

Abstract
BRAF V600E mutation and homozygous deletion of CDKN2A (p16) are frequent molecular alterations in pleomorphic xanthoastrocytomas (PXAs). We investigated 49 PXAs for clinical, histological and immunohistochemical characteristics related to BRAF mutation status. BRAF mutation was detected by immunohistochemical assay and DNA sequencing in 38/49 (78%) tumors. All but one PXA located in the temporal lobe harbored a BRAF V600E mutation (23/24; 96%) compared with 10/19 nontemporal PXAs (53%; P = 0.0009). Histological and immunohistochemical analysis demonstrated increased reticulin deposition (76% vs. 27%; P = 0.003) and a more frequent expression of CD34 in BRAF‐mutant PXAs (76% vs. 27%; P = 0.003).
We further investigated the utility of combined BRAF V600E (VE1) and p16 analysis by immunohistochemistry to distinguish PXAs from relevant histological mimics like giant‐cell glioblastoma. Among PXAs, 38/49 (78%) were VE1‐positive, and 30/49 (61%) had a loss of p16 expression. The combined features (VE1 positivity/p16 loss) were observed in 25/49 PXAs (51%) but were not observed in giant‐cell glioblastoma (VE1 0/28, p16 loss 14/28). We demonstrate that temporal location, reticulin deposition and CD34 expression are associated with BRAF mutation in PXA. Combined VE1 positivity and p16 loss represents a frequent immunoprofile of PXA and may therefore constitute an additional diagnostic tool for its differential diagnosis.
Keywords: brain tumor, BRAF V600E, CDKN2A, CD34, immunohistochemistry, pleomorphic xanthoastrocytoma, p16, VE1
Introduction
Pleomorphic xanthoastrocytoma (PXA) is a rare glial tumor mostly affecting children and adolescents. The tumor is often localized in superficial regions of the neocortex and typically presents with a well‐circumscribed growth 15. The histological appearance of PXA is characterized by the presence of pleomorphic giant tumor cells with bizarre shape, foamy/xanthomatous cytoplasm, multinucleation and nuclear atypia in absence of mitotic activity 16, 17. Furthermore, a dense reticulin network frequently surrounds these tumor cells 15. The predominant expression of glial fibrillary acidic protein (GFAP) indicates a mostly astrocytic differentiation, but expression of neuronal markers has also been detected in a considerable fraction of PXAs 17. Therefore, a sharp demarcation between PXA and glioneuronal tumors, especially ganglioglioma, may be challenging in some instances.
Despite their pleomorphic appearance, PXAs often have a more favorable clinical outcome compared with high‐grade astrocytomas with similar histological features 27. Such cases correspond to WHO grade II and are designated as PXA II in this article 15. Nevertheless, malignant histological features have been observed in up to 20% of PXA cases 16, 18, 26, 35. Such cases, designated as PXA with anaplastic features (PXA‐AF), are prone to recurrence and have a less favorable prognosis 16.
The molecular biology of PXA is currently under investigation. Recently, v‐Raf murine sarcoma viral oncogene homolog B1 (BRAF) mutations have been detected in up to 70% of PXAs 12, 13, 33, 34 and have become recognized as a hallmark genetic alteration in these tumors. In two recent exome and extensive panel sequencing studies of a total of 20 PXAs, cases without BRAF mutations (n = 7) either had no detectable mutations (n = 4) or harbored mutations of TSC2 (n = 1) or NF1 (n = 1) or an ETV6–NTRK3 fusion (n = 1) 2, 40. Further, several case reports have previously pointed out a possible association of NF1 germ‐line mutation carriers with the development of PXA 1. Another frequent alteration in PXA is the loss of chromosome locus 9p21, resulting in homozygous deletion of CDKN2A (coding for p16) in up to 60% of cases 39.
A second predominantly pediatric brain tumor with frequent BRAF V600E mutations is ganglioglioma. In a previous project we sought to further characterize ganglioglioma in relation to BRAF mutation status and were able to demonstrate strong associations of BRAF mutation status with expression of synaptophysin, presence of dysplastic neurons, presence of lymphocytic cuffs and younger patient age 24. An immunohistochemical assay using BRAF V600E mutation‐specific antibody VE1 further demonstrated that dysplastic neurons were frequent target cells of BRAF mutation 10, 24.
In PXA, detection of BRAF V600E‐mutated protein by VE1 immunohistochemistry has recently been investigated by two independent studies, both demonstrating high sensitivity and specificity 8, 19.
Although CDKN2A deletions are frequent in PXA, loss of p16 protein expression in PXA has so far not been further investigated. For glioblastoma, an immunohistochemical approach for assessing p16 status had a high sensitivity (81.5%) for combined homozygous and hemizygous deletions (sensitivity for homozygous alone was 95%), although specificity was not optimal (70%), with 30% of cases without CDKN2A deletions showing negative p16 immunohistochemistry 29.
Here we investigated a large series of 49 PXAs for clinical, histological and immunohistochemical characteristics in relation to BRAF mutation status. Further, we investigated the utility of combined VE1 and p16 immunohistochemistry to separate PXA from the clinically relevant differential diagnoses ganglioglioma and giant‐cell glioblastoma.
Materials and Methods
Tissue samples, patient characteristics and histology
In total, 49 PXAs (32 PXA II and 17 PXA‐AF; Supporting Information Table S1) were included in this study. Tissue samples were retrieved from the archives of the Department of Neuropathology of Ruprecht‐Karls‐Universität Heidelberg, the Department of Neuropathology of the University of Hannover, the Institute for Neuropathology of the University of Münster, the Department of Neuropathology of the University of Jena, the Institute of Neuropathology of Justus Liebig University Giessen, the Department of Pathology of the Klinikum Stuttgart, the Institute for Pathology and Neuropathology of the University of Tübingen, the Department of Neuropathology of the University of Bonn, the Institute of Neurology of the Medical University of Vienna and the Institute of Neurology (Edinger‐Institut) of Goethe University in Frankfurt. All tumors were reviewed by members of the Department of Neuropathology at Heidelberg (CK, FS, AvD, DC) and diagnosed according to the revised WHO 2007 classification of brain tumors. Additional patient characteristics are given in Supporting Information Table S1. Hematoxylin‐and‐eosin (HE)‐ and reticulin‐stained slides of all cases were evaluated for the presence of dysplasia in neurons, multinucleated cells, giant cells, xanthomatous cells, perivascular cuffs of lymphocytes, eosinophilic granular bodies, nuclear inclusions, Rosenthal fibers, angiocentric growth pattern, calcifications and extensive hemosiderin deposition. Further assessment was made of structure of tumor matrix, proliferation (mitoses per 10 high‐power fields, HPFs), microvascular proliferation, prominence of capillary network and extent of argyrophilic fibers. These parameters were assessed in 10 suitable HPFs. Intercellular reticulin deposition (argyrophilic fibers) was scored as absent if confined to vessel walls. Furthermore, 28 giant‐cell glioblastomas were analyzed for VE1, p16 and CD34 immunohistochemistry and extent of argyrophilic fibers (Supporting Information Table S2).
Immunohistochemistry
Prior to additional investigations, all cases were tested to ensure they were negative for IDH1 R132H mutation, either by immunohistochemistry (clone H09) or by IDH1 Sanger sequencing as previously described 6. Sections cut to 4 μm were dried at 80°C for 15 minutes and stained with BRAF V600E‐specific clone VE1 on a Ventana BenchMark XT immunostainer (Ventana Medical Systems, Tucson, AZ, USA). The Ventana staining procedure included pretreatment with cell conditioner 1 (pH 8) for 64 minutes, followed by incubation with VE1 hybridoma supernatant (monoclonal, dilution 1:5) at 37°C for 32 minutes. Antibody incubation was followed by incubation with OptiView HQ Universal Linker (Ventana) for 12 minutes, incubation with OptiView HRP Multimer (Ventana) for 12 minutes, signal amplification using the Ventana OptiView Amplification Kit (Ventana, catalogue number 760‐099), and counterstaining with one drop of hematoxylin for 4 minutes and one drop of bluing reagent for 4 minutes. As positive and negative controls, a tissue microarray consisting of four melanoma samples of known BRAF V600 status (two wild‐type, two BRAF V600E) was included in every staining run.
Immunohistochemistry was also performed with antibodies for detecting GFAP (polyclonal, code Z0334, 1:1000, Dako, Glostrup, Denmark), CD34 (clone QBEnd10, 1:2, Thermo Scientific, Waltham, MA, USA), synaptophysin (clone SY38, 1:20, Dako), Ki‐67 (clone SP6, 1:100, Cell Marque Corporation, Rocklin, CA, USA) and phosphorylated extracellular signal‐regulated kinase (pERK; polyclonal, 1:200, Cell Signaling, Danvers, MA, USA); polyclonal, 1:200, Cell Signaling, Danvers, MA, USA) using standard staining procedures, employing the UltraView Universal DAB Detection Kit (Ventana). Anti‐p16 antibody (clone G175–405, 1:200, Becton Dickinson Biosciences, Franklin Lakes, NJ, USA) was visualized with the Ventana OptiView kit under standard conditions.
For evaluation of pERK and p16, tumors with at least 5% of tumor cell nuclei stained were scored positive. Staining of vessels or clearly reactive glia was not considered. VE1 and IDH1 were scored as either positive or negative. For both antibodies, nonspecific staining of macrophages, eosinophilic granular bodies and calcified deposits was observed and was not considered for scoring. For Ki‐67 analysis, tumor areas with the highest Ki‐67 labeling index were evaluated for the fraction of positive cells by counting all cells, excluding vascular cells and lymphocytes, in one 200× microscopic field using a counting grid. The semiquantitative immunoreactive score (IRS) for synaptophysin, GFAP and CD34 31 was calculated as previously described 24. Photographs were taken with a BX53 microscope (Olympus, Tokyo, Japan) equipped with an SC30 digital color camera (Olympus) and Olympus cellSens software.
Sequencing and statistics
BRAF sequencing results of parts of this series were previously reported by Schindler et al 34. For the remaining cases, PCR amplification and sequencing for codon 600 of BRAF were performed as described 34. For DNA extraction, areas of the tumor with the highest available DNA content were chosen and macrodissected. For VE1‐positive cases, areas with the highest concentration of VE1‐positive tumor cells were selected for DNA extraction. In one PXA, Sanger sequencing failed to confirm a BRAF V600E mutation detected by immunohistochemistry, but its presence was confirmed by additional pyrosequencing. Pyrosequencing was performed as previously described 23. All sequencing reactions were carried out in forward and reverse directions. Mutations were identified by visual analysis of the sequence chromatograms using Sequence Pilot software, version 3.1 (JSI‐Medisys, Kippenheim, Germany). GenBank sequence NM_004333 for BRAF (National Center for Biotechnology Information, Bethesda, MD, USA) was used as reference.
A χ2‐test (Pearson) was used to examine associations between nominal variables. Student's t‐test was used to examine the association between nominal and continuous variables. Significance was defined as P < 0.05. JMP 9.0 was used for statistical analysis.
Results
VE1 immunohistochemistry and BRAF mutations in PXA
Mutated BRAF V600E protein was detected in 38/49 (78%) of PXAs. The vast majority of PXA tumor cells were stained by the VE1 immunohistochemical assay, irrespective of their highly variant shapes (Figures 1A and 2A). However, immunohistochemical staining was frequently more intense in the ballooned and xanthomatous cell types (Supporting Information Figure S1). DNA sequencing (Sanger sequencing and, in one case, pyrosequencing) could be performed in 45 cases and confirmed the immunohistochemically detected presence or absence of the BRAF V600E mutation in all cases (Supporting Information Table S1). Sequencing revealed a single PXA with a BRAF 599insT mutation not detected by VE1. In one case with low tumor burden, the BRAF mutation could only be confirmed by pyrosequencing (data not shown).
Figure 1.
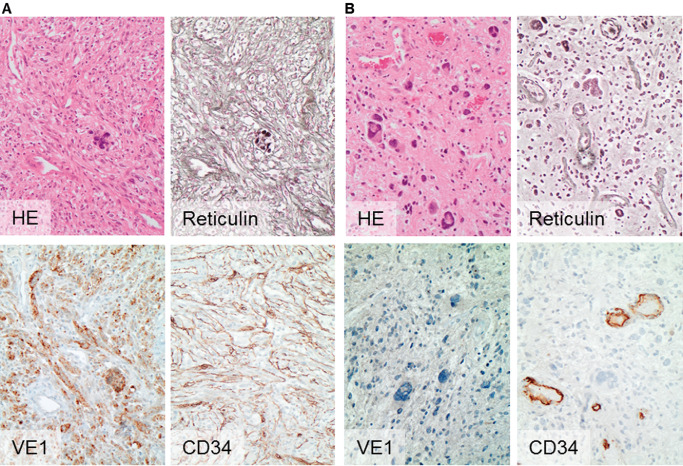
Histology and immunohistochemistry of PXA in relation to BRAF status. PXA (id 60350) of temporal location with BRAF V600E mutation (A) compared with a PXA (id 61998) of nontemporal location of BRAF wild‐type status and without VE1 binding (B). Note the abundance of pericellular reticulin fiber deposits in and the CD34 expression of tumor cells in A, whereas PXAs of BRAF wild‐type status are more likely to be associated with absence of pericellular reticulin deposits and restriction of CD34 expression to the vasculature, as exemplified in case B. Magnification: 200×. HE, hematoxylin and eosin.
Figure 2.
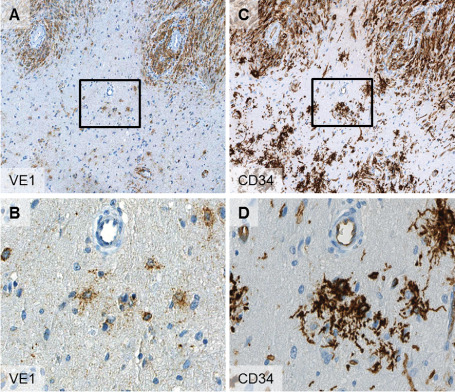
Serial sections of border zone of PXA (id 60358) with solid tumor areas (A,C upper areas) and infiltration zone (A,C lower areas). Note the high congruence of VE1 and CD34 expression in the solid tumor areas as well as in adjacent “satellite” cells in the infiltration zone (B,D). The boxes in (A) and (B) indicate the field of higher magnification seen in (B) and (D). Magnification: 100× (A,C); 400× (B,D).
No differences in the BRAF mutation rates were observed between PXA II (26/33; 79%) and PXA‐AF (12/16; 75%).
Clinical, immunohistochemical and histological features of PXA in relation to BRAF status
The available clinical data for this study comprised tumor location, patient age at operation and gender. Tumor location was available in 43 cases, with 24 tumors originating in the temporal lobe and 19 in other parts of the brain (Table 1). Analysis of BRAF mutation status revealed that 23/24 (96%) of temporally located tumors were BRAF V600E‐mutated, whereas in other locations only 10/19 (53%) harbored this genetic alteration (P = 0.0009). The association of BRAF mutation with location was significant for both PXA II (P = 0.004) and PXA‐AF (P = 0.05). Patients with BRAF‐mutated PXA were younger at operation, but this did not reach statistical significance (P = 0.06; Table 1). No associations between gender and BRAF status were observed (Table 1).
Table 1.
Correlation of BRAF V600E status with clinical data, histology and immunohistochemical features in pleomorphic xanthoastrocytomas
| Parameter | BRAF V600E | BRAF wild type | P value |
|---|---|---|---|
| Patient age (years), median (range) (n = 49) | 21 (5–74) | 27 (18–65) | 0.06 |
| Patient sex (male, female) (n = 49) | 20, 18 | 8, 3 | 0.326 |
| Tumor location (temporal, other) (n = 43) | 23, 10 | 1, 9 | 0.0009 |
| Growth pattern (fibrillary, other) (n = 48) | 24, 13 | 6, 5 | 0.54 |
| Immunohistochemical characteristics | |||
| Synaptophysin‐positive (n = 49), n (%) | 8/38 (21) | 2/11 (18) | 0.84 |
| GFAP‐positive (n = 49), n (%) | 34/38 (94) | 11/11 (100) | 0.26 |
| CD34‐positive (n = 49), n (%) | 29/38 (76) | 3/11 (27) | 0.003 |
| pERK‐positive (n = 49), n (%) | 24/38 (63) | 9/11 (82) | 0.25 |
| p16 loss (n = 49), n (%) | 25/38 (66) | 5/11 (45) | 0.22 |
| Ki‐67‐positive (%), mean (95% CI) (n = 48) | 5.0 (2.9–7.0) | 6.1 (2.3–9.9) | 0.6 |
| Histological characteristics, n (%) | |||
| Eosinophilic granular bodies | 33/38 (87) | 8/11 (73) | 0.26 |
| Reticulin fibers | 29/38 (76) | 3/11 (27) | 0.003 |
| Lymphocytic cuffs | 10/38 (26) | 4/11 (36) | 0.52 |
| Xanthomatous tumor cells | 22/38 (58) | 7/11 (63) | 0.73 |
| Dysplastic neurons | 3/38 (8) | 0/11 (0) | 0.34 |
| Pleomorphic/multinucleated cells | 32/38 (84) | 11/11 (100) | 0.16 |
| Giant cells | 13/36 (36) | 3/9 (33) | 0.88 |
| Rosenthal fibers | 11/38 (29) | 2/11 (18) | 0.48 |
| Tumoral calcifications | 7/38 (18) | 1/11 (9) | 0.46 |
| Extensive hemosiderin deposition | 3/38 (8) | 0/11 (0) | 0.34 |
| Nuclear inclusions | 30/38 (79) | 8/11 (72) | 0.66 |
| Angiocentric growth | 4/38 (11) | 2/11 (18) | 0.5 |
| Prominent capillary network | 0/38 (0) | 0/11 (0) | 1 |
| Endothelial proliferation | 12/38 (32) | 4/11 (36) | 0.77 |
| Necrosis | 9/38 (24) | 3/11 (27) | 0.81 |
| Mitoses (at least 1 in 10 high‐power fields) | 15/38 (39) | 5/11 (45) | 0.72 |
Immunohistochemical analysis of the series was carried out for synaptophysin, GFAP, CD34, pERK, p16 and Ki‐67 (Table 1). Of these, only CD34 was differentially expressed between BRAF wild‐type and BRAF mutant PXA. CD34 expression was present in 29/38 PXAs (76%) with the BRAF V600E mutation and only 3/11 PXAs (27%) without it (P = 0.003) (Figure 1A,B). CD34 expression was more frequently detected in PXA II (70%) than in PXA‐AF (56%). The staining pattern of most CD34‐positive tumor cells was either cytoplasmic (Figure 2C) or a more pericellular, stroma‐like binding (Figure 1A). Adjacent to solid tumor areas we frequently observed isolated CD34‐positive cells, sometimes with the previously described “bushy” staining 11 or reminiscent of “satellite” cells 3 (Figure 2D). On serial sectioning, these isolated CD34‐positive cells were often also found to be VE1‐positive (Figure 2). Results of the other immunohistochemical staining assays are summarized in Table 1.
Among the diverse histological features investigated in PXA (Table 1), only the presence of reticulin fibers was significantly associated with BRAF mutation (Figure 1). This feature was present in 29/38 BRAF‐mutated cases (76%) but in only 3/11 (27%) without BRAF mutation. Additional investigated histological parameters and results are compiled in Table 1; unprocessed data can be reviewed in Supporting Information Table S1.
Combined analysis of BRAF VE1 and p16 expression for PXA differential diagnosis
PXAs are characterized by a high frequency of BRAF V600E mutation and deletions of CDKN2A (p16). As both features can readily be assessed by immunohistochemistry, we further investigated the utility of combined VE1 and p16 analysis to separate PXA from the clinically relevant differential diagnoses giant‐cell glioblastoma and ganglioglioma (Table 2). For p16 analysis, we employed an antibody that has previously demonstrated high specificity 14.
Table 2.
VE1 and p16 immunohistochemistry in pleomorphic xanthoastrocytomas, giant‐cell glioblastomas and gangliogliomas
| Pleomorphic xanthoastrocytoma (n = 49) | Giant‐cell glioblastoma (n = 28) | Ganglioglioma (n = 71)* | |
|---|---|---|---|
| VE1 positivity, n (%) | 38/49 (78) | 0/28 (0) | 41/71 (58) |
| Loss of p16 expression, n (%) | 30/49 (61) | 14/28 (50) | 6/63 (10) |
| Both (%) | 51 | 0 | 5 |
The combination of BRAF V600E mutation and loss of p16 expression is encountered in half of pleomorphic xanthoastrocytomas but is rarely observed in gangliogliomas and absent in giant‐cell glioblastomas.
*Koelsche et al 24.
In PXA, p16 loss was observed in 30/49 tumors (61%). As noted above, VE1 was positive in 38/49 cases (78%). No statistical association between VE1 and p16 was observed in PXA (P = 0.22). The combined features of VE1 positivity and p16 loss were observed in 25/49 PXAs (51%) (Table 2). We additionally analyzed 28 cases of giant‐cell glioblastoma and observed no case positive for VE1, whereas 14/28 cases showed a loss of p16 (Table 2). None of the cases presented with the combined features of VE1 positivity and p16 loss (Table 2, Supporting Information Table S2).
We previously investigated a large cohort of ganglioglioma for both VE1 and p16 immunohistochemistry 24. In that study, VE1 positivity was frequent (41/71; 58%), whereas p16 loss was only observed in 6/63 (10%) gangliogliomas, and only 3/63 (5%) cases showed combined VE1 positivity and p16 loss (Table 2; Supporting Information Table S3). Typical staining patterns for VE1 and p16 of PXA, giant‐cell glioblastoma and ganglioglioma are compiled in Figure 3.
Figure 3.
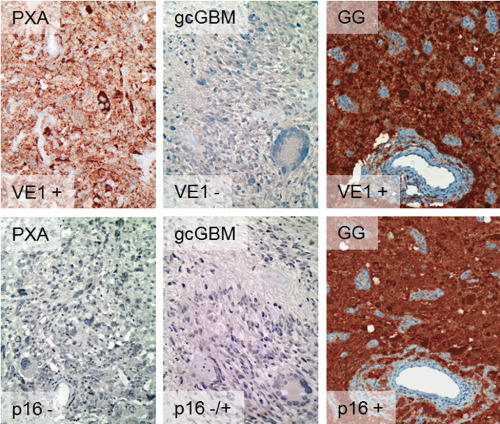
Typical VE 1 and p 16 immunohistochemistry profiles of PXA , giant‐cell glioblastoma ( gcGBM ) and ganglioglioma ( GG ). Among PXAs, 51% of cases were both VE1‐immunopositive (+) and p16‐immunonegative (−). This combination was not observed among 28 giant‐cell glioblastomas, which were VE1‐negative in all cases and either p16‐negative or p16‐positive. Among gangliogliomas, combined VE1‐positive and p16‐negative results were observed in only 3/63 cases (5%), the majority being VE1‐positive and p16‐positive. Data on ganglioglioma from Koelsche et al 24. Magnification: 200×.
Discussion
Our investigation confirms exceedingly high BRAF V600E mutation rates in PXA. The incidence of BRAF V600E mutation in this series is slightly higher than in previous reports relying on DNA‐based methods for mutation detection 12, 13, 34. This is most likely a result of the capability of VE1 immunohistochemistry to detect the mutation in tissues with a low tumor burden and of the rigorous macrodissection approach we applied in obtaining DNA for sequence confirmation. As BRAF V600E‐targeted therapy may in future become an additional therapeutic option for recurrent PXA or PXA undergoing malignant progression 7, implementation of such stringent assays for diagnostic detection of BRAF mutations may soon become essential.
On the other hand, one case with a rare BRAF 599insT mutation was not detected by VE1 immunohistochemistry. Thus, our findings confirm previous observations in other tumor entities that VE1 immunohistochemistry is of a slightly higher sensitivity compared with Sanger sequencing but may fail to detect the rarer types of BRAF mutations 5, 9, 23, 24, 25, 36. However, in contrast to melanoma, with its considerable frequencies of BRAF V600K and other BRAF mutations, alterations other than BRAF V600E seem to be exceedingly rare in PXA.
BRAF mutation rates were surprisingly close for PXA II (79%) and PXA‐AF (75%). This demonstrates the expected close relationship between PXA II and PXA‐AF; on the other hand, this indicates that BRAF V600E mutation is likely an early event in the tumorigenesis of PXA and is likely not a driver of malignant progression to PXA‐AF.
Our analysis of associations between BRAF mutation status and clinicopathological characteristics revealed temporal location as the most significant factor. Our series indicates that temporally located PXAs almost certainly harbor BRAF mutations and adds to the growing body of evidence for specific relationships between genotype and location of brain tumors, as recently demonstrated for mutations of H3F3A 37. Interestingly, such a relationship between location and BRAF status was not observed in our previous series of gangliogliomas, which otherwise showed a similar predilection for the temporal lobe 24. This indicates that such associations between mutation and location may be tumor type‐specific.
In the same study we observed a significant association between younger age and BRAF mutation for ganglioglioma 24. While patients with BRAF‐mutated PXA were also younger (median 21 vs. 27 years) the difference did not reach statistical significance (P = 0.06).
A prominent network of reticulin fibers has been recognized as a hallmark histological feature of PXA 20. Here we demonstrate that the presence of reticulin fibers is strongly associated with the presence of BRAF mutations. An association of BRAF mutation with a more mesenchymal differentiation and prominent intercellular reticulin deposition was also observed in a smaller series of PXAs by Dias‐Santagata et al 12, further corroborating our observation. It is not clear whether BRAF plays a functional role in this mesenchymal shift. In our previous series of gangliogliomas, reticulin fiber deposition was not associated with BRAF mutation—a result not supporting, if not ruling out, a functional connection 24. Interestingly, in thyroid cancer a link between BRAF mutation and epithelial–mesenchymal transition has been established. In a mouse model of thyroid cancer, overexpression of BRAF V600E renders thyroid cells susceptible to transforming growth factor β (TGF‐β)‐induced epithelial–mesenchymal transition 22. In a different model of thyroid cancer, BRAF V600E even directly induced TGF‐β secretion 32. It would be interesting to investigate if mutated BRAF also has a functional interaction with TGF‐β‐mediated signaling in brain tumors.
CD34 is a typical diagnostic marker for PXA and was found in up to 73% of cases in a series by Reifenberger et al 30. This was confirmed in the present series (32/49 PXA, 65%); in line with previous reports, CD34 expression was more frequently detected in PXA II (70%) than in PXA‐AF (56%). CD34 was the only immunohistochemical marker in our series showing significant associations with BRAF mutation. The cause of this association is not clear, but a recent study by Prabowo et al demonstrated associations between BRAF V600E and CD34 expression in both ganglioglioma and dysembryoplastic neuroepithelial tumors 28, indicating that our observation does not apply only to PXA. The CD34 staining pattern was concordant with patterns observed in epilepsy‐associated tumors 3, 8, 11, 38, and samples with infiltration zone and adjacent cortex available for investigation repeatedly presented with the previously described “bushy” CD34 staining in adjacent tissue and more distant CD34‐positive “satellite” cells 3. The nature of these “satellite” cells is unclear, but they have been considered as dysplastic and/or neoplastic transformed precursor cells 4. In our observations, CD34‐positive “satellite‐like” cells in PXA frequently coexpressed BRAF V600E‐mutated protein (Figure 2), thus identifying this cell population as an integral part of the transformed cell population in PXA.
Diagnostic differentiation of PXA and other primary brain tumors, especially ganglioglioma and giant‐cell glioblastoma, may be challenging. We therefore investigated the application of combined VE1 and p16 immunohistochemistry for this diagnostic question.
We observed p16 loss in 61% of PXAs, which is well in line with the expected 50–60% rate of homozygous CDKN2A deletions in these tumors 39. The combined features of VE1 positivity and p16 loss were observed in 51% of PXAs. All giant‐cell glioblastomas were VE1‐negative, confirming our previous genetic investigation of 15 giant‐cell glioblastomas where we did also not detect BRAF mutations 34. The combined features (VE1 negativity and p16 loss) were accordingly not observed in giant‐cell glioblastoma. Very recently, the exceedingly rare epithelioid glioblastoma has shown a high prevalence of BRAF V600E mutations 21. These tumors would probably have a higher occurrence of combined VE1 positivity and p16 loss. Future confirmation of this hypothesis is currently required, possibly together with a more precise definition of epithelioid glioblastoma and delineation from PXA‐AF.
In ganglioglioma we observed the combined features of VE1 positivity and p16 loss in as few as 5% of cases 24. Thus, combined analyses may prove helpful in delineating PXA from giant‐cell glioblastoma and ganglioglioma, especially in small‐sized specimens. In clinical routine, the need may be for delineation either from ganglioglioma or from giant‐cell glioblastoma, but usually not for both at the same time. Thus, in many cases, performing only the VE1 assay for the differentiation of PXA from giant‐cell glioblastoma and only the p16 assay for differentiation from ganglioglioma may also be sufficient.
Conclusion
BRAF V600E mutations are very frequent in both PXA II and PXA‐AF and nearly always present in PXAs located in the temporal lobe. BRAF V600E mutation is further associated with CD34 positivity and abundant formation of reticulin fibers. The combination of VE1 positivity and loss of p16 expression is seen in approximately half of PXAs but very rarely in the important differential diagnoses giant‐cell glioblastoma and ganglioglioma.
Supporting information
Figure S1. VE1 overview staining (A) of a BRAF V600E‐mutated pleomorphic xanthoastrocytoma (ID 59196) with weak to moderate cytoplasmic staining in the vast majority of tumor cells. Pleomorphic multinucleated giant cells presented with a strong cytoplasmic binding. The box indicates the area of higher magnification shown in (B). Magnification: (A) 25×, (B) 200×.
Table S1. Clinical, histological, immunohistochemical and molecular characteristics of the studied Pleomorphic Xanthoastrocytomas. ID = internal patient identifier; Loc = location; Seq = BRAF codon 600 sequencing; wt = wild type; V600E = substitution of valine (V) by a glutamic acid (E); 599insT = 3‐bp insertion at codon position 599 resulting in an additional threonine (T); y = yes; n = no; + = positive; − = negative; VE1 = BRAF V600E mutation‐specific antibody clone VE1; Diag = diagnosis; PXA II = pleomorphic xanthoastrocytoma, WHO grade II; PXA waf = pleomorphic xanthoastrocytoma with anaplastic features; reti = reticulin deposits; EGB = eosinophilic granular bodies; PC/MC = pleomorphic cells/multinucleated cells; XC = xanthomatous cells; TM = tumor matrix; Ro = Rosenthal fibers; Ca = calcium deposits; Hm = hemosiderin; Mi = mitoses; DN = dysplastic neurons; LC = lymphocytic cuffs; PCN = prominent capillary network; EP = endothelial proliferation; Nec = necrosis; GC = giant cells; NI = nuclear inclusions; AG = angiocentric growth; IRS = semiquantitative immunoreactive score; NA = information/data not available.
Table S2. Clinical, histological, immunohistochemical and molecular characteristics of the studied Giant Cell Glioblastomas. ID = internal patient identifier; Loc = location; Seq = BRAF codon 600 sequencing; wt = wild type; y = yes; n = no; + = positive; − = negative; VE1 = BRAF V600E mutation‐specific antibody clone VE1; Diag = diagnosis; gcGBM = giant‐cell glioblastoma, WHO grade IV; Reti = reticulin.
Table S3. p16, VE1 and CD34 staining status (+ = positive; − = negative) according to temporal/nontemporal location in pleomorphic xanthoastrocytoma (A), giant‐cell glioblastoma (B) and ganglioglioma (C).
Acknowledgments
We thank Tanja Göck and Katja Böhmer for excellent technical assistance.
Conflict of interest: David Capper and Andreas von Deimling have applied for a patent on the diagnostic use of BRAF V600E mutant‐specific antibody VE1. All terms are being managed by the German Cancer Research Center in accordance with its conflict of interest policies. The other authors do not have any conflicts of interest to disclose.
References
- 1. Adeleye AO, Okolo CA, Akang EE, Adesina AM (2012) Cerebral pleomorphic xanthoastrocytoma associated with NF1: an updated review with a rare atypical case from Africa. Neurosurg Rev 35:313–319, discussion 9. [DOI] [PubMed] [Google Scholar]
- 2. Bettegowda C, Agrawal N, Jiao Y, Wang Y, Wood LD, Rodriguez FJ et al (2013) Exomic sequencing of four rare central nervous system tumor types. Oncotarget 4:572–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Blumcke I, Giencke K, Wardelmann E, Beyenburg S, Kral T, Sarioglu N et al (1999) The CD34 epitope is expressed in neoplastic and malformative lesions associated with chronic, focal epilepsies. Acta Neuropathol 97:481–490. [DOI] [PubMed] [Google Scholar]
- 4. Blumcke I, Lobach M, Wolf HK, Wiestler OD (1999) Evidence for developmental precursor lesions in epilepsy‐associated glioneuronal tumors. Microsc Res Tech 46:53–58. [DOI] [PubMed] [Google Scholar]
- 5. Bosmuller H, Fischer A, Pham DL, Fehm T, Capper D, von Deimling A et al (2013) Detection of the BRAF V600E mutation in serous ovarian tumors: a comparative analysis of immunohistochemistry with a mutation‐specific monoclonal antibody and allele‐specific PCR. Hum Pathol 44:329–335. [DOI] [PubMed] [Google Scholar]
- 6. Capper D, Weissert S, Balss J, Habel A, Meyer J, Jager D et al (2010) Characterization of R132H mutation‐specific IDH1 antibody binding in brain tumors. Brain Pathol 20:245–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chamberlain MC (2013) Salvage therapy with BRAF inhibitors for recurrent pleomorphic xanthoastrocytoma: a retrospective case series. J Neurooncol 114:237–240. [DOI] [PubMed] [Google Scholar]
- 8. Chappe C, Padovani L, Scavarda D, Forest F, Nanni‐Metellus I, Loundou A et al (2013) Dysembryoplastic neuroepithelial tumors share with pleomorphic xanthoastrocytomas and gangliogliomas BRAF(V600E) mutation and expression. Brain Pathol 23:574–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Colomba E, Helias‐Rodzewicz Z, Von Deimling A, Marin C, Terrones N, Pechaud D et al (2013) Detection of BRAF p.V600E mutations in melanomas: comparison of four methods argues for sequential use of immunohistochemistry and pyrosequencing. J Mol Diagn 15:94–100. [DOI] [PubMed] [Google Scholar]
- 10. Dahiya S, Haydon DH, Alvarado D, Gurnett CA, Gutmann DH, Leonard JR (2013) BRAF(V600E) mutation is a negative prognosticator in pediatric ganglioglioma. Acta Neuropathol 125:901–910. [DOI] [PubMed] [Google Scholar]
- 11. Deb P, Sharma MC, Tripathi M, Sarat Chandra P, Gupta A, Sarkar C (2006) Expression of CD34 as a novel marker for glioneuronal lesions associated with chronic intractable epilepsy. Neuropathol Appl Neurobiol 32:461–468. [DOI] [PubMed] [Google Scholar]
- 12. Dias‐Santagata D, Lam Q, Vernovsky K, Vena N, Lennerz JK, Borger DR et al (2011) BRAF V600E mutations are common in pleomorphic xanthoastrocytoma: diagnostic and therapeutic implications. PLoS ONE 6:e17948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Dougherty MJ, Santi M, Brose MS, Ma C, Resnick AC, Sievert AJ et al (2010) Activating mutations in BRAF characterize a spectrum of pediatric low‐grade gliomas. Neuro‐Oncol 12:621–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Geradts J, Hruban RH, Schutte M, Kern SE, Maynard R (2000) Immunohistochemical p16INK4a analysis of archival tumors with deletion, hypermethylation, or mutation of the CDKN2/MTS1 gene. A comparison of four commercial antibodies. Appl Immunohistochem Mol Morphol 8:71–79. [DOI] [PubMed] [Google Scholar]
- 15. Giannini C, Paulus W, Louis DN, Liberski PP (2007) Pleomorphic xanthoastrocytoma. Chapter 1. In: WHO Classification of Tumours of the Central Nervous System. Louis DN, Ohgaki H, Wiestler OD, Cavenee WK (eds), pp. 22–24. International Agency for Research on Cancer: Lyons, France. [Google Scholar]
- 16. Giannini C, Scheithauer BW, Burger PC, Brat DJ, Wollan PC, Lach B, O'Neill BP (1999) Pleomorphic xanthoastrocytoma: what do we really know about it? Cancer 85:2033–2045. [PubMed] [Google Scholar]
- 17. Giannini C, Scheithauer BW, Lopes MB, Hirose T, Kros JM, VandenBerg SR (2002) Immunophenotype of pleomorphic xanthoastrocytoma. Am J Surg Pathol 26:479–485. [DOI] [PubMed] [Google Scholar]
- 18. Hirose T, Ishizawa K, Sugiyama K, Kageji T, Ueki K, Kannuki S (2008) Pleomorphic xanthoastrocytoma: a comparative pathological study between conventional and anaplastic types. Histopathology 52:183–193. [DOI] [PubMed] [Google Scholar]
- 19. Ida CM, Vrana JA, Rodriguez FJ, Jentoft ME, Caron AA, Jenkins SM, Giannini C (2013) Immunohistochemistry is highly sensitive and specific for detection of BRAF V600E mutation in pleomorphic xanthoastrocytoma. Acta Neuropathologica Communications 1:20. doi: 10.1186/2051-5960-1-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kepes JJ, Rubinstein LJ, Eng LF (1979) Pleomorphic xanthoastrocytoma: a distinctive meningocerebral glioma of young subjects with relatively favorable prognosis. A study of 12 cases. Cancer 44:1839–1852. [DOI] [PubMed] [Google Scholar]
- 21. Kleinschmidt‐DeMasters BK, Aisner DL, Birks DK, Foreman NK (2013) Epithelioid GBMs show a high percentage of BRAF V600E mutation. Am J Surg Pathol 37:685–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Knauf JA, Sartor MA, Medvedovic M, Lundsmith E, Ryder M, Salzano M et al (2011) Progression of BRAF‐induced thyroid cancer is associated with epithelial–mesenchymal transition requiring concomitant MAP kinase and TGFβ signaling. Oncogene 30:3153–3162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Koelsche C, Sahm F, Paulus W, Mittelbronn M, Giangaspero F, Antonelli M et al (2013) BRAF V600E expression and distribution in desmoplastic infantile astrocytoma/ganglioglioma. Neuropathol Appl Neurobiol doi: 10.1111/nan.12072. [DOI] [PubMed] [Google Scholar]
- 24. Koelsche C, Wohrer A, Jeibmann A, Schittenhelm J, Schindler G, Preusser M et al (2013) Mutant BRAF V600E protein in ganglioglioma is predominantly expressed by neuronal tumor cells. Acta Neuropathol 125:891–900. [DOI] [PubMed] [Google Scholar]
- 25. Long GV, Wilmott JS, Capper D, Preusser M, Zhang YE, Thompson JF et al (2013) Immunohistochemistry is highly sensitive and specific for the detection of V600E BRAF mutation in melanoma. Am J Surg Pathol 37:61–65. [DOI] [PubMed] [Google Scholar]
- 26. Okazaki T, Kageji T, Matsuzaki K, Horiguchi H, Hirose T, Watanabe H et al (2009) Primary anaplastic pleomorphic xanthoastrocytoma with widespread neuroaxis dissemination at diagnosis—a pediatric case report and review of the literature. J Neurooncol 94:431–437. [DOI] [PubMed] [Google Scholar]
- 27. Perkins SM, Mitra N, Fei W, Shinohara ET (2012) Patterns of care and outcomes of patients with pleomorphic xanthoastrocytoma: a SEER analysis. J Neurooncol 110:99–104. [DOI] [PubMed] [Google Scholar]
- 28. Prabowo AS, Iyer AM, Veersema TJ, Anink JJ, Schouten‐van Meeteren AY, Spliet WG et al (2013) BRAF V600E mutation is associated with mTOR signalling activation in glioneuronal tumors. Brain Pathol 24:52–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Purkait S, Jha P, Sharma MC, Suri V, Sharma M, Kale SS, Sarkar C (2013) CDKN2A deletion in pediatric versus adult glioblastomas and predictive value of p16 immunohistochemistry. Neuropathology 33:405–412. [DOI] [PubMed] [Google Scholar]
- 30. Reifenberger G, Kaulich K, Wiestler OD, Blumcke I (2003) Expression of the CD34 antigen in pleomorphic xanthoastrocytomas. Acta Neuropathol 105:358–364. [DOI] [PubMed] [Google Scholar]
- 31. Remmele W, Hildebrand U, Hienz HA, Klein PJ, Vierbuchen M, Behnken LJ et al (1986) Comparative histological, histochemical, immunohistochemical and biochemical studies on oestrogen receptors, lectin receptors, and Barr bodies in human breast cancer. Virchows Arch A Pathol Anat Histopathol 409:127–147. [DOI] [PubMed] [Google Scholar]
- 32. Riesco‐Eizaguirre G, Rodriguez I, De la Vieja A, Costamagna E, Carrasco N, Nistal M, Santisteban P (2009) The BRAFV600E oncogene induces transforming growth factor beta secretion leading to sodium iodide symporter repression and increased malignancy in thyroid cancer. Cancer Res 69:8317–8325. [DOI] [PubMed] [Google Scholar]
- 33. Schiffman JD, Hodgson JG, VandenBerg SR, Flaherty P, Polley MY, Yu M et al (2010) Oncogenic BRAF mutation with CDKN2A inactivation is characteristic of a subset of pediatric malignant astrocytomas. Cancer Res 70:512–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Schindler G, Capper D, Meyer J, Janzarik W, Omran H, Herold‐Mende C et al (2011) Analysis of BRAF V600E mutation in 1320 nervous system tumors reveals high mutation frequencies in pleomorphic xanthoastrocytoma, ganglioglioma and extra‐cerebellar pilocytic astrocytoma. Acta Neuropathol 121:397–405. [DOI] [PubMed] [Google Scholar]
- 35. Schmidt Y, Kleinschmidt‐Demasters BK, Aisner DL, Lillehei KO, Damek D (2013) Anaplastic PXA in adults: case series with clinicopathologic and molecular features. J Neurooncol 111:59–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Skorokhod A, Capper D, von Deimling A, Enk A, Helmbold P (2012) Detection of BRAF V600E mutations in skin metastases of malignant melanoma by monoclonal antibody VE1. J Am Acad Dermatol 67:488–491. [DOI] [PubMed] [Google Scholar]
- 37. Sturm D, Witt H, Hovestadt V, Khuong‐Quang DA, Jones DT, Konermann C et al (2012) Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell 22:425–437. [DOI] [PubMed] [Google Scholar]
- 38. Sung CO, Suh YL, Hong SC (2011) CD34 and microtubule‐associated protein 2 expression in dysembryoplastic neuroepithelial tumours with an emphasis on dual expression in non‐specific types. Histopathology 59:308–317. [DOI] [PubMed] [Google Scholar]
- 39. Weber RG, Hoischen A, Ehrler M, Zipper P, Kaulich K, Blaschke B et al (2007) Frequent loss of chromosome 9, homozygous CDKN2A/p14(ARF)/CDKN2B deletion and low TSC1 mRNA expression in pleomorphic xanthoastrocytomas. Oncogene 26:1088–1097. [DOI] [PubMed] [Google Scholar]
- 40. Zhang J, Wu G, Miller CP, Tatevossian RG, Dalton JD, Tang B et al (2013) Whole‐genome sequencing identifies genetic alterations in pediatric low‐grade gliomas. Nat Genet 45:602–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. VE1 overview staining (A) of a BRAF V600E‐mutated pleomorphic xanthoastrocytoma (ID 59196) with weak to moderate cytoplasmic staining in the vast majority of tumor cells. Pleomorphic multinucleated giