

Abstract
Age‐related neurodegenerative diseases such as Alzheimer's disease (AD) are distressing conditions causing countless levels of suffering for which treatment is often insufficient or inexistent. Considered to be the most common cause of dementia and an incurable, progressive neurodegenerative disorder, the intricate pathogenic mechanisms of AD continue to be revealed and, consequently, an effective treatment needs to be developed. Among the diverse hypothesis that have been proposed to explain AD pathogenesis, the one concerning mitochondrial dysfunction has raised as one of the most discussed with an actual acceptance in the field. It posits that manipulating mitochondrial function and understanding the deficits that result in mitochondrial injury may help to control and/or limit the development of AD. To achieve such goal, the concept of mitochondrial medicine places itself as a promising gathering of strategies to directly manage the major insidious disturbances of mitochondrial homeostasis as well as attempts to directly or indirectly manage its consequences in the context of AD. The aim of this review is to summarize the evolution that occurred from the establishment of mitochondrial homeostasis perturbation as masterpieces in AD pathogenesis up until the development of mitochondrial medicine. Following a brief glimpse in the past and current hypothesis regarding the triad of aging, mitochondria and AD, this manuscript will address the major mechanisms currently believed to participate in above mentioned events. Both pharmacological and lifestyle interventions will also be reviewed as AD‐related mitochondrial therapeutics.
Keywords: Alzheimer's disease, mitochondrial anomalies, mitochondrial targets
Introduction
It has been a long road since the first case of Alzheimer's disease (AD) was observed by Alois Alzheimer in 1901. Back then, the young physician observed a patient complaining from deteriorating memory, even of recent events, disorientation, decreasing speech abilities and lack of judgment of the different surrounding situations 132. Later, in patient post‐mortem brain investigation, it was visible the thinning of the cerebral cortex together with two characteristic lesions, neurofibrillary tangles (NFTs) and extracellular deposits that would be later named senile plaques 154.
Beginning with memory loss of recent events (short‐term memory impairment) that proceed depriving the patients of their sense of self, AD represents about 50%–70% of all dementia cases affecting 4%–8% of the elderly population worldwide 4. Due to its age‐related incidence, as population ages, the prevalence of AD is expected to be more than triple by 2050, reaching over 115 million, what will represent a great threat to older individuals and their families, becoming a serious social problem with increasing longevity 171. Often diagnosed in people aged 65 (>95%) and older, the disease is typically referred to as late‐onset AD, occurring due to a sporadic component; in contrast to early‐onset AD, where initial symptoms can be observed between 30 and 65 years of age and occurs due to a familiar genetic cause involving mutations in the amyloid‐β protein precursor (APP) and presenilins 1 and 2 (PS1 and PS2) genes 69. Clinically, AD is characterized by a progressive memory and cognitive impairment caused by the extensive death of neurons. Starting in the entorhinal cortex and hippocampus and later proceeding to other parts of the brain cortex and subcortical gray matter, neuronal loss gradually compromises brain health of the patients, culminating in the complete need for care within several years after clinical diagnosis 69. The main pathological hallmarks found in affected parts of the AD brain concerns two neurodegenerative processes: accumulation of amyloid β‐peptide (Aβ), leading to the presence of extracellular Aβ deposits; and neurofibrillary degeneration, corresponding to the formation of intracellular deposits of the microtubule‐associated protein tau as NFTs 185, 192. Of note, AD has an insidious onset and it has been estimated that neurodegeneration begins 20–30 years before the clinical manifestations become evident 139. However, and despite the progresses in basic and clinical research that occurred after the discovery of Alois Alzheimer, until now AD still lacks disease‐modifying treatments that can influence the underlying pathogenic phenotypes of the disease 65. A condition that is not facilitated by the fact that AD is largely idiopathic, with many proposed hypothesis to explain its pathophysiology. In this regard, one of the most debated theory implies mitochondrial dysfunction and oxidative stress as early events in AD development and potential therapeutic targets 207.
As postulated in an impressive body of evidence, neuronal survival critically depends on the integrity and functionality of mitochondria. In order to achieve the well‐being of such vital organelles, a hierarchical system of cellular surveillance mechanisms protects mitochondria against stress, monitors mitochondrial damage and ensures the selective removal of dysfunctional mitochondrial proteins or organelles 181. However, in situations that a failure of this system occurs, damaged mitochondria will emerge as a central piece in cell energy deficiency and disruption of cell function, which will eventually compromise neuronal health leading to neurogeneration 20. In this scenario, with its proved involvement in aging and in the early pathogenesis of sporadic AD, mitochondria represent attractive targets for treatment strategies. As so, as the incessant search for new compounds and therapeutic tools for AD continues, nowadays it can include pharmacological and lifestyle interventions, both with the rational of directly manage mitochondrial dysfunction as well as attempts to directly or indirectly manage its consequences 205. In this manuscript, we will start with a brief contextualization of the past and current hypothesis regarding the triad of aging, mitochondria and AD. Later we will summarize key aspects of mitochondrial abnormalities in AD and review mitochondrial medicine approaches to target and/or manipulate such disturbances in order to re‐establish mitochondrial homeostasis.
Insights into Mitochondrial Homeostasis Perturbation in AD
It has been some years since mitochondrial structural and functional perturbations were noted in AD and it was agreed that mitochondria from AD subjects differ from those of age‐matched, non‐demented subjects 206. Beginning in the 70s, electron microscopy images revealed an abnormal mitochondria morphology in the brains of AD subjects 86, a condition that was corroborated years later and postulated to perhaps even precede dendritic degeneration 186. Later, systemic alterations in calcium homeostasis in AD patient fibroblasts suggested for a systemic bioenergetics defect 165. Also in the 80s, a possible major role for energy metabolism and mitochondria defects in AD was proposed, when it was noted that AD brains possess reduced activities of several mitochondria‐localized enzymes, including α‐ketoglutarate dehydrogenase (α‐KGDH) and pyruvate dehydrogenase (PDH) enzymatic complexes, suggesting AD as a disease of perturbed brain energy metabolism 17, 63, 64, 199. Nowadays it is widely accepted that mitochondrial dysfunctions involved in AD pathophysiology may include disturbances in oxidative phosphorylation (OXPHOS), and impaired energy metabolism as well as excess generation of reactive oxygen species (ROS), altered mitochondrial biogenesis, transport and dynamics 82. The next subsections briefly discuss the interrelation of aging, mitochondria and AD and, then, an update of the body of research that describes the pathophysiological effects of mitochondrial dysfunctions in AD will be presented.
The aging process, mitochondria and AD: A glimpse into the main hypothesis
Aging, an unavoidable biological process, is characterized by a general decline in physiological function that leads to morbidity and mortality 222. Due to the exponential increase of world aging population, the increase in life expectancy and, the consequent social burden of health care and pharmacoeconomic systems, the study of aging has become one of the major fields of basic and clinical research 193. As featured in a compelling body of proof, some aging paradigms place mitochondrial DNA (mtDNA) mutations and oxidative damage as main contributors to the aging process 76. As stated in the “free radical theory” of aging, proposed by Harman in the 50s 74, the gradual accumulation of damage within cellular macromolecules (eg, nuclear and mitochondrial DNA, lipids and proteins) with age could be attributed to deleterious effects of free radicals produced by aerobic metabolism. Thus, although mitochondria are responsible for energy production, they also have another, less beneficial legacy in the cell, the continuous production of potentially harmful ROS, which has become an essential focus of aging research 12. Using animal models such as the senescence‐accelerated mice (SAM) strain, researchers could explore the mechanisms of the age‐related mitochondrial decline 98, 196, 233. Among the results observed, behavioral studies showed that learning and memory deficits already started as early as 6 months and worsened with aging in SAMP8 (accelerated senescence‐prone 8) mice 158. Further, it was demonstrated that aging is also connected with an unbalance of the protective antioxidant machinery inside mitochondria. For instance, age‐related changes in levels of antioxidant enzymes, such as copper/zinc superoxide dismutase (Cu/Zn‐SOD) and manganese SOD (Mn‐SOD), have been found in liver and cortex of SAMP8 mice when compared with age‐matched SAMR1 (accelerated senescence‐resistant 1) mice, supporting increased oxidative stress as a key mechanism involved in the aging process 97. Of note, besides the progressive mitochondrial decline and increased oxidative stress, this mice model also presented tau hyperphosphorylation at an early age 3, 204 and, an age‐related increase in mRNA and protein levels of APP. In particular, the cleavage product Aβ was significantly increased at 9 months in SAMP8 and amyloid plaques started to form at around 16 months of age 143, 211. So, the understanding that mitochondrial dysfunction can act as a prominent and early, chronic oxidative stress‐associated event that contributes to synaptic abnormalities in aging and, ultimately, increased susceptibility to age‐related disorders including AD, conducted Harman to rename his previous hypothesis to “mitochondrial theory of aging” 75.
By definition, AD is an age‐related progressive neurodegenerative disorder mainly affecting elderly individuals and, as previously suggested, the manifest common end point of brain aging and AD is the impairment of memory and cognition, and at the molecular and cellular level the similarity is very striking between these two conditions 206. Nonetheless, one question that remains without an absolute answer is why the aging brain progresses to AD with extensive neurodegeneration in some cases but not in others. The first clues, that are in the basis of the “amyloid cascade hypothesis,” came with the identification of familial AD mutations in APP, PS1 and PS2 genes, which have given a big lift to the hypothesis of proteotoxic mechanisms mediated by the oligomers of Aβ in explaining the neurodegeneration of the sporadic AD 71, 73. Briefly, the amyloid cascade hypothesis, which has prevailed for more than 20 years in the AD field, speculates that in the common, late‐onset AD variants, mutations or polymorphisms in genes that regulate Aβ production or removal directly control Aβ levels, oligomers formation and AD itself 206. From this point, AD‐related research uncovered a plethora of mechanistic pathways triggered by Aβ oligomers that culminate in the apoptotic or autophagic death of neurons 72. Despite that, most of the investigational drugs available targeting APP and Aβ have consistently failed 56. Also, new evidence revealed some inconsistencies in the amyloid hypothesis, namely that the amount of Aβ deposition in the brain of sporadic AD patients does not always correlate with cognitive impairment and, inversely, Aβ deposition can also be found in cognitively normal individuals 32, 78, 144. In fact, the underlying process that leads to familial AD appears to be distinct from that leading to late‐onset AD and so, at this stage, another hypothesis has been raised. Knowing that APP processing is a highly regulated event directly affected by bioenergetics metabolism and, having in mind the previous assumption that with advancing age mitochondrial function changes, investigators hypothesized that the accumulation of Aβ could be a consequence of mitochondria damage during aging rather than the cause of the neuropathological cascade in sporadic AD 208. As so, all of those concepts were brought together in the “mitochondrial cascade hypothesis” 209. In brief, this hypothesis assumes that inheritance defines an individual's baseline mitochondrial function that is affected by environmental factors determining the speed at which age‐associated mitochondrial changes appear, and along with this influence, mitochondria may accumulate damage resulting in both the symptoms and neuropathology found in AD 208. Even though this hypothesis is still actual with several arguments in favor, as investigation proceeds, a new neuroenergetics perspective has been recently postulated in AD field, the “Inverse Warburg hypothesis” 45. Proposed by Demetrius and Simon 46, this hypothesis tries to explain the pathogenesis of sporadic AD cases, and postulate that AD is a metabolic disease initiated by an age‐related mitochondrial deficit 44, 45, 46. Concisely, this hypothesis is based on the mitochondrial cascade hypothesis and implicates energy and age, as the critical elements in the origin of neurodegenerative diseases. In this regard, it posits that the primary cause of sporadic forms of AD is an age‐induced energy deficit in the mitochondrial activity of neurons, and the increased ROS production and oxidative stress that surpasses a threshold after the failure of compensatory mechanism (the upregulation of oxidative phosphorylation), this will trigger the amyloidogenic pathway leading to Aβ accumulation, death of susceptible neurons and dementia 45, 66. Of note, it cannot be neglected that similar to genetic risk factors, environmental and lifestyle risk factors for AD can also be implicated in the acceleration of aging, morbidity and mortality 77.
Overall, a never‐ending number of studies emphasize that mitochondrial dysfunction, and its consequences, have a primary role in the neurodegenerative processes and, may indeed represent the missing link between aging and sporadic AD. Thus, presently, the goal is to find alternative approaches and/or future directions in understanding the neurodegenerative process from the known mechanisms of cellular aging that may eventually provide a rationale to new therapeutic possibilities in confronting the brain deficits of aging and AD.
Maternally inherited, mitochondria are considered the most complex and metabolically active organelles in the cell, being often denominated as the “powerhouses” of cells 11. Mitochondria play a paramount function in cell survival and death by responding to physiological and environmental signs in order to meet cellular energy and metabolic demands 133. Their role is even more important in neurons that need a large amount of ATP for the synthesis and secretion of neurotransmitters, to enhance the synaptic plasticity and also to maintain the neuronal membrane potential 131. As a result, impaired mitochondrial function inevitably leads to a pathological state, ranging from subtle alterations in neuronal function to cell death and neurodegeneration. As mentioned before, an age‐dependent decrease of brain bioenergetics metabolism together with an impaired redox homeostasis occur during the aging process in individuals with a history of a normal and healthy life 13, 40, 66, 103. In this situation, mitochondrial dysfunction is usually latent until a threshold of perturbations is reached, which eventually results in the disruption of cellular function. On the other hand, this scenario may be accelerated in a context of AD or the threshold of cell impairment in response to abnormal mitochondria may be lower in AD patients 11. In this regard, dysfunctional mitochondria are presumed to compromise neuronal plasticity and neuronal response to metabolic challenges, physiological and environmental cues and the encoding of new memories by lowering the energy charge in neurons 105. In the next subsections, it will be made a survey among the major insidious disturbances of mitochondrial homeostasis in the context of AD (ie, oxidative stress, energy hypometabolism and defects in mitochondrial dynamics, transport and quality control).
