

Abstract
Arboviruses (Arthropod‐borne viruses) include several families of viruses (Flaviviridae, Togaviradae, Bunyaviradae, Reoviradae) that are spread by arthropod vectors, most commonly mosquitoes, ticks and sandflies. The RNA genome allows these viruses to rapidly adapt to ever‐changing host and environmental conditions. Thus, these virus families are largely responsible for the recent expansion in geographic range of emerging viruses including West Nile virus (WNV), dengue virus and Chikungunya virus. This review will focus on WNV, especially as it has progressively spread westward in North America since its introduction in New York in 1999. By 2003, WNV infections in humans had reached almost all lower 48 contiguous United States (US) and since that time, fluctuations in outbreaks have occurred. Cases decreased between 2008 and 2011, followed by a dramatic flair in 2012, with the epicenter in the Dallas‐Fort Worth region of Texas. The 2012 outbreak was associated with an increase in reported neuroinvasive cases. Neuroinvasive disease continues to be a problem particularly in the elderly and immunocompromised populations, although WNV infections also represented the second most frequent cause of pediatric encephalitis in these same years. Neuropathological features in cases from the 2012 epidemic highlight the extent of viral damage that can occur in the CNS.
Keywords: arbovirus, epidemic, infection, neuropathology
Introduction
Epidemiology of West Nile virus (WNV) outbreaks
WNV originated from African lineages less than 2000 years ago 14, 45 and was dispersed out of Africa into many European and Asian countries via migratory birds 14. WNV was originally isolated in 1937 from a native woman from the West Nile district of Uganda who was suffering from a mild febrile illness 37. However, major clinical disease was not recognized until the 1950s when epidemics occurred in Israel and Egypt 17. Serological studies published 12 years later demonstrated that neutralizing antibodies were widely distributed in native populations of Uganda, Kenya, the Belgian Congo and the Sudan 17. As reviewed by Mann et al, “sporadic outbreaks of febrile disease occurred throughout the 1950s–1980s in Africa, Australia and the Middle East with larger outbreaks in Israel (1951–1952, 1957, and 1962), France (1962–1965) and South Africa (1974 and 1983–1984) which were self‐limited with few confirmed cases of neuroinvasive or other clinical disease” 25. Additional epidemics occurred in Algeria (1994 and 1997), Morocco (1996), Romania (1996), Tunisia (1997), Russia (1999), Israel (1998–2000) and France (2000). The 1996 Romanian epidemic marked the first human epidemic associated with significant incidence of encephalitic disease, according to Mann et al 25.
WNV in North America appeared in 1999 and spread westward across the United States (US) and into Canada. Interestingly, as WNV spread south, it was able to establish endemic infection only in temperate southern portions of South America. Indeed, little or no morbidity or mortality occurred in humans in tropical regions of Latin America or South America 22. By 2000, in the US, human infections were found in Connecticut and New Jersey, by 2001, also in Pennsylvania, Massachusetts, Maryland, Florida, Georgia, Alabama and Louisiana, and by 2002 throughout all states east of the Rocky Mountain region. By 2003, human infections had reached 47 of the lower 48 states (http://www.cdc.gov/ncidod/dvbid/westnile/background.htm). In each year, the virus was identified in avian, animal or mosquito vectors in several additional nearby states, usually immediately south or west of the states with reported human cases.
In the US, ArboNET, the Arboviral Diseases Branch, Centers for Disease Control and Prevention tracks cases by year, state, county, including neuroinvasive vs. non‐neuroinvasive disease and deaths (http://www.cdc.gov/westnile/statsMaps/). Cases of neuroinvasive disease jumped astronomically between 2001 (64) and 2002 (2946)/2003 (2866), but then diminished progressively between 2004 and 2007, with 1227 in 2007. An even sharper decline occurred thereafter with 689 (2008), 386 (2009), 629 (2010) and 486 (2011) cases in following years.
However, in 2012, an unprecedented outbreak occurred when the US experienced 5674 confirmed WNV disease cases, with 2873 reports of neuroinvasive disease and 270 deaths (http://www.cdc.gov/westnile/statsMaps/). Hardest hit was the Dallas‐Fort Worth, Texas metropolitan area, which represented >29% of the US public health burden and >50% of all reported neurologic cases (http://www.dshs.state.tx.us/idcu/disease/arboviral/westNile/).
In 2013, US cases leveled off to 2007 levels (1267 neuroinvasive cases). For the 2014 season, as of January 13, 2015, 47 states and the District of Columbia reported 2122 human cases of WNV (http://www.cdc.gov/westnile/statsMaps/). Of the 2014 reported cases, 1283 (60%) were classified as neuroinvasive disease (meningitis or encephalitis), with 839 (40%) classified as non‐neuroinvasive disease. Thus, the recorded disease for 2014 nears the record numbers of 2012. While there is no doubt that more compliance with reporting, especially of severe cases, has occurred over the last decade, the peak outbreak years of 2002/2003 and 2012 clearly exist.
A few studies have specifically tried to directly compare clinical variations that might be associated with different seasonal outbreaks within a given geographical region. A study out of Oklahoma comparing their three outbreak years of 2003, 2007 and 2012 showed differences in numbers of cases per year (79, 107 and 180 cases, respectively), median ages of affected patients (48, 58 and 59 years), and differences in case numbers by county 20. Race, gender and symptomatology did not differ during these separate outbreaks. There was no dramatic difference in death rates between the three peak outbreaks, either during the hospitalization or in the first 2 weeks post‐discharge (13%, 15%, 9%, respectively). Neuroinvasive disease was not recorded in death certificates during the 2003 outbreak, but by 2012, with greater recognition of the meningitic, encephalitic and flaccid paralysis associated with infection, “West Nile encephalitis” was documented in 81% of death certificates from WNV patients 20.
In comparison, similar databases from Europe show vastly fewer numbers of affected individuals. As of 20 November 2014, for the 2014 season, 74 human cases of West Nile fever (including 66 neuroinvasive infections) were reported in the EU; one case in Austria, 15 in Greece, 11 in Hungary, 24 in Italy and 23 in Romania. In neighboring countries, 136 cases were detected in Bosnia and Herzegovina, Israel, Palestine, Serbia and the Russian Federation. In the European Union, the highest numbers of cases were reported in Italy and Romania (http://www.ecdc.europa.eu/en/healthtopics/west_nile_fever/).
Interestingly, in the US ArboNet database, the death rates have not changed significantly over the 16 years since WNV first appeared in North America, varying only from 8% to 12% of neuroinvasive cases. However, this is still considerable in comparison with the cited death rates of <1% for non‐neuroinvasive West Nile infection.
To put WNV in perspective, the epidemics of WNV in 2003 and 2012 were the largest outbreaks of neuroinvasive viral infections ever reported in the Western Hemisphere 22, 30. When reviewing data from the Nationwide Inpatient Sample for cases of hospitalization for “encephalitis” in the US during the years 2000–2010, 1.5% of all encephalitis hospitalizations were found to be due to WNV 13.
Mosquito Transmission
WNV is no different than most arboviruses in that it maintains an enzootic (animal) cycle that does not involve human infection. For WNV, passerine (perching) bird species such as jays and finches are the reservoir host, mosquitoes are the bridging vectors, and humans, horses and other animals are incidental hosts 20. As noted by Johnson et al, “WNV has been able to disperse successfully throughout North America because of the abundance of competent mosquito vectors, a flavivirus‐naïve bird reservoir and a susceptible host population” 20.
In both Europe and North America, the virus is most frequently associated with Culex spp. mosquitoes which can amplify the virus 14. While greater than 40 mosquito species have been reported to carry WNV, in the US the most important vectors are Culex tarsalis in the northern Great Plains, C. pipiens in the Midwest, and C. quinquefasciatus in the south central regions 41. Attesting to the adaptability of the virus is the fact that it utilizes different mosquito species to cope with different climatic conditions. In harsh winter climates, the virus is found in mosquito species that have adaption mechanisms of entering diapause (“physiologically enforced dormancy between periods of activity” 22, ie, mosquito “hibernation”) to survive cold temperatures. In contrast, other species in warmer regions such as C. quinquefasciatus never need to enter diapause and remain active throughout the winter 41.
After an infected mosquito bites a human, the virus replicates in dendritic cells and macrophages in local tissue and lymph nodes, resulting in primary viremia that seeds the spleen and reticuloendothelial system. Following growth in the spleen, patients develop a secondary viremia with dissemination to the central nervous system 28. Of note, West Nile viremia in humans is not high enough or sustained enough to support subsequent transmission back to mosquitoes, so humans are dead‐end hosts. The ability of an arbovirus to invade the CNS (neuroinvasiveness) is determined by multiple viral and host factors 7, 39.
Climate and rainfall are important influences on the timing and distribution of WNV outbreaks. Elevated temperature may accelerate virus replication and shorten the time span of the reproductive feeding cycle 20, 41. Drought can cause birds (the hosts) to come into closer contact with the vector (mosquitoes) and one of the reasons the 2012 outbreak may have been so severe was that it coincided with the highest average annual temperature and one of the lowest annual precipitations rates in one of the outbreak states, Oklahoma, in over 100 years (20, http://www.ncdc.noaa.gov/climate‐us). However, the influence of climate in differing areas of the country may differ. For the 2012 outbreak, three epicenters of disease, the upper Midwest (Illinois, Indian, Michigan, Ohio, Wisconsin), northern Great Plains (Minnesota, Nebraska, North and South Dakota) and south central (Arkansas, Louisiana, Mississippi, Oklahoma, Texas) were studied for temperature and precipitation at three different time points (January, March, July) 41. From that study, it was apparent that the 2012 WNV outbreak coincided with areas of the US with the greatest climate fluctuations, ie, higher‐than‐average temperature, especially in December and January, and lower‐than‐average precipitation in January. This suggests that the vulnerable time period for influencing mosquito vectors might be during the winter months when mosquitoes “overwinter” 41. Wimberly et al state that the “overwintering phase of the WNV enzootic cycle is a climatically sensitive period that has the potential to influence virus amplification and ultimately, the risk to humans during the subsequent transmission season” 41. However, the same study found that climactic influence was not as strong a factor in the areas of the country with less harsh winters, ie, the south central regions of the US.
Viral Strain Evolution
Two main lineages of WNV exist, types I and II, with the first most commonly involved in human infection 16, 22. However, phenotypic strain evolution has occurred over time 25. The original New York (NY99) genotype was a type I lineage with relationship to strains isolated in Israel 16. The original NY99 replaced by the North America/West Nile 2002 (NA/WN02) genotype in 2002, and the latter continues to be the primary circulating strain causing human infections in the US. A new genotype was found in 2003 in isolates collected from the southwestern US (SW/WN03 genotype) and “both genotypes co‐circulate to date” 25. Some additional genotypes in the US “have become extinct due to lack of a selective advantage or stochastic effect; however, the dynamic emergence, displacement, and extinction of multiple WNV genotypes in the US from 1999–2012 indicates the continued evolution of WNV in North America” 22. For example, WNV isolates gathered in the coastal region of southeast Texas in 2002 (termed the Southeastern Coastal Texas genotype) showed five unique amino acid substitutions, but since that time no isolates have been identified elsewhere in the US that belong to this genotype, suggesting extinction. The dominant genotype is the North American (NA/WN02) genotype although a second frequent type Southwestern (SW/WN03) remains relatively restricted to the southwestern US, first appearing in Colorado and Arizona in 2003 and still co‐circulating with the NA/WN02 22. A Midwest type (MW/WN06 cluster) was isolated from human blood donors and birds from Idaho and North Dakota during 2006–2011 but is considered to be within the SW/WN03 genotype 22.
In the 2012 outbreak in the Dallas‐Fort Worth/Houston area, all isolates clustered within the NA/WN02 genotype, although there had been a 0.41–0.72% nucleotide divergence from NY99 10, 22. However, the authors could find no correlation between the changes in WNV genetics in the Texas epidemic and the increased neuroinvasive features 10. In other words, factors in addition to genetic drift appeared to account for the severity and increased incidence of encephalitis in the 2012 outbreak.
Transmission of WNV Infection to Humans
Almost all cases of WNV disease are due to mosquito transmission following a bite. Occasional cases of WNV disease also occur following organ transplantation of WNV‐infected organs 18, breast‐feeding, and blood transfusions from asymptomatic, WNV‐infected individuals 5.
Currently, donor organs and blood products are screened for WNV nucleic acids to prevent iatrogenic transmission. Basavaraju et al recently reviewed encephalitis caused by pathogens transmitted through blood products and organ transplants in the US from 2002 to 2013 4. As of their 2014 review, there had been 12 published cases of transfusion‐transmitted WNV since the time of nucleic acid testing of pooled donation lots, but none since individual blood samples have undergone the testing. It is unusual for WNV to be transmitted through the blood of a WNV IgM‐positive donor, as it is assumed that the host has already cleared the virus from blood, but this may not be true for solid organ transplant recipients 4.
When WNV is transmitted via organ transplantation, the risk for neuroinvasive disease is significantly greater (40–70%) than in the general population (1%) 22, 42. Fortunately, the raw numbers of such cases are relatively small. Organ transplant‐transmitted WNV reported to the Centers for Disease control between the years 2002 and 2013 consisted of six clusters of transmission and WNV infection developed in 12 of 16 recipients. Nine of the 12 developed encephalitis and 4 of these succumbed to infection. In comparison, two clusters each of rabies and Balamuthia mandrillaris and three clusters of transplant‐related lymphocytic choriomeningitis virus were identified, underscoring the preeminence of WNV infection in transplant‐related infection, but the overall rarity of the events 4.
We reported our experience with naturally‐acquired WNV encephalomyelitis in transplant recipients in 2004 21, and these six cases with kidney/pancreas transplants, along with 18 others reported in the literature, have recently been reviewed in light of the 2012 outbreak 44. Most of the transplant recipients survived, usually following immunosuppression reduction or discontinuation, but the minority (11/27) had received treatment with either interferon‐a, IVIg (or both) or Omr‐IgG‐am 44. Among the 89 deaths in Texas following the 2012 outbreak, 30 were in the Dallas‐Fort Worth area and one death occurred in a case of fulminant WNV encephalitis in a kidney/pancreas transplant recipient, although two other recipients survived 42. In a recent report of four WNV infections in solid organ transplant recipients all from a common donor, 2 of the 4 patients died 41. When lung, heart and liver recipients were included, Winston et al calculated a 30% mortality rate from published cases 42.
There is no question that some immunocompromised hosts can experience particularly severe neuropathological damage at autopsy, as evidenced by our report of a patient with alpha‐1‐antitrypsin deficiency who underwent single lung transplantation and was maintained on tacrolimus, Cytoxan, and prednisone, before receiving two courses of rituximab for recurrent rejection and presented 6 months later with rapid, fulminant WNV meningoencephalitis and succumbed 23. This patient was found to have severe damage of the bilateral thalami, basal ganglia, substantia nigra, red nuclei, hippocampi, molecular layer of cerebellum and proximal anterior motor nerve roots as well as the entire cross section of the anterior horn regions at spinal cord levels 23.
Immunosuppressed patients may also experience more widespread distribution of virus. In a study by Armah et al from six patients with encephalitis, WNV immunohistochemistry was able to detected virus in neurons of CNS (all six cases), kidney (four cases), lungs (two cases), pancreas (two cases), thyroid (two cases), intestine (two cases), stomach (one case), esophagus (one case), bile duct (one case), skin (one case), prostate (one case) and testis (one case) 2. In systemic organs, epithelial cells were infected. All cases in which viral antigens were identified in systemic organs in addition to CNS were severely immunocompromised transplant recipients. With the exception of testis and brain, most foci of infection were not associated with inflammation, suggesting that when compartmentalized in epithelial cells, WNV may escape immune recognition 2.
Review of Clinical Features of WNV Infection
Following transmission of WNV, most (∼80%) infections are asymptomatic. Approximately 20% of infected individuals develop an acute febrile flu‐like illness (West Nile fever) characterized by fever, headache, fatigue, anorexia, nausea, myalgia and lymphadenopathy. A maculopapular rash involving the trunk and limbs occurs in 25% to 50% 39. Less than 1% of WNV‐infected individuals develop neuroinvasive disease, which can manifest as meningitis, encephalitis, acute flaccid paralysis or a combination of these clinical syndromes 6, 22. An estimated 30% to 40% of patients with neuroinvasive WNV infection develop meningitis, 50% to 60% develop encephalitis, and 5% to 10% develop acute flaccid paralysis 34, 35. Other reported syndromes include chorioretinitis 3, myositis 36 and autonomic nerve dysfunction 11.
Neuroinvasive disease most commonly occurs in older individuals (>60 years old). In one study, the odds ratio (95% CI) of developing encephalitis was 2.2 (1.6 to 3.1) in individuals older than 64 years 19. Additional identified risk factors for encephalitis include hypertension and diabetes 19, 27.
However, greater recognition of the role of WNV infection in children has emerged. In a study of neuroinvasive arboviral disease in children <18 years between 2003 and 2012 in the US. 1217 cases and 22 deaths were reported in the lower 48 states 12. While the majority were caused by La Crosse virus (665 cases, 55%), the second most common etiology was WNV (505 cases, 41%). WNV was more common in older children and adolescents and occurred throughout the country while La Crosse, and other arboviral infections, were more regionally restricted 12.
Specific genetic factors in humans shown to enhance susceptibility to serious WNV disease include single nucleotide polymorphisms in the oligoadenylate synthetase gene, which encodes an interferon inducible enzyme involved in antiviral innate immunity 43, and a genetic deficiency of the chemokine receptor CCR5, which may inhibit trafficking of WNV‐specific CD8+ T‐cells into the CNS 24. Increased expression of adhesion molecules (ICAM‐1, CAM‐1, E‐selectin) on endothelial cells with WNV infection may contribute to white cell infiltration and blood–brain barrier disruption 31.
Proposed models of WNV neuroinvasion include the hematogenous, transneural (including peripheral nervous system to CNS or olfactory nerve to olfactory bulb), or multiple routes 38. However, much of the work on WNV neuroinvasion has admittedly been conducted in murine models, which have limitations 38.
Greater Recognition of Post‐Infectious Sequelae
Mortality from WNV neuroinvasive disease is approximately 12%, and occurs almost exclusively in the subsets of patients with severe encephalitis or severe acute flaccid paralysis. The frequency and severity of sequelae are still not well understood 26, 33, 40. Six months after the acute infection, 40% of patients with movement disorders such as myoclonus, parkinsonism or tremors have residual symptoms, and 20% have ongoing symptoms at 18 months of follow‐up 33. Up to 50–60% of WNV encephalitis survivors report cognitive problems, decreased motor speed, and diminished dexterity 3 months after the initial infection 33, 40.
Long‐term follow‐up by Murray et al of 157 WNV‐infected persons in the Houston, Texas, area most of whom had had neuroinvasive disease (62%) is one of the larger studies 26. Of participants in the study, 74% had been discharged to home, 15% to a long‐term care or rehabilitation facility and 7% to the home of a family member or friend, with 3% dying in hospital 26. Patient‐reported sequelae data following symptomatic WNV infection at 1, 5 and 8 years post‐onset were gathered from 103, 73 and 45 participants, respectively, of whom 60% (1 year), 40% (5 years) and 40% (8 years) still self‐reported symptoms 26. Fatigue, weakness and depression were the top three difficulties on self‐report, followed by difficulty walking/ataxia and memory loss 26. Thus, subjective symptoms particularly predominate in long‐term survivors of WNV infection.
Neuropathological Features of Cases from Recent Outbreaks 2012–2014
Although Colorado was not at the epicenter of the 2012 WNV outbreak, we had several deaths from severe WNV encephalomyelitis that allowed us to further examine the histological features of the disease. Three autopsies were seen at our tertiary care center in 2012, with one each in 2013 and 2014; 5 of the 6 had full autopsy permission for examination of brain and spinal cord. All six deaths had occurred in patients with major immunosuppressive conditions, but only 1 of 6 was over age 65 years. Specifically, fatal cases were identified in a 20‐year‐old female with liver transplant, a 45‐year‐old male with pancreatic/renal transplant, a 51‐year‐old female with rheumatoid arthritis on immunosuppressive medications, a 53‐year‐old female myasthenia gravis on prednisone and rituximab, a 62‐year‐old male with cardiac, pulmonary and neuro‐sarcoidosis and a 75‐year‐old male with mantle cell lymphoma, status post‐bone marrow transplant.
This cohort is similar in terms of risk factors to those seen in previous reports 15, 21. From the early days of the WNV epidemic in the US, it became apparent that the subset of patients coming to autopsy often had significant comorbidities such as congestive heart failure 32, cancer 32, transplantation 21, acquired immunodeficiency syndrome 15, diabetes or chronic obstructive pulmonary disease, although occasional elderly patients without these conditions still succumbed 15. While autopsy cases clearly exist at the extreme end of the WNV infection spectrum, they serve to illustrate which anatomical areas and cells are targeted by the virus in CNS and PNS.
The gross appearance of the brain of a patient succumbing to WNV neuroinvasive disease is usually either normal or shows non‐specific cerebral edema 21. Leptomeningeal opacification is absent. Histologically, almost all patients with encephalitis are found to have glial nodules, but the degree of perivascular cuffing with mononuclear cells is variable, as is the extent of neuronal loss 15, 21. Neuronophagia and, occasionally, focal areas of brain necrosis occasionally can be seen 15, 23. In general, inflammation and loss of neurons is usually most prominent in the gray matter of medulla, pons and midbrain 15, as well as anterior horn cell areas of spinal cord, thalamus and basal ganglia, with lesser involvement in cerebral and cerebellar cortices.
In a 2004 report of 23 autopsy cases from the early years of the epidemic, inflammation was noted in brainstem in 100% (20/20, usually at 3 + level), in cortex in 69% (11/16, usually at 1 + level) and in the cerebellum in 60% (6/10, usually at 1 + level) 15. Patchy brainstem involvement was noted with involvement of various brainstem nuclei including substantia nigra, nucleus of the 12th cranial nerve, dorsal motor nucleus and inferior olivary nucleus. Inflammatory changes were also found in 100% of spinal cords examined (17/17, 1–3 + levels) 15. Within the spinal cord, inflammatory changes were more prominent in the anterior horn in 53% and equal in anterior and posterior horns in 47%. Twenty‐two percent of patients (5/23) had lymphocytic infiltrates involving cranial or spinal nerves, ie, neuritis 15.
WNV antigen, often sparse or focal, was detected by immunohistochemistry in 60% of brainstem, 12.5% of cortex and 41% of spinal cords examined 15.The intensity of antigen staining was maximal in patients dying during the first week of encephalitis, but positive staining was seen as late as 3 weeks after onset in two patients who were immunosuppressed. Antigen was usually detected in neurons and neuronal processes, usually those associated with glial nodules 15. While WNV antigen was often seen only in 1–2 neurons per section in a case, in four patients (two transplant recipients, two patients with malignancies), extensive antigen could be detected in neurons from the pons, medulla, midbrain, cerebellum and cerebral cortex 15. Thus, very severely immunocompromised patients coming to autopsy allows appreciation of the extent of involvement possible in the CNS and PNS.
Five of the six severely immunocompromised patients with WNVE seen at autopsy at our institution from 2012–2014 had examination of the brain. The same CNS and PNS anatomical sites were affected as had been described in many of the original studies 11, 15, 32, but the difference was simply in the severity and widespread extent of the damage. These severely immunocompromised patients (pancreatic/renal transplant, cardiac/pulmonary/neuro‐sarcoidosis, rheumatoid arthritis, mantle cell lymphoma, all on immunosuppressive medications) cases manifested multifocal ovoid areas of tissue necrosis. All cases showed involvement of substantia nigra compacta (Figure 1A–C), encompassing large volumes of the area (Figure 1A), with intense microglial influx and neuronal loss (Figure 1B) and necrosis (Figure 1C). Basal ganglia (Figure 1D) and especially thalamus (Figure 1E,F) showed diffuse microgliosis (Figure 1D) or multifocal ovoid small areas of tissue necrosis (Figure 1F) highlighted by CD68 immunostaining (Figure 1E). Varying degrees of perivascular lymphocytic cuffing (Figure 1D) were present from case to case, with some showing only acellular, acute, patchy ovoid areas of pallor, vacuolization and tissue necrosis (Figure 1F). Collections of macrophages could be seen in areas of longer standing tissue damage (Figure 2A).
Figure 1.
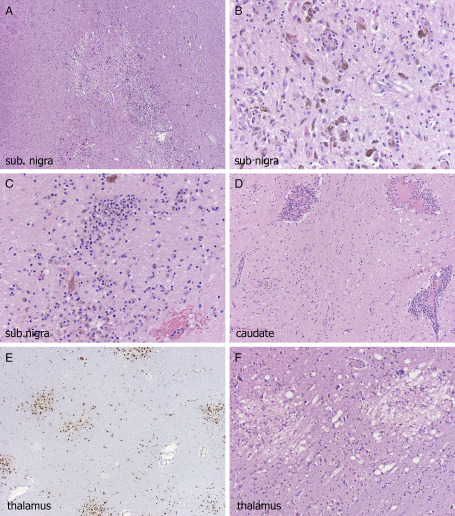
A. Severe zonal cohesive necrosis occupying almost the entire substantia nigra (sub. nigra), with B, profuse microglial influx, and C, foci of necrosis; all cases were severely affected at this site; three different cases illustrated. Caudate (D) was often involved, with diffuse microgliosis and, in this case, profuse perivascular lymphocytic cuffing. Thalamus was usually severely affected along with brainstem and multifocal disease could be highlighted at lower power by CD68 immunohistochemistry (E); acutely ischemic foci showed rarefaction, necrotic neurons, and sometimes minimal cellular response (F).
Figure 2.
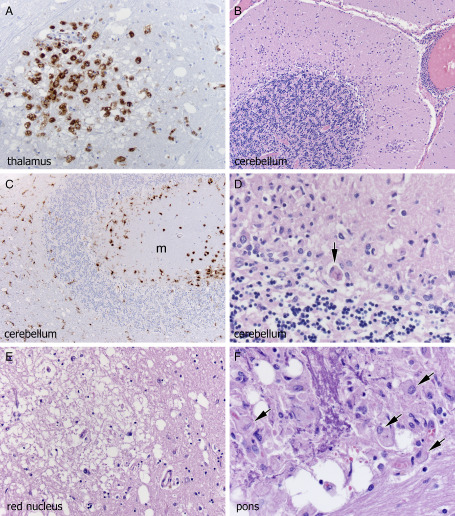
(A) Thalamus here shows necrotic foci with CD68 positive macrophages. Cerebellum was often severely affected by Purkinje cell, but not granule cell, neuronal loss (B–D), microgliosis of the molecular layer (m) (B–D), neuronophagia (D, arrow). Red nucleus could manifest microglial clusters or overt pallor, vacuolization and necrosis, as seen here (E). Basis pontis was frequently severely affected and in this case, multifocal ovoid areas of necrosis contained dystrophic basophilic axonal mineralization (F, upper left); without the admixed macrophages (F, arrows) and more typical non‐mineralized foci elsewhere, this might be construed as pontine leukodystrophy.
The cerebellum was particularly severely affected in 4 of 5 severely immunocompromised patients, compared to early reports 15, 32, with extensive microglial clusters in molecular layer (Figure 2B), widespread Purkinje cell (but not granule cell) neuronal loss (Figure 2B), numerous microglial cells in the Purkinje cell layer as well as white matter (Figure 2C) and near‐granuloma‐like aggregates of histiocytes in a few foci (Figure 2D). Interestingly, despite the severity of damage in cerebellar cortex, the cerebral cortex was relatively minimally affected in all five cases.
Red nucleus (Figure 2E) could be significantly affected, as we illustrated in our 2003 transplant cohort 21 as was basis pontis (Figure 2F) although one of our cases additionally showed dystrophic axonal mineralization in addition to tissue necrosis and macrophage influx (Figure 2F); the cellular response and more typical necrosis elsewhere in the pons distinguished this from pontine leukodystrophy. A few less‐common sites showed microglial influx in these cases including the lateral medullary tegmentum (Figure 3A), inferior olivary nucleus (Figure 3B), optic chiasm and cranial nerve nucleus XII.
Figure 3.
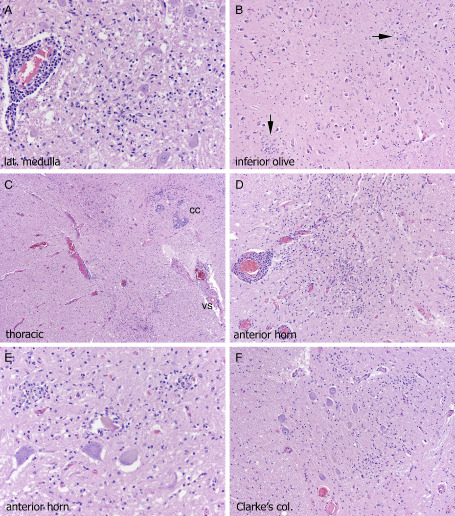
The same anatomical CNS/PNS sites are involved in severely immunocompromised patients as in patients with less severe comorbidities, but they are simply of larger volume and more necrotic; however, occasionally additional sites can be affected such as lateral medullary tegmentum (A), inferior olivary nucleus (B, arrows on microglial clusters), or dorsal gray matter of spinal cord (c, cc = central canal, vs = ventral sulcus). Near‐total cross‐sectional diameter of anterior horn (C,D), with neuronal loss, microglial clusters and neuronophagia (E) can be seen. The extension of inflammation in continuity from dorsal gray matter to anterior horn cell region, with involvement of Clarke's column nucleus (F) lends histological support to theories of possible spread from PNS nerve roots to CNS.
As noted by Guarner et al 15, in the most severe cases, inflammation extended from the dorsal horn in continuity to the anterior horn cell region (Figure 3C), suggesting at least the possibility of viral access route being transneural, in particular PNS to CNS (as reviewed in 38). Near‐total cross‐sectional area of anterior horn could be involved (Figure 3D), with neuronal loss and neuronophagia in anterior horn (Figure 3E) and Clarke's column nucleus (Figure 3F). Neuritis, as illustrated in the original description of the disease by Sampson et al 32 was focally present in both dorsal nerve roots adjacent to spinal cord (Figure 4A) and rarely cranial nerves (Figure 4B). Lymphocytes were generally small and cytologically bland (Figure 4B) but in one patient (pancreatic/renal transplant), more activated inflammatory cells were identified (Figure 4C).
Figure 4.
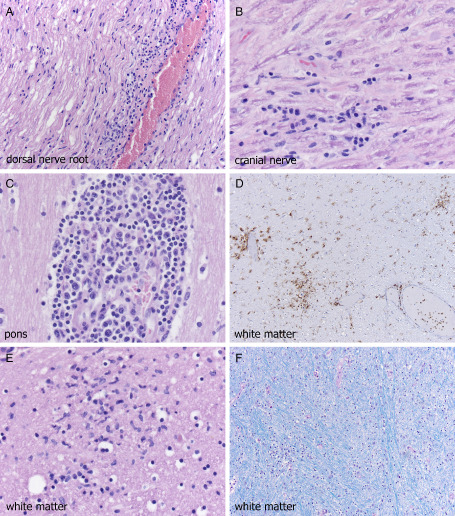
Dorsal nerve root of spinal cord (A) from the mid‐thoracic region shows neuritis, as was noted in the original cases of WNVE, but severely immunocompromised patients may also show cranial nerve neuritis (B). Extent of lymphocytic response is variable from patient to patient and sometimes even site to site, but at times can consist of very activated lymphocytes (E); however small cytologically bland lymphocytes (A,B) are the rule. Despite the very minimal involvement of cerebral cortical gray matter even in these severely affected patients, cerebral white matter can show microgliosis (D,E), the extent of which is highlighted by CD68 immunohistochemistry on low power (D); myelin pallor, however, is usually minimal (F, Luxol fast blue‐periodic acid Schiff).
We have been particularly interested in white matter involvement, given the complaints of cognitive problems 33, and fatigue, weakness, depression and memory loss 26 as post‐infectious sequelae in so many patients who have suffered from WNV infection. Magnetic resonance imaging has clearly shown white matter signal abnormalities in persons infected with WNV 1, 8, 21, 29 and cerebral white matter microgliosis was identified in our 2003 cohort 21. Not surprisingly, several of these recent cases also manifested multifocal white matter CD68 positive microglial infiltrates (Figure 4D) and clusters (Figure 4E) but relatively mild myelin pallor (Figure 4F).
A final unresolved question is whether different WNV strains are responsible for differing severity of CNS tissue damage. Using a mouse model, Donadieu et al investigated phylogenetically distant WNV strains of lineage 1, neuroinvasive WNV(IS98) and a lineage II, non‐neuroinvasive WNV(KUN35 911) strain, The lineage I strain caused more rapid death and was associated with increased level of apoptosis. However, inflammation was higher in the lineage II, WNV(KUN35 911)‐ than in WNV(IS98)‐infected mice 9. While the two strains showed similar tropism in cortex, striatum, brainstem, and cerebellum, they exhibit different tropisms in the hippocampus 9. Thus, there may be different histological features seen not only based on the severity of underlying immunosuppression but on the exact strain that the patient was infected with, although this awaits further validation.
Acknowledgments
The authors thank Mrs. Diane Hutchinson for expert manuscript preparation and Ms. Lisa Litzenberger for photographic expertise.
The authors have nothing to disclose.
References
- 1. Ali M, Safriel Y, Sohi J, Llave A, Weathers S (2005) West Nile virus infection: MR imaging findings in the nervous system. AJNR Am J Neuroradiol 26:289–297. [PMC free article] [PubMed] [Google Scholar]
- 2. Armah HB, Wang G, Omalu BI, Tesh RB, Gyure KA, Chute DJ et al (2007) Systemic distribution of West Nile virus infection: postmortem immunohistochemical study of six cases. Brain Pathol 17:354–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bains HS, Jampol LM, Caughron MC, Parnell JR (2003) Vitritis and chorioretinitis in a patient with West Nile virus infection. Arch Ophthalmol 121:205–207. [DOI] [PubMed] [Google Scholar]
- 4. Basavaraju SV, Kuehnert MJ, Zaki SR, Sejvar JJ (2014) Encephalitis caused by pathogens transmitted through organ transplants, United States, 2002–2013. Emerg Infect Dis 20:1443–1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Biggerstaff BJ, Peterson LR (2002) Estimated risk of West Nile virus transmission through blood transfusion during an epidemic in Queens, New York City. Transfusion 42:1019–1026. [DOI] [PubMed] [Google Scholar]
- 6. Bode AV, Sejvar JJ, Pape WJ, Campbell GL, Marfin AA (2006) West Nile virus disease: a descriptive study of 228 patients hospitalized in a 4‐county region of Colorado in 2003. Clin Infect Dis 42:1234–1240. [DOI] [PubMed] [Google Scholar]
- 7. Dai J, Wang P, Bai F, Town T, Fikrig E (2008) Icam‐1 participates in the entry of West Nile virus into the nervous system. J Virol 82:4164–4168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. DeBiasi RL, Parsons JA, Grabert BE (2005) West Nile virus meningoencephalitis in an immunocompetent adolescent. Pediatr Neurol 33:217–219. [DOI] [PubMed] [Google Scholar]
- 9. Donadieu E, Lowenski S, Servely JL, Laloy E, Lilin T, Nowotny N et al (2013) Comparison of the neuropathology induced by two West Nile virus strains. PLoS ONE 8:e84473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Duggal NK, D'Anton M, Xiang J, Seiferth R, Day J, Nasci R, Brault AC (2013) Sequence analyses of 2012 West Nile virus isolates from Texas fail to associate viral genetic factors with outbreak magnitude. Am J Trop Med Hyg 89:205–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Fratkin JD, Leis AA, Stokic DS, Slavinski SA, Geiss RW (2004) Spinal cord neuropathology in human West Nile virus infection. Arch Pathol Lab Med 128:533–537. [DOI] [PubMed] [Google Scholar]
- 12. Gaensbauer JT, Lindsey NP, Messacar K, Staples JE, Fischer M (2014) Neuroinvasive arboviral disease in the United States: 2003 to 2012. Pediatrics 134:e642–e650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. George BP, Schneider EB, Venkatesan A (2014) Encephalitis hospitalization rates and inpatient mortality in the United States, 2000–2010. PLoS ONE 9:e104169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gould EA, Higgs S (2009) Impact of climate change and other factors on emerging arbovirus diseases. Trans R Soc Trop Med Hyg 103:109–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Guarner J, Shieh WJ, Hunter S, Paddock CD, Morken T, Campbell GL et al (2004) Clinicopathologic study and laboratory diagnosis of 23 cases with West Nile virus encephalomyelitis. Hum Pathol 35:983–990. [DOI] [PubMed] [Google Scholar]
- 16. Huang YJ, Higgs S, Horne KM, Vanlandingham DL (2014) Flavivirus–mosquito interactions. Viruses 6:4703–4730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hurlbut HS, Rizk F, Taylor RM, Work TH (1956) A study of the ecology of West Nile virus in Egypt. Am J Trop Med Hyg 5:579–620. [DOI] [PubMed] [Google Scholar]
- 18. Iwamoto M, Jernigan DB, Guasch A, Trepka MJ, Blackmore CG, Hellinger WC et al (2003) Transmission of West Nile virus from an organ donor to four transplant recipients. NEJM 348:2196–2203. [DOI] [PubMed] [Google Scholar]
- 19. Jean CM, Honarmand S, Louie JK, Glaser CA (2007) Risk factors for West Nile virus neuroinvasive disease, California, 2005. Emerg Infect Dis 13:1918–1920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Johnson MG, Adams J, McDonald‐Hamm C, Wendelboe A, Bradley KK (2015) Seasonality and survival associated with three outbreak seasons of West Nile virus disease in Oklahoma‐2003, 2007, and 2012. J Med Virol. doi: 10.1002/jmv.24235; [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 21. Kleinschmidt‐DeMasters BK, Marder BA, Levi ME, Laird SP, McNutt JT, Escott EJ et al (2004) Naturally acquired West Nile virus encephalomyelitis in transplant recipients: clinical, laboratory, diagnostic, and neuropathological features. Arch Neurol 61:1210–1220. [DOI] [PubMed] [Google Scholar]
- 22. Kramer LD, Li J, Shi PY (2007) West Nile virus. Lancet Neurol 6:171–181. [DOI] [PubMed] [Google Scholar]
- 23. Levi ME, Quan D, Ho JT, Kleinschmidt‐DeMasters BK, Tyler KL, Grazia TJ (2010) Impact of rituximab‐associated B‐cell defects on West Nile virus meningoencephalitis in solid organ transplant recipients. Clin Transplant 24:223–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lim JK, Louie CY, Glaser C, Jean C, Johnson B, Johnson H et al (2008) Genetic deficiency of chemokine receptor CCR5 is a strong risk factor for symptomatic West Nile virus infection: a meta‐analysis of 4 cohorts in the US epidemic. J Infect Dis 197:262–265. [DOI] [PubMed] [Google Scholar]
- 25. Mann BR, McMullen AR, Swetnam DM, Barrett AD (2013) Molecular epidemiology and evolution of West Nile virus in North America. Int J Environ Res Public Health 10:5111–5129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Murray K, Garcia M, Rahbar M, Martinez D, Khuwaja S, Arafat R, Rossmann S (2014) Survival analysis, long‐term outcomes, and percentage of recovery up to 8 years post‐infection among the Houston West Nile virus cohort. PLoS ONE 9:e102953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Nash D, Mostashari F, Fine A, Miller J, O'Leary D, Murray K et al (1999) West Nile Outbreak Response Working Group (2001) The outbreak of West Nile virus infection in the New York City area in 1999. N Engl J Med 344:1807–1814. [DOI] [PubMed] [Google Scholar]
- 28. Petersen LR, Brault AC, Nasci RS (2013) West Nile virus: review of the literature. JAMA 310:308–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Petropoulou KA, Gordon SM, Prayson RA, Ruggierri PM (2005) West Nile virus meningoencephalitis: MR imaging findings. AJNR Am J Neuroradiol 26:1986–1995. [PMC free article] [PubMed] [Google Scholar]
- 30. Rasca L, Gander R, Chung W, Southern P, Le J, Beal S et al (2014) Clinical features of West Nile virus epidemic in Dallas, Texas, 2012. Diagn Microbiol Infect Dis 78:132–136. [DOI] [PubMed] [Google Scholar]
- 31. Roe K, Orillo B, Verma S (2014) West Nile virus‐induced cell adhesion molecules on human brain microvascular endothelial cells regulate leukocyte adhesion and modulate permeability of the in vitro blood‐brain barrier model. PLoS ONE 9:e102598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sampson BA, Ambrosi C, Charlot A, Reiber K, Veress JF, Armbrustmacher V (2000) The pathology of human West Nile virus infection. Hum Pathol 31:527–531. [DOI] [PubMed] [Google Scholar]
- 33. Sejvar JJ (2007) The long‐term outcomes of human West Nile virus infection. Clin Infect Dis 44:1617–1624. [DOI] [PubMed] [Google Scholar]
- 34. Sejvar JJ, Haddad MB, Tierney BC, Campbell GL, Marfin AA, Van Gerpen JA et al (2003) Neurologic manifestations and outcome of West Nile virus infection. JAMA 290:511–515. [DOI] [PubMed] [Google Scholar]
- 35. Sejvar JJ, Leis AA, Stokic DS, Van Gerpen JA, Marfin AA, Webb R et al (2003) Acute flaccid paralysis and West Nile virus infection. Emerg Infect Dis 9:788–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Smith RD, Konoplev S, DeCourten‐Myers G, Brown T (2004) West Nile virus encephalitis with myositis and orchitis. Hum Pathol 35:254–258. [DOI] [PubMed] [Google Scholar]
- 37. Smithburn KC, Hughes TP, Burke AW, Paul JH (1940) A neurotropic virus isolated from the blood of a native of Uganda. Am J Trop Med Hyg 20:471–492. [Google Scholar]
- 38. Suen WW, Prow NA, Hall RA, Bielefeldt‐Ohmann H (2014) Mechanism of West Nile virus neuroinvasion: a critical appraisal. Viruses 6:2796–2825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wang P, Bai F, Zenewicz LA, Dai J, Gate D, Cheng G et al (2012) IL‐22 signaling contributes to West Nile encephalitis pathogenesis. PLoS ONE 7:e44153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Watson JT, Pertel PE, Jones RC, Siston AM, Paul WS, Austin CC, Gerber SI (2004) Clinical characteristics and functional outcomes of West Nile Fever. Ann Intern Med 141:360–365. [DOI] [PubMed] [Google Scholar]
- 41. Wimberly MC, Lamsal A, Giacomo P, Chuang TW (2014) Regional variation of climatic influences on West Nile virus outbreaks in the United States. Am J Trop Med Hyg 91:677–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Winston DJ, Vikram HR, Rabe IB, Dhillon G, Mulligan D, Hong JC et al (2014) Donor‐derived West Nile virus infection in solid organ transplant recipients: report of four additional cases and review of clinical, diagnostic, and therapeutic features. Transplantation 97:881–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Yakub I, Lillibridge KM, Moran A, Gonzalez OY, Belmont J, Gibbs RA, Tweardy DJ (2005) Single nucleotide polymorphisms in genes for 2′‐5′‐oligoadenylate synthetase and RNase L inpatients hospitalized with West Nile virus infection. J Infect Dis 192:1741–1748. [DOI] [PubMed] [Google Scholar]
- 44. Yango AF, Fischbach BV, Levy M, Chandrakantan A, Tan V, Spak C et al (2014) West Nile Virus infection in kidney and pancreas transplant recipients in the Dallas‐Fort Worth Metroplex during the 2012 Texas epidemic. Transplantation 97:953–957. [DOI] [PubMed] [Google Scholar]
- 45. Zanotto PM, Gould EA, Gao GF, Harvey PH, Holmes EC (1996) Population dynamics of flaviviruses revealed by molecular phylogenies. Proc Natl Acad Sci U S A 93:548–553. [DOI] [PMC free article] [PubMed] [Google Scholar]