

Abstract
RRP22 (Ras‐related protein on chromosome 22) has been suggested as a candidate tumor suppressor in human cancers. Investigating a panel of 70 human gliomas, we found a frequent decrease in the RRP22 mRNA expression levels (67%), preferentially in high‐grade gliomas [World Health Organization (WHO) grades III and IV] as compared with low‐grade gliomas (WHO grade II). Moreover, reduced RRP22 mRNA expression was associated with shorter overall survival in 180 glioblastoma patients included in the National Institutes of Health Repository for Molecular Brain Neoplasia Data (NIH REMBRANDT) database. Decreased RRP22 expression levels were in part explained by 5'‐CpG island hypermethylation and increased by the treatment with the demethylating agent 5‐aza‐2'‐deoxycytidine in glioblastoma cell lines. In addition, the in vitro treatment with the histone deacetylase inhibitor trichostatin A alone resulted in RRP22 reexpression as well as a significant increase in the levels of RRP22 promoter DNA bound to pan‐acetylated histone H3 and H4. Moreover, in primary human glioblastomas, we observed an increase of H3K9me3‐bound and a decrease of pan‐Ac‐H3‐bound RRP22 in comparison with non‐neoplastic brain tissue, consistent with a heterochromatinization of the RRP22 promoter. Taken together, our findings demonstrate that both 5'‐CpG island hypermethylation and histone modifications contribute to the frequent and prognostically unfavorable transcriptional downregulation of RRP22 in malignant gliomas.
Keywords: astrocytoma, brain tumor, chromatin immunoprecipitation, glioblastoma, methylation, RASL10A
INTRODUCTION
Ras belongs to a superfamily of small GTPases that all share pronounced sequence homologies 2, 14. While most of the Ras family members possess oncogenic properties, a number of Ras‐related genes exhibit characteristics of tumor suppressors, such as the DIRAS3 gene that is frequently hypermethylated and transcriptionally silenced in oligodendroglial tumors with 1p loss (15). A further Ras‐related gene with tumor suppressor properties, which may be involved in the pathogenesis of human gliomas, is the small GTPase gene RRP22 (Ras‐related protein on chromosome 22) (21). RRP22 (syn. RASL10A) was first identified in 1996 and maps to the chromosomal band 22q12, which often shows allelic losses in human tumors, including gliomas (21). Its expression appears strictly limited to the central nervous system (21). While further bibliographic data on the role of RRP22 in human tumors is sparse (16), two studies in gliomas reported that RRP22 may suppress tumor cell growth, promote caspase‐independent cell death, decrease invasiveness and inhibit glioma cell growth in soft agar, thereby supporting a tumor suppressor function 3, 4. Moreover, Chen et al (3) showed that the mRNA expression level decreases with increasing malignancy, and Elam et al (4) pointed to a role of promoter hypermethylation as a potential cause for the inactivation of RRP22 in glioblastoma cell lines. However, the relevance of RRP22 and its potential inactivation mechanisms in primary human glioma tissue samples in vivo were not investigated.
To further address the role of RRP22 in glioma pathogenesis, we investigated primary gliomas and glioma cell lines for expression, mutation, 5'‐CpG island methylation and histone modifications of RRP22. In addition, we used the National Institutes of Health Repository for Molecular Brain Neoplasia Data (NIH REMBRANDT) database to validate the correlation between RRP22 gene expression, malignancy grade and patient survival. We conclude that RRP22 is frequently downregulated in gliomas because of both aberrant promoter methylation and/or heterochromatinization, and that inactivation of RRP22 is a negative prognostic factor in glioma patients.
MATERIALS AND METHODS
Cell lines, patients and controls
Human glioma cell lines U87MG, T98G, U138MG and A172 were obtained from the American Type Culture Collection (Manassas, VA, USA) and TP365MG cells were kindly provided by Prof V. P. Collins (Cambridge, UK). Tumors were selected from the archives of the Department of Neuropathology, Heinrich Heine University, Düsseldorf, Germany and investigated according to protocols approved by the institutional review board. All tumors were classified according to the criteria of the World Health Organization (WHO) 2007 classification of tumors of the nervous system (12). Parts of each tumor were snap‐frozen immediately after operation and stored at −80°C. Only tissue samples with an estimated tumor cell content of 80% or more were used for molecular analyses. The tumor series was composed of the following: 70 human gliomas, including 16 primary glioblastomas, WHO grade IV (GB); six anaplastic astrocytomas, WHO grade III (AA); seven diffuse astrocytomas, WHO grade II (A); 11 anaplastic oligoastrocytomas, WHO grade III (AOA); 10 oligoastrocytomas, WHO grade II (OA); 12 anaplastic oligodendrogliomas, WHO grade III (AO); and eight oligodendrogliomas, WHO grade II (O) (Figure 1). For chromatin immunoprecipitation (ChIP) analyses, we additionally prepared nuclei from glioblastoma tissue specimens of seven patients (T1–T7, Figure 5). Five non‐neoplastic brain samples from different individuals (NB1–NB5) were used as reference (four male, one female; median age: 69 years, age range: 43–76 years). As a positive control for the methylation studies, we used commercially available hypermethylated DNA (CpG Genome™ Universal Methylated DNA, Cat. No. S7821; Millipore, Billerica, MA).
Figure 1.
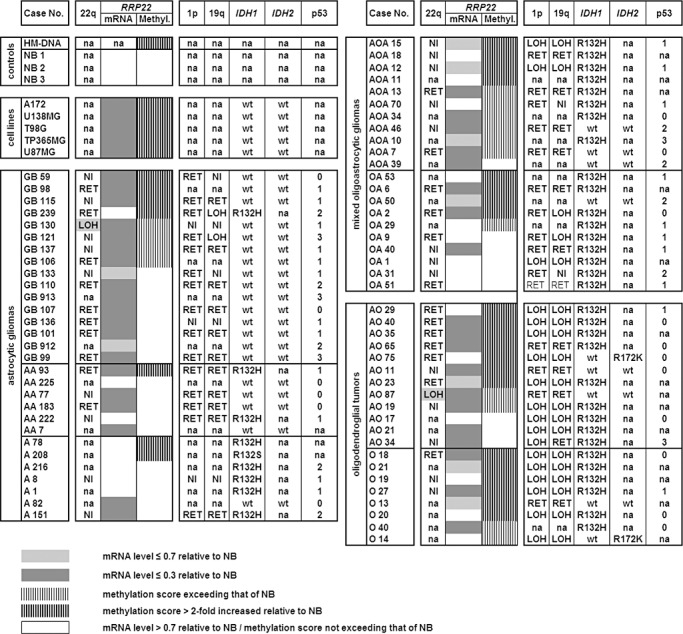
Molecular genetic aberrations of RRP22 in synopsis with other common molecular alterations in human gliomas. Note that RRP22 transcript levels (mRNA) are decreased relative to non‐neoplastic brain tissue in all glioblastoma cell lines and the majority of the investigated tumors. Transcriptional downregulation of RRP22 is only, in part, explained by RRP22 5'‐CpG island hypermethylation (methyl.). NB1‐3, 3 different samples of non‐neoplastic brain tissue; HM‐DNA, in vitro hypermethylated DNA control; LOH, loss of heterozygosity on 22q12 (spanning the RRP22 locus), 1p and 19q as assessed by microsatellite analyses; RET, retention of heterozygosity at all investigated informative loci on these chromosomal arms; NI, not informative at the investigated loci; na, not analyzed. IDH1/2 mutations were assessed by pyrosequencing (wt, wild‐type sequence) and p53 protein expression by semiquantitative immunohistochemistry (0, no immunopositive tumor cells; 1, <10%; 2, 10%–49%; 3, 50%–90% and 4, >90% tumor cells with immunopositive nuclei).
Figure 5.
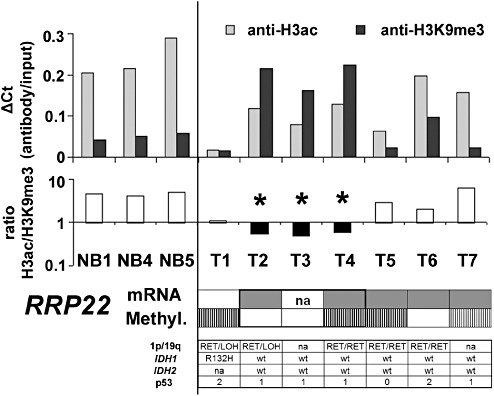
Chromatin immunoprecipitation confirms heterochromatinization of the RRP22 promoter in human glioma tissues. Shown are ΔCt values (antibody/input) for chromatin immunoprecipitation with antibodies against H3ac (associated with euchromatin) and H3K9me3 (associated with heterochromatin) as well as the H3ac/H3K9me3 ratios for three different non‐neoplastic brain tissues (NB1, 4, 5) and seven glioblastomas. The numbers correspond to: T1, GB239; T2, GB1060; T3, GB1061; T4, GB1062; T5, GB1063; T6, GB1064; T7, GB1065. Note that in non‐neoplastic brain tissues, the ratio between anti‐H3ac and anti‐H3K9me3 is invariably exceeding 1, indicating a prevailing euchromatic stage of the H3‐bound promoters. Three glioblastomas (T2, T3 and T4) show a shift toward a heterochromatinization of H3‐bound RRP22 promoter DNA (ratio H3ac/H3K9me3 <1, ▾). One of these three patients has a concomitant RRP22 hypermethylation, while the other two patients lack RRP22 methylation indicating that histone modifications together with promoter hypermethylation or alone contribute to the frequent transcriptional downregulation of RRP22 in human gliomas (color‐code for visualization of mRNA expression and methylation status is the same as introduced in Figure 1). Also shown is the allelic status on 1p and 19q, the mutational status of IDH1 and IDH2 as well as the p53 protein expression score for all seven glioblastomas.
DNA/RNA extraction
Extraction of DNA and RNA from frozen tumor tissue was performed by ultracentrifugation. In brief, tumor samples were homogenized in 6 mL 4 mol/L guanidine isothiocyanate solution. The homogenate was then layered over 4 mL CsCl and ultracentrifuged at 170 000 × g for 16 h. The RNA was recovered as a pellet and dissolved in diethylpyrocarbonate‐treated water containing the RNase inhibitor RNasin (Promega, Madison, WI). The DNA was purified from the CsCl phase using proteinase K digestion followed by phenol/chloroform extraction. Total DNA and RNA extraction from cell culture and peripheral blood leukocytes was performed according to a standard protocol.
Real‐time reverse transcription (RT) PCR analysis
Three micrograms of total RNA from each tumor or cell line were reverse transcribed into cDNA with superscript reverse transcriptase (Invitrogen, Carlsbad, CA). Expression of RRP22 transcripts was determined by SybrGreen‐based real‐time PCR using the StepOnePlus™ sequence detection system (Applied Biosystems, Foster City, CA) as previously described (1). Fold expression changes relative to non‐neoplastic brain tissue were calculated with the ΔΔCT method (11) using ARF1 (ADP‐ribosylation factor 1) as the reference transcript (for primer sequences, see Supporting Table S1).
Microsatellite analysis
Peripheral blood samples were available from 47 of the 70 patients. To investigate the respective tumors for allelic losses spanning the RRP22 locus on 22q12, we employed loss of heterozygosity (LOH) analysis at the following four microsatellite markers [nucleotide (nt) numbering according to the University of California, Santa Cruz (UCSC) genome browser at http://www.genome.ucsc.edu, primers supplied in Supporting Table S1]: D22S1176 (nt 32 226 567–32 226 714) and D22S531 (nt 30 699 219–30 699 650) located telomerically as well as D22S1150 (nt 29 501 326–29 501 658) and D22S689 (nt 28 856 340–28 856 833) located centromerically to RRP22 (nt 29 708 923–29 711 748).
Mutational analysis
Single‐strand conformation polymorphism (SSCP)/heteroduplex analysis was performed to screen for mutations in the RRP22 coding sequences. In brief, PCR products (primer sequences listed in Supporting Table S1) were separated by electrophoresis on 10%–12% non‐denaturing polyacrylamide gels at room temperature and at 4°C. After electrophoresis, the SSCP/heteroduplex band patterns were visualized by silver staining of the gels. In case of aberrant band patterns, PCR products were sequenced using the BigDye Cycle Sequencing Kit and an ABI PRISM 377 semi‐automated DNA sequencer (Applied Biosystems, Foster City, CA).
Methylation analysis using sodium bisulfite sequencing
Sodium bisulfite treatment of 1 µg DNA was performed overnight (16 h) according to a standard protocol (13). PCR fragments were then amplified from sodium bisulfite‐modified DNA using the primers as reported in Supporting Table S1. The amplified fragment of the RRP22 5′‐CpG island covered a total of 36 CpG sites (nt 29 711 182–29 711 416 according to UCSC, CpGs +53 to +88 relative to the transcription start site), and for 24 of these CpG sites overlapped with a region that had been shown to be methylated in glioblastoma cell lines (4). Direct sequencing and semiquantitative calculation of a promoter methylation score were carried out as described (19). In short, the methylation status at each of the analyzed CpG sites was rated using the following scale: 0, completely unmethylated; 1, a weak methylated signal detectable in the sequence; 2, methylated signal approximately equal to unmethylated signal; 3, methylated signal markedly stronger than unmethylated signal. Based on this rating, a cumulative promoter methylation score was calculated for each tumor. Tumors with methylation scores exceeding that of non‐neoplastic brain tissue were regarded as hypermethylated.
5‐Aza‐2'‐deoxycytidine (AZA) and trichostatin A (TSA) treatment of malignant glioma cell lines
Two glioma cell lines (A172 and U87MG) were grown under standard conditions or under two different treatment conditions with either the demethylating agent AZA (1 µM for 72 h, AZA) or the histone deacetylase inhibitor TSA (1 µM for 36 h, TSA). After harvesting the cells and extracting the mRNA, expression of RRP22 transcripts under the different treatment conditions compared with the untreated controls was assessed by real‐time reverse transcription PCR analysis as described earlier (Figure 3). Results obtained were reproduced in at least three independent biological experiments.
Figure 3.
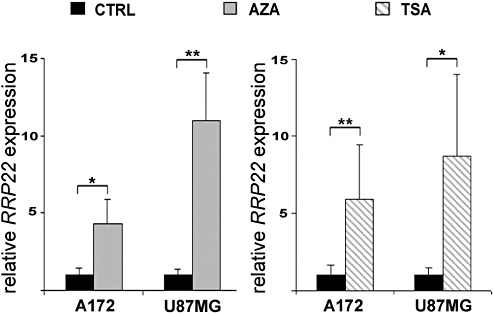
Increased expression of RRP22 transcripts in human malignant glioma cell lines A172 and U87MG after treatment with 5‐aza‐2′‐deoxycytidine (AZA) or trichostatin A (TSA). Real‐time RT‐PCR analysis shows increased expression of RRP22 transcripts relative to the untreated control (CTRL, cells grown under standard conditions) both when applying AZA or TSA. These findings corroborate a causal relationship between RRP22 promoter hypermethylation and decreased mRNA expression but also suggest histone modifications as an additional cause of RRP22 downregulation in glioma cells.
Chromatin immunoprecipitation (ChIP) assays
ChIP was performed from TSA‐treated glioblastoma cells as well as from fresh frozen glioblastoma tissues. DNA and proteins were cross‐linked with 1% formaldehyde for 10 minutes and then resuspended in swelling buffer to isolate nuclei. Purification of nuclei from fresh frozen tissues was accomplished via sucrose gradient ultracentrifugation as described (9). Shortly, frozen tissues were homogenized by douncing in nuclear extraction buffer, nuclei were then fixed by adding 1% formaldehyde for 10 minutes and the tissue homogenates transferred on top of a sucrose cushion and ultracentrifuged at 25 000 rpm for 3 h at 4°C (SW41 Ti Rotor, Beckman Coulter).
Nuclei prepared from cells or tumor tissues were further processed using a commercial ChIP assay kit (Upstate, Charlottesville, VA) according to the manufacturer's recommendations. After resuspension in SDS‐lysis buffer, genomic DNA was sheared to 200–800 bp fragments by sonication with an ultrasonic processor (Vibracell 75022; Novodirect, Kehl, Germany). Sonicated samples were then precleared with protein A agarose/salmon sperm DNA (Upstate) for 30 minutes at 4°C to reduce nonspecific binding. A 5% sample volume was saved as input control while the rest was used for immunoprecipitation at 4°C overnight. In case of cell lines, immunoprecipitation was performed with anti‐H3ac and anti‐H4ac antibodies (Figure 4). In the tumors, we employed antibodies against H3ac and H3K9me3 (Figure 5) for immunoprecipitation. In both assays, rabbit antihuman IgG fraction served as a negative isotype control (all antibodies from Upstate). Antibody/histone/DNA complexes were collected using protein A agarose/salmon sperm DNA (1 h at 4°C) and the histone/DNA complexes were then eluted from the antibodies (2 × 15 minutes. incubation at room temperature in freshly prepared elution buffer). After reversing the histone–DNA crosslink (NaCl for 4–6 h at 65°C), the DNA was extracted by a standard proteinase K digest and subsequent phenol/chloroform extraction.