Oxidative stress, bioenergetics and energy metabolism in AD
It is recognized that AD has a long latent period before symptoms appear and a diagnosis can be made 227. Further, recent studies demonstrate that the onset of AD is commonly preceded by a transitional state known as mild cognitive impairment (MCI), when there is no significant increase of senile plaques and NFTs 124, 227. As demonstrated, MCI subjects often present increased levels of oxidative stress markers and decreased levels of non‐enzymatic antioxidants 22, 94, 170, 180. Likewise, evidence shows that such oxidative stress imbalance directly correlates with severity of cognitive impairment as well as with increasing age 221 and symptomatic progression from MCI to AD 10. In this scenario, oxidative modifications have been proposed as one of the biochemical changes possibly leading to the neuropathology and neuronal dysfunctions and death, generally found in AD 107. As described, ROS‐mediated injury, mainly through increased levels of lipid peroxidation markers, namely thiobarbituric acid reactive substances and 4‐hydroxynonenal, is found in the brains of AD patients compared to controls 21, 123 Noteworthy, a previous study elegantly demonstrated that mitochondria‐derived ROS themselves trigger Aβ generation by enhancing the amyloidogenic pathway 104. As authors described, this situation will start a vicious cycle of enhanced Aβ production in the progression of sporadic AD, since Aβ itself accelerates mitochondrial dysfunction and its own production via enhanced β‐site APP cleaving enzyme 1 (BACE1) activity due to increased ROS levels 104. In fact, as previously proposed, there is an intrinsic correlation between mitochondrial Aβ levels and mitochondrial dysfunction in different brain regions and also between these parameters and cognitive impairment in AD transgenic mice 51. In this regard, mitochondrial Aβ levels strongly influence mitochondrial respiratory function, ROS production rates and membrane potential in different brain regions such as frontal cortex and hippocampus 51. Within mitochondria, Aβ can bind and inhibit Aβ‐binding alcohol dehydrogenase (ABAD) interfering with the respiratory chain function (namely complex IV), altering mitochondrial efficiency in the modulation of cellular properties and increasing ROS production and damage 113. Furthermore, Aβ was also shown to interact with the mitochondrial protein import machinery and block TOM40 and TIM23 from importing subunits of complex IV into the mitochondria 70, 113, thereby also resulting in affected complex IV activity and in dysfunctional mitochondria.
Using the triple transgenic mouse model of AD (3xTg‐AD), investigators were able to demonstrate that, at least in this mice model, mitochondrial dysfunction in frontal cortex is characterized by decreased mitochondrial respiration and by a significant reduction in ATP synthesis in the hippocampus, compared with control 193. Likewise, studies from our laboratory show that brain mitochondrial anomalies present in the 3xTg‐AD mice culminate in energy deficits, increased ROS production and susceptibility to mitochondrial permeability transition pore opening 29, 178. Interestingly, data revealed that the 3xTg‐AD mice exhibit stronger defects on OXPHOS, synthesis of ATP and ROS production when compared with the age‐matched double transgenic littermates (APPxPS2). In particular, authors observed that a decrease in mitochondrial membrane potential is already detected at 8 months of age in triple AD mice compared to 12 months of age in their double AD mice 179. As suggested, such results are indicative that Aβ and abnormally hyperphosphorylated tau protein may act synergistically to trigger mitochondrial dysfunction in AD 179, 189. Also, others have observed that in 3xTg‐AD at 3 months of age, mitochondrial dysfunction occurs prior to the development of amyloid plaque 239. In this work, mitochondrial impairment in brain tissue from 3xTg‐AD was characterized by an increase in hydrogen peroxide production and lipid peroxidation as well by a decrease in mitochondrial respiration, and PDH activity, one of the most important enzyme of oxidative metabolism 239.
Energy hypometabolism in association with reduced neuronal expression of several key enzymes of oxidative metabolism in the diseased AD brain are among the best documented abnormalities in AD 227. In fact, low glucose metabolism at baseline and longitudinal glucose metabolism decline are viewed as sensitive measures useful for monitoring change in cognition and functionality in AD and MCI, and are being increasingly adopted to assist diagnosis and used to predict future cognitive decline 11. In this scenario, consistent data demonstrate that several key respiratory mitochondrial enzymes including isocitrate dehydrogenase, PDH, α‐KDH and cytochrome oxidase (COX) present a significant reduced expression and/or activity in the AD brain, occurring prior to the onset of memory deficits and the appearance of the two histopathological culprits of the disease 2, 118, 162, 219. As established, the impairment of the respiratory chain, in connection with AD, is mainly due to the decrease in complex IV (COX) activity 19, 28. Such impairment is often correlated with the early manifestation of Aβ toxicity and can have as immediate consequences excess oxygen radical production, which damages COX and surrounding structures decreasing ATP synthesis 190, 214. Further, others have demonstrated that energy deficiency causes an increase in the levels of intracellular amyloidogenic APP fragments in vitro 59, 62; and leads to an increase in the amyloidogenic APP processing in vivo 220. In further support of the importance of metabolic abnormalities in AD pathogenesis, previous studies have demonstrated that using a pharmacological model of energy metabolism inhibition in APP overexpressing transgenic mice (Tg2576), β‐secretase‐1 (BACE‐1) and Aβ levels become elevated, suggesting that energy deprivation may be amyloidogenic in vivo 156. Likewise, in vitro glucose deprivation has been shown to induce AD‐like changes in hippocampal neurons and in a neuronal cell line by influencing tau phosphorylation 35, 100. Further, starvation‐induced hypoglycemia was earlier suggested to be a simple model to study in vivo AD‐like tau hyperphosphorylation 235. Thus, in line with those observations and, contrary to common concepts, the AD brain “does not follow a suicide but a rescue program” 77. According to this concept, the brain during aging and in the initial stages of AD actively adapts to the progressive energy deprivation through neuronal compensatory responses and downstream adaptations, such as oxidative utilization of ketone bodies, activation of stress‐activated protein kinase pathways, Aβ deposition and tau protein hyperphosphorylation 41, 77, 142. In fact, data show that the most extensive Aβ deposits and neurons containing NFTs can have an inverse correlation with markers of oxidative stress, despite an obvious history of oxidative damage 153. This suggests that, in early stages of AD, both Aβ and NFTs may be cellular compensations to the energy crises, a process that eventually culminates in the exhaustion of cell's defense response consequently leading to neuronal degeneration and death 142.
Mitochondrial dynamics in AD
Through the action of mitochondrial “shaping proteins,” mitochondria have the ability to rapidly change morphology and complex networks within the cell in order to better answer the facing demand. In this context, under physiological conditions, mitochondria assume a thread‐like or tubular morphology that change to small round organelles through the rapid and reversible coordination of fusion/fission events 16. Being a process extremely organized and balanced in the normal cell, the disruption of this balance results in morphological changes and abnormal function of mitochondria, resulting in cell death 30. In fact, defects in mitochondrial dynamics events, particularly fission, have been identified in a number of human pathologies, including AD 226. In the first study that implicated the involvement of abnormal mitochondrial dynamics in AD, authors observed a pattern of structurally damaged mitochondria, characterized by broken cristae and partial or near complete loss of the internal structure in biopsied AD brain 79. In particular, authors observed a slight but significant increase in mitochondrial size along with a significant decrease in mitochondrial number in these neurons 79. Further studies demonstrated significant changes in the expression of the mitochondrial proteins involved in the fission/fusion processes including dynamin‐like protein 1 (DLP1), optic atrophy protein 1 (OPA1), mitofusins (Mfn) 1 and 2 and mitochondrial fission protein 1 (Fis1) in postmortem AD brains 36, 116, 224. Using the well‐known cybrid cell model, which lacks their own mtDNA and in which exogenous mtDNA from control, AD and MCI patients were introduced, it was found that AD cybrid cells had significant changes in morphology and function, with mitochondria fragmented, misshapen, bleb‐like and collapsed away from the mitochondrial network, such changes being associated with altered expression and distribution of Drp1 and Mfn2 60. Concurrently, others found that cybrids from MCI and AD patients develop bioenergetics and mitochondrial mass adaptations changes and present a mitochondrial fission–fusion balance shifted toward increased fission characterized by increased levels of mitochondrial Drp1 197. Also, from a plethora of in vitro studies it is becoming possible to underscore the dynamic balance of fission and fusion in AD in different experimental models 225. For instance, a recent study in Neuro‐2a (N2a) cells overexpressing APP, demonstrated that Aβ production triggers mitochondrial fragmentation through a decrease in the levels of Mfn1 and Mfn2 without changing the levels of Drp1 161. In this same context, a previous study revealed that the impaired mitochondrial dynamics in AD seems to result from the co‐localization of Drp1 and Aβ and subsequent increased production of free radicals in AD brains 116. In turn, this elevation of free radicals activates fission proteins Drp1 and Fis1 and causes excessive mitochondrial fragmentation, defective transport of mitochondria to synapses, low synaptic ATP levels and synaptic dysfunction in AD neurons 177. Of note, more recently, Zhang et al. 240, using a new technology of 3‐dimensional electron microscopy (3D EM) reconstruction to visualize mitochondrial structure in the brain tissue from patients and mouse models of AD, identified a previously unknown mitochondrial fission arrest phenotype that results in elongated interconnected organelles, “mitochondria‐on‐a‐string” (MOAS). Accordingly to this study, instead of the expected excessive mitochondrial fragmentation, AD mouse models presented this MOAS phenotype that increased with disease progression and was more pronounced in animals with multiple FAD mutations 240. Such results prompted authors to suggest that the MOAS phenotype may represent a sustained transition state dynamics allowing for a compensatory adaptation of mitochondria to bioenergetics stress, since it was also observed in animal models and in humans in response to energetics stress associated with AD, hypoxia and aging 240. Taken together, the aforementioned studies highlight the importance of mitochondrial dynamics in AD pathogenesis.
Mitochondrial autophagy and biogenesis in AD
When neurons accumulate dysfunctional mitochondria or face increased metabolic or bioenergetics demands, they must pursue one or a combination of several strategies to ensure sustained function. One response includes mitophagy, a form of autophagy in which defective mitochondria are selectively degraded in double membrane autophagosomes to ensure the maintenance of a healthy mitochondrial pool 184. In brief, autophagic vesicles formed in neurites must be transported back to the cell body by retrograde transport where they fuse with lysosomes. In normal conditions, the autophagic process in neurons is constitutively active and very efficient so autophagosome accumulation is rarely observed 149. In fact, previous studies suggest that in the early stages of AD, the upregulation of autophagy 83 and the increased expression of lysosomal enzymes 33 can act as a protective mechanism to fight cell degeneration 184. However, the presence of Aβ increases the number of autophagome‐engulfed mitochondria present in neurites and becomes a major intracellular reservoir of toxic peptides in AD brains 39, 150. This is likely due to an increased rate of autophagic sequestration of organelles coupled with a decreased rate of fusion of autophagic vesicles with lysosomes 39. This disrupted autophagosomal and lysosomal function may play an important role in mitochondrial dysfunction and intracellular Aβ accumulation in AD 190. As firstly described by Hirai et al. 79, postmortem AD brain tissue possesses decreased number of mitochondria in vulnerable neurons but increased cytosolic accumulation of mitochondrial markers such as mtDNA and subunit I of COX, suggesting an impaired autophagic lysosomal proteolytic degradation, and a possible leak of sequestered material from the autophagosome vacuoles. Also, elevated levels of mitochondrial components, namely COX and lipoic acid, within autophagosomes were detected in human postmortem brain tissue, suggesting an increase in the rate of mitochondrial degradation by autophagy 140, 141. So, it seems that in AD, the combination of increased autophagy induction and defective clearance of Aβ‐generating autophagic vacuoles creates conditions favorable for Aβ accumulation in AD 151.
Another major link between mitochondria and neuronal (dys)function concerns mitochondrial biogenesis and disturbances in its major regulator, the peroxisome proliferator‐activated receptor gamma (PPARγ) coactivator 1α (PGC‐1α), which is the coactivator of nuclear transcription factors (NRFs) 1 and 2 and mitochondrial transcription factor A (TFAM) 202. As pinpointed in literature, brains of patients with neurodegenerative diseases present low levels of PGC‐1α which may underscore mitochondrial dysfunction and oxidative stress 227. Also in AD patients 172 and cell models 195, a downregulation of PGC‐1α and its major target genes has been documented. For instance, data from our laboratory show a decrease in NRF1 and NRF2 levels in 3xTg‐AD brain, which may underlie the decrease in ND1 levels, a mitochondrial DNA encoded protein, which is suggestive of a decreased mitochondrial biogenesis 31. Following this line of thought, an increased expression of PGC1α was considered a compensatory response against Aβ toxicity in N2a cells treated with Aβ 24 and in APP/PS1 mice 241. As so, overexpression of TFAM protected SH‐SY5Y cells from Aβ‐induced mitochondrial dysfunction 234 whereas PGC‐1α overexpression in N2a neuroblastoma cells promoted a decrease in secreted Aβ and increased the levels of non‐amyloidogenic soluble Aβ 93. However, in opposite, the cross of Tg19959‐AD mice with mice overexpressing human PGC‐1α worsened Aβ and tau accumulation, led to mitochondrial abnormalities, neuronal cell death and an exacerbation of behavioral hyperactivity in mice 53. As authors discussed, preserving the exquisite balance of PGC‐1α expression and function may be critical to achieve beneficial effects, which may be of relevance when designing therapeutic strategies 53.
Mitochondrial trafficking in AD
Neurons, that are long, excitable and, highly compartmentalized cells, have an intrinsic dependence in the proper distribution of mitochondria to sustain the spatial and temporal demand of energy that differs within the axons and synapses compared to dendrites and the cell body 58, 81, 114. Both synaptic and nonsynaptic mitochondria are synthesized in the neuronal soma and consequently have to be transported to the nerve terminals where the bioenergetics demands are high in order to fulfill their physiological functions 114. A number of observations, such as a reduced dendritic mitochondrial coverage, a selective and increased degradation of mitochondria by mitophagy, a reduction of synaptic vesicle number at nerve terminals, as well as mitochondria accumulation in the neuronal soma, implicate disturbances in axonal transport in the progression of AD 228. In brief, mitochondrial long‐distance transport in axons, to reach all the places that need energy, is driven by kinesin, the primary anterograde mitochondrial motor, and by dynein, primary retrograde mitochondrial motor, powered by ATP hydrolysis that shuttle mitochondria along microtubules 188. As shown in previous studies, an impairment in kinesin‐based axonal anterograde transport including the transport of mitochondria is implicated in in vitro and in mouse models of AD 102, 167. For instance, a brief treatment of Aβ monomers and fibrils was demonstrated to be enough to induce significant reduction in motile mitochondria in hippocampal neurons 182; a process likely to occur through the activation of glycogen synthase kinase 3β (GSK3β) 182 and consequent increased phosphorylation of kinesin light chains (KLC) 167. Similarly, a study using real‐time analysis of vesicle motility demonstrated that isolated axoplasms perfused with soluble intracellular oligomeric Aβ exhibit inhibition of bidirectional axonal transport as a result of increased phosphorylation of KLC and subsequent release of kinesin from its cargoes (e.g. mitochondria) 166. Others have shown that hippocampal neurons treated with Aβ‐derived diffusible ligands (ADDLs) develop an impaired axonal transport of mitochondria in both anterograde and retrograde directions 226. Likewise, Aβ overexpression in Drosophila decreased mitochondrial velocity in both directions, which is associated with axonal depletion of mitochondria 244. In the same animal model of AD, Folwell et al. 57 found that the coexpression of Aβ with phosphorylated human tau increases tau phosphorylation and exacerbates axonal transport dysfunction. In turn, Du et al. 52 found that low levels of Aβ promote an impairment in anterograde but enhance the retrograde axonal transport of mitochondria, whereas Calkins et al. 24 demonstrated a specific impairment in anterograde transport of mitochondria without changes in the retrograde direction in primary neurons from Tg2576 APP transgenic mice. So, from the studies discussed above and others 26, 42, 215, it can be stated that axonal transport defects strongly impact AD‐linked pathologic factors. Importantly, studies in 3xTg‐AD mice showed that the deficits in axonal transport and axonal swelling precede Aβ deposition or filamentous tau aggregation, suggesting that such deficits might be early events in AD 136. As the machineries mediating axonal transport of mitochondria and mitochondrial fusion are closely interconnected 138, alterations in the morphology as well as decreased ATP levels per se will interfere with axonal trafficking of mitochondria and result in depletion of functional mitochondria in the synapses causing defects in synaptic function and neurodegeneration. Thus, maintaining a healthy mitochondrial network is a major actual goal 91.
Mitochondrial Medicine for AD: Mitochondria as a Therapeutic Target
Albeit the question whether the mitochondrial‐related changes contribute to the development of or are a consequence of the disease process remains open, an undoubted link between mitochondrial dysfunction and AD has been extensively demonstrated. As previously mentioned, in general, mitochondrial perturbation can arise in several ways, e.g. alterations in mitochondrial function, morphology and dynamics associated with Aβ accumulation and/or ROS production are among the earliest observed pathogenic alterations observed in AD, preceding the formation of amyloid plaques 168. Thus, understanding the mechanisms behind the occurrence of such mitochondrial alterations will likely offer new avenues for preventive or therapeutic strategies. Since the first report of a disease characterized by mitochondrial dysfunction 112 and the emergence of the “mitochondrial medicine” concept 111, medicine has evolved and nowadays it include pharmacological and lifestyle interventions, both with the rational of directly or indirectly target mitochondrial dysfunction and its consequences 205.
Pharmacological interventions to target mitochondria in AD
One of the critical goals of pharmacological science is the development of novel and safe drugs for the prevention and treatment of age‐related diseases. While AD‐related hypotheses postulate several causes and therapeutic targets, no clearly effective disease‐modifying interventions are currently recognized 30. With the increasing understanding of the mechanisms underlying mitochondrial dysfunction and its key role in AD pathogenesis, several preventive and therapeutic strategies have emerged.
From antioxidants to TPP+ and SS conjugated compounds
Being widely discussed in both the lay press and the scientific literature as health‐promoting agents that may protect against various age‐related diseases, antioxidants were the first form of mitochondrial medicine intended to treat and/or delay AD progression. In general, antioxidant strategies can be divided into three main categories: 1) free radical scavengers, e.g., vitamins C and E, β‐carotene; 2) preventive antioxidants such as metal chelators, glutathione peroxidases and superoxide dismutase (SOD) enzymes; and 3) de novo and repair enzymes such as lipases, proteases and DNA repair enzymes 152. Also, nonspecific antioxidants can include melatonin 55, omega‐3 polyunsaturated fatty acid (docosahexaenoic acid) 14, curcumin 237, ubiquinone 38 and α‐lipoic acid 174. Focused in reducing oxidative stress and/or preventing ROS damage to mitochondria, the use of most of those antioxidants in a preclinical step culminated in very promising results and, for example low‐molecular weight antioxidants, such as vitamin E and N‐acetylcysteine were soon considered as wannabe candidates for clinical trials in AD. However, human clinical experience with such antioxidant neuroprotectants quickly showed some caveats and did not matched the expectations 89. For instance, even though numerous cellular and animal models of AD have been developed, one of the major challenges in translating a therapy from a preclinical to a clinical setting is the inability to find an animal model that completely mimic the human nature of the disease 43. Further, another possibility relates with the fact that in the human clinical trials, treatment is often initiated too late in the course of the disease 89. Importantly, as pinpointed elsewhere, when it comes to designing potential antioxidant approaches, cell compartmentalization issues require consideration. Antioxidants that only access the cytosol may have a limited impact on mitochondrial ROS 203. As so, antioxidant therapy become blunted by the inability to enhance antioxidant levels in mitochondria mainly due to the low permeability of the blood–brain barrier to most of the antioxidants currently used and the difficulty encountered in surpass multiple barriers such as the cell membrane and the outer and inner mitochondrial membranes 135. To circumvent this concern and to better assess whether antioxidant approaches may be valuable therapeutic treatments, researchers have developed strategies to direct antioxidants into mitochondria. One of those strategies is the conjugation of the lipophilic triphenylphosphonium cation (TPP+) to antioxidants such as vitamin E, coenzyme Q (MitoQ) and α‐lipoic acid. Since these molecules enter into mitochondria several hundred‐fold more than natural antioxidants, they will rapidly neutralize free radicals at their source with an improved therapeutic potential 146. In fact, as expected, MitoQ demonstrated effectiveness in attenuating Aβ‐induced neurotoxicity, ROS production and loss of mitochondrial membrane potential (Δψm) in cortical neurons 134 as well as in preventing the loss of spatial memory and to delay early neuropathological changes in young 3xTg‐AD females 134. Likewise, using a Caenorhabditis elegans model overexpressing human Aβ, Ng et al. 148 evidenced that the early administration of MitoQ exerts protective effects on lifespan and Aβ‐induced paralysis, observations that were related with the protection of complexes I and IV of the electron transport chain (ETC) 148. Further, MitoQ also revealed to be capable of preventing Aβ‐induced mitochondrial fragmentation, to increase neurite outgrowth in N2a neuroblastoma cells treated with Aβ, and to enhance synaptic branching and connectivity in primary neurons from AD and wild‐type mice 117.
More recently, to overcome the logistics of safe drug delivery to the mitochondria in sufficient amounts and the toxicity associated with high doses, mitochondria‐targeted polymeric nanoparticle system (NPs) started to be used 229. Because of its ease of production, flexibility with respect to surface modification and tunable drug release profiles, NPs are becoming an attractive mitochondria‐targeting drug delivery system 155. In this context, the conjugation of NPs to TPP+, allowed researchers to formulate a targeted curcumin‐loaded NPs 127. Previously tested in clinical trials to slow AD progression but without improvements in cognitive decline, curcumin is a well described inhibitor of Aβ toxicity with antioxidant and anti‐inflammatory properties 127. Being mitochondrial‐targeted, curcumin‐loaded NPs exhibited significantly higher protective effects against Aβ in human neuroblastoma cells when compared with nontargeted particles or free curcumin 127, revealing the importance of targeting the antioxidant agents to the mitochondria.
Later on, as an alternative to the TPP+ strategy, researchers developed the SS peptides, a family of small mitochondria‐targeted antioxidant molecules 210. By having a sequence motif that allows them to target mitochondria, even in depolarized mitochondria, these SS peptides display mitochondrial accumulation and ability to scavenge H2O2 and inhibit lipid peroxidation 210. SS31, the most extensively studied SS peptide, in particular, is capable of entering mitochondria and of concentrating in the inner mitochondrial membrane, protecting mitochondria against mitochondrial permeability transition pore (mPTP) formation, swelling and cytochrome c release 243. Using SS31 in different AD models as N2a neuroblastoma cells treated with Aβ, primary neurons from Tg2576 mice and aged Tg2576 mice, a range of effects on mitochondria were observed 25, 117. In particular, SS31 decreased the levels of mitochondrial fission proteins (Drp1, Fis1) and matrix protein, CypD and reduced mitochondrial dysfunction in neurons affected by AD. Further, SS31 enhanced the number of healthy and intact mitochondria, and increased synaptic outgrowth and neuronal branching 117. Meanwhile, SS31 also proved its efficacy by restoring mitochondrial transport and synaptic viability, and decreasing the percentage of defective mitochondria in primary neurons from Tg2576 mice 25, thus indicating that SS31 protects mitochondria and synapses from Aβ toxicity.
In general, even though the application of these mitochondria‐targeted agents to AD is at its early stages and is mainly focused on animal models of AD, the outcomes are positive and worth to follow as potential drugs to enter in clinical trials with AD patients.
Strategies to manipulate mitochondrial quality control and dynamics in AD
Through studies that aim to elucidate the role of mitochondria in AD onset and development it is clear that abnormal mitochondrial dynamics and quality control play an important role in AD neurodysfunction and neurodegeneration. In this scenario, mitochondrial fission inhibitors, such as mdivi‐1, ranks itself as a new candidate for AD treatment 60. Using AD cybrids as an in vitro AD model, researchers observed that this inhibitor of Drp1 GTPase activity, protected against mitochondrial fragmentation and mitochondrial functional defects, including deficits in complex IV activity, observed in AD cybrid cells 60. Simultaneously, others also reported that mdivi‐1 treatment markedly reverses mitochondrial fission and mitochondrial membrane potential loss, cytochrome c release and caspase‐3 activation caused by Aβ treatment in BV‐2 and primary microglial cells 232.
As the search for novel and potentially effective agents for the treatment of AD, as well as selected promising treatment strategies lasts, researchers strive for new mitochondrial targets. For instance, using an animal model with a genetically manipulated voltage dependent anion channel (VDAC1) protein, Manczak et al. 120 showed that a reduction in VDAC levels is associated with a decrease in the activity of Drp1 in neuronal cells and concomitant decrease in mitochondrial fission and prevention of neuronal death, thus suggesting that compounds to target VDAC protein may constitute a novel therapeutic target 119. More recently, others reported that CR6‐interacting factor 1 (Crif1), a MIM protein that is key for the translation of mitochondrial OXPHOS subunits and their insertion into the MIM 95, is a key player in Aβ‐induced mitochondrial dysfunction 23. In particular, authors observed that Crif1 is decreased in the brains of AD patients and mouse models and its reduction is associated with massive mitochondrial fission and loss of cristae in an in vitro model whereas its overexpression rescues Aβ‐induced disruption of mitochondrial morphology 23. Such promising results prompted authors to propose that Crif1 may serve as a novel therapeutic target in the treatment of AD 23. Using a different strategy, in a recent study, Zhang et al. 241 observed that the impairments in mitochondrial homeostasis and the cognitive deficits present in an AD mice model can be reverted by a neural stem cell (NSC) transplantation method. Specifically, NSC transplantation upregulated mitochondrial biogenesis, thus generating more mitochondria with normal function to rescue the mitochondrial dysfunction and cognitive deficits found in AD 241. Supported by these and by other similar observations 18, 242, authors point out that a NSC‐based therapy targeting mitochondrial biogenesis could be considered a promising therapeutic strategy in AD 241.
Overall, research performed in in vitro and animal models of AD highlights several therapeutic targets and pharmacological strategies aimed to reestablish mitochondrial homeostasis (Figure 1). Even though more in‐depth studies are needed before translation to human medicine, future studies will hopefully define more clearly which of those strategies are worth to follow.
Figure 1.
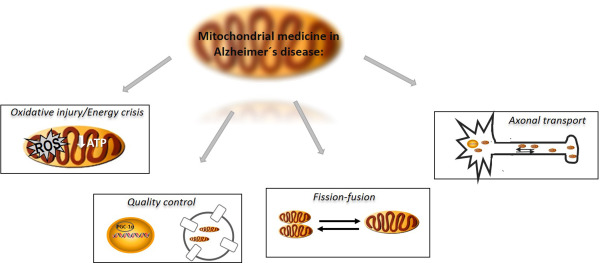
Mitochondrial targets of intervention in Alzheimer's disease. Being actively engaged in a plethora of neurodegenerative diseases, including Alzheimer's disease (AD), the actual compromise of mitochondrial medicine is to find strategies to target and/or manipulate the major insidious disturbances of mitochondrial homeostasis and to directly or indirectly manage their consequences in the context of AD in order to reestablish mitochondrial homeostasis. With this goal, mitochondrial medicine point to reset the mitochondrial interconnected features of quality control (autophagy and mitochondrial biogenesis), mitochondrial axonal transport, fission/fusion processes, bioenergetics (energy production) and reactive oxygen species production.
Lifestyle interventions
It is widely accepted that diet strongly influences the incidence and outcome of major age‐related diseases including diabetes, obesity and vascular disease. As posited by a growing body of evidence, brain aging and neurodegeneration are tightly linked with metabolic and energy balance with recent findings extending influences of diet to AD 54, 68. As the main source of energy for the normal function of brain cells, the decline of mitochondria function has been proposed to be a major factor in the loss of brain function with aging and in age‐related neurodegenerative disorders 176. As so, strategies to preserve brain mitochondrial integrity and metabolism during aging are often regarded as critical for maintaining healthy brain function, often referred to as healthspan, and for extending lifespan 129. While current therapeutics only temporarily ameliorate the symptoms of AD, but very few affect the underlying disease mechanism 191, a number of epidemiological studies, however, suggest that simple lifestyle changes may be sufficient to slow the onset and progression of AD 169. In this setting, evidence from antioxidant‐based trials and pre‐clinical studies have shed light to the need of concomitant dietary and lifestyle factors in refining efficacy of antioxidant therapies 37, 164. As so, non‐pharmacological interventions, such as calorie restriction, and/or physical activity have been gaining recognition as a very effective means to extend lifespan and delay the appearance of age‐related pathological conditions, notably those associated with brain functional decline 5.
Calorie restriction and calorie restriction mimetics in AD
“Eat less, age well, and remember well,” the mantra that is becoming principal in cutting‐edge research on aging and human health, highlights for the importance of dietary nutrition intervention in human aging and age‐related diseases, including AD 106, 110. Defined as a moderate reduction in calorie intake of 20%–40% in the absence of malnutrition, calorie restriction (CR) or dietary restriction (DR) is nowadays the only nongenetical experimental manipulation that is known to preserve metabolism in aging process and extend the lifespan of a broad range of species, spanning from yeast to rodents and non‐human primates 5, 84, 200. By activating a class of enzymes known as sirtuins, CR posits itself as having remarkable neuroprotective properties 173. For instance, in AD mouse models, CR has been found to diminish AD symptoms 130, 145, 163 whereas in a primate model, CR increased neurotrophic factors and attenuated behavioral deficits 128. In a close way, others show that CR ameliorates neurodegenerative phenotypes assessed by object recognition and contextual fear conditioning tests and reduces tau hyperphosphorylation in cDKO (conditional double knockout) AD mice 231, suggesting a potential therapeutic value of CR for patients with AD. In this context, even though an epidemiological study by Luchsinger et al. indicates that individuals with a low‐calorie intake may have a reduced risk of developing AD 110, the utility of CR as a strategy for humans is somehow hampered by the degree and length of restriction required and the will of patients to go through a reduction in food ratio 9. Thus, it become of great interest to understand the mechanisms by which CR modifies organismal physiology, particularly in light of current efforts to develop CR mimetic (CRM) compounds 34, 85, 198. By definition, CRM are alternative chemical compounds that can “mimic” the biological effects of CR without significantly reducing calorie intake. And so, the ideal CRM would be an agent consumed in food or water that would delay death and age‐associated diseases without requiring a change in calorie intake 84. Of note, even though these compounds could also be included in the pharmacological subsection of this manuscript, their effects will be discussed here in the context of its CR‐related effects. Due to its ability to mimic the metabolic, hormonal and physiological effects of CR, activate stress response pathways and reduce the incidence of age‐related diseases, CRM are often associated with an improvement in the overall health‐ and lifespan of the organism 200.
One of the most popular compounds with CR properties is resveratrol 15. With a previous epidemiological link to longer life in humans 109, 160, resveratrol by activating sirtuins was found to decrease aging‐dependent cognitive decline and pathology in AD animal models 92, 122. Further, this CR mimetic was also able to protect cells against Aβ‐induced ROS production and DNA damage in vitro 85, 187. Also, in a rat model of sporadic AD, resveratrol was found to prevent cognitive impairment induced by an intracerebroventricular injection of streptozotocin 194. More recently, a randomized, placebo controlled, double‐blind, multicenter 52‐week phase 2 trial of resveratrol in individuals with mild to moderate AD revealed that resveratrol consumption is safe, well‐tolerated and alters some AD biomarker trajectories (216; clinicaltrials.gov ID NCT01504854).
Nevertheless, even having a well‐documented neuroprotective role in several models of neurodegenerative diseases 200, the exact mechanism by which CR and its mimetics influences aging and/or AD remains unclear. Still, one of such mechanisms that is currently accepted relates with mitochondria and mitochondria function, namely with the prevention of oxidative damage and mitochondrial ROS production, improvement of metabolic parameters, increased mitochondrial biogenesis through activation of the SIRT1‐PGC1α pathway and resistance to cellular stress 47, 121. As demonstrated, by activating SIRT1, resveratrol reduces defects in electron transfer in ETC, decreases mitochondrial ROS, increases oxygen consumption and maintains ATP production in neurons 8, 9. As nicely described by López‐Lluch et al. 108, mitochondria under CR conditions show less oxygen consumption, reduced membrane potential and generate less ROS than controls, but remarkably they are able to maintain their critical ATP production. A process that, accordingly to the authors, critically relies in the activation of diverse regulatory pathways greatly enhances stress resistance via the SIRT1 pathway and markedly improves bioenergetics through the activation of the PGC‐1α pathway 108. Being responsible for the transcription of cellular programs regulating mitochondrial respiration, oxidative stress defense and adaptive thermogenesis, PGC‐1α is considered a master regulator of mitochondrial biogenesis and metabolism. So, it can be suggested that CR and/or CRM through its direct effects in PGC1α promote an adaptive response in the cell that accounts for an increase in the number and size of mitochondria as well as an enhancement in the respiratory rate. Overall, those changes will elevate the oxidative buffer capacity of the cell, augmenting its resistance to conditions of stress 121. As mentioned before, mitochondrial dysfunction, especially a defect in mitochondrial biogenesis, is an early and prominent feature of AD 172, so it seems that compounds and/or interventions that reverse the PGC1α deficit, or otherwise enhance PGC1α activity or expression, constitutes a feasible AD therapeutic target 6.
Considering that energy sensing pathways appear critical in regulating aging in a number of model systems, in a general way, CRM are targeted to energy pathways that mimic the physiological responses of CR and to enhance stress responses 84. As so, besides resveratrol, to date, several candidate CRM have been proposed and studied to treat and/or delay AD, among others are included, 2‐deoxy‐d‐glucose (2‐DG), sirtuin activators and the inhibitors of the mammalian target of rapamycin (mTOR) 212. Due to the structural similarity between 2‐DG and glucose, 2‐DG is transported by glucose transporters into the cell where it binds to, but since it cannot be phosphorylated by hexokinase, it will induces a compensatory rise in alternative substrates, primarily ketone bodies by the liver and, activates an alternative energetic pathway in brain 238. As previously demonstrated, in a similar way to CR, the 2‐DG treatment was shown to mediate neuroprotection in models of AD 67. For instance, Yao et al. 238 using 3xTgAD mice fed with 2‐DG, observed that this dietary intervention was responsible for an improvement in brain mitochondrial bioenergetics paralleled with a reduction in oxidative stress. Also, dietary 2‐DG promoted a reduction in Aβ generation and increased mechanisms of Aβ clearance, further suggesting dietary 2‐DG as a disease‐modifying intervention to delay progression of bioenergetics deficits in brain and associated amyloid burden 238.
One key sensor of nutrient availability in higher organisms is mTOR. With previous evidence that CR inhibits mTOR signaling in multiple species including mice 90, 230, mTOR has become a candidate mediator of at least some of CR's beneficial effects 137. In this set, strategies for the inhibition of mTOR as a therapeutic strategy in AD have been tested in animal models of the disease 99, 236. For instance, some studies offer strong evidence that rapamycin, the well‐known inhibitor of mTOR, and its derivatives can decrease amyloid burden in APP‐overexpressing mouse models, when administration is performed in early stages of the disease 115 and ameliorate tau pathology in the 3xTg‐AD and P301S tau transgenic mice models 157. Further, other compounds with structural similarities to resveratrol, RSVA314 and RSVA405, were found to inhibit mTOR activity, and to promote the degradation of Aβ by the autophagic‐lysosomal machinery 223.
Physical exercise in AD
Besides the widely accepted beneficial effects of exercise in enhancing a range of physical indices from balance, bone density, strength and endurance to lipid profiles, blood pressure and cardiovascular health 87, 213, it is now recognized that regular exercise holds important benefits for both affective experience and cognitive performance regardless of age 80. In fact, physical inactivity and a sedentary lifestyle are nowadays considered significant risk factors to develop dementia and neurodegeneration 101, 175. In this scenario, over the past years, retrospective and prospective epidemiological studies documented brain health benefits of exercise on the development of AD and dementia of any type 159. In concordance with those studies, data obtained from several animal models of AD show that physical exercise is a simple behavioral intervention sufficient to inhibit the development of AD‐like neuropathology and to improve cognitive behavior 1, 37, 61, 218. Of note, at least in AD mouse models, the age of the mouse and thus level of pathology must be considered when selecting an appropriate exercise regime, with maximal effects on AD pathology observed when exercise is initiated prior to the appearance of Aβ plaques or at an early‐mild stage of plaque deposition 183.
Albeit the significant outcomes regarding cognitive function reported in both epidemiological and animal studies, the cellular and molecular mechanisms underlying this exercise‐induced protective phenotype in the brain are still elusive 125. For instance, even though the brain is a non‐contractile tissue, neuronal function seems to be indirectly influenced by an increase in energy metabolism 50. In this setting, brain mitochondrial metabolism seems to be central in the cross‐tolerance phenomena by which physical exercise confers neuroprotection 125. As reported, exercise induces important brain mitochondrial adaptations in order to sustain increased metabolic demands 48. Among those, are included an increased content and/or activity of several enzymes involved in aerobic energy production 48, 49, 96, increased activity of mitochondrial complexes I, III and IV 147, decreased expression/activation of several pro‐apoptotic proteins 217, increased mitochondrial biogenesis 201 and antioxidant capacity 27, as well as, alterations in proteins involved in mitochondrial dynamics, apoptosis and autophagic signaling 126. As so, previous data confirm that endurance training attenuates neuronal cell apoptosis involved in the pathogenesis of AD by promoting reductions in brain cytochrome c, Bax, caspase 3 and 9 levels, and an elevation of heat shock protein 70 (HSP70) 37, 218. Further, as suggested by others, other important mitochondrial bioenergetics adaptations associated with voluntary exercise concerns an increase in the mitochondrial uncoupling protein 2 (UCP2) gene expression 48. Previously reported as exerting an important protection against Aβ toxicity and oxidative stress 88, UCPs, mainly UCP2, are often regarded as an effective strategy in the regulation of mitochondrial biogenesis by decreasing ROS overproduction, increasing ATP generation and improving calcium homeostasis 7.
From the aforementioned studies, one can hypothesize that strategies aimed to modulate mitochondrial function and trigger neuroprotective mechanisms represent effective strategies against brain aging and several age‐related neurodegenerative diseases (Figure 1). However, further investigation is required to elucidate the real impact of these strategies in AD patients 7, 48, 125.
Conclusions
As outlined in the studies discussed above, it is clear that mitochondria are at the heart of the neurodegenerative process occurring in AD. Studies reveal that mitochondria play a major role in several stages of AD progression; they can be considered triggers as well as targets of the neurodegenerative cascade that characterizes AD. The ongoing research in this field continues to shed light on the mechanisms involved in the maintenance of mitochondrial integrity and their relevance for disease, making them interesting and valuable targets for therapeutic interventions. Until now, due to the intricate mechanisms involved in disease pathogenesis most of the clinical trials carried out with the investigational drugs available terminated without being successful and so, there is an urge to develop strategies aimed to delay the onset or to slow down AD progression. Due to the deep involvement of mitochondria in AD, a great expectation is being posed in mitochondrial medicine to directly manage the major insidious disturbances of mitochondrial homeostasis occurring in AD. Nevertheless, and considering AD a multifactorial pathology, a mono‐target approach like those currently used is insufficient to foster positive results, and so it must be stressed out that clinical trials should try to combine efforts to include multiple intervention arms, such as co‐administration of mitochondrial‐directed antioxidants with other promising therapeutic options (e.g. lifestyle interventions). Even though more in‐depth studies are needed before pharmaceutical industry can apply this knowledge to human medicine, the continuous advances in AD research field facilitate and accelerate the development of more effective preventive or therapeutic strategies to fight this devastating disease.
Acknowledgments
The authors’ work is supported by FEDER funds through the Operational Programme Competitiveness Factors—COMPETE and national funds by FCT—Foundation for Science and Technology under the project (PEst‐C/SAU/LA0001/2013‐2014) and strategic project UID/NEU/04539/2013. Susana Cardoso is recipient of a PostDoc fellowship from the Foundation for Science and Technology (FCT) (SFRH/BPD/95770/2013). The authors of the manuscript have no conflicts of interest to declare.
References
- 1. Adlard PA, Perreau VM, Pop V, Cotman CW (2005) Voluntary exercise decreases amyloid load in a transgenic model of Alzheimer's disease. J Neurosci 25:4217–4221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Aksenov MY, Tucker HM, Nair P, Aksenova MV, Butterfield DA, Estus S et al (1999) The expression of several mitochondrial and nuclear genes encoding the subunits of electron transport chain enzyme complexes, cytochrome c oxidase, and NADH dehydrogenase, in different brain regions in Alzheimer's disease. Neurochem Res 24:767–774. [DOI] [PubMed] [Google Scholar]
- 3. Alvarez‐Garcia O, Vega‐Naredo I, Sierra V, Caballero B, Tomas‐Zapico C, Camins A et al (2006) Elevated oxidative stress in the brain of senescence‐accelerated mice at 5 months of age. Biogerontology 7:43–52. [DOI] [PubMed] [Google Scholar]
- 4. Amemori T, Jendelova P, Ruzicka J, Urdzikova LM, Sykova E (2015) Alzheimer's disease: mechanism and approach to cell therapy. Int J Mol Sci 16:26417–26451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Amigo I, Kowaltowski AJ (2014) Dietary restriction in cerebral bioenergetics and redox state. Redox Biol 2:296–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Andreux PA, Houtkooper RH, Auwerx J (2013) Pharmacological approaches to restore mitochondrial function. Nat Rev Drug Discov 12:465–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Andrews ZB, Diano S, Horvath TL (2005) Mitochondrial uncoupling proteins in the CNS: in support of function and survival. Nat Rev Neurosci 6:829–840. [DOI] [PubMed] [Google Scholar]
- 8. Anekonda TS (2006) Resveratrol—a boon for treating Alzheimer's disease? Brain Res Rev 52:316–326. [DOI] [PubMed] [Google Scholar]
- 9. Anekonda TS, Reddy PH (2006) Neuronal protection by sirtuins in Alzheimer's disease. J Neurochem 96:305–313. [DOI] [PubMed] [Google Scholar]
- 10. Ansari MA, Scheff SW (2010) Oxidative stress in the progression of Alzheimer disease in the frontal cortex. J Neuropathol Exp Neurol 69:155–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Atamna H, Frey WH II (2007) Mechanisms of mitochondrial dysfunction and energy deficiency in Alzheimer's disease. Mitochondrion 7:297–310. [DOI] [PubMed] [Google Scholar]
- 12. Balaban RS, Nemoto S, Finkel T (2005) Mitochondria, oxidants, and aging. Cell 120:483–495. [DOI] [PubMed] [Google Scholar]
- 13. Barja G (2002) Endogenous oxidative stress: relationship to aging, longevity and caloric restriction. Ageing Res Rev 1:397–411. [DOI] [PubMed] [Google Scholar]
- 14. Bascoul‐Colombo C, Guschina IA, Maskrey BH, Good M, O'Donnell VB, Harwood JL (2016) Dietary DHA supplementation causes selective changes in phospholipids from different brain regions in both wild type mice and the Tg2576 mouse model of Alzheimer's disease. Biochim Biophys Acta 1861:524–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Baur JA (2010) Resveratrol, sirtuins, and the promise of a DR mimetic. Mech Ageing Dev 131:261–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Benard G, Karbowski M (2009) Mitochondrial fusion and division: regulation and role in cell viability. Semin Cell Dev Biol 20:365–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Blass JP (2002) Alzheimer's disease and Alzheimer's dementia: distinct but overlapping entities. Neurobiol Aging 23:1077–1084. [DOI] [PubMed] [Google Scholar]
- 18. Blurton‐Jones M, Kitazawa M, Martinez‐Coria H, Castello NA, Muller FJ, Loring JF et al (2009) Neural stem cells improve cognition via BDNF in a transgenic model of Alzheimer disease. Proc Natl Acad Sci USA 106:13594–13599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bosetti F, Brizzi F, Barogi S, Mancuso M, Siciliano G, Tendi EA et al (2002) Cytochrome c oxidase and mitochondrial F1F0‐ATPase (ATP synthase) activities in platelets and brain from patients with Alzheimer's disease. Neurobiol Aging 23:371–376. [DOI] [PubMed] [Google Scholar]
- 20. Burte F, Carelli V, Chinnery PF, Yu‐Wai‐Man P (2015) Disturbed mitochondrial dynamics and neurodegenerative disorders. Nat Rev Neurol 11:11–24. [DOI] [PubMed] [Google Scholar]
- 21. Butterfield DA, Castegna A, Lauderback CM, Drake J (2002) Evidence that amyloid beta‐peptide‐induced lipid peroxidation and its sequelae in Alzheimer's disease brain contribute to neuronal death. Neurobiol Aging 23:655–664. [DOI] [PubMed] [Google Scholar]
- 22. Butterfield DA, Reed T, Perluigi M, De Marco C, Coccia R, Cini C et al (2006) Elevated protein‐bound levels of the lipid peroxidation product, 4‐hydroxy‐2‐nonenal, in brain from persons with mild cognitive impairment. Neurosci Lett 397:170–173. [DOI] [PubMed] [Google Scholar]
- 23. Byun J, Son SM, Cha MY, Shong M, Hwang YJ, Kim Y et al (2015) CR6‐interacting factor 1 is a key regulator in Abeta‐induced mitochondrial disruption and pathogenesis of Alzheimer's disease. Cell Death Differ 22:959–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Calkins MJ, Manczak M, Mao P, Shirendeb U, Reddy PH (2011) Impaired mitochondrial biogenesis, defective axonal transport of mitochondria, abnormal mitochondrial dynamics and synaptic degeneration in a mouse model of Alzheimer's disease. Hum Mol Genet 20:4515–4529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Calkins MJ, Manczak M, Reddy PH (2012) Mitochondria‐targeted antioxidant SS31 prevents amyloid beta‐induced mitochondrial abnormalities and synaptic degeneration in Alzheimer's disease. Pharmaceuticals 5:1103–1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Calkins MJ, Reddy PH (2011) Amyloid beta impairs mitochondrial anterograde transport and degenerates synapses in Alzheimer's disease neurons. Biochim Biophys Acta 1812:507–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Camiletti‐Moiron D, Aparicio VA, Aranda P, Radak Z (2013) Does exercise reduce brain oxidative stress? A systematic review. Scand J Med Sci Sports 23:e202–e212. [DOI] [PubMed] [Google Scholar]
- 28. Cardoso SM, Santana I, Swerdlow RH, Oliveira CR (2004) Mitochondria dysfunction of Alzheimer's disease cybrids enhances Abeta toxicity. J Neurochem 89:1417–1426. [DOI] [PubMed] [Google Scholar]
- 29. Carvalho C, Cardoso S, Correia SC, Santos RX, Santos MS, Baldeiras I et al (2012) Metabolic alterations induced by sucrose intake and Alzheimer's disease promote similar brain mitochondrial abnormalities. Diabetes 61:1234–1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Carvalho C, Correia SC, Cardoso S, Placido AI, Candeias E, Duarte AI et al (2015) The role of mitochondrial disturbances in Alzheimer, Parkinson and Huntington diseases. Expert Rev Neurother 15:867–884. [DOI] [PubMed] [Google Scholar]
- 31. Carvalho C, Santos MS, Oliveira CR, Moreira PI (2015) Alzheimer's disease and type 2 diabetes‐related alterations in brain mitochondria, autophagy and synaptic markers. Biochim Biophys Acta 1852:1665–1675. [DOI] [PubMed] [Google Scholar]
- 32. Castello MA, Soriano S (2014) On the origin of Alzheimer's disease. Trials and tribulations of the amyloid hypothesis. Ageing Res Rev 13:10–12. [DOI] [PubMed] [Google Scholar]
- 33. Cataldo AM, Barnett JL, Berman SA, Li J, Quarless S, Bursztajn S et al (1995) Gene expression and cellular content of cathepsin D in Alzheimer's disease brain: evidence for early up‐regulation of the endosomal‐lysosomal system. Neuron 14:671–680. [DOI] [PubMed] [Google Scholar]
- 34. Chen D, Guarente L (2007) SIR2: a potential target for calorie restriction mimetics. Trends Mol Med 13:64–71. [DOI] [PubMed] [Google Scholar]
- 35. Cheng B, Mattson MP (1992) Glucose deprivation elicits neurofibrillary tangle‐like antigenic changes in hippocampal neurons: prevention by NGF and bFGF. Exp Neurol 117:114–123. [DOI] [PubMed] [Google Scholar]
- 36. Cho DH, Nakamura T, Fang J, Cieplak P, Godzik A, Gu Z et al (2009) S‐nitrosylation of Drp1 mediates beta‐amyloid‐related mitochondrial fission and neuronal injury. Science 324:102–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Cho JY, Um HS, Kang EB, Cho IH, Kim CH, Cho JS et al (2010) The combination of exercise training and alpha‐lipoic acid treatment has therapeutic effects on the pathogenic phenotypes of Alzheimer's disease in NSE/APPsw‐transgenic mice. Int J Mol Med 25:337–346. [DOI] [PubMed] [Google Scholar]
- 38. Choi H, Park HH, Koh SH, Choi NY, Yu HJ, Park J et al (2012) Coenzyme Q10 protects against amyloid beta‐induced neuronal cell death by inhibiting oxidative stress and activating the P13K pathway. Neurotoxicology 33:85–90. [DOI] [PubMed] [Google Scholar]
- 39. Correia SC, Resende R, Moreira PI, Pereira CM (2015) Alzheimer's disease‐related misfolded proteins and dysfunctional organelles on autophagy menu. DNA Cell Biol 34:261–273. [DOI] [PubMed] [Google Scholar]
- 40. Cortopassi GA, Wong A (1999) Mitochondria in organismal aging and degeneration. Biochim Biophys Acta 1410:183–193. [DOI] [PubMed] [Google Scholar]
- 41. Cunnane SC, Courchesne‐Loyer A, St‐Pierre V, Vandenberghe C, Pierotti T, Fortier M et al (2016) Can ketones compensate for deteriorating brain glucose uptake during aging? Implications for the risk and treatment of Alzheimer's disease. Ann NY Acad Sci 1367:12–20. [DOI] [PubMed] [Google Scholar]
- 42. Dai J, Buijs RM, Kamphorst W, Swaab DF (2002) Impaired axonal transport of cortical neurons in Alzheimer's disease is associated with neuropathological changes. Brain Res 948:138–144. [DOI] [PubMed] [Google Scholar]
- 43. De Felice FG, Munoz DP (2016) Opportunities and challenges in developing relevant animal models for Alzheimer's disease. Ageing Res Rev 26:112–114. [DOI] [PubMed] [Google Scholar]
- 44. Demetrius LA, Driver J (2013) Alzheimer's as a metabolic disease. Biogerontology 14:641–649. [DOI] [PubMed] [Google Scholar]
- 45. Demetrius LA, Magistretti PJ, Pellerin L (2014) Alzheimer's disease: the amyloid hypothesis and the Inverse Warburg effect. Front Physiol 5:522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Demetrius LA, Simon DK (2012) An inverse‐Warburg effect and the origin of Alzheimer's disease. Biogerontology 13:583–594. [DOI] [PubMed] [Google Scholar]
- 47. Desquiret‐Dumas V, Gueguen N, Leman G, Baron S, Nivet‐Antoine V, Chupin S et al (2013) Resveratrol induces a mitochondrial complex I‐dependent increase in NADH oxidation responsible for sirtuin activation in liver cells. J Biol Chem 288:36662–36675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Dietrich MO, Andrews ZB, Horvath TL (2008) Exercise‐induced synaptogenesis in the hippocampus is dependent on UCP2‐regulated mitochondrial adaptation. J Neurosci 28:10766–10771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ding Q, Vaynman S, Souda P, Whitelegge JP, Gomez‐Pinilla F (2006) Exercise affects energy metabolism and neural plasticity‐related proteins in the hippocampus as revealed by proteomic analysis. Eur J Neurosci 24:1265–1276. [DOI] [PubMed] [Google Scholar]
- 50. Dishman RK, Berthoud HR, Booth FW, Cotman CW, Edgerton VR, Fleshner MR et al (2006) Neurobiology of exercise. Obesity 14:345–356. [DOI] [PubMed] [Google Scholar]
- 51. Dragicevic N, Mamcarz M, Zhu Y, Buzzeo R, Tan J, Arendash GW et al (2010) Mitochondrial amyloid‐beta levels are associated with the extent of mitochondrial dysfunction in different brain regions and the degree of cognitive impairment in Alzheimer's transgenic mice. J Alzheimer's Dis 20 (Suppl. 2):S535–S550. [DOI] [PubMed] [Google Scholar]
- 52. Du H, Guo L, Yan S, Sosunov AA, McKhann GM, Yan SS (2010) Early deficits in synaptic mitochondria in an Alzheimer's disease mouse model. Proc Natl Acad Sci USA 107:18670–18675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Dumont M, Stack C, Elipenahli C, Jainuddin S, Launay N, Gerges M et al (2014) PGC‐1alpha overexpression exacerbates beta‐amyloid and tau deposition in a transgenic mouse model of Alzheimer's disease. FASEB J 28:1745–1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Engelhart MJ, Geerlings MI, Ruitenberg A, Van Swieten JC, Hofman A, Witteman JC et al (2002) Diet and risk of dementia: does fat matter? The Rotterdam Study. Neurology 59:1915–1921. [DOI] [PubMed] [Google Scholar]
- 55. Feng Z, Qin C, Chang Y, Zhang JT (2006) Early melatonin supplementation alleviates oxidative stress in a transgenic mouse model of Alzheimer's disease. Free Radic Biol Med 40:101–109. [DOI] [PubMed] [Google Scholar]
- 56. Folch J, Petrov D, Ettcheto M, Pedros I, Abad S, Beas‐Zarate C et al (2015) Masitinib for the treatment of mild to moderate Alzheimer's disease. Expert Rev Neurother 15:587–596. [DOI] [PubMed] [Google Scholar]
- 57. Folwell J, Cowan CM, Ubhi KK, Shiabh H, Newman TA, Shepherd D et al (2010) Abeta exacerbates the neuronal dysfunction caused by human tau expression in a Drosophila model of Alzheimer's disease. Exp Neurol 223:401–409. [DOI] [PubMed] [Google Scholar]
- 58. Friedman JR, Nunnari J (2014) Mitochondrial form and function. Nature 505:335–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Gabuzda D, Busciglio J, Chen LB, Matsudaira P, Yankner BA (1994) Inhibition of energy metabolism alters the processing of amyloid precursor protein and induces a potentially amyloidogenic derivative. J Biol Chem 269:13623–13628. [PubMed] [Google Scholar]
- 60. Gan X, Huang S, Wu L, Wang Y, Hu G, Li G et al (2014) Inhibition of ERK‐DLP1 signaling and mitochondrial division alleviates mitochondrial dysfunction in Alzheimer's disease cybrid cell. Biochim Biophys Acta 1842:220–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Garcia‐Mesa Y, Lopez‐Ramos JC, Gimenez‐Llort L, Revilla S, Guerra R, Gruart A et al (2011) Physical exercise protects against Alzheimer's disease in 3xTg‐AD mice. J Alzheimer's Dis 24:421–454. [DOI] [PubMed] [Google Scholar]
- 62. Gasparini L, Gouras GK, Wang R, Gross RS, Beal MF, Greengard P et al (2001) Stimulation of beta‐amyloid precursor protein trafficking by insulin reduces intraneuronal beta‐amyloid and requires mitogen‐activated protein kinase signaling. J Neurosci 21:2561–2570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Gibson GE, Sheu KF, Blass JP, Baker A, Carlson KC, Harding B et al (1988) Reduced activities of thiamine‐dependent enzymes in the brains and peripheral tissues of patients with Alzheimer's disease. Arch Neurol 45:836–840. [DOI] [PubMed] [Google Scholar]
- 64. Gibson GE, Zhang H, Sheu KF, Bogdanovich N, Lindsay JG, Lannfelt L et al (1998) Alpha‐ketoglutarate dehydrogenase in Alzheimer brains bearing the APP670/671 mutation. Ann Neurol 44:676–681. [DOI] [PubMed] [Google Scholar]
- 65. Giulietti A, Vignini A, Nanetti L, Mazzanti L, Primio RD, Salvolini E (2016) Alzheimer's disease risk and progression: the role of nutritional supplements and their effect on drug therapy outcome. Curr Neuropharmacol 14:177–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Grimm A, Friedland K, Eckert A (2016) Mitochondrial dysfunction: the missing link between aging and sporadic Alzheimer's disease. Biogerontology 17:281–296. [DOI] [PubMed] [Google Scholar]
- 67. Guo ZH, Mattson MP (2000) In vivo 2‐deoxyglucose administration preserves glucose and glutamate transport and mitochondrial function in cortical synaptic terminals after exposure to amyloid beta‐peptide and iron: evidence for a stress response. Exp Neurol 166:173–179. [DOI] [PubMed] [Google Scholar]
- 68. Gustafson D, Rothenberg E, Blennow K, Steen B, Skoog I (2003) An 18‐year follow‐up of overweight and risk of Alzheimer disease. Arch Intern Med 163:1524–1528. [DOI] [PubMed] [Google Scholar]
- 69. Hampel H, Prvulovic D, Teipel S, Jessen F, Luckhaus C, Frolich L et al (2011) The future of Alzheimer's disease: the next 10 years. Prog Neurobiol 95:718–728. [DOI] [PubMed] [Google Scholar]
- 70. Hansson Petersen CA, Alikhani N, Behbahani H, Wiehager B, Pavlov PF, Alafuzoff I et al (2008) The amyloid beta‐peptide is imported into mitochondria via the TOM import machinery and localized to mitochondrial cristae. Proc Natl Acad Sci USA 105:13145–13150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Hardy J, Allsop D (1991) Amyloid deposition as the central event in the aetiology of Alzheimer's disease. Trends Pharmacol Sci 12:383–388. [DOI] [PubMed] [Google Scholar]
- 72. Hardy J, Selkoe DJ (2002) The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science 297:353–356. [DOI] [PubMed] [Google Scholar]
- 73. Hardy JA, Higgins GA (1992) Alzheimer's disease: the amyloid cascade hypothesis. Science 256:184–185. [DOI] [PubMed] [Google Scholar]
- 74. Harman D (1956) Aging: a theory based on free radical and radiation chemistry. J Gerontol 11:298–300. [DOI] [PubMed] [Google Scholar]
- 75. Harman D (1972) The biologic clock: the mitochondria? J Am Geriatr Soc 20:145–147. [DOI] [PubMed] [Google Scholar]
- 76. Harman D (2003) The free radical theory of aging. Antioxid Redox Signal 5:557–561. [DOI] [PubMed] [Google Scholar]
- 77. Heininger K (2000) A unifying hypothesis of Alzheimer's disease. III. Risk factors. Hum Psychopharmacol 15:1–70. [DOI] [PubMed] [Google Scholar]
- 78. Herrup K (2015) The case for rejecting the amyloid cascade hypothesis. Nat Neurosci 18:794–799. [DOI] [PubMed] [Google Scholar]
- 79. Hirai K, Aliev G, Nunomura A, Fujioka H, Russell RL, Atwood CS et al (2001) Mitochondrial abnormalities in Alzheimer's disease. J Neurosci 21:3017–3023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Hogan CL, Mata J, Carstensen LL (2013) Exercise holds immediate benefits for affect and cognition in younger and older adults. Psychol Aging 28:587–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Hollenbeck PJ, Saxton WM (2005) The axonal transport of mitochondria. J Cell Sci 118:5411–5419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Hroudova J, Singh N, Fisar Z, Ghosh KK (2016) Progress in drug development for Alzheimer's disease: an overview in relation to mitochondrial energy metabolism. Eur J Med Chem doi: 10.1016/j.ejmech.2016.03.084 [DOI] [PubMed] [Google Scholar]
- 83. Hung SY, Huang WP, Liou HC, Fu WM (2009) Autophagy protects neuron from Abeta‐induced cytotoxicity. Autophagy 5:502–510. [DOI] [PubMed] [Google Scholar]
- 84. Ingram DK, Roth GS, Lane MA, Ottinger MA, Zou S, de Cabo R et al (2006) The potential for dietary restriction to increase longevity in humans: extrapolation from monkey studies. Biogerontology 7:143–148. [DOI] [PubMed] [Google Scholar]
- 85. Jang JH, Surh YJ (2003) Protective effect of resveratrol on beta‐amyloid‐induced oxidative PC12 cell death. Free Radic Biol Med 34:1100–1110. [DOI] [PubMed] [Google Scholar]
- 86. Johnson AB, Blum NR (1970) Nucleoside phosphatase activities associated with the tangles and plaques of alzheimer's disease: a histochemical study of natural and experimental neurofibrillary tangles. J Neuropathol Exp Neurol 29:463–478. [DOI] [PubMed] [Google Scholar]
- 87. Judge JO (1993) Exercise programs for older persons: writing an exercise prescription. Connecticut Med 57:269–275. [PubMed] [Google Scholar]
- 88. Jun Z, Ibrahim MM, Dezheng G, Bo Y, Qiong W, Yuan Z (2015) UCP2 protects against amyloid beta toxicity and oxidative stress in primary neuronal culture. Biomed Pharmacother 74:211–214. [DOI] [PubMed] [Google Scholar]
- 89. Kamat CD, Gadal S, Mhatre M, Williamson KS, Pye QN, Hensley K (2008) Antioxidants in central nervous system diseases: preclinical promise and translational challenges. J Alzheimer's Dis 15:473–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Kapahi P, Zid B (2004) TOR pathway: linking nutrient sensing to life span. Sci Aging Knowledge Environ 2004:PE34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Karbowski M, Neutzner A (2012) Neurodegeneration as a consequence of failed mitochondrial maintenance. Acta Neuropathol 123:157–171. [DOI] [PubMed] [Google Scholar]
- 92. Karuppagounder SS, Pinto JT, Xu H, Chen HL, Beal MF, Gibson GE (2009) Dietary supplementation with resveratrol reduces plaque pathology in a transgenic model of Alzheimer's disease. Neurochem Int 54:111–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Katsouri L, Parr C, Bogdanovic N, Willem M, Sastre M (2011) PPARgamma co‐activator‐1alpha (PGC‐1alpha) reduces amyloid‐beta generation through a PPARgamma‐dependent mechanism. J Alzheimer's Dis 25:151–162. [DOI] [PubMed] [Google Scholar]
- 94. Keller JN, Schmitt FA, Scheff SW, Ding Q, Chen Q, Butterfield DA et al (2005) Evidence of increased oxidative damage in subjects with mild cognitive impairment. Neurology 64:1152–1156. [DOI] [PubMed] [Google Scholar]
- 95. Kim SJ, Kwon MC, Ryu MJ, Chung HK, Tadi S, Kim YK et al (2012) CRIF1 is essential for the synthesis and insertion of oxidative phosphorylation polypeptides in the mammalian mitochondrial membrane. Cell Metab 16:274–283. [DOI] [PubMed] [Google Scholar]
- 96. Kirchner L, Chen WQ, Afjehi‐Sadat L, Viidik A, Skalicky M, Hoger H et al (2008) Hippocampal metabolic proteins are modulated in voluntary and treadmill exercise rats. Exp Neurol 212:145–151. [DOI] [PubMed] [Google Scholar]
- 97. Kurokawa T, Asada S, Nishitani S, Hazeki O (2001) Age‐related changes in manganese superoxide dismutase activity in the cerebral cortex of senescence‐accelerated prone and resistant mouse. Neurosci Lett 298:135–138. [DOI] [PubMed] [Google Scholar]
- 98. Kuro‐o M (2001) Disease model: human aging. Trends Mol Med 7:179–181. [DOI] [PubMed] [Google Scholar]
- 99. Lai AY, McLaurin J (2012) Inhibition of amyloid‐beta peptide aggregation rescues the autophagic deficits in the TgCRND8 mouse model of Alzheimer disease. Biochim Biophys Acta 1822:1629–1637. [DOI] [PubMed] [Google Scholar]
- 100. Lauretti E, Pratico D (2015) Glucose deprivation increases tau phosphorylation via P38 mitogen‐activated protein kinase. Aging Cell 14:1067–1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Laurin D, Verreault R, Lindsay J, MacPherson K, Rockwood K (2001) Physical activity and risk of cognitive impairment and dementia in elderly persons. Arch Neurol 58:498–504. [DOI] [PubMed] [Google Scholar]
- 102. Lazarov O, Morfini GA, Pigino G, Gadadhar A, Chen X, Robinson J et al (2007) Impairments in fast axonal transport and motor neuron deficits in transgenic mice expressing familial Alzheimer's disease‐linked mutant presenilin 1. J Neurosci 27:7011–7020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Lee HC, Wei YH (1997) Role of mitochondria in human aging. J Biomed Sci 4:319–326. [DOI] [PubMed] [Google Scholar]
- 104. Leuner K, Schutt T, Kurz C, Eckert SH, Schiller C, Occhipinti A et al (2012) Mitochondrion‐derived reactive oxygen species lead to enhanced amyloid beta formation. Antioxid Redox Signal 16:1421–1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Li Z, Okamoto K, Hayashi Y, Sheng M (2004) The importance of dendritic mitochondria in the morphogenesis and plasticity of spines and synapses. Cell 119:873–887. [DOI] [PubMed] [Google Scholar]
- 106. Lin AL, Coman D, Jiang L, Rothman DL, Hyder F (2014) Caloric restriction impedes age‐related decline of mitochondrial function and neuronal activity. J Cereb Blood Flow Metab 34:1440–1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Lin MT, Beal MF (2006) Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 443:787–795. [DOI] [PubMed] [Google Scholar]
- 108. Lopez‐Lluch G, Hunt N, Jones B, Zhu M, Jamieson H, Hilmer S et al (2006) Calorie restriction induces mitochondrial biogenesis and bioenergetic efficiency. Proc Natl Acad Sci USA 103:1768–1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Luchsinger JA, Mayeux R (2004) Dietary factors and Alzheimer's disease. Lancet Neurol 3:579–587. [DOI] [PubMed] [Google Scholar]
- 110. Luchsinger JA, Tang MX, Shea S, Mayeux R (2002) Caloric intake and the risk of Alzheimer disease. Arch Neurol 59:1258–1263. [DOI] [PubMed] [Google Scholar]
- 111. Luft R (1994) The development of mitochondrial medicine. Proc Natl Acad Sci USA 91:8731–8738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Luft R, Ikkos D, Palmieri G, Ernster L, Afzelius B (1962) A case of severe hypermetabolism of nonthyroid origin with a defect in the maintenance of mitochondrial respiratory control: a correlated clinical, biochemical, and morphological study. J Clin Investig 41:1776–1804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Lustbader JW, Cirilli M, Lin C, Xu HW, Takuma K, Wang N et al (2004) ABAD directly links Abeta to mitochondrial toxicity in Alzheimer's disease. Science 304:448–452. [DOI] [PubMed] [Google Scholar]
- 114. MacAskill AF, Kittler JT (2010) Control of mitochondrial transport and localization in neurons. Trends Cell Biol 20:102–112. [DOI] [PubMed] [Google Scholar]
- 115. Majumder S, Richardson A, Strong R, Oddo S (2011) Inducing autophagy by rapamycin before, but not after, the formation of plaques and tangles ameliorates cognitive deficits. PloS One 6:e25416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Manczak M, Calkins MJ, Reddy PH (2011) Impaired mitochondrial dynamics and abnormal interaction of amyloid beta with mitochondrial protein Drp1 in neurons from patients with Alzheimer's disease: implications for neuronal damage. Hum Mol Genet 20:2495–2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Manczak M, Mao P, Calkins MJ, Cornea A, Reddy AP, Murphy MP et al (2010) Mitochondria‐targeted antioxidants protect against amyloid‐beta toxicity in Alzheimer's disease neurons. J Alzheimer's Dis 20 (Suppl. 2):S609–S631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Manczak M, Park BS, Jung Y, Reddy PH (2004) Differential expression of oxidative phosphorylation genes in patients with Alzheimer's disease: implications for early mitochondrial dysfunction and oxidative damage. Neuromol Med 5:147–162. [DOI] [PubMed] [Google Scholar]
- 119. Manczak M, Reddy PH (2012) Abnormal interaction of VDAC1 with amyloid beta and phosphorylated tau causes mitochondrial dysfunction in Alzheimer's disease. Hum Mol Genet 21:5131–5146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Manczak M, Sheiko T, Craigen WJ, Reddy PH (2013) Reduced VDAC1 protects against Alzheimer's disease, mitochondria, and synaptic deficiencies. J Alzheimer's Dis 37:679–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Manzanero S, Gelderblom M, Magnus T, Arumugam TV (2011) Calorie restriction and stroke. Exp Transl Stroke Med 3:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Marambaud P, Zhao H, Davies P (2005) Resveratrol promotes clearance of Alzheimer's disease amyloid‐beta peptides. J Biol Chem 280:37377–37382. [DOI] [PubMed] [Google Scholar]
- 123. Marcus DL, Thomas C, Rodriguez C, Simberkoff K, Tsai JS, Strafaci JA et al (1998) Increased peroxidation and reduced antioxidant enzyme activity in Alzheimer's disease. Exp Neurol 150:40–44. [DOI] [PubMed] [Google Scholar]
- 124. Markesbery WR, Schmitt FA, Kryscio RJ, Davis DG, Smith CD, Wekstein DR (2006) Neuropathologic substrate of mild cognitive impairment. Arch Neurol 63:38–46. [DOI] [PubMed] [Google Scholar]
- 125. Marques‐Aleixo I, Oliveira PJ, Moreira PI, Magalhaes J, Ascensao A (2012) Physical exercise as a possible strategy for brain protection: evidence from mitochondrial‐mediated mechanisms. Prog Neurobiol 99:149–162. [DOI] [PubMed] [Google Scholar]
- 126. Marques‐Aleixo I, Santos‐Alves E, Balca MM, Rizo‐Roca D, Moreira PI, Oliveira PJ et al (2015) Physical exercise improves brain cortex and cerebellum mitochondrial bioenergetics and alters apoptotic, dynamic and auto(mito)phagy markers. Neuroscience 301:480–495. [DOI] [PubMed] [Google Scholar]
- 127. Marrache S, Dhar S (2012) Engineering of blended nanoparticle platform for delivery of mitochondria‐acting therapeutics. Proc Natl Acad Sci USA 109:16288–16293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Maswood N, Young J, Tilmont E, Zhang Z, Gash DM, Gerhardt GA et al (2004) Caloric restriction increases neurotrophic factor levels and attenuates neurochemical and behavioral deficits in a primate model of Parkinson's disease. Proc Natl Acad Sci USA 101:18171–18176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Mattson MP (2002) Contributions of mitochondrial alterations, resulting from bad genes and a hostile environment, to the pathogenesis of Alzheimer's disease. Int Rev Neurobiol 53:387–409. [DOI] [PubMed] [Google Scholar]
- 130. Mattson MP, Duan W, Guo Z (2003) Meal size and frequency affect neuronal plasticity and vulnerability to disease: cellular and molecular mechanisms. J Neurochem 84:417–431. [DOI] [PubMed] [Google Scholar]
- 131. Mattson MP, Gleichmann M, Cheng A (2008) Mitochondria in neuroplasticity and neurological disorders. Neuron 60:748–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Maurer K, Volk S, Gerbaldo H (1997) Auguste D and Alzheimer's disease. Lancet 349:1546–1549. [DOI] [PubMed] [Google Scholar]
- 133. McBride HM, Neuspiel M, Wasiak S (2006) Mitochondria: more than just a powerhouse. Curr Biol 16:R551–R560. [DOI] [PubMed] [Google Scholar]
- 134. McManus MJ, Murphy MP, Franklin JL (2011) The mitochondria‐targeted antioxidant MitoQ prevents loss of spatial memory retention and early neuropathology in a transgenic mouse model of Alzheimer's disease. J Neurosci 31:15703–15715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Mecocci P, Polidori MC (2012) Antioxidant clinical trials in mild cognitive impairment and Alzheimer's disease. Biochim Biophys Acta 1822:631–638. [DOI] [PubMed] [Google Scholar]
- 136. Millecamps S, Julien JP (2013) Axonal transport deficits and neurodegenerative diseases. Nat Rev Neurosci 14:161–176. [DOI] [PubMed] [Google Scholar]
- 137. Minor RK, Allard JS, Younts CM, Ward TM, de Cabo R (2010) Dietary interventions to extend life span and health span based on calorie restriction. J Gerontol Ser A Biol Sci Med Sci 65:695–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Misko A, Jiang S, Wegorzewska I, Milbrandt J, Baloh RH (2010) Mitofusin 2 is necessary for transport of axonal mitochondria and interacts with the Miro/Milton complex. J Neurosci 30:4232–4240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Mistur R, Mosconi L, Santi SD, Guzman M, Li Y, Tsui W et al (2009) Current challenges for the early detection of Alzheimer's disease: brain imaging and CSF studies. J Clin Neurol 5:153–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Moreira PI, Harris PL, Zhu X, Santos MS, Oliveira CR, Smith MA et al (2007) Lipoic acid and N‐acetyl cysteine decrease mitochondrial‐related oxidative stress in Alzheimer disease patient fibroblasts. J Alzheimer's Dis 12:195–206. [DOI] [PubMed] [Google Scholar]
- 141. Moreira PI, Siedlak SL, Wang X, Santos MS, Oliveira CR, Tabaton M et al (2007) Autophagocytosis of mitochondria is prominent in Alzheimer disease. J Neuropathol Exp Neurol 66:525–532. [DOI] [PubMed] [Google Scholar]
- 142. Moreira PI, Zhu X, Lee HG, Honda K, Smith MA, Perry G (2006) The (un)balance between metabolic and oxidative abnormalities and cellular compensatory responses in Alzheimer disease. Mech Ageing Dev 127:501–506. [DOI] [PubMed] [Google Scholar]
- 143. Morley JE, Kumar VB, Bernardo AE, Farr SA, Uezu K, Tumosa N et al (2000) Beta‐amyloid precursor polypeptide in SAMP8 mice affects learning and memory. Peptides 21:1761–1767. [DOI] [PubMed] [Google Scholar]
- 144. Morris GP, Clark IA, Vissel B (2014) Inconsistencies and controversies surrounding the amyloid hypothesis of Alzheimer's disease. Acta Neuropathol Commun 2:135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Mouton PR, Chachich ME, Quigley C, Spangler E, Ingram DK (2009) Caloric restriction attenuates amyloid deposition in middle‐aged dtg APP/PS1 mice. Neurosci Lett 464:184–187. PubMed PMID: 19699265. Pubmed Central PMCID: 2748166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Murphy MP, Smith RA (2000) Drug delivery to mitochondria: the key to mitochondrial medicine. Adv Drug Deliv Rev 41:235–250. PubMed PMID: 10699318. [DOI] [PubMed] [Google Scholar]
- 147. Navarro A, Gomez C, Lopez‐Cepero JM, Boveris A (2004) Beneficial effects of moderate exercise on mice aging: survival, behavior, oxidative stress, and mitochondrial electron transfer. Am J Physiol Regul Integr Comp Physiol 286:R505–R511. [DOI] [PubMed] [Google Scholar]
- 148. Ng LF, Gruber J, Cheah IK, Goo CK, Cheong WF, Shui G et al (2014) The mitochondria‐targeted antioxidant MitoQ extends lifespan and improves healthspan of a transgenic Caenorhabditis elegans model of Alzheimer disease. Free Radic Biol Med 71:390–401. [DOI] [PubMed] [Google Scholar]
- 149. Nixon RA (2007) Autophagy, amyloidogenesis and Alzheimer disease. J Cell Sci 120:4081–4091. PubMed PMID: 18032783. [DOI] [PubMed] [Google Scholar]
- 150. Nixon RA, Wegiel J, Kumar A, Yu WH, Peterhoff C, Cataldo A et al (2005) Extensive involvement of autophagy in Alzheimer disease: an immuno‐electron microscopy study. J Neuropathol Exp Neurol 64:113–122. [DOI] [PubMed] [Google Scholar]
- 151. Nixon RA, Yang DS, Lee JH (2008) Neurodegenerative lysosomal disorders: a continuum from development to late age. Autophagy 4:590–599. [DOI] [PubMed] [Google Scholar]
- 152. Nunomura A, Castellani RJ, Zhu X, Moreira PI, Perry G, Smith MA (2006) Involvement of oxidative stress in Alzheimer disease. J Neuropathol Exp Neurol 65:631–641. [DOI] [PubMed] [Google Scholar]
- 153. Nunomura A, Perry G, Pappolla MA, Friedland RP, Hirai K, Chiba S et al (2000) Neuronal oxidative stress precedes amyloid‐beta deposition in Down syndrome. J Neuropathol Exp Neurol 59:1011–1017. [DOI] [PubMed] [Google Scholar]
- 154. O'Brien C, Auguste D (1996) Alzheimer's disease. Science 273:28. [DOI] [PubMed] [Google Scholar]
- 155. Obulesu M, Jhansilakshmi M (2016) Neuroprotective role of nanoparticles against Alzheimer's disease. Curr Drug Metab 17:142–149. [DOI] [PubMed] [Google Scholar]
- 156. O'Connor T, Sadleir KR, Maus E, Velliquette RA, Zhao J, Cole SL et al (2008) Phosphorylation of the translation initiation factor eIF2alpha increases BACE1 levels and promotes amyloidogenesis. Neuron 60:988–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Ozcelik S, Fraser G, Castets P, Schaeffer V, Skachokova Z, Breu K et al (2013) Rapamycin attenuates the progression of tau pathology in P301S tau transgenic mice. PLoS One 8:e62459. 23667480 [Google Scholar]
- 158. Pallas M, Camins A, Smith MA, Perry G, Lee HG, Casadesus G (2008) From aging to Alzheimer's disease: unveiling “the switch” with the senescence‐accelerated mouse model (SAMP8). J Alzheimer's Dis 15:615–624. [DOI] [PubMed] [Google Scholar]
- 159. Palleschi L, Vetta F, De Gennaro E, Idone G, Sottosanti G, Gianni W et al (1996) Effect of aerobic training on the cognitive performance of elderly patients with senile dementia of Alzheimer type. Arch Gerontol Geriatr 22 (Suppl 1):47–50. [DOI] [PubMed] [Google Scholar]
- 160. Panza F, Solfrizzi V, Colacicco AM, D'Introno A, Capurso C, Torres F et al (2004) Mediterranean diet and cognitive decline. Public Health Nutr 7:959–963. [DOI] [PubMed] [Google Scholar]
- 161. Park J, Choi H, Min JS, Kim B, Lee SR, Yun JW et al (2015) Loss of mitofusin 2 links beta‐amyloid‐mediated mitochondrial fragmentation and Cdk5‐induced oxidative stress in neuron cells. J Neurochem 132:687–702. [DOI] [PubMed] [Google Scholar]
- 162. Parker WD Jr, Filley CM, Parks JK (1990) Cytochrome oxidase deficiency in Alzheimer's disease. Neurology 40:1302–1303. [DOI] [PubMed] [Google Scholar]
- 163. Patel NV, Gordon MN, Connor KE, Good RA, Engelman RW, Mason J et al (2005) Caloric restriction attenuates Abeta‐deposition in Alzheimer transgenic models. Neurobiol Aging 26:995–1000. [DOI] [PubMed] [Google Scholar]
- 164. Petersen RC, Thomas RG, Grundman M, Bennett D, Doody R, Ferris S et al (2005) Vitamin E and donepezil for the treatment of mild cognitive impairment. N Engl J Med 352:2379–2388. [DOI] [PubMed] [Google Scholar]
- 165. Peterson C, Goldman JE (1986) Alterations in calcium content and biochemical processes in cultured skin fibroblasts from aged and Alzheimer donors. Proc Natl Acad Sci USA 83:2758–2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Pigino G, Morfini G, Atagi Y, Deshpande A, Yu C, Jungbauer L et al (2009) Disruption of fast axonal transport is a pathogenic mechanism for intraneuronal amyloid beta. Proc Natl Acad Sci USA 106:5907–5912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Pigino G, Morfini G, Pelsman A, Mattson MP, Brady ST, Busciglio J (2003) Alzheimer's presenilin 1 mutations impair kinesin‐based axonal transport. J Neurosci 23:4499–4508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Pinho CM, Teixeira PF, Glaser E (2014) Mitochondrial import and degradation of amyloid‐beta peptide. Biochim Biophys Acta 1837:1069–1074. [DOI] [PubMed] [Google Scholar]
- 169. Pope SK, Shue VM, Beck C (2003) Will a healthy lifestyle help prevent Alzheimer's disease? Annu Rev Public Health 24:111–132. [DOI] [PubMed] [Google Scholar]
- 170. Pratico D, Clark CM, Liun F, Rokach J, Lee VY, Trojanowski JQ (2002) Increase of brain oxidative stress in mild cognitive impairment: a possible predictor of Alzheimer disease. Arch Neurol 59:972–976. [DOI] [PubMed] [Google Scholar]
- 171. Prince M, Bryce R, Albanese E, Wimo A, Ribeiro W, Ferri CP (2013) The global prevalence of dementia: a systematic review and metaanalysis. Alzheimer's Dementia 9:63–75 e2. [DOI] [PubMed] [Google Scholar]
- 172. Qin W, Haroutunian V, Katsel P, Cardozo CP, Ho L, Buxbaum JD et al (2009) PGC‐1alpha expression decreases in the Alzheimer disease brain as a function of dementia. Arch Neurol 66:352–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Qin W, Yang T, Ho L, Zhao Z, Wang J, Chen L et al (2006) Neuronal SIRT1 activation as a novel mechanism underlying the prevention of Alzheimer disease amyloid neuropathology by calorie restriction. J Biol Chem 281:21745–21754. [DOI] [PubMed] [Google Scholar]
- 174. Quinn JF, Bussiere JR, Hammond RS, Montine TJ, Henson E, Jones RE et al (2007) Chronic dietary alpha‐lipoic acid reduces deficits in hippocampal memory of aged Tg2576 mice. Neurobiol Aging 28:213–225. [DOI] [PubMed] [Google Scholar]
- 175. Radak Z, Hart N, Sarga L, Koltai E, Atalay M, Ohno H et al (2010) Exercise plays a preventive role against Alzheimer's disease. J Alzheimer's Dis 20:777–783. [DOI] [PubMed] [Google Scholar]
- 176. Reddy PH (2008) Mitochondrial medicine for aging and neurodegenerative diseases. Neuromol Med 10:291–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Reddy PH, Tripathi R, Troung Q, Tirumala K, Reddy TP, Anekonda V et al (2012) Abnormal mitochondrial dynamics and synaptic degeneration as early events in Alzheimer's disease: implications to mitochondria‐targeted antioxidant therapeutics. Biochim Biophys Acta 1822:639–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Resende R, Moreira PI, Proenca T, Deshpande A, Busciglio J, Pereira C et al (2008) Brain oxidative stress in a triple‐transgenic mouse model of Alzheimer disease. Free Radic Biol Med 44:2051–2057. [DOI] [PubMed] [Google Scholar]
- 179. Rhein V, Song X, Wiesner A, Ittner LM, Baysang G, Meier F et al (2009) Amyloid‐beta and tau synergistically impair the oxidative phosphorylation system in triple transgenic Alzheimer's disease mice. Proc Natl Acad Sci USA 106:20057–20062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Rinaldi P, Polidori MC, Metastasio A, Mariani E, Mattioli P, Cherubini A et al (2003) Plasma antioxidants are similarly depleted in mild cognitive impairment and in Alzheimer's disease. Neurobiol Aging 24:915–919. [DOI] [PubMed] [Google Scholar]
- 181. Rugarli EI, Langer T (2012) Mitochondrial quality control: a matter of life and death for neurons. EMBO J 31:1336–1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Rui Y, Tiwari P, Xie Z, Zheng JQ (2006) Acute impairment of mitochondrial trafficking by beta‐amyloid peptides in hippocampal neurons. J Neurosci 26:10480–10487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Ryan SM, Kelly AM (2016) Exercise as a pro‐cognitive, pro‐neurogenic and anti‐inflammatory intervention in transgenic mouse models of Alzheimer's disease. Ageing Res Rev 27:77–92. [DOI] [PubMed] [Google Scholar]
- 184. Santos RX, Correia SC, Cardoso S, Carvalho C, Santos MS, Moreira PI (2011) Effects of rapamycin and TOR on aging and memory: implications for Alzheimer's disease. J Neurochem 117:927–936. [DOI] [PubMed] [Google Scholar]
- 185. Santos RX, Correia SC, Wang X, Perry G, Smith MA, Moreira PI et al (2010) Alzheimer's disease: diverse aspects of mitochondrial malfunctioning. Int J Clin Exp Pathol 3:570–581. [PMC free article] [PubMed] [Google Scholar]
- 186. Saraiva AA, Borges MM, Madeira MD, Tavares MA, Paula‐Barbosa MM (1985) Mitochondrial abnormalities in cortical dendrites from patients with Alzheimer's disease. J Submicrosc Cytol 17:459–464. [PubMed] [Google Scholar]
- 187. Savaskan E, Olivieri G, Meier F, Seifritz E, Wirz‐Justice A, Muller‐Spahn F (2003) Red wine ingredient resveratrol protects from beta‐amyloid neurotoxicity. Gerontology 49:380–383. [DOI] [PubMed] [Google Scholar]
- 188. Saxton WM, Hollenbeck PJ ( 2012) The axonal transport of mitochondria. J Cell Sci 125:2095–2104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Schmitt K, Grimm A, Kazmierczak A, Strosznajder JB, Gotz J, Eckert A (2012) Insights into mitochondrial dysfunction: aging, amyloid‐beta, and tau‐A deleterious trio. Antioxid Redox Signal 16:1456–1466. [DOI] [PubMed] [Google Scholar]
- 190. Selfridge JE, EL, Lu J, Swerdlow RH (2013) Role of mitochondrial homeostasis and dynamics in Alzheimer's disease. Neurobiol Dis 51:3–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. Selkoe DJ, Schenk D (2003) Alzheimer's disease: molecular understanding predicts amyloid‐based therapeutics. Annu Rev Pharmacol Toxicol 43:545–584. [DOI] [PubMed] [Google Scholar]
- 192. Serrano‐Pozo A, Frosch MP, Masliah E, Hyman BT (2011) Neuropathological alterations in Alzheimer disease. Cold Spring Harbor Perspect Med 1:a006189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193. Serviddio G, Romano AD, Cassano T, Bellanti F, Altomare E, Vendemiale G (2011) Principles and therapeutic relevance for targeting mitochondria in aging and neurodegenerative diseases. Curr Pharm Design 17:2036–2055. [DOI] [PubMed] [Google Scholar]
- 194. Sharma M, Gupta YK (2002) Chronic treatment with trans resveratrol prevents intracerebroventricular streptozotocin induced cognitive impairment and oxidative stress in rats. Life Sci 71:2489–2498. [DOI] [PubMed] [Google Scholar]
- 195. Sheng B, Wang X, Su B, Lee HG, Casadesus G, Perry G et al (2012) Impaired mitochondrial biogenesis contributes to mitochondrial dysfunction in Alzheimer's disease. J Neurochem 120:419–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196. Shi Y, Buffenstein R, Pulliam DA, Van Remmen H (2010) Comparative studies of oxidative stress and mitochondrial function in aging. Integr Comp Biol 50:869–879. [DOI] [PubMed] [Google Scholar]
- 197. Silva DF, Selfridge JE, Lu J, EL, Roy N Hutfles L et al (2013) Bioenergetic flux, mitochondrial mass and mitochondrial morphology dynamics in AD and MCI cybrid cell lines. Hum Mol Genet 22:3931–3946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198. Smith DL Jr, Nagy TR, Allison DB (2010) Calorie restriction: what recent results suggest for the future of ageing research. Eur J Clin Investig 40:440–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199. Sorbi S, Bird ED, Blass JP (1983) Decreased pyruvate dehydrogenase complex activity in Huntington and Alzheimer brain. Ann Neurol 13:72–78. [DOI] [PubMed] [Google Scholar]
- 200. Srivastava S, Haigis MC (2011) Role of sirtuins and calorie restriction in neuroprotection: implications in Alzheimer's and Parkinson's diseases. Curr Pharm Design 17:3418–3433. [DOI] [PubMed] [Google Scholar]
- 201. Steiner JL, Murphy EA, McClellan JL, Carmichael MD, Davis JM (2011) Exercise training increases mitochondrial biogenesis in the brain. J Appl Physiol 111:1066–1071. [DOI] [PubMed] [Google Scholar]
- 202. Stotland A, Gottlieb RA (2015) Mitochondrial quality control: easy come, easy go. Biochim Biophys Acta 1853:2802–2811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203. Su B, Wang X, Bonda D, Perry G, Smith M, Zhu X (2010) Abnormal mitochondrial dynamics—a novel therapeutic target for Alzheimer's disease? Mol Neurobiol 41:87–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204. Sureda FX, Gutierrez‐Cuesta J, Romeu M, Mulero M, Canudas AM, Camins A et al (2006) Changes in oxidative stress parameters and neurodegeneration markers in the brain of the senescence‐accelerated mice SAMP‐8. Exp Gerontol 41:360–367. [DOI] [PubMed] [Google Scholar]
- 205. Swerdlow RH (2009) The neurodegenerative mitochondriopathies. J Alzheimer's Dis 17:737–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206. Swerdlow RH (2011) Brain aging, Alzheimer's disease, and mitochondria. Biochim Biophys Acta 1812:1630–1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207. Swerdlow RH, Burns JM, Khan SM (2014) The Alzheimer's disease mitochondrial cascade hypothesis: progress and perspectives. Biochim Biophys Acta 1842:1219–1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208. Swerdlow RH, Burns JM, Khan SM (2010) The Alzheimer's disease mitochondrial cascade hypothesis. J Alzheimer's Dis 20 (Suppl. 2):S265–S279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209. Swerdlow RH, Khan SM (2004) A “mitochondrial cascade hypothesis” for sporadic Alzheimer's disease. Med Hypotheses 63:8–20. [DOI] [PubMed] [Google Scholar]
- 210. Szeto HH (2008) Development of mitochondria‐targeted aromatic‐cationic peptides for neurodegenerative diseases. Ann NY Acad Sci 1147:112–121. [DOI] [PubMed] [Google Scholar]
- 211. Takemura M, Nakamura S, Akiguchi I, Ueno M, Oka N, Ishikawa S et al (1993) Beta/A4 proteinlike immunoreactive granular structures in the brain of senescence‐accelerated mouse. Am J Pathol 142:1887–1897. [PMC free article] [PubMed] [Google Scholar]
- 212. Testa G, Biasi F, Poli G, Chiarpotto E (2014) Calorie restriction and dietary restriction mimetics: a strategy for improving healthy aging and longevity. Curr Pharm Design 20:2950–2977. [DOI] [PubMed] [Google Scholar]
- 213. Thompson PD (2003) Exercise and physical activity in the prevention and treatment of atherosclerotic cardiovascular disease. Arterioscler Thromb Vasc Biol 23:1319–1321. [DOI] [PubMed] [Google Scholar]
- 214. Tillement L, Lecanu L, Papadopoulos V (2011) Alzheimer's disease: effects of beta‐amyloid on mitochondria. Mitochondrion 11:13–21. [DOI] [PubMed] [Google Scholar]
- 215. Trimmer PA, Borland MK (2005) Differentiated Alzheimer's disease transmitochondrial cybrid cell lines exhibit reduced organelle movement. Antioxid Redox Signal 7:1101–1109. [DOI] [PubMed] [Google Scholar]
- 216. Turner RS, Thomas RG, Craft S, van Dyck CH, Mintzer J, Reynolds BA et al (2015) A randomized, double‐blind, placebo‐controlled trial of resveratrol for Alzheimer disease. Neurology 85:1383–1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217. Um HS, Kang EB, Koo JH, Kim HT, Jin L, Kim EJ et al (2011) Treadmill exercise represses neuronal cell death in an aged transgenic mouse model of Alzheimer's disease. Neurosci Res 69:161–173. [DOI] [PubMed] [Google Scholar]
- 218. Um HS, Kang EB, Leem YH, Cho IH, Yang CH, Chae KR et al (2008) Exercise training acts as a therapeutic strategy for reduction of the pathogenic phenotypes for Alzheimer's disease in an NSE/APPsw‐transgenic model. Int J Mol Med 22:529–539. [PubMed] [Google Scholar]
- 219. Valla J, Schneider L, Niedzielko T, Coon KD, Caselli R, Sabbagh MN et al (2006) Impaired platelet mitochondrial activity in Alzheimer's disease and mild cognitive impairment. Mitochondrion 6:323–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220. Velliquette RA, O'Connor T, Vassar R (2005) Energy inhibition elevates beta‐secretase levels and activity and is potentially amyloidogenic in APP transgenic mice: possible early events in Alzheimer's disease pathogenesis. J Neurosci 25:10874–10883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221. Venkateshappa C, Harish G, Mahadevan A, Srinivas Bharath MM, Shankar SK (2012) Elevated oxidative stress and decreased antioxidant function in the human hippocampus and frontal cortex with increasing age: implications for neurodegeneration in Alzheimer's disease. Neurochem Res 37:1601–1614. [DOI] [PubMed] [Google Scholar]
- 222. Vina J, Borras C, Miquel J (2007) Theories of ageing. IUBMB Life 59:249–254. [DOI] [PubMed] [Google Scholar]
- 223. Vingtdeux V, Chandakkar P, Zhao H, d'Abramo C, Davies P, Marambaud P (2011) Novel synthetic small‐molecule activators of AMPK as enhancers of autophagy and amyloid‐beta peptide degradation. FASEB J 25:219–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224. Wang X, Su B, Lee HG, Li X, Perry G, Smith MA et al (2009) Impaired balance of mitochondrial fission and fusion in Alzheimer's disease. J Neurosci 29:9090–9103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225. Wang X, Su B, Siedlak SL, Moreira PI, Fujioka H, Wang Y et al (2008) Amyloid‐beta overproduction causes abnormal mitochondrial dynamics via differential modulation of mitochondrial fission/fusion proteins. Proc Natl Acad Sci USA 105:19318–19323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226. Wang X, Su B, Zheng L, Perry G, Smith MA, Zhu X (2009) The role of abnormal mitochondrial dynamics in the pathogenesis of Alzheimer's disease. J Neurochem 109 (Suppl. 1):153–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227. Wang X, Wang W, Li L, Perry G, Lee HG, Zhu X (2014) Oxidative stress and mitochondrial dysfunction in Alzheimer's disease. Biochim Biophys Acta 1842:1240–1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228. Wang ZX, Tan L, Yu JT (2015) Axonal transport defects in Alzheimer's disease. Mol Neurobiol 51:1309–1321. [DOI] [PubMed] [Google Scholar]
- 229. Wongrakpanich A, Geary SM, Joiner ML, Anderson ME, Salem AK (2014) Mitochondria‐targeting particles. Nanomedicine 9:2531–2543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230. Wu P, Jiang C, Shen Q, Hu Y (2009) Systematic gene expression profile of hypothalamus in calorie‐restricted mice implicates the involvement of mTOR signaling in neuroprotective activity. Mech Ageing Dev 130:602–610. [DOI] [PubMed] [Google Scholar]
- 231. Wu P, Shen Q, Dong S, Xu Z, Tsien JZ, Hu Y (2008) Calorie restriction ameliorates neurodegenerative phenotypes in forebrain‐specific presenilin‐1 and presenilin‐2 double knockout mice. Neurobiol Aging 29:1502–1511. [DOI] [PubMed] [Google Scholar]
- 232. Xie N, Wang C, Lian Y, Wu C, Zhang H, Zhang Q (2014) Inhibition of mitochondrial fission attenuates Abeta‐induced microglia apoptosis. Neuroscience 256:36–42. [DOI] [PubMed] [Google Scholar]
- 233. Xu J, Shi C, Li Q, Wu J, Forster EL, Yew DT (2007) Mitochondrial dysfunction in platelets and hippocampi of senescence‐accelerated mice. J Bioenergetics Biomembr 39:195–202. [DOI] [PubMed] [Google Scholar]
- 234. Xu S, Zhong M, Zhang L, Wang Y, Zhou Z, Hao Y et al (2009) Overexpression of Tfam protects mitochondria against beta‐amyloid‐induced oxidative damage in SH‐SY5Y cells. FEBS J 276:3800–3809. [DOI] [PubMed] [Google Scholar]
- 235. Yanagisawa M, Planel E, Ishiguro K, Fujita SC (1999) Starvation induces tau hyperphosphorylation in mouse brain: implications for Alzheimer's disease. FEBS Lett 461:329–333. [DOI] [PubMed] [Google Scholar]
- 236. Yang DS, Stavrides P, Mohan PS, Kaushik S, Kumar A, Ohno M et al (2011) Therapeutic effects of remediating autophagy failure in a mouse model of Alzheimer disease by enhancing lysosomal proteolysis. Autophagy 7:788–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237. Yang F, Lim GP, Begum AN, Ubeda OJ, Simmons MR, Ambegaokar SS et al (2005) Curcumin inhibits formation of amyloid beta oligomers and fibrils, binds plaques, and reduces amyloid in vivo. J Biol Chem 280:5892–5901. [DOI] [PubMed] [Google Scholar]
- 238. Yao J, Chen S, Mao Z, Cadenas E, Brinton RD (2011) 2‐Deoxy‐D‐glucose treatment induces ketogenesis, sustains mitochondrial function, and reduces pathology in female mouse model of Alzheimer's disease. PLoS One 6:e21788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239. Yao J, Irwin RW, Zhao L, Nilsen J, Hamilton RT, Brinton RD (2009) Mitochondrial bioenergetic deficit precedes Alzheimer's pathology in female mouse model of Alzheimer's disease. Proc Natl Acad Sci USA 106:14670–14675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240. Zhang L, Trushin S, Christensen TA, Bachmeier BV, Gateno B, Schroeder A et al (2016) Altered brain energetics induces mitochondrial fission arrest in Alzheimer's Disease. Sci Rep 6:18725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241. Zhang W, Gu GJ, Shen X, Zhang Q, Wang GM, Wang PJ (2015) Neural stem cell transplantation enhances mitochondrial biogenesis in a transgenic mouse model of Alzheimer's disease‐like pathology. Neurobiol Aging 36:1282–1292. [DOI] [PubMed] [Google Scholar]
- 242. Zhang W, Wang PJ, Sha HY, Ni J, Li MH, Gu GJ (2014) Neural stem cell transplants improve cognitive function without altering amyloid pathology in an APP/PS1 double transgenic model of Alzheimer's disease. Mol Neurobiol 50:423–437. [DOI] [PubMed] [Google Scholar]
- 243. Zhao K, Zhao GM, Wu D, Soong Y, Birk AV, Schiller PW et al (2004) Cell‐permeable peptide antioxidants targeted to inner mitochondrial membrane inhibit mitochondrial swelling, oxidative cell death, and reperfusion injury. J Biol Chem 279:34682–34690. [DOI] [PubMed] [Google Scholar]
- 244. Zhao XL, Wang WA, Tan JX, Huang JK, Zhang X, Zhang BZ et al (2010) Expression of beta‐amyloid induced age‐dependent presynaptic and axonal changes in Drosophila. J Neurosci 30:1512–1522. [DOI] [PMC free article] [PubMed] [Google Scholar]