (2014) Layer‐specific intracortical connectivity revealed with diffusion MRI. Cereb Cortex 24:328–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mani N, Khaibullina A, Krum JM, Rosenstein JM (2005) Astrocyte growth effects of vascular endothelial growth factor (VEGF) application to perinatal neocortical explants: receptor mediation and signal transduction pathways. Exp Neurol 192:394–406. [DOI] [PubMed] [Google Scholar]
- 25. Manley GT, Fujimura M, Ma T, Noshita N, Filiz F, Bollen AW et al (2000) Aquaporin‐4 deletion in mice reduces brain edema after acute water intoxication and ischemic stroke. Nat Med 6:159–163. [DOI] [PubMed] [Google Scholar]
- 26. Mayer SA, Lignelli A, Fink ME, Kessler DB, Thomas CE, Swarup R et al (1998) Perilesional blood flow and edema formation in acute intracerebral hemorrhage: a SPECT study. Stroke 29:1791–1798. [DOI] [PubMed] [Google Scholar]
- 27. Nagelhus EA, Mathiisen TM, Ottersen OP (2004) Aquaporin‐4 in the central nervous system: cellular and subcellular distribution and coexpression with KIR4.1. Neuroscience 129:905–913. [DOI] [PubMed] [Google Scholar]
- 28. Nielsen S, Nagelhus EA, Amiry‐Moghaddam M, Bourque C, Agre P, Ottersen OP (1997) Specialized membrane domains for water transport in glial cells: high‐resolution immunogold cytochemistry of aquaporin‐4 in rat brain. J Neurosci 17:171–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Okubo Y, Sekiya H, Namiki S, Sakamoto H, Iinuma S, Yamasaki M et al (2010) Imaging extrasynaptic glutamate dynamics in the brain. Proc Natl Acad Sci U S A 107:6526–6531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Okuno K, Taya K, Marmarou CR, Ozisik P, Fazzina G, Kleindienst A et al (2008) The modulation of aquaporin‐4 by using PKC‐activator (phorbol myristate acetate) and V1a receptor antagonist (SR49059) following middle cerebral artery occlusion/reperfusion in the rat. Acta Neurochir Suppl 102:431–436. [DOI] [PubMed] [Google Scholar]
- 31. Papadopoulos MC, Manley GT, Krishna S, Verkman AS (2004) Aquaporin‐4 facilitates reabsorption of excess fluid in vasogenic brain edema. FASEB J 18:1291–1293. [DOI] [PubMed] [Google Scholar]
- 32. Papadopoulos MC, Verkman AS (2013) Aquaporin water channels in the nervous system. Nat Rev Neurosci 14:265–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Pirici D, Mogoanta L, Kumar‐Singh S, Pirici I, Margaritescu C, Simionescu C et al (2009) Antibody elution method for multiple immunohistochemistry on primary antibodies raised in the same species and of the same subtype. J Histochem Cytochem 57:567–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Qureshi AI, Suarez JI, Yahia AM, Mohammad Y, Uzun G, Suri MF et al (2003) Timing of neurologic deterioration in massive middle cerebral artery infarction: a multicenter review. Crit Care Med 31:272–277. [DOI] [PubMed] [Google Scholar]
- 35. Ribeiro MC, Hirt L, Bogousslavsky J, Regli L, Badaut J (2006) Time course of aquaporin expression after transient focal cerebral ischemia in mice. J Neurosci Res 83:1231–1240. [DOI] [PubMed] [Google Scholar]
- 36. Ridet JL, Malhotra SK, Privat A, Gage FH (1997) Reactive astrocytes: cellular and molecular cues to biological function. Trends Neurosci 20:570–577. [DOI] [PubMed] [Google Scholar]
- 37. Roger VL, Go AS, Lloyd‐Jones DM, Adams RJ, Berry JD, Brown TM et al (2011) Heart disease and stroke statistics—2011 update: a report from the American Heart Association. Circulation 123:e18–e209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Saadoun S, Papadopoulos MC, Watanabe H, Yan D, Manley GT, Verkman AS (2005) Involvement of aquaporin‐4 in astroglial cell migration and glial scar formation. J Cell Sci 118:5691–5698. [DOI] [PubMed] [Google Scholar]
- 39. Sakamaki M, Igarashi H, Nishiyama Y, Hagiwara H, Ando J, Chishiki T et al (2003) Effect of glycerol on ischemic cerebral edema assessed by magnetic resonance imaging. J Neurol Sci 209:69–74. [DOI] [PubMed] [Google Scholar]
- 40. Satoh J, Tabunoki H, Yamamura T, Arima K, Konno H (2007) Human astrocytes express aquaporin‐1 and aquaporin‐4 in vitro and in vivo . Neuropathology 27:245–256. [DOI] [PubMed] [Google Scholar]
- 41. Seal RP, Amara SG (1999) Excitatory amino acid transporters: a family in flux. Annu Rev Pharmacol Toxicol 39:431–456. [DOI] [PubMed] [Google Scholar]
- 42. Tanaka K, Watase K, Manabe T, Yamada K, Watanabe M, Takahashi K et al (1997) Epilepsy and exacerbation of brain injury in mice lacking the glutamate transporter GLT‐1. Science 276:1699–1702. [DOI] [PubMed] [Google Scholar]
- 43. Verkman AS (2012) Aquaporins in clinical medicine. Annu Rev Med 63:303–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Verkman AS, Binder DK, Bloch O, Auguste K, Papadopoulos MC (2006) Three distinct roles of aquaporin‐4 in brain function revealed by knockout mice. Biochim Biophys Acta 1758:1085–1093. [DOI] [PubMed] [Google Scholar]
- 45. Viegas MS, Martins TC, Seco F, do Carmo A (2007) An improved and cost‐effective methodology for the reduction of autofluorescence in direct immunofluorescence studies on formalin‐fixed paraffin‐embedded tissues. Eur J Histochem 51:59–66. [PubMed] [Google Scholar]
- 46. Wolburg‐Buchholz K, Mack AF, Steiner E, Pfeiffer F, Engelhardt B, Wolburg H (2009) Loss of astrocyte polarity marks blood–brain barrier impairment during experimental autoimmune encephalomyelitis. Acta Neuropathol 118:219–233. [DOI] [PubMed] [Google Scholar]
- 47. Xie L, Kang H, Xu Q, Chen MJ, Liao Y, Thiyagarajan M et al (2013) Sleep drives metabolite clearance from the adult brain. Science 342:373–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Yang J, Li MX, Luo Y, Chen T, Liu J, Fang P et al (2013) Chronic ceftriaxone treatment rescues hippocampal memory deficit in AQP4 knockout mice via activation of GLT‐1. Neuropharmacology 75C:213–222. [DOI] [PubMed] [Google Scholar]
- 49. Zeng XN, Sun XL, Gao L, Fan Y, Ding JH, Hu G (2007) Aquaporin‐4 deficiency down‐regulates glutamate uptake and GLT‐1 expression in astrocytes. Mol Cell Neurosci 34:34–39. [DOI] [PubMed] [Google Scholar]
- 50. Zhang F, Guo A, Liu C, Comb M, Hu B (2013) Phosphorylation and assembly of glutamate receptors after brain ischemia. Stroke 44:170–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Zinchuk V, Zinchuk O, Okada T (2007) Quantitative colocalization analysis of multicolor confocal immunofluorescence microscopy images: pushing pixels to explore biological phenomena. Acta Histochem Cytochem 40:101–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Immunohistochemistry controls. While specific and as expected glial‐like, neuronal and vascular patterns were obtained for the individual antibodies utilized in this study (A), skipping the primary antibodies and adding only the detection systems (anti‐mouse and anti‐rabbit polymer‐HRP and, respectively, anti‐goat‐biotinylated + streptavidin HRP), ensured no false‐positive immunostaining (B). For double immunohistochemistry on two primary rabbit antibodies, after detecting collagen IV (rabbit anti‐human) with anti‐rabbit polymer‐HRP and Alexa‐488 tyramide, slides were subjected (C and D) or not (E and F) to a low pH SDS antibody elution step. Adding another anti‐rabbit Alexa 594 antibody shows no labeling for collagen IV when using the elution buffer (C), but reveals strong cross‐reactivity when not utilizing the elution step (E). Moreover, continuing the immunodetection sequence with the anti‐AQP4 antibody (also rabbit anti‐human) and an anti‐rabbit Alexa 594 for labeling shows besides the new glial signal, again, no cross‐reactivity with collagen IV when utilizing the elution method (D), and a strong cross‐reactivity when not using it (F), proving that PBS washing steps during the second immunoprocedure are not enough to wash away the first primary antibody, which is still well detectable. Scale bars represent 50 μm.