Figure 4.
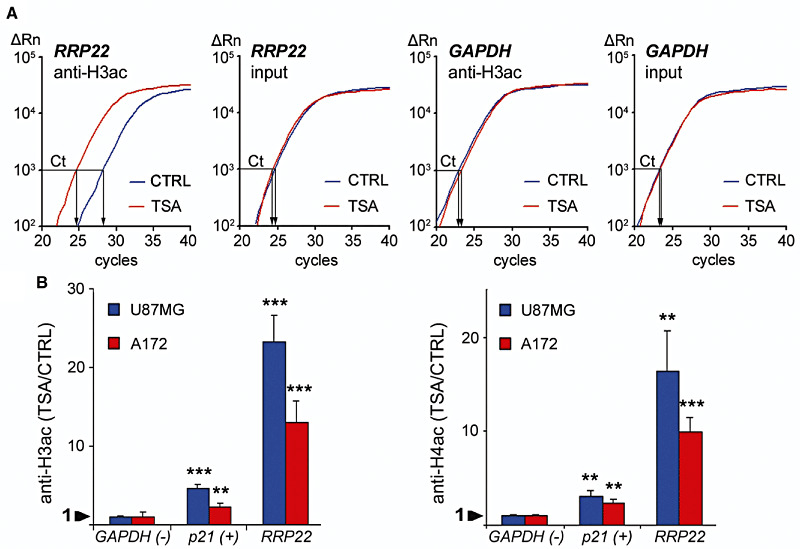
In vitro treatment with the histone deactylase inhibitor trichostatin A (TSA) causes euchromatinization of the RRP22 promoter in the glioblastoma cell lines U87MG and A172. Quantitative real‐time PCR analysis of RRP22 promoter DNA binding to anti‐H3ac and anti‐H4ac after euchromatinizing treatment with TSA. (A) Note that in the glioblastoma cell line, A172 the TSA (red) curve for anti‐H3ac‐bound RRP22 is shifted to the left relative to the CTRL (blue) curve, while the reference curves (input) for TSA and CTRL pass the threshold (Ct) at an approximately equal cycle number. This corresponds to markedly increased RRP22 promoter DNA levels bound to H3ac after treatment with TSA. Abscissa, cycle number; ordinate, relative amount of PCR product. (B) After TSA treatment, an increase of promoter DNA binding of RRP22 to H3ac (left) and H4ac (right) is observed in both cell lines (ratio TSA/CTRL >1). p21, positive control gene (CDKN1A/p21WAF1) known to be inactivated by histone modifications in gliomas; GAPDH, negative control gene not regulated by histone modifications.
Immunoprecipitated DNA was assessed by using real‐time PCR analysis with primers targeting the promoter sequences of RRP22 and normalized to the respective input fraction as a reference. GAPDH was used as a negative control gene associated with euchromatin and not regulated by histone modifications. p21 served as a positive control gene previously shown to be inactivated by histone modifications in human glioblastoma cells (20) (for primers, compare Supporting Table S1). Results obtained from glioblastoma cells were reproduced in three independent biological experiments. For fresh frozen glioblastoma tissues, the results are based on the measurement of three technical replicates caused by the restricted amount of immunoprecipitated DNA. Tricine‐SDS‐PAGE (17) was performed to control for the efficacy of TSA treatment and the specificity of ChIP antibody reactions (data not shown).
Assessment of the prognostic role of RRP22 in the NIH REMBRANDT database
The NIH REMBRANDT database (URL: https://caintegrator.nci.nih.gov/rembrandt/home.do) was used to correlate RRP22 gene expression with tumor malignancy grade as well as patient overall survival (Kaplan–Meier survival plot for RRP22 gene expression in gliomas of different WHO grades as well as glioblastomas of WHO grade IV; Figure 6). Patients were divided into three groups based on the RRP22 expression levels (high, ≥twofold; intermediate, 0.5–twofold; and low, ≤0.5‐fold compared with non‐neoplastic brain tissue) using the means of all probe sets annotated for RRP22 (Figure 6). The log rank P‐value was calculated using the Mantel–Haenszel algorithm and indicated the significance of survival differences between any two groups of samples segregated based on RRP22 gene expression.
Figure 6.
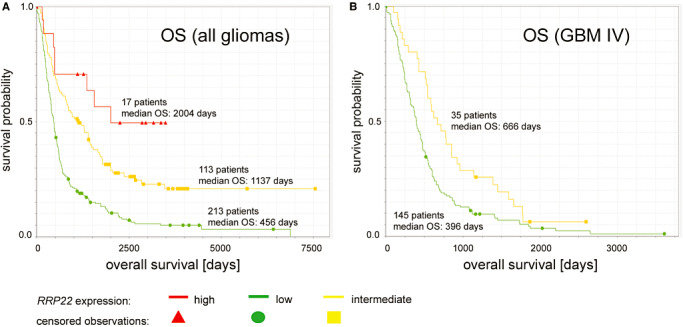
Kaplan–Meier survival plots of glioma patients in the REMBRANDT data set stratified for RRP22 expression levels. (A) Note that overall survival (OS) is positively associated with the level of RRP22 mRNA expression in a group of 343 patients diagnosed with diffusely infiltrating gliomas of different WHO grades. (B) Univariate survival correlation of 180 glioblastoma patients stratified for differences in RRP22 expression levels revealed that higher RRP22 transcripts levels are associated with longer OS. As only a single glioblastoma patient fell into the group of “high RRP22 expression”, for statistical comparisons we only utilized survival curves for low and intermediate RRP22 expression levels.
Assessment of other molecular alterations common in gliomas
Microsatellite analyses were used to assess the allelic status on chromosome arms 1p (microsatellite markers D1S211, D1S468, D1S507, D1S2629) and 19q (microsatellite markers D19S572, D19S1182, D19S210 and D19S219) as reported elsewhere (5). IDH1 and IDH2 mutational status was assessed by pyrosequencing as previously described (7). IDH2 mutations were only assessed in case of the detection of a wild‐type sequence for IDH1. p53 protein expression was assessed by immunohistochemistry (clone DO‐7; Dako, Hamburg, Germany) scoring nuclear staining with the following semiquantitative score: 0, no immunopositive tumor cells; 1, <10%; 2, 10%–49%; 3, 50%–90%; and 4, >90% tumor cells with immunopositive nuclei.
Statistical methods
Two‐sided Student's t‐test analyses were employed to compare RRP22 mRNA expression levels between the different WHO grades and to compare RRP22 mRNA expression levels between tumors with and without RRP22 5′‐CpG island hypermethylation (relative to non‐neoplastic brain tissue). One‐sided Student's t‐test analyses were used to evaluate the increase of RRP22 expression levels in glioblastoma cell lines after treatment with AZA and TSA. One‐sided Student's t‐test analyses were also used to assess the increase of RRP22 immunoprecipitated DNA bound to acetylated histones after TSA treatment. P‐values of <0.05 (*), <0.01 (**) and <0.001 (***) were considered as significant. For the correlation of the RRP22 promoter methylation status with IDH1 and IDH2 mutations as well as with the allelic status on 1p and 19q, we used two‐tailed Fisher's exact test. Correlation between the RRP22 promoter methylation status and the p53 protein expression score was assessed by using Mann–Whitney U‐test analysis.
RESULTS
RRP22 is transcriptionally downregulated in the majority of the investigated gliomas
When investigating a series of 70 glioma patients by means of real‐time reverse transcription PCR analysis, we found decreased RRP22 mRNA levels (<0.7‐fold relative to non‐neoplastic brain tissue) in 47 out of 70 gliomas (67%) (Figure 1). Interestingly, when comparing RRP22 expression levels between the different WHO grades, RRP22 expression decreased with higher malignancy grade. Highest RRP22 expression levels were observed in WHO grade II lesions (mean: 1.7; standard deviation, SD: 1.7), intermediate expression levels in WHO grade III gliomas (mean: 0.8; SD: 1.4) and lowest expression levels in glioblastomas of WHO grade IV (mean: 0.3; SD: 0.8). The expression differences between the grades were statistically significant, particularly when comparing high‐grade (malignant) gliomas (WHO grade III and IV) with low‐grade gliomas of WHO grade II (Student's t‐test, P = 0.003; Figure 2A). All five investigated glioblastoma cell lines (A172, U138MG, T98G, TP365MG and U87MG) exhibited a nearly complete loss of expression (<0.1) of RRP22 transcripts (Figure 1).
Figure 2.
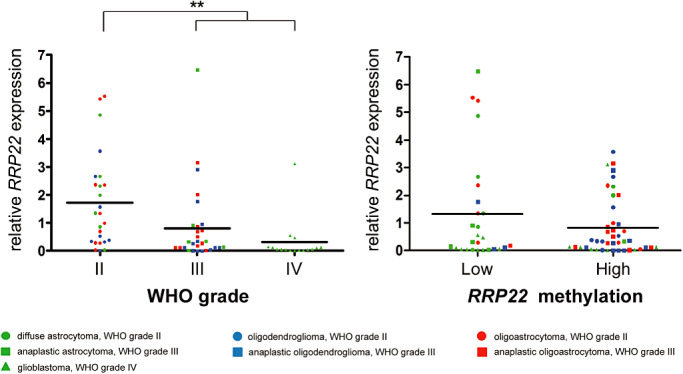
RRP22 mRNA expression in human gliomas stratified for WHO grade and methylation status. Note that RRP22 mRNA expression is significantly decreased in high‐grade (WHO grade III and IV) as compared with low‐grade (WHO grade II) gliomas (A). Tumors with increased RRP22 5'‐CpG island methylation scores (relative to that of non‐neoplastic brain tissue) had a lower mean RRP22 mRNA expression level (0.8) as compared with tumors with absent or low RRP22 methylation (1.3); however, these expression differences missed the significance threshold (B).
Lack of detectable RRP22 mutations and low frequencies of allelic deletions on 22q12
Mutational analysis of the entire coding regions of RRP22 by SSCP/heteroduplex analysis did not detect somatic mutations in any of the 70 gliomas (data not shown). As RRP22 is located in a chromosomal region that is often affected by allelic losses in human tumors, including gliomas (8), we analyzed a set of microsatellite markers spanning the RRP22 locus on 22q12 and identified two patients (GB130, AO87) with allelic losses in the tumor tissue (Figure 1). Both patients with RRP22 allelic deletions exhibited 5′‐CpG island hypermethylation of the second allele (see later) resulting in strongly decreased RRP22 mRNA expression levels of 0.1 relative to non‐neoplastic brain tissue.
Hypermethylation of the RRP22 5'‐CpG island partially accounts for the downregulation of RRP22 expression in human gliomas
Direct bisulfite sequencing revealed RRP22 5′‐CpG island hypermethylation in 43 out of 70 glioma patients (61%) and in all five investigated glioblastoma cell lines. In vitro treatment with the demethylating agent 5‐aza‐2′‐deoxycytidine induced a significant increase of RRPP2 transcripts in two selected glioblastoma cell lines (A172: fourfold, P = 0.013; U87MG: 11‐fold, P = 0.003) relative to untreated control cells (CTRL), thus corroborating a causal relationship between RRP22 5′‐CpG island methylation and mRNA expression (Figure 3). In the primary gliomas, we found lower mean RRP22 mRNA expression levels in tumors with increased RRP22 methylation (mean: 0.8; SD: 1.1) compared with tumors with low or absent RRP22 methylation (mean: 1.3; SD: 2.0). However, these expression differences did not reach statistical significance (Student's t‐test, P = 0.15; Figure 2B) and only 30 out of the 47 gliomas with decreased RRP22 expression demonstrated a concomitant 5′‐CpG island hypermethylation, suggesting that additional mechanisms contribute to the reduced expression of RRP22 in a subgroup of human gliomas.
In vitro treatment of glioblastoma cell lines with the histone deacetylase inhibitor TSA suggests a role for histone modifications in RRP22 downregulation
We then treated two glioblastoma cell lines (A172 and U87MG) with the histone deacetylase inhibitor TSA. Relative to the untreated controls, we found a significant mRNA expression increase for both genes in A172 (RRP22: sixfold, P = 0.004) and U87MG (RRP22: ninefold, P = 0.013) glioblastoma cells (Figure 3). Consequently, we next employed ChIP analyses in the same two cell lines to assess a potential euchromatinization of the RRP22 promoter after TSA treatment. Quantitative real‐time PCR analysis following ChIP analysis revealed a significant increase of RRP22 promoter DNA bound to acetylated H3 (H3ac; 23‐fold for U87MG, P = 0.0002; and 13‐fold for A172, P = 0.0007) and H4 (H4ac; 16‐fold for U87MG, P = 0.002; and 10‐fold for A172, P = 0.0003) when comparing TSA‐treated and untreated cells (Figure 4).
ChIP confirms heterochromatinization of the RRP22 promoter in a fraction of primary glioblastomas
To further address the relevance of histone modifications for the regulation of RRP22 in primary glioblastoma tissues, we optimized our ChIP protocol for the use on tissue samples and prepared the chromatin of three non‐neoplastic brain tissues (NB1, 4 and 5) and seven glioblastomas ( T1–T7, Figure 5). With antibodies against acetylated histone H3 (associated with euchromatin) and histone H3 trimethylated at lysine residue 9 (H3K9me3, which is associated with heterochromatin), we found that in the three non‐neoplastic brain tissues, the ratio between H3ac‐ and H3K9me3‐bound DNA invariably exceeded one, indicating a prevailing euchromatic stage of the H3‐bound RRP22 promoter (Figure 5). A similar euchromatic pattern was observed for the promoter of the control gene GAPDH (data not shown) in the non‐neoplastic tissue samples as well as in all investigated tumors. For RRP22, in contrast, three out of the seven investigated glioblastomas (T2, T3 and T4) exhibited a shift toward heterochromatinization of the H3‐bound RRP22 promoter (ratio H3ac/H3K9me3 <1, Figure 5A, ▾) in comparison with non‐neoplastic brain tissue, thus indicating promoter inactivation by histone modifications. Notably, heterochromatinization of the RRP22 promoter was observed alone (two out of three patients) as well as in conjunction with RRP22 hypermethylation (one out of three patients), and RRP22 mRNA expression was strongly decreased in both of these settings (Figure 5).
Correlation of RRP22 gene expression data to tumor malignancy grade and patient overall survival in the NIH REMBRANDT database
Our experimental findings of a decreased RRP22 expression with higher malignancy grade can be retraced in the NIH REMBRANDT data set (data not shown). More interestingly, correlation of RRP22 expression and patient survival revealed a longer overall survival for patients with increased RRP22 expression (Figure 6). A total of 343 patients with diffusely infiltrating gliomas of different WHO grades were divided into three groups according to RRP22 mRNA expression relative to non‐neoplastic brain tissue (high, intermediate and low RRP22 expression levels). The NIH REMBRANDT database reveals significant differences when comparing the group of tumors with low RRP22 transcript levels (213 patients, median overall survival: 456 days) with tumors with high RRP22 expression (17 patients, median overall survival: 2004 days; log‐rank test: P = 5.95 × 10−5; Figure 6A) as well as with tumors with intermediate RRP22 transcript levels (113 patients; median overall survival: 1137 days; log‐rank test: P = 1.27 × 10−8; Figure 6A). In addition, in a cohort of 180 glioblastoma patients (WHO grade IV), individuals with intermediate RRP22 expression (35 patients; median survival: 666 days) exhibited a significantly better overall survival than patients with low RRP22 expression levels (145 patients; median overall survival: 396 days; log‐rank test: P = 0.005; Figure 6B). As the group of glioblastoma patients with a high RRP22 expression was composed of only a single individual, it was not usable for meaningful statistical comparisons.
Correlation of RRP22 epigenetic inactivation with other molecular alterations common in gliomas
We correlated the RRP22 methylation status in our tumor panel to other common molecular alterations in gliomas, namely IDH1 and IDH2 mutations, allelic losses on chromosome arms 1p and 19q and p53 protein expression (results for the individual tumors are shown in Figure 1). For the microsatellite analyses on 1p and 19q, we were restricted to the patients from whom we had peripheral blood samples available (51 out of 70 patients of our tumor panel). None of the assessed correlations proved statistically significant (RRP22 promoter methylation status vs. IDH1/2 mutations, Fisher's exact test, P = 0.08; vs. 1p deletions, Fisher's exact test, P = 0.21; vs. 19q deletions, Fisher's exact test, P = 0.06; vs. 1p/19q deletions, Fisher's exact test, P = 0.09; vs. p53 protein expression, Mann–Whitney U‐test, P = 0.14). Also, the three out of the seven glioblastomas with histone modifications impacting RRP22 did not exhibit a strikingly unique pattern of associated molecular alterations (Figure 5). Here, statistical analyses were not performed because of the restricted number of tumors analyzed by the ChIP method.
DISCUSSION
The RAS‐related gene RRP22 has been suggested as a tumor suppressor in glioma cells 3, 4, but its relevance and inactivation mechanisms in glioma patient samples had not been fully assessed so far. We performed a comprehensive molecular analysis of RRP22 in a panel of 70 human gliomas and found a frequent reduction of RRP22 (67%) transcript levels. RRP22 mRNA expression levels were significantly decreased in high‐grade (WHO grade III and IV) as compared with low‐grade (WHO grade II) gliomas. These findings are in accordance with a recently published report on RRP22 transcriptional downregulation with increasing WHO grade in astrocytomas (3).
RRP22 methylation analysis by means of direct bisulfite sequencing revealed hypermethylation of the RRP22 5′‐CpG island in a subset of gliomas with decreased RRP22 mRNA expression levels. This finding is in line with a previous report on hypermethylation of RRP22 in selected immortalized glioblastoma cell lines (4). Furthermore, when treating A172 and U87MG glioblastoma cell lines with the demethylating agent 5‐aza‐2′‐deoxycytidine in vitro, we found a significant increase in RRP22 transcript levels, arguing for a causal relationship between RRP22 5′‐CpG island methylation and decreased expression. In our primary tumor panel, there was only a trend toward significance for the correlation between RRP22 methylation status and RRP22 mRNA expression levels. In fact, several RRP22 hypermethylated gliomas did not exhibit reduced RRP22 transcript levels, which might be explained by the often high degree of cellular and molecular heterogeneity of these tumors. Given that RRP22 hypermethylation in such cases may be restricted to a subset of tumor cells, expression might still be sustained from unmethylated tumor cell subpopulations or contaminating non‐neoplastic cell populations, such as, for example, microglial cells. Similar pitfalls in the correlation of methylation and expression data when using lsyate‐base approaches have been described for other hypermethylated genes, such as the O6‐methylguanine‐DNA‐methyltransferase (MGMT) gene (6).
More importantly, however, we found that RRP22 hypermethylation could only explain for the reduced mRNA expression in a subset of tumor samples (30 out of 47 patients), suggesting that mechanisms in addition to CpG island methylation might contribute to the frequent silencing of RRP22 in human gliomas. Our search for tumor‐associated structural genetic alterations yielded only low frequencies of allelic losses at the RRP22 locus on chromosome 22q12.2, while tumor‐associated mutations were absent.
As histone modifications may serve as an important alternative mechanism of gene inactivation 10, 18, we treated glioblastoma cell lines in vitro with the histone deactylase inhibitor TSA, leading to an euchromatinization of gene promoters. Indeed, TSA treatment significantly increased the mRNA expression levels of RRP22, suggesting that histone modifications might contribute to the silencing of RRP22 in gliomas. Chromatin‐immunoprecipitation after TSA treatment corroborated a significant increase of RRP22 promoter DNA bound to the acetylated histones H3 (H3ac) and H4 (H4ac). This finding was confirmed by ChIP in a separate subset of glioblastoma tissue samples. Three out of seven investigated primary glioblastomas showed a shift toward a heterochromatinization of the RRP22 promoter relative to non‐neoplastic brain tissue. This heterochromatinization of RRP22 was found alone or in coincidence with RRP22 5'‐CpG island methylation and in both settings associated with a marked downregulation of RRP22 transcripts. Thus, histone modifications might serve as an additional cause of RRP22 inactivation and either alone or in conjunction with RRP22 5'‐CpG island hypermethylation account for the gene's frequent transcriptional downregulation in human gliomas.
RRP22 epigenetic inactivation did not reveal a significant association to other molecular alterations common in gliomas, namely IDH1/2 mutations, allelic losses on 1p and 19q as well as p53 protein accumulation. This does not appear surprising given the fact that RRP22 epigenetic inactivation neither spares or prefers primary glioblastomas nor occurs strikingly more often in either oligodendroglial or astrocytic neoplasms. Gliomas with RRP22 epigenetic inactivation by promoter hypermethylation or histone modifications are thus not associated with a distinct group of gliomas characterized by one of the three investigated “classic” molecular alterations (IDH1/2 mutation, 1p/19q deletion, p53 protein expression).
Publicly available data from the NIH REMBRANDT database are in accordance with our finding that RRP22 expression decreases with an increasing malignancy grade of gliomas. More interestingly, Kaplan–Meier survival plots indicate a worse overall survival for patients whose tumors demonstrate low RRP22 expression levels, both in a group of glioma patients of different WHO grades as well as in glioblastoma patients alone. These data strongly suggest that RRP22 inactivation represents a negative prognostic factor in glioma patients.
In summary, we here present the first study systematically assessing the molecular aberrations and inactivation mechanisms of RRP22 in primary human tumors. We uncovered that RRP22 5′‐CpG island hypermethylation and/or histone modifications may underlie the frequent and prognostically unfavorable transcriptional downregulation of the RRP22 gene in human gliomas.
CONFLICT OF INTEREST STATEMENT
The authors have no conflict of interest to declare.
Supporting information
Table S1. Primers used for expression, mutation, methylation and ChIP analysis of RRP22 and microsatellite analyses of the 22q12 locus spanning RRP22.
Supporting info item
ACKNOWLEDGMENTS
The authors would like to thank Nina Graffmann (Institute for Transplantation Diagnostics and Cell Therapeutics, Düsseldorf), Dr. Melanie Ruppel (German Cancer Research Center, Heidelberg) and Dr. Daniela Karra (Department of Neuropathology, Düsseldorf) for their valuable support in establishing the ChIP‐Assays. Britta Friedensdorf is acknowledged for her skillful technical assistance and Joerg Felsberg for his support in scoring the p53 immunostains. This work was financially supported by grants from the German Cancer Aid (Max‐Eder Junior Research Group Program, grant no. 107709 and 109426), the Research Commission of the Medical Faculty of the Heinrich Heine University Düsseldorf (grant no. 9772307) and the Academy of Sciences of Northrhine‐Westfalia/Mercator Foundation (“Junges Kolleg”) (all to Markus J. Riemenschneider).
REFERENCES
- 1. Barski D, Wolter M, Reifenberger G, Riemenschneider MJ (2010) Hypermethylation and transcriptional downregulation of the TIMP3 gene is associated with allelic loss on 22q12.3 and malignancy in meningiomas. Brain Pathol 20:623–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bos JL (1989) Ras oncogenes in human cancer: a review. Cancer Res 49:4682–4689. [PubMed] [Google Scholar]
- 3. Chen R, Yang L, Fang J, Huo L, Zhang M, Chen F et al (2011) RRP22: a novel neural tumor suppressor for astrocytoma. Med Oncol 2011 Jan 25. [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 4. Elam C, Hesson L, Vos MD, Eckfeld K, Ellis CA, Bell A et al (2005) RRP22 is a farnesylated, nucleolar, Ras‐related protein with tumor suppressor potential. Cancer Res 65:3117–3125. [DOI] [PubMed] [Google Scholar]
- 5. Felsberg J, Erkwoh A, Sabel MC, Kirsch L, Fimmers R, Blaschke B et al (2004) Oligodendroglial tumors: refinement of candidate regions on chromosome arm 1p and correlation of 1p/19q status with survival. Brain Pathol 14:121–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Felsberg J, Rapp M, Loeser S, Fimmers R, Stummer W, Goeppert M et al (2009) Prognostic significance of molecular markers and extent of resection in primary glioblastoma patients. Clin Cancer Res 15:6683–6693. [DOI] [PubMed] [Google Scholar]
- 7. Felsberg J, Wolter M, Seul H, Friedensdorf B, Goppert M, Sabel MC, Reifenberger G (2010) Rapid and sensitive assessment of the IDH1 and IDH2 mutation status in cerebral gliomas based on DNA pyrosequencing. Acta Neuropathol 119:501–507. [DOI] [PubMed] [Google Scholar]
- 8. Ino Y, Silver JS, Blazejewski L, Nishikawa R, Matsutani M, von Deimling A, Louis DN (1999) Common regions of deletion on chromosome 22q12.3‐q13.1 and 22q13.2 in human astrocytomas appear related to malignancy grade. J Neuropathol Exp Neurol 58:881–885. [DOI] [PubMed] [Google Scholar]
- 9. Jiang Y, Matevossian A, Huang HS, Straubhaar J, Akbarian S (2008) Isolation of neuronal chromatin from brain tissue. BMC Neurosci 9:42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kouzarides T (2007) Chromatin modifications and their function. Cell 128:693–705. [DOI] [PubMed] [Google Scholar]
- 11. Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real‐time quantitative PCR and the 2(‐delta delta C(T)) method. Methods 25:402–408. [DOI] [PubMed] [Google Scholar]
- 12. Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Burger PC, Jouvet A et al (2007) The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol 114:97–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mueller W, Nutt CL, Ehrich M, Riemenschneider MJ, von Deimling A, van den Boom D, Louis DN (2007) Downregulation of RUNX3 and TES by hypermethylation in glioblastoma. Oncogene 26:583–593. [DOI] [PubMed] [Google Scholar]
- 14. Newton HB (2003) Molecular neuro‐oncology and development of targeted therapeutic strategies for brain tumors. Part 1: growth factor and Ras signaling pathways. Expert Rev Anticancer Ther 3:595–614. [DOI] [PubMed] [Google Scholar]
- 15. Riemenschneider MJ, Reifenberger J, Reifenberger G (2008) Frequent biallelic inactivation and transcriptional silencing of the DIRAS3 gene at 1p31 in oligodendroglial tumors with 1p loss. Int J Cancer 122:2503–2510. [DOI] [PubMed] [Google Scholar]
- 16. Sayagues JM, Tabernero MD, Maillo A (2007) [Cytogenetic alterations in meningioma tumors and their impact on disease outcome]. Med Clin (Barc) 128:226–232. [DOI] [PubMed] [Google Scholar]
- 17. Schagger H (2006) Tricine‐SDS‐PAGE. Nat Protoc 1:16–22. [DOI] [PubMed] [Google Scholar]
- 18. Schones DE, Zhao K (2008) Genome‐wide approaches to studying chromatin modifications. Nat Rev Genet 9:179–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tepel M, Roerig P, Wolter M, Gutmann DH, Perry A, Reifenberger G, Riemenschneider MJ (2008) Frequent promoter hypermethylation and transcriptional downregulation of the NDRG2 gene at 14q11.2 in primary glioblastoma. Int J Cancer 123:2080–2086. [DOI] [PubMed] [Google Scholar]
- 20. Yin D, Ong JM, Hu J, Desmond JC, Kawamata N, Konda BM et al (2007) Suberoylanilide hydroxamic acid, a histone deacetylase inhibitor: effects on gene expression and growth of glioma cells in vitro and in vivo . Clin Cancer Res 13:1045–1052. [DOI] [PubMed] [Google Scholar]
- 21. Zucman‐Rossi J, Legoix P, Thomas G (1996) Identification of new members of the Gas2 and Ras families in the 22q12 chromosome region. Genomics 38:247–254. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Primers used for expression, mutation, methylation and ChIP analysis of RRP22 and microsatellite analyses of the 22q12 locus spanning RRP22.
Supporting info item