

Abstract
Progressive supranuclear palsy (PSP) is a four‐repeat tauopathy with tau‐positive, argyrophilic tuft‐shaped astrocytes (TAs). We performed a pathological and clinical investigation in 40 consecutive autopsied Japanese patients with pathological diagnoses of PSP or PSP‐like disease. Unequivocal TAs were present in 22 cases, all of which were confirmed to be PSP. Such TAs were hardly detected in the other 18 cases, which instead exhibited tau‐positive, argyrophilic astrocytes, appearing as comparatively small clusters with central nuclei of irregularly shaped, coarse structures (equivocal TAs). Cluster analysis of the distribution pattern of tau‐related pathology for these 18 cases identified two subgroups, pallido‐nigro‐luysian atrophy (PNLA) Type 1 (n = 9) and Type 2 (n = 9), the former being distinguished from the latter by the presence of tau‐related lesions in the motor cortex, pontine nucleus and cerebellar dentate nucleus in addition to the severely affected PNL system. The duration from symptom onset until becoming wheelchair‐bound was significantly longer in PNLA Type 1. Immunoblotting of samples from the three disease conditions revealed band patterns of low‐molecular‐mass tau fragments at ∼35 kDa. These findings shed further light on the wide pathological and clinical spectrum of four‐repeat tauopathy, representing PSP in the broad sense rather than classical PSP.
Keywords: glial tau pathology, pallido‐nigro‐luysian atrophy, progressive supranuclear palsy, tauopathy, tuft‐shaped astrocyte
Introduction
Progressive supranuclear palsy (PSP), which was originally described by Steele, Richardson and Olszewski in 1964 44, is a rare clinical syndrome comprising supranuclear gaze palsy, severe postural instability leading to falls, axial rigidity, and cognitive dysfunction 26, 27. PSP is known to be a representative four‐repeat (4R) tauopathy, showing widespread degeneration with tau‐positive neurofibrillary and glial tangles: the pathologic hallmark of the disease is the presence of the so‐called tuft‐shaped (or tufted) astrocytes (TAs) 8, 23, 50, 57. From a clinical viewpoint, PSP can be classified into several types: PSP‐Richardson's syndrome (PSP‐RS) 51, PSP‐Parkinsonism (PSP‐P) 51, PSP‐pure akinesia with gait freezing (PSP‐PAGF) 52, PSP‐cerebellum (PSP‐C) 21, PSP‐corticobasal syndrome (PSP‐CBS) 25 and PSP‐frontotemporal dementia (PSP‐FTD) 20, 22. However, cases representing new clinicopathological variants of PSP have also been reported in recent years 14, 18, 19.
In Japan, it has long been known that some cases exhibiting clinical features similar to those of PSP also show severe systemic degeneration involving the globus pallidus, substantia nigra and subthalamic nucleus 45, the neuropathology corresponding to that of pallido‐nigro‐luysian atrophy (PNLA) first described by Contamin et al 6. Autopsy reports of pure akinesia 17 have suggested that PSP and PNLA might be part of a disease spectrum 29, 56. Shimoda et al studied three cases of PNLA and conducted a literature review of 11 similar cases reported previously 40. They concluded that PNLA, in which the cardinal symptom was predominantly or purely akinesia, was characterized by a comparatively smaller number of argyrophilic, tau‐positive neurofibrillary and glial tangles in limited brain regions, suggesting that the disease can be differentiated pathologically from PSP 40. These observations were followed by two case reports of PNLA in which no convincing tau‐positive or argyrophilic TAs characteristics of PSP were evident 24, 30.
Recently, among a pathological series of over 400 cases of PSP, Ahmed et al reported that eight cases showed features similar to those of PNLA; pathological examination revealed severe degeneration of the PNL system with apparently less tau‐related pathology in the motor cortex, striatum, pontine nuclei and cerebellum 1. Consequently, these cases were referred to as PSP‐PNLA and considered to be a variant of PSP 1. Importantly, with reference to observations by Nishimura et al 31, it was pointed out that all of these cases showed TAs in the caudate nucleus 1. Unfortunately, no mention was made of the morphology of the TAs in PSP‐PNLA, and no illustrations of the TAs were provided.
In the light of the reports mentioned earlier, we re‐examined autopsy cases that had been diagnosed as PSP or PSP‐like disease, and registered at our institution. All of the autopsied patients were Japanese. Our aim in this study was to clarify the pathological and clinical features of PSP and PSP‐like disease, and discuss possible overlap between the two conditions, paying special attention to the morphologic features of the astrocytic tau pathology.
Materials and Methods
Subjects
Using our institutional database of autopsy cases, we selected 50 consecutive cases that had been diagnosed as PSP or PSP‐like disease. All of the cases were from our institutional files covering the period between 1983 and 2013, and included four cases that had been referred from Sagamihara National Hospital.
In each case, we confirmed the pathologic diagnosis and the absence of a family history of parkinsonism or other neurological disease by reviewing the autopsy records. PSP‐like cases included no examples of corticobasal degeneration (CBD), either typical 48 or atypical 39, 46, or globular glial tauopathies 11 we have experienced so far. In 10 of the 50 cases, pathologic features indicative of complications arising from other neurodegenerative diseases were evident (7 of the 10 patients also had Parkinson's disease; PD), and therefore, these 10 cases were excluded from the present study, leaving 40 cases for analysis. The final clinical diagnosis included PSP/PSP syndrome (n = 28), atypical parkinsonism (n = 5), CBD/CB syndrome (n = 3), spinocerebellar degeneration (n = 2) and multiple system atrophy (n = 2).
Neuropathological study
Multiple formalin‐fixed, paraffin‐embedded tissue blocks for all cases were available for the present study. Histological examinations were performed on newly prepared 4‐μm‐thick sections cut from various regions of the brain using two stains: hematoxylin–eosin (H‐E) and Klüver–Barrera (K‐B). All of the prepared sections were immunostained with a mouse monoclonal antibody against hyperphosphorylated tau (clone AT8, Innogenetics, Ghent, Belgium; 1:200). Selected sections were also immunostained with a mouse monoclonal antibody against phospho‐PHF‐tau pThr212/pSer214 (AT100, Thermo Scientific, Cergy Pontoise, France; 1:200) and a rabbit polyclonal antibody against phospho‐tau pThr212 (T212, Abcam plc, Cambridge, UK; 1:50; autoclave at 120°C in 0.05 M citrate buffer for 10 minutes). Immunolabeling was performed using the avidin‐biotin‐peroxidase complex (ABC) method with a Vectastain ABC kit (Vector, Burlingame, CA, USA), and visualized with diaminobenzidine (DAB) as the chromogen. In addition, Gallyas–Braak silver impregnation (G‐B) was carried out to reveal the presence of argyrophilia in tau‐related pathology.
For all the anatomical regions examined, sections immunostained with AT8 and stained with G‐B were used for morphological assessment of astrocytic tau pathology and those stained with H‐E and K‐B were used for semiquantitative analysis (0, none; 1, mild; 2, moderate; 3, severe) of neuronal cell loss. The distribution and severity of tau‐related pathology (neuronal tangles/pretangles, neuropil threads and glial tangles) were also similarly assessed (0, absent; 1, sparse; 2, moderate; 3, numerous) using AT8‐immunostained sections (Figure 1). Moreover, the occurrence of axonal spheroids was similarly assessed (0, absent; 1, sparse; 2, moderate; 3, numerous) in the globus pallidus and substantia nigra using the H‐E‐stained sections. When necessary, sections immunostained with a mouse monoclonal antibody against phosphorylated neurofilament protein (SMI31, Sternberger Monoclonals, Baltimore, MD, USA; 1:10 000) were also used. All studies were carried out by one of the authors (Y.Y.), and reviewed by the other investigators (Y.T. and H.T.) to ensure evaluation consistency.
Figure 1.
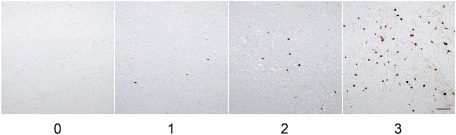
Grading scale for tau‐related pathology. The occurrence of tau‐related lesions was assessed using a 4‐point rating scale: 0, absent; 1, sparse; 2, moderate; 3, numerous. Pontine nuclei immunostained with AT8. Bar = 200 μm.
After assessment of astrocytic tau pathology [the presence of unequivocal TAs (uTAs) and equivocal TAs (eTAs) and the possible morphological distinction between the two, as described below], further observation was conducted on the following items.
Three‐dimensional structural analysis of astrocytic tau pathology
Three‐dimensional (3D) structural analysis of astrocytic tau pathology was performed using formalin‐fixed tissues from the putamen of cases showing uTAs and eTAs (three each). Multiple consecutive sections, approximately 50 μm thick, were cut from the agarose‐embedded tissues using a vibratome and then immunostained with AT8 (1:100). The second antibody used was Alexa Fluor 488 goat anti‐mouse IgG (Molecular Probes, Eugene, OR, USA; 1:1000). Nuclei were stained with 4,6‐diamidino‐2‐phenylindole (DAPI). The sections were then treated with Autofluorescence Eliminator Reagent (Millipore, Billerica, MA, USA).
Confocal images were collected using a laser‐scanning microscope (LSM700, Carl Zeiss Co., Ltd., Oberkochen, Germany). 3D images were then prepared using LSM software (ZEN 2011): every image was collected in the same field of vision of the LSM700. For comparison of morphometrical differences between uTAs and eTAs, 10 AT8‐positive astrocytes in each case were analyzed using IMARIS (Carl Zeiss Co., Ltd.).
Ultrastructural study of astrocytic tau pathology
Based on the features of the astrocytic tau pathology, the study cases were divided into uTA and eTA groups, from which two cases each were selected for conventional and immunoelectron microscopy studies. Formalin‐fixed small tissue blocks of the precentral cortex and putamen were post‐fixed with 1% osmium tetroxide, dehydrated through a graded ethanol series and embedded in Epon 812. Ultrathin sections were then cut and stained with uranyl acetate and lead citrate. For pre‐embedded immunoelectron microscopy, 50‐μm‐thick sections were cut from agarose‐embedded tissue blocks of the putamen using a vibratome, immunostained with AT8 (1:100) using DAB as the chromogen, embedded in Epon 812, and then prepared into ultrathin sections. These sections, starting with those for immunoelectron microscopy, were examined with a Hitachi (Tokyo, Japan) HT7700 electron microscope at 75 kV.
Reliability of case classification based on the two types of astrocytic tau pathology
To confirm the reliability of the morphological distinction between the two types of astrocytic tau pathology, ie, uTAs and eTAs, 4‐μm‐thick sections of the neostriatum (caudate nucleus and putamen) and motor cortex from each case were prepared and subjected to G‐B staining. Two experienced neurologists with more than 4 years of neuropathological training reviewed these specimens in a blinded manner and independently classified the examined cases into two groups (uTA and eTA groups).
Cluster analysis of the tau‐related lesion distribution pattern
Analytical estimation of the tau‐related lesion distribution pattern was performed in cases belonging to the eTA group (a total of 18 cases, as described below). In each case, tau‐related lesions in various regions of the brain were recorded as absent (0) or present 3. The presence of neuropil threads in each region was not taken into consideration; predominant occurrence of neuropil threads without neuronal or glial tangles in each region was not a feature in individual cases. A dataset of pathological findings for tau‐related lesions, each coded as “0” or “1,” was obtained from the 18 cases. Briefly, Ward's method, hierarchical cluster analysis using squared Euclidean distance measures, was performed to investigate how the cases were classified into subgroups dependent on the distribution of tau‐related lesions. Cluster analysis was performed using IBM SPSS Statistics version 20.0 (IBM, Chicago, IL, USA).
The results obtained eventually allowed us to classify the examined cases into three pathological subtypes (PSP, and PNLA Type 1 and Type 2), as described below.
Clinical analysis
Clinical features were analyzed retrospectively in the three groups—PSP (n = 22), and PNLA Type 1 (n = 9) and Type 2 (n = 9)—by reviewing the available medical records. In each case, the presence or absence of major clinical symptoms of PSP 21, 51, as well as cognitive or psychiatric dysfunction, within the initial 2 years of the disease course and the entire disease course, was examined carefully.
Statistical analysis
The data from the 3D images of the two groups (uTA and eTA) were analyzed using the Mann–Whitney test. Pathological (neuronal loss and tau‐related pathology) and demographic (age at onset/death and disease duration) data for cases from the three groups were analyzed using Kruskal–Wallis test with post‐hoc Dunn's multiple comparison test. An inter‐rater agreement statistic (Cohen's Kappa) was calculated to evaluate the agreement between the two pathological diagnoses of the uTA and eTA groups. To compare the proportions of clinical (gender, diagnosis and clinical symptoms) data, Ryan's multiple comparison test was used to detect differences. In addition, for comparison of overall survival and the duration from symptom onset until becoming wheelchair‐bound, the Kaplan–Meier method and log‐rank test were used to detect differences. These statistical analyses were performed using GraphPad Prism version 5.0 (GraphPad Software, San Diego, CA, USA), IBM SPSS Statistics version 20.0 (IBM) and R version 3.1.2 (http://www.r‐project.org/). Differences were considered statistically significant at P < 0.05.
Biochemical analysis of tau
Sarkosyl‐insoluble, guanidine‐soluble fractions were extracted from post‐mortem frozen tissue of the globus pallidus from PSP (n = 2), and PNLA Type 1 (n = 5) and Type 2 (n = 2) cases, as well as from CBD and normal control cases (one each), as described previously 11. The samples before and after dephosphorylation and a mixture of recombinant human tau containing the six isoforms were subjected to sodium dodecyl sulfate–polyacrylamide gel electrophoresis. Sarkosyl‐insoluble tau was visualized using a phosphorylation‐independent anti‐tau antibody (T46, Zymed, South San Francisco, CA, USA; 1:2000), which recognizes the C‐terminal regions of human tau.
Sequencing of the MAPT gene
We selected five patients aged less than 60 years at disease onset (PSP, n = 2; PNLA Type 1, n = 2; PNLA Type 2, n = 1) for genetic study. Genomic DNAs were extracted from frozen samples of the cerebral cortex. Mutation analysis for MAPT genes was carried out in all exons (one non‐coding and 14 coding exons), as described previously 16. Unfortunately, one patient with PNLA Type 2, who was 43 years old—the youngest patient—at disease onset, and who had a disease duration of 15 years, was not included because no frozen tissue samples were available for analysis.
Results
Morphology of astrocytic tau pathology
TAs have been recognized to be a characteristic feature of PSP 8, 11, 24, 33, 50, 57. With regard to tau‐related pathology, we first recognized two types of astrocytic AT8‐positive tau pathology, ie, unequivocal TA (uTA) (11, see Figure 2) and an unusual type of astrocyte, designated “equivocal TA (eTA)” here for convenience. In comparison with uTA (Figure 2A,C), eTA was apparently small in size and was characterized by irregularly shaped, coarse deposits of tau mainly in the perikarya (Figure 2B); the profiles were also reproduced with G‐B staining (Figure 2D). uTAs was observed in 22 cases, in which eTAs were not a feature. eTAs were observed in the other 18 cases, in which uTAs were hardly detected.
Figure 2.
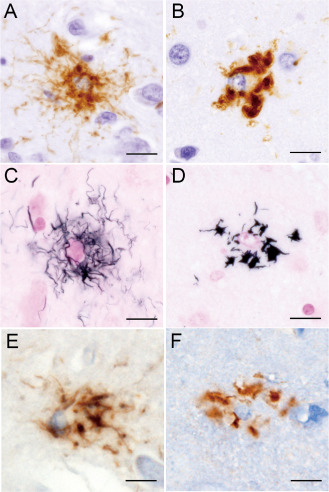
A–D. Tau‐positive, argyrophilic unequivocal tuft‐shaped (uTA) and equivocal tuft‐shaped (eTA) astrocytes. In uTAs, tau appears to have accumulated mainly in the cell processes, showing a radiating, fine, long curvilinear pattern of tau positivity (A) and argyrophilia (C). On the other hand, in eTAs, tau appears to have accumulated mainly in the perikarya, showing a thick and coarse pattern of tau positivity (B) and argyrophilia (D). Note that uTAs (A, C) show larger expanses of tau immunoreactivity and argyrophilia than eTAs (B, D). UTAs (E) and eTAs (F) are also recognizable with another phosphorylation‐dependent anti‐tau antibody. (A, B) AT8 immunostaining; (C, D) Gallyas–Braak staining. (E, F) T212 immunostaining. Bars = 10 μm in A–F.
T212 immunostaining also depicted these two types of astrocytic tau pathology (Figure 2E,F). However, AT100 immunostaining revealed an apparently smaller number of neuronal and glial tau lesions than AT8 immunostaining, showing occasional astrocytic tau lesions, which were hardly recognizable as uTAs or eTAs (data not shown).
3D reconstruction of astrocytic tau lesions revealed that AT8‐positve uTAs and eTAs were aggregates of numerous particles: the former showed round clusters of fine and radiating long process‐like structures (Figure 3A,C) and the latter were also seen to be round clusters, but were smaller and composed of coarse, thicker and shorter structures (Figure 3B,D). The total volumes of the AT8‐positive uTAs and eTAs in the same voxels measured by IMARIS were significantly larger in uTAs than in eTAs (Figure 3E). Furthermore, there were significant differences in the total numbers of particles and the mean volume per particle between uTAs and eTAs: in other words, uTAs were composed of a larger number of smaller particles, whereas eTAs were composed of a smaller number of larger particles (Figure 3E).
Figure 3.
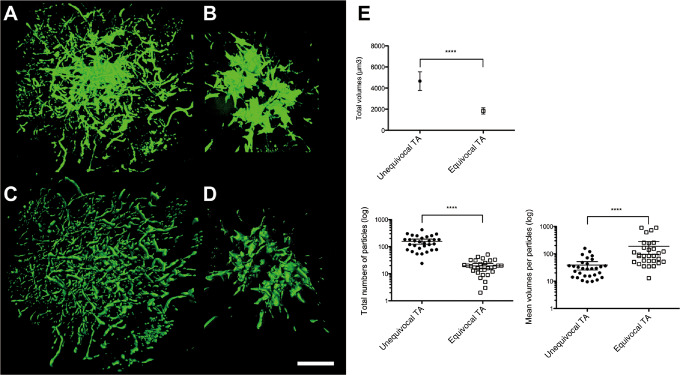
A–D. Three‐dimensional (3D) reconstruction of the astrocytic tau pathology: a AT8‐positive uTA (A, C) and an AT8‐positive eTA (B, D). 3D images obtained by LSM software (in the ZEN transparent mode) (A, B) and by IMARIS software based on fluorescence intensity for morphometric analysis (C, D). E. Morphometric analysis using IMARIS indicated significant differences in the volumes, the total numbers of particles and the mean volumes per particle between uTAs and eTAs (****P < 0.0001). The values shown as vertical lines indicate the mean with 95% confidence interval (CI). Note that with regard to each parameter, there is no overlap in the 95% CI between uTAs and eTAs.
Ultrastructurally, uTAs were recognized as central nuclei surrounded by radiating fine processes containing AT8‐positive products (Figure 4A,C) 5. eTAs were also shown to have central nuclei, but coarse masses of AT8‐positive products were present mainly in the perikarya (Figure 4B,D). Under conventional electron microscopy, occasional bundles of tubules were found in all cases examined. These tubules were mostly straight in appearance and approximately 10–13 nm in diameter (Figure 4E,F). Our impression was that in comparison to uTAs (Figure 4E), eTAs tended to show bundles of tubules that were arranged in a looser manner (Figure 4F).
Figure 4.
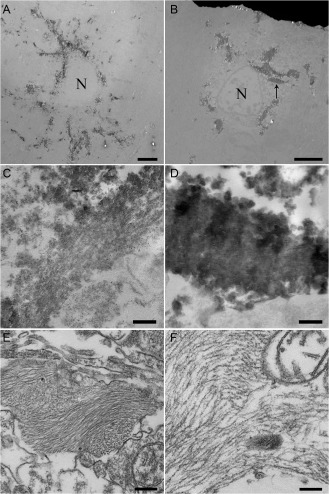
A–D. Pre‐embedding immunoelectron microscopy with AT8. In an uTA, AT8‐positive products can be seen as radiating slender structures extending from the small perinuclear area (A); in an area of another TA at higher magnification, although indistinct, a bundle of straight tubule‐like structures can be seen (C). In an eTA, AT8‐positive products are evident as thick and coarse structures located mainly in the relatively large cytoplasm (B); higher magnification view of the area indicated by the arrow in B, showing an aggregate of AT8‐positive products; similar straight tubule‐like structures are dimly evident (D). E, F. Conventional electron microscopy shows bundles of tubular structures in the cell process of an uTA (E) and the cell body of an eTA (F). (A–F) Putamen. Bars: (A, B) 5 μm; (C, D) 500 nm; (E, F) 200 nm.
Neuropathological classification of cases
Based on the astrocytic tau pathology with argyrophilia, we were able to classify these 40 cases into two groups, ie, uTA (n = 22, 13 males and 9 females) and eTA (n = 18, 8 males and 10 females). With regard to the classification of the examined cases into uTA and eTA groups, two raters agreed in 92.5% of cases (Kappa = 0.84). This result confirmed that inter‐rater reproducibility was excellent.
All cases belonging to the uTA group (22/22) had pathologically definite PSP, also showing tau‐related pathology in many brain regions 8, 9. On the other hand, all cases belonging to the eTA group showed severe PNLA‐type systemic degeneration and comparatively less conspicuous tau pathology in the affected brains 24, 30, 40.
Accordingly, we performed cluster analysis of tau‐related lesion distribution in these 18 cases belonging to the eTA group. This identified two subgroups designated as PNLA Type 1 (n = 9) and Type 2 (n = 9) (Figure 5). PNLA Type 1 was distinguished from PNLA Type 2 by the presence of tau‐related lesions in the motor and frontal cortices, pontine nucleus and cerebellar dentate nucleus (Figure 6). Therefore, we eventually classified with certainty these 40 cases into three groups, namely PSP, and PNLA Type 1 and Type 2.
Figure 5.
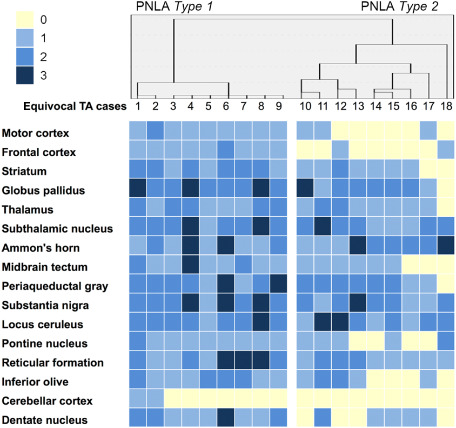
Cluster analysis and distribution of AT8‐positive tau lesions in 18 cases belonging to the eTA group. Dendrogram of cluster analysis of the distribution of AT8‐positive tau lesions (upper panel). Distribution of AT8‐positive tau lesions (lower panel). The severity is also shown in each case and in each region.
Figure 6.
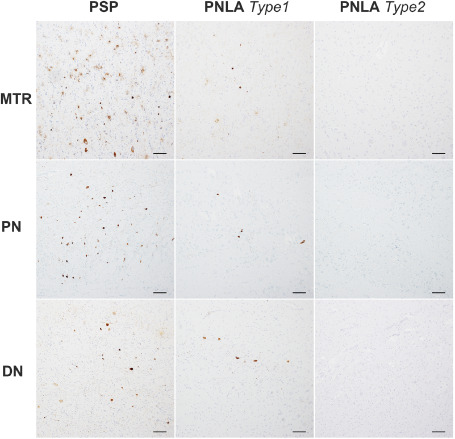
Representative densities of AT8‐positive tau lesions seen in the motor cortex (MTR), pontine nuclei (PN) and cerebellar dentate nucleus (DN) in PSP, and PNLA Type 1 and Type 2 cases. In these regions, the scores for tau‐related pathology were generally 2–3 in PSP, 1–2 in PNLA Type 1 and 0–1 in PNLA Type 2. Bars = 100 μm.
Retrospectively, the 3D structural analysis of astrocytic tau pathology included PSP (n = 3), and PNLA Type 1 (n = 1) and Type 2 (n = 2) cases and the ultrastructural study also included PSP (n = 2), and PNLA Type 1 and Type 2 cases (one each).
Neuropathological features
There was no significant difference in brain weight among the three groups: PSP (n = 22, 1106 ± 151.1 g), and PNLA Type 1 (n = 9, 1120 ± 139.2 g) and Type 2 (n = 9, 1222 ± 127.8 g). The regional severities of neuronal loss and tau‐related pathology in the three pathological phenotypes are summarized in Table 1. When statistical comparisons were made between the three disease conditions, significant differences in neuronal loss and tau‐related pathology were evident in several brain regions, including the frontal and motor cortices, subthalamic nucleus, midbrain tectum, pontine nucleus and cerebellar dentate nucleus (Table 1).
Table 1.
Neuropathological findings in the PSP and PNLA Type 1 and Type 2 groups. Abbreviation: TA = tuft‐shaped astrocyte
| Regions | Neuronal loss (mean ± SD) | Tau‐related pathology (mean ± SD) | ||||
|---|---|---|---|---|---|---|
| PSP (n = 22) | PNLA type 1 (n = 9) | PNLA type 2 (n = 9) | PSP (n = 22) | PNLA type 1 (n = 9) | PNLA type 2 (n = 9) | |
| Motor cortex | 1.36 ± 0.90 | 0.67 ± 0.71 | 0.22 ± 0.44* | 2.19 ± 0.75 | 1.00 ± 0.50* | 0.44 ± 0.53** |
| Frontal cortex | 1.91 ± 1.02 | 1.22 ± 1.09 | 0.78 ± 0.67* | 2.10 ± 0.70 | 1.00 ± 0.50* | 0.33 ± 0.50*** |
| Striatum | 0.73 ± 0.77 | 0.67 ± 0.50 | 0.11 ± 0.33 | 1.64 ± 0.66 | 0.44 ± 0.73 | 1.11 ± 0.60 |
| Globus pallidus | 2.27 ± 0.70 | 2.56 ± 0.53 | 2.44 ± 1.01 | 2.32 ± 0.57 | 1.89 ± 0.93 | 2.11 ± 0.60 |
| Thalamus | 1.14 ± 0.56 | 0.67 ± 0.50 | 0.56 ± 0.73 | 1.68 ± 0.57 | 1.44 ± 0.73 | 1.22 ± 0.44 |
| Subthalamic nucleus | 1.91 ± 0.61 | 2.78 ± 0.44** | 2.56 ± 0.53* | 2.14 ± 0.71 | 2.22 ± 0.44 | 1.67 ± 0.71 |
| Ammon's horn | 1.18 ± 0.91 | 2.00 ± 1.89 | 1.89 ± 0.93 | 1.75 ± 0.85 | 1.89 ± 0.93 | 1.67 ± 0.71 |
| Midbrain tectum | 1.74 ± 0.92 | 1.33 ± 0.71 | 0.67 ± 0.71** | 2.09 ± 0.75 | 1.33 ± 0.87 | 0.75 ± 0.46* |
| Periaqueductal gray | 1.52 ± 0.95 | 1.67 ± 0.71 | 0.89 ± 0.60 | 1.95 ± 0.72 | 1.89 ± 0.93 | 1.67 ± 0.50 |
| Substantia nigra | 2.14 ± 0.83 | 2.56 ± 0.73 | 2.78 ± 0.44 | 2.46 ± 0.51 | 2.11 ± 0.60 | 1.89 ± 0.78 |
| Locus ceruleus | 1.73 ± 0.77 | 1.33 ± 0.50 | 1.11 ± 0.33* | 2.23 ± 0.69 | 2.11 ± 0.33 | 1.78 ± 0.83 |
| Pontine nucleus | 1.18 ± 0.66 | 0.33 ± 0.50** | 0.33 ± 0.71** | 2.18 ± 0.73 | 1.22 ± 0.44 | 0.56 ± 0.53** |
| Reticular formation | 1.82 ± 0.80 | 1.56 ± 0.73 | 1.11 ± 0.60 | 2.14 ± 0.56 | 1.89 ± 0.78 | 1.44 ± 0.73 |
| Inferior olive | 0.86 ± 0.64 | 0.89 ± 0.60 | 0.78 ± 0.83 | 1.41 ± 0.50 | 1.33 ± 0.71 | 0.89 ± 0.78 |
| Cerebellar cortex | 0.32 ± 0.48 | 0.44 ± 0.726 | 0.00 ± 0.00 | 0.55 ± 0.51 | 0.22 ± 0.44 | 0.00 ± 0.00 |
| Cerebellar dentate nucleus | 2.41 ± 0.67 | 1.56 ± 0.73 | 0.56 ± 0.53*** | 1.91 ± 0.68 | 1.56 ± 0.88 | 0.67 ± 0.50** |
| Axonal spheroid (mean ± SD) | ||||||
| Globus pallidus | 0.82 ± 0.96 | 1.33 ± 1.22 | 0.44 ± 0.73 | |||
| Substantia nigra | 1.36 ± 01.09 | 1.44 ± 1.33 | 0.89 ± 1.05 | |||
Neuronal loss: 0, none; 1, mild; 2, moderate; 3, severe. Tau pathology: 0, absent; 1, sparse; 2, moderate; 3, numerous. *P < 0.05; **P < 0.01; ***P < 0.001 vs. PSP (Kruskal–Wallis test with post‐hoc Dunn's test).
In general, PNLA Type 2 was characterized by severe neuronal loss, but less conspicuous tau pathology, restricted to the globus pallidus, substantia nigra and subthalamic nucleus. On the other hand, PNLA Type 1 showed mild neuronal loss and tau pathology in other areas, such as the motor cortex, pontine nuclei and cerebellar dentate nucleus, in addition to the severely affected PNL system (Figure 5, Table 1).
Clinical features
The clinical demographics are summarized in Table 2. Among the cases examined, diagnosis of PSP in the initial clinical stage was rare. There was no evident difference in gender or age at death between patients with PSP, and those with PNLA Type 1 and Type 2. However, statistical comparison among the three groups revealed a significant difference in disease duration between PSP and PNLA Type 1 (P < 0.01), and in the frequency of initial diagnosis of PD between PSP and PNLA Type 1 as well as between PSP and PNLA Type 2 (P < 0.05) (Table 2). In this connection, a clinical phenotype of pure or predominant akinesia, which has been noted to be a feature of PNLA 40, was evident only in PNLA Type 1 (3/9) and Type 2 (2/9); there were significant differences between PNLA Type 1 and PSP (P < 0.01), and PNLA Type 2 and PSP (P < 0.05).
Table 2.
Clinical demographic data in the PSP and PNLA Type 1 and Type 2 groups. Abbreviations: PD = Parkinson's disease; PNLA = pallido‐nigro‐luysian atrophy; PSP = progressive supranuclear palsy
| All (n = 40) | PSP (n = 22) | PNLA type1 (n = 9) | PNLA type 2 (n = 9) | |
|---|---|---|---|---|
| Gender (male/female) | 21/19 (male, 52.5%) | 13/9 (male, 59.1%) | 3/6 (male, 33.3%) | 5/4 (male, 55.6%) |
| Age at onset (years: mean ± SD) | 65.9 ± 9.22 | 69.1 ± 8.42 | 61.1 ± 4.29 | 62.4 ± 11.7 |
| Age at death (years: mean ± SD) | 76.2 ± 8.44 | 76.9 ± 8.90 | 77.9 ± 8.08 | 72.8 ± 7.50 |
| Disease duration (years: mean ± SD) | 9.3 ± 5.24 | 7.29 ± 4.22 | 14.9 ± 4.32** | 9.06 ± 4.94 |
| Initial diagnosis of PSP | 4/32 (12.5%) | 3/16 (18.8%) | 0/8 (0%) | 1/8 (12.5%) |
| Diagnosis of PSP at death | 27/40 (67.5%) | 14/22 (63.6%) | 7/9 (77.8%) | 6/9 (66.7%) |
| Initial diagnosis of PD | 14/32 (43.8%) | 2/16 (12.5%) | 6/8 (75.0%)* | 6/8 (75.0%)* |
This “Table” was prepared according to Ahmed et al 1. *P < 0.05 vs. PSP (Ryan's multiple comparison test); **P < 0.01 vs. PSP (Kruskal–Wallis test with post‐hoc Dunn's test).
The clinical findings are summarized in Table 3. The rates of occurrence of limb rigidity and nuchal/axial rigidity in the early stage were higher in PNLA Type 2 than in PNLA Type 1 (P < 0.05). The rate of occurrence of difficulty in eye‐opening, including apraxia, during the entire disease course was higher in PNLA Type 1 (P < 0.01) than in PSP and PNLA Type 2; on the other hand, those of postural instability and nuchal/axial rigidity during the entire disease course were higher in PNLA Type 2 (P < 0.05) than in PSP and PNLA Type 1.
Table 3.
Clinical findings in the PSP and PNLA Type 1 and Type 2 groups. Abbreviations: PNLA = pallido‐nigro‐luysian atrophy; PSP = progressive supranuclear palsy
| Symptoms and signs | Initial 2 years of disease course | Entire disease course | ||||
|---|---|---|---|---|---|---|
| PSP (n = 22) | PNLA Type 1 (n = 9) | PNLA Type 2 (n = 9) | PSP (n = 22) | PNLA Type 1 (n = 9) | PNLA Type 2 (n = 9) | |
| Vertical gaze palsy | 7/22 (31.8%) | 0/9 (0.0%) | 3/9 (33.3%) | 16/22 (72.7%) | 8/9 (88.9%) | 8/9 (88.9%) |
| Falls | 8/22 (36.4%) | 1/9 (11.1%) | 2/9 (22.2%) | 16/22 (72.7%) | 5/9 (55.6%) | 7/9 (77.8%) |
| Postural instability | 7/22 (31.8%) | 0/9 (0.0%) | 2/9 (22.2%) | 12/22 (54.5%) | 5/9 (55.6%) | 9/9 (100%)† |
| Cognitive decline | 8/22 (36.4%) | 0/9 (0.0%) | 1/9 (11.1%) | 18/22 (81.8%) | 4/9 (44.4%) | 6/9 (66.7%) |
| Asymmetry | 7/22 (31.8%) | 2/9 (22.2%) | 1/9 (11.1%) | 8/22 (31.8%) | 2/9 (22.2%) | 1/9 (11.1%) |
| Tremor | 6/22 (27.3%) | 5/9 (55.6%) | 4/9 (44.4%) | 8/22 (31.8%) | 5/9 (55.6%) | 4/22 (18.2%) |
| L‐Dopa response | 4/22 (18.2%) | 1/9 (11.1%) | 4/9 (44.4%) | 5/22 (22.7%) | 2/9 (22.2%) | 6/9 (66.7%) |
| Ataxia | 3/22 (13.6%) | 0/9 (0.0%) | 0/9 (0.0%) | 4/22 (18.2%) | 0/9 (0.0%) | 2/9 (22.2%) |
| Limb rigidity | 8/22 (36.4%) | 0/9 (0.0%) | 6/9 (66.7%)* | 14/22 (63.6%) | 5/9 (55.6%) | 8/9 (88.9%) |
| Gait disturbance | 14/22 (63.6%) | 5/9 (55.6%) | 6/9 (66.7%) | 20/22 (90.9%) | 8/9 (88.9%) | 8/9 (88.9%) |
| Freezing gait | 2/22 (9.1%) | 1/9 (11.1%) | 2/9 (22.2%) | 4/22 (18.2%) | 5/9 (55.6%) | 2/9 (22.2%) |
| Bradykinesia | 10/22 (45.5%) | 1/9 (11.1%) | 5/9 (55.6%) | 15/22 (68.2%) | 7/9 (77.8%) | 8/9 (88.9%) |
| Psychiatric symptom | 2/22 (9.1%) | 0/9 (0.0%) | 2/9 (22.2%) | 4/22 (18.2%) | 3/9 (33.3%) | 5/9 (55.6%) |
| Speech problem | 9/22 (40.9%) | 5/9 (55.6%) | 5/9 (55.6%) | 19/22 (86.4%) | 7/9 (77.8%) | 9/9 (100%) |
| Dysphagia | 4/22 (18.2%) | 2/9 (22.2%) | 2/9 (22.2%) | 16/22 (72.7%) | 7/9 (77.8%) | 9/9 (100%) |
| Nuchal/axial rigidity | 3/22 (13.6%) | 0/9 (0.0%) | 4/9 (44.4%)* | 13/22 (59.1%) | 4/9 (44.4%) | 9/9 (100%)† |
| Writing disturbance | 1/22 (4.5%) | 0/9 (0.0%) | 2/9 (22.2%) | 2/22 (9.1%) | 1/9 (11.1%) | 3/9 (33.3%) |
| Difficulty in eye‐opening | 0/22 (0.0%) | 1/9 (11.1%) | 0/9 (0.0%) | 1/22 (4.5%) | 5/9 (55.6%)** | 0/9 (0.0%) |
*P < 0.05 vs. PNLA Type 1; †P < 0.05 vs. PSP or vs. PNLA Type 1; **P < 0.01 vs. PSP or vs. PNLA Type 2 (Ryan's multiple comparison test).
As shown in Figure 7, the duration from symptom onset until becoming wheelchair‐bound was significantly longer (P < 0.05) in PNLA Type 1 cases (median, 12.0 years) than in PSP (median, 5.5 years) and PNLA Type 2 (median, 8.0 years) cases. The overall survival time was also significantly longer (P < 0.05) in PNLA Type 1 cases (median, 14.0 years) than in PSP cases (median, 6.0 years); there was no significant difference in the overall survival time between PNLA Type 1 and Type 2 (median, 10 years) cases (compare the disease duration shown in Table 2).
Figure 7.
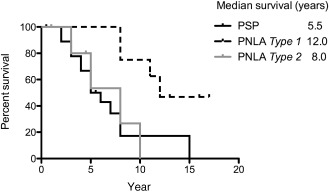
Kaplan–Meier survival analysis of the period from symptom onset until the patient becoming wheelchair‐bound. Survival time was significantly longer in PNLA Type 1 than in PSP and PNLA Type 2 (P < 0.05).
Sarkosyl‐insoluble tau
The data for immunoblot analysis of sarkosyl‐insoluble fractions are shown in Figure 8. Dephosphorylated samples revealed two major bands that aligned with the recombinant 4R tau 412‐ (1N, 4R) and 383‐ (0N, 4R) amino acid isoforms in cases of PSP (PSP 1, 2), and PLNA Type 1 (PNLA 1. 1–4) and Type 2 (PNLA 2. 1), as well as CBD (Figure 8A). Immunoblot analysis using non‐dephosphorylated samples was performed for the globus pallidus. The samples in the cases of PSP (PSP 2), and PLNA Type 1 (PNLA 1. 1–3, 5) and Type 2 (PNLA 2. 1, 2) revealed band patterns of low‐molecular‐mass tau fragments at ∼35 kDa (arrowhead); the sample obtained from the case of CBD revealed a band pattern of low‐molecular‐mass tau fragments at ∼40 kDa (Figure 8B).
Figure 8.
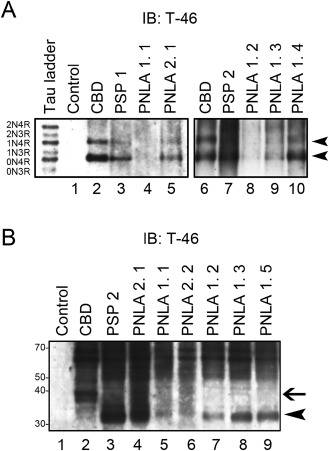
A. Immunoblot analysis of the sarkosyl‐insoluble fraction after dephosphorylation in two PSP (PSP 1, 2: lanes 3, 7) cases, and four PNLA Type 1 (PNLA 1. 1–4: lanes 4, 8, 9, 10) and one Type 2 (PNLA 2. 1: lane 5) cases, as well as control (lane 1) and CBD (lanes 2, 6) cases. Sarkosyl‐insoluble tau in cases of PSP and PNLA Type 1 and Type 2, as well as CBD consists predominantly of 4R tau isoforms (0N4R and 1N4R, arrowheads). Six recombinant human tau isoforms are indicated on the left (tau ladder). B. Immunoblot analysis of the sarkosyl‐insoluble fraction before dephosphorylation. The pattern of low‐molecular‐mass tau fragments in a PSP (PSP 2: lane 3) case, and 4 PNLA Type 1 (PNLA 1. 1–3, 5: lanes 5, 7, 8, 9) and 2 Type 2 (PNLA 2. 1, 2: lanes 4, 6) cases shows a prominent band of 33 kDa (arrowhead), while that in a CBD (lane 2) case shows a prominent band of 37 kDa (arrow).
Detection of MAPT gene mutation
No mutations in any of MAPT exons were detected in any of the patients.
Discussion
In 2008, Ahmed et al reported that cases of PSP rarely demonstrated pathology consistent with PNLA and showed many axonal spheroids in the globus pallidus and substantia nigra; all of these cases showed TAs in the caudate nucleus 1.
What is a TA in terms of PSP‐related astrocytic tau pathology? Oshima and Dickson defined TAs as star‐like tufts of tau‐positive abnormal fibers 33, whereas Yoshida described them as a radial arrangement of thin, long, branching accumulated tau protein from the cytoplasm to the proximal processes of astrocytes 57. In the present study, we first focused on the morphology of astrocytic tau lesions and recognized two types: uTAs and eTAs. eTAs sometimes appeared to resemble tau‐positive globular astrocytic inclusions (GAIs), which have been described recently as an important feature for classification of 4R tauopathies 3, 11.
As individual eTAs appeared to be smaller than TAs in the histological sections examined, we performed morphometric analysis using a 3D reconstruction method to clarify the difference. This revealed that the volumes of eTAs in a voxel of the same size were significantly smaller than those of uTAs, and that when compared with each other, uTAs were composed of a larger number of smaller particles, whereas eTAs were composed of a smaller number of larger particles. These morphological differences might reflect the way in which the process of tau protein polymerization has been perturbed during formation of the two types of astrocytic lesions. Ultrastructurally, however, bundles of straight tubules 5, 31 were a common feature of both uTAs and eTAs.
In the present study, 22 cases in the uTA group were confirmed to be PSP. On the other hand, 18 cases in the eTA group were further divided into 9 cases of PNLA Type 1 and 9 cases of PNLA Type 2. Most importantly, based on the severity of neuronal loss and the density of tau‐related pathology described by Ahmed et al 1, the entity of PSP‐PNLA they studied corresponded unequivocally to all of our present eTA cases (PNLA Type 1 and Type 2). Ahmed et al reported that in comparison with typical cases of PSP, PSP‐PNLA showed earlier onset and slower disease progression 1. They also stated that eyelid apraxia was present only in PSP‐PNLA cases 1. In the present study, in cases of PNLA Type 1 as a subset of PSP‐PNLA 1, a long disease duration with relatively good mobility was a notable feature. From a pathological viewpoint, it was difficult to explain such a long disease duration for PNLA Type 1: there were no significant differences in regional neuronal loss and tau‐related pathology between PNLA Type 1 and Type 2. From a clinical viewpoint, development of difficulty in eye‐opening, including eyelid apraxia, during the entire disease course may predict a diagnosis of PNLA Type 1 with a favorable prognosis. It was also noteworthy that an initial diagnosis of PD was significantly more frequent in both PNLA Type 1 and Type 2 cases than in PSP cases.
Interestingly, there was a marked difference between the present study and that of Ahmed et al in the disease frequency of PSP‐PNLA. Ahmed et al found 8 cases of PSP‐PNLA in their pathological series of over 400 cases of PSP (<2%) 1. On the other hand, our series of 40 cases of PSP and PSP‐like disease included 18 cases of PSP‐PNLA, ie, 9 cases of PNLA Type 1 (22.5%) and 9 cases of PNLA Type 2 (22.5%). This surprising result might have been attributable to bias in the clinical focus and selection of cases for autopsy examination. However, considering that reports of PNLA from Japan have been comparatively frequent 24, 29, 30, 40, 56, 57, it is likely that certain unknown risk factors may be linked to PSP‐PNLA (PNLA Type 1/Type 2) as a variant of PSP in the Japanese population, whose ethnic background differs from that of Caucasians.
Returning to the pathological findings, Ahmed et al suggested that the regional density of tau‐related pathology in the motor cortex and caudate nucleus would be helpful for differentiating PSP from PSP‐PNLA, and that the presence of many axonal spheroids in the globus pallidus and substantia nigra was a feature of PSP‐PNLA 1. The findings of the present study suggest that not only the density of the tau‐related pathology but also the morphology of astrocytic tau lesions are important for differentiating PSP from PSP‐PNLA. Very recently, Yoshida described the significant occurrence of astrocytic tau lesions designated “atypical TAs” morphologically very similar, if not identical, to eTAs observed in the present study in cases of PNLA as a PSP subtype, and stated that in the PNLA subtype, astrocytic tau lesions in the striatum showed marked proximally dominant tau accumulation 57. With regard to the axonal spheroids, we consider that they are not a characteristic feature of PSP‐PNLA (PNLA Type 1/Type 2). Unusual cases of PNLA with spheroids have recently been reported 3, 13; PNLA, which is characterized by degeneration of the globus pallidus, substantia nigra and subthalamic nucleus, does not always involve tauopathy 54.
In the present study, immunoblotting of insoluble tau after dephosphorylation revealed predominantly 4R tau in PSP, and in PNLA Type 1 and Type 2 cases. It was notable that in these cases, immunoblotting of insoluble tau before dephosphorylation revealed prominent C‐terminal tau fragments at ∼35 kDa. To our knowledge, this biochemical feature detectable in a 4R tauopathy, PSP 4, has not yet been demonstrated in cases of PNLA as a representative tauopathy. Mori et al performed immunoblot analysis of tau protein before and after dephosphorylation in a case of PNLA, and found that the features of 4R tauopathy were similar to those seen in PSP and CBD 30.
Several familial cases with MAPT gene mutations have been reported to show astrocytic tau lesions similar to TAs 7, 10, 34, 41, 42, 43. However, PSP is a rare manifestation of MAPT gene mutations 36, and routine screening of sporadic PSP for mutations in this gene is not recommended because of low yield 53. Recently, Ogaki et al reported MAPT‐positive Japanese patients with the PSP phenotype, who were younger than 50 years at disease onset 32. It is also noteworthy that a familial case of pallido‐ponto‐nigral degeneration with MAPT gene mutation has been reported 41; however, the overall clinicopathological picture was apparently different from those seen in the present cases. In our present series of 40 cases, including one of early onset PNLA Type 2 (mentioned previously), none of the patients had a family history of neurological disease. Moreover, there were no mutations in any of the sequenced exons of the MAPT gene in five examined patients who were less than 60 years of age at disease onset. Thus, it appears very unlikely that the astrocytic tau lesions in the present cases, both uTAs and eTAs, were attributable to a genetic factor.
Finally, it is also important to note that LRRK2 gene mutations, which are the most common cause of familial and sporadic PD, may be associated with PSP‐like pathological features 35, 37, 38, 49, 55, 58. We did not sequence the LRRK2 gene in the present patients, none of whom had a family history or clinical diagnosis of PD. The LRRK2 gene has already been screened in clinically 15, 28, 47 and pathologically confirmed cases of PSP 12, 35, 37, suggesting that mutations of this gene do not constitute a risk factor for PSP. Moreover, to our knowledge, no autopsy cases with LRRK2 gene mutations associated with neurodegeneration recognized as 4R tauopathy, PSP or PNLA have been reported so far.
In conclusion, using the morphologic features of astrocytic tau pathology as an initial indicator, we have been able to classify autopsy cases of PSP or PSP‐like disease into three groups: PSP and PNLA Type 1 and Type 2. Our pathological, clinical and biochemical findings strongly suggest that PSP and PNLA Type 1 and Type 2 are part of a single 4R tauopathy, ie, PSP in the broad sense. However, it is also important to note that a pathological distinction between classical PSP (PSP in the narrow sense) 8, 9, 50, 57 and PSP‐PNLA 1, or between classical PSP and PNLA Type 1 and Type 2 shown in the present study, would be possible based on the morphology of the astrocytic tau pathology and the regional density of the tau‐related pathology.
Conflict of interest
The authors have no conflicts of interest to declare.
Acknowledgments
We thank C. Tanda, J. Takasaki, H. Saito, T. Fujita, S. Nigorikawa and S. Egawa for their technical assistance, and M. Machida and Y. Ueda for secretarial assistance. This work was supported by Grants‐in‐Aid, 26430052 (to Y. T.) and 26250017 (to H. T.), for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology, Japan.
References
- 1. Ahmed Z, Josephs KA, Gonzalez J, DelleDonne A, Dickson DW (2008) Clinical and neuropathologic features of progressive supranuclear palsy with severe pallido‐nigro‐luysial degeneration and axonal dystrophy. Brain 131:460–472. [DOI] [PubMed] [Google Scholar]
- 2. Ahmed Z, Tabrizi SJ, Li A, Houlden H, Sailer A, Lees AJ et al (2010) A Huntington's disease phenocopy characterized by pallido‐nigro‐luysian degeneration with brain iron accumulation and p62‐positive glial inclusions. Neuropathol Appl Neurobiol 36:551–557. [DOI] [PubMed] [Google Scholar]
- 3. Ahmed Z, Bigio EH, Budka H, Dickson DW, Ferrer I, Ghetti B et al (2013) Globular glial tauopathies (GGT): consensus recommendations. Acta Neuropathol 126:537–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Arai T, Ikeda K, Akiyama H, Nonaka T, Hasegawa M, Ishiguro K et al (2004) Identification of amino‐terminally cleaved tau fragments that distinguish progressive supranuclear palsy from corticobasal degeneration. Ann Neurol 55:72–79. [DOI] [PubMed] [Google Scholar]
- 5. Arima K (2006) Ultrastructural characteristics of tau filaments in tauopathies: immuno‐electron microscopic demonstration of tau filaments in tauopathies. Neuropathology 26:475–483. [DOI] [PubMed] [Google Scholar]
- 6. Contamin F, Escourolle R, Nick J, Mignot B (1971) Atrophy of the globus pallidus, substantia nigra, and nucleus subthalamicus. Akinetic syndrome with palilalia, oppositional rigidity and catatonia. Rev Neurol 124:107–120. [PubMed] [Google Scholar]
- 7. Delisle MB, Murrell JR, Richardson R, Trofatter JA, Rascol O, Soulages X et al (1999) A mutation at codon 279 (N279K) in exon 10 of the Tau gene causes a tauopathy with dementia and supranuclear palsy. Acta Neuropathol 98:62–77. [DOI] [PubMed] [Google Scholar]
- 8. Dickson D, Hauw J‐J, Agid Y, Litvan I (2011) Progressive supranuclear palsy and corticobasal degeneration. In: Neurodegeneration: the Molecular Pathology of Dementia and Movement Disorders, 2nd edn. Dickson DW, Weller RO (eds), pp. 135–155. Blackwell: Oxford. [Google Scholar]
- 9. Dickson DW (1999) Neuropathologic differentiation of progressive supranuclear palsy and corticobasal degeneration. J Neurol 246 (Suppl. 2):II6–II15. [DOI] [PubMed] [Google Scholar]
- 10. Ferrer I, López‐González I, Carmona M, Arregui L, Dalfó E, Torrejón‐Escribano B et al (2014) Glial and neuronal tau pathology in tauopathies: characterization of disease‐specific phenotypes and tau pathology progression. J Neuropathol Exp Neurol 73:81–97. [DOI] [PubMed] [Google Scholar]
- 11. Fu YJ, Nishihira Y, Kuroda S, Toyoshima Y, Ishihara T, Shinozaki M et al (2010) Sporadic four‐repeat tauopathy with frontotemporal lobar degeneration, Parkinsonism, and motor neuron disease: a distinct clinicopathological and biochemical disease entity. Acta Neuropathol 120:21–32. [DOI] [PubMed] [Google Scholar]
- 12. Gaig C, Ezquerra M, Martí MJ, Valldeoriola F, Muñoz E, Lladó A et al (2008) Screening for the LRRK2 G2019S and codon‐1441 mutations in a pathological series of parkinsonian syndromes and frontotemporal lobar degeneration. J Neurol Sci 270:94–98. [DOI] [PubMed] [Google Scholar]
- 13. Hashimoto T, Matsubara S, Mochizuki Y, Tsuji S, Mizutani T, Oyanagi K (2008) Forme fruste or incipient form of widespread‐type amyotrophic lateral sclerosis, or motor neuron disease with pallido‐nigro‐luysian atrophy? An autopsy case report. Neuropathology 28:309–316. [DOI] [PubMed] [Google Scholar]
- 14. Hassan A, Parisi JE, Josephs KA (2012) Autopsy‐proven progressive supranuclear palsy presenting as behavioral variant frontotemporal dementia. Neurocase 18:478–488. [DOI] [PubMed] [Google Scholar]
- 15. Hernandez D, Paisan Ruiz C, Crawley A, Malkani R, Werner J, Gwinn‐Hardy K et al (2005) The dardarin G 2019 S mutation is a common cause of Parkinson's disease but not other neurodegenerative diseases. Neurosci Lett 389:137–139. [DOI] [PubMed] [Google Scholar]
- 16. Ikeuchi T, Kaneko H, Miyashita A, Nozaki H, Kasuga K, Tsukie T et al (2008) Mutational analysis in early‐onset familial dementia in the Japanese population. The role of PSEN1 and MAPT R406W mutations. Dement Geriatr Cogn Disord 26:43–49. [DOI] [PubMed] [Google Scholar]
- 17. Imai H, Narabayashi H, Sakata E (1987) “Pure akinesia” and the later added supranuclear ophthalmoplegia. Adv Neurol 45:207–212. [PubMed] [Google Scholar]
- 18. Josephs KA, Boeve BF, Duffy JR, Smith GE, Knopman DS, Parisi JE et al (2005) Atypical progressive supranuclear palsy underlying progressive apraxia of speech and nonfluent aphasia. Neurocase 11:283–296. [DOI] [PubMed] [Google Scholar]
- 19. Josephs KA, Katsuse O, Beccano‐Kelly DA, Lin WL, Uitti RJ, Fujino Y et al (2006) Atypical progressive supranuclear palsy with corticospinal tract degeneration. J Neuropathol Exp Neurol 65:396–405. [DOI] [PubMed] [Google Scholar]
- 20. Kaat LD, Boon AJ, Kamphorst W, Ravid R, Duivenvoorden HJ, van Swieten JC (2007) Frontal presentation in progressive supranuclear palsy. Neurology 69:723–729. [DOI] [PubMed] [Google Scholar]
- 21. Kanazawa M, Shimohata T, Toyoshima Y, Tada M, Kakita A, Morita T et al (2009) Cerebellar involvement in progressive supranuclear palsy: a clinicopathological study. Mov Disord 24:1312–1318. [DOI] [PubMed] [Google Scholar]
- 22. Kobilecki C, Jones M, Thompson JC, Richardson AM, Neary D, Mann DM et al (2015) Cognitive‐behavioural features of progressive supranuclear palsy syndrome with frontotemporal dementia. J Neurol 262:916–922. [DOI] [PubMed] [Google Scholar]
- 23. Komori T, Arai N, Oda M, Nakayama H, Mori H, Yagishita S et al (1998) Astrocytic plaques and tufts of abnormal fibers do not coexist in corticobasal degeneration and progressive supranuclear palsy. Acta Neuropathol 96:401–408. [DOI] [PubMed] [Google Scholar]
- 24. Konishi Y, Shirabe T, Katayama S, Funakawa I, Terao A (2005) Autopsy case of pure akinesia showing pallidonigro‐luysian atrophy. Neuropathology 25:220–227. [DOI] [PubMed] [Google Scholar]
- 25. Ling H, de Silva R, Massey LA, Courtney R, Hondhamuni G, Bajaj N et al (2014) Characteristics of progressive supranuclear palsy presenting with corticobasal syndrome: a cortical variant. Neuropathol Appl Neurobiol 40:149–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Litvan I, Agid Y, Calne D, Campbell G, Dubois B, Duvoisin RC et al (1996) Clinical research criteria for the diagnosis of progressive supranuclear palsy (Steele‐Richardson‐Olszewski syndrome): report of the NINDS‐SPSP international workshop. Neurology 47:1–9. [DOI] [PubMed] [Google Scholar]
- 27. Litvan I, Campbell G, Mangone CA, Verny M, McKee A, Chaudhuri KR et al (1997) Which clinical features differentiate progressive supranuclear palsy (Steele‐Richardson‐Olszewski syndrome) from related disorders? A clinicopathological study. Brain 120:65–74. [DOI] [PubMed] [Google Scholar]
- 28. Madzar D, Schulte C, Gasser T (2009) Screening for LRRK2 R1441 mutations in a cohort of PSP patients from Germany. Eur J Neurol 16:1230–1232. [DOI] [PubMed] [Google Scholar]
- 29. Matsuo H, Takashima H, Kishikawa M, Kinoshita I, Mori M, Tsujihata M, Nagataki S (1991) Pure akinesia: an atypical manifestation of progressive supranuclear palsy. J Neurol Neurosurg Psychiatry 54:397–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mori H, Motoi Y, Kobayashi T, Hasegawa M, Yamamura A, Iwatsubo T, Mizuno Y (2001) Tau accumulation in a patient with pallidonigroluysian atrophy. Neurosci Lett 309:89–92. [DOI] [PubMed] [Google Scholar]
- 31. Nishimura M, Namba Y, Ikeda K, Oda M (1992) Glial fibrillary tangles with straight tubules in the brains of patients with progressive supranucelar paksy. Neurosci Lett 143:35–38. [DOI] [PubMed] [Google Scholar]
- 32. Ogaki K, Li Y, Takanashi M, Ishikawa K, Kobayashi T, Nonaka T et al (2013) Analyses of the MAPT, PGRN, and C9orf72 mutations in Japanese patients with FTLD, PSP, and CBS. Parkinsonism Relat Disord 19:15–20. [DOI] [PubMed] [Google Scholar]
- 33. Oshima K, Dickson DW (2009) Cortical Alzheimer type pathology does not influence tau pathology in progressive supranuclear palsy. Int J Clin Exp Pathol 2:399–406. [PMC free article] [PubMed] [Google Scholar]
- 34. Poorkaj P, Muma NA, Zhukareva V, Cochran EJ, Shannon KM, Hurtig H et al (2002) An R5L tau mutation in a subject with a progressive supranuclear palsy phenotype. Ann Neurol 52:511–516. [DOI] [PubMed] [Google Scholar]
- 35. Rajput A, Dickson DW, Robinson CA, Ross OA, Dächsel JC, Lincoln SJ et al (2006) Parkinsonism, Lrrk2 G2019S, and tau neuropathology. Neurology 67:1506–1508. [DOI] [PubMed] [Google Scholar]
- 36. Rohrer JD, Paviour D, Vandrovcova J, Hodges J, de Silva R, Rossor MN (2011) Novel L284R MAPT mutation in a family with an autosomal dominant progressive supranuclear palsy syndrome. Neurodegener Dis 8:149–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ross OA, Whittle AJ, Cobb SA, Hulihan MM, Lincoln SJ, Toft M et al (2006) Lrrk2 R1441 substitution and progressive supranuclear palsy. Neuropathol Appl Neurobiol 32:23–25. [DOI] [PubMed] [Google Scholar]
- 38. Ruffmann C, Giaccone G, Canesi M, Bramerio M, Goldwurm S, Gambacorta M et al (2012) Atypical tauopathy in a patient with LRRK2‐G2019S mutation and tremor‐dominant Parkinsonism. Neuropathol Appl Neurobiol 38:382–386. [DOI] [PubMed] [Google Scholar]
- 39. Sakai K, Piao YS, Kikugawa K, Ohara S, Hasegawa M, Takano H et al (2006) Corticobasal degeneration with focal, massive tau accumulation in the subcortical white matter astrocytes. Acta Neuropathol 112:341–348. [DOI] [PubMed] [Google Scholar]
- 40. Shimoda M, Hosoda Y, Kato S, Kaneto D, Takahashi K, Yen SH, Ohama E (1996) Pallidonigroluysian atrophy: clinicopathological and immunohistochemical studies. Neuropathology 16:21–28. [Google Scholar]
- 41. Slowinski J, Dominik J, Uitti RJ, Ahmed Z, Dickson DD, Wszolek ZK (2007) Frontotemporal dementia and Parkinsonism linked to chromosome 17 with the N279K tau mutation. Neuropathology 27:73–80. [DOI] [PubMed] [Google Scholar]
- 42. Spina S, Farlow MR, Unverzagt FW, Kareken DA, Murrell JR, Fraser G et al (2008) The tauopathy associated with mutation +3 in intron 10 of Tau: characterization of the MSTD family. Brain 131:72–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Stanford PM, Halliday GM, Brooks WS, Kwok JB, Storey CE, Creasey H et al (2000) Progressive supranuclear palsy pathology caused by a novel silent mutation in exon 10 of the tau gene: expansion of the disease phenotype caused by tau gene mutations. Brain 123:880–893. [DOI] [PubMed] [Google Scholar]
- 44. Steele JC, Richardson JC, Olszewski J (1964) Progressive supranuclear palsy. A heterogeneous degeneration involving the brain stem, basal ganglia and cerebellum with vertical gaze and pseudobulbar palsy, nuchal dystonia and dementia. Arch Neurol 10:333–359. [DOI] [PubMed] [Google Scholar]
- 45. Takahashi K, Nakashima R, Takao T, Nakamura H (1977) Pallido‐nigro‐luysial atrophy associated with degeneration of the centrum medianum. A clinicopathologic and electron microscopic study. Acta Neuropathol 37:81–85. [DOI] [PubMed] [Google Scholar]
- 46. Tan CF, Piao YS, Kakita A, Yamada M, Takano H, Tanaka M et al (2005) Frontotemporal dementia with co‐occurrence of astrocytic plaques and tufted astrocytes, and severe degeneration of the cerebral white matter: a variant of corticobasal degeneration? Acta Neuropathol 109:329–338. [DOI] [PubMed] [Google Scholar]
- 47. Tan EK, Skipper L, Chua E, Wong MC, Pavanni R, Bonnard C et al (2006) Analysis of 14 LRRK2 mutations in Parkinson's plus syndromes and late‐onset Parkinson's disease. Mov Disord 21:997–1001. [DOI] [PubMed] [Google Scholar]
- 48. Tatsumi S, Mimuro M, Iwasaki Y, Takahash R, Kakita A, Takahashi H, Yoshida M (2014) Argyrophilic grains are reliable disease‐specific features of corticobasal degeneration. J Neuropathol Exp Neurol 73:30–38. [DOI] [PubMed] [Google Scholar]
- 49. Ujiie S, Hatano T, Kubo S, Imai S, Sato S, Uchihara T et al (2012) LRRK2 I2020T mutation is associated with tau pathology. Parkinsonism Relat Disord 18:819–823. [DOI] [PubMed] [Google Scholar]
- 50. Wakabayashi K, Takahashi H (2004) Pathological heterogeneity in progressive supranuclear palsy and corticobasal degeneration. Neuropathology 24:79–86. [DOI] [PubMed] [Google Scholar]
- 51. Williams DR, de Silva R, Paviour DC, Pittman A, Watt HC, Kilford L et al (2005) Characteristics of two distinct clinical phenotypes in pathologically proven progressive supranuclear palsy: Richardson's syndrome and PSP‐parkinsonism. Brain 128:1247–1258. [DOI] [PubMed] [Google Scholar]
- 52. Williams DR, Holton JL, Strand K, Revesz T, Lees AJ (2007) Pure akinesia with gait freezing: a third clinical phenotype of progressive supranuclear palsy. Mov Disord 22:2235–2241. [DOI] [PubMed] [Google Scholar]
- 53. Williams DR, Pittman AM, Revesz T, Lees AJ, de Silva R (2007) Genetic variation at the tau locus and clinical syndromes associated with progressive supranuclear palsy. Mov Disord 22:895–897. [DOI] [PubMed] [Google Scholar]
- 54. Wong JC, Armstrong MJ, Lang AE, Hazrati LN (2013) Clinicopathological review of pallidonigroluysian atrophy. Mov Disord 28:274–281. [DOI] [PubMed] [Google Scholar]
- 55. Wszolek ZK, Pfeiffer RF, Tsuboi Y, Uitti RJ, McComb RD, Stoessl AJ et al (2004) Autosomal dominant parkinsonism associated with variable synuclein and tau pathology. Neurology 62:1619–1622. [DOI] [PubMed] [Google Scholar]
- 56. Yamamoto T, Kawamura J, Hashimoto S, Nakamura M, Iwamoto H, Kobashi Y, Ichijima K (1991) Pallido‐nigro‐luysian atrophy, progressive supranuclear palsy and adult onset Hallervorden‐Spatz disease: a case of akinesia as a predominant feature of parkinsonism. J Neurol Sci 101:98–106. [DOI] [PubMed] [Google Scholar]
- 57. Yoshida M (2014) Astrocytic inclusions in progressive supranuclear palsy and corticobasal degeneration. Neuropathology 34:555–570. [DOI] [PubMed] [Google Scholar]
- 58. Zimprich A, Biskup S, Leitner P, Lichtner P, Farrer M, Lincoln S et al (2004) Mutations in LRRK2 cause autosomal‐dominant parkinsonism with pleomorphic pathology. Neuron 44:601–607. [DOI] [PubMed] [Google Scholar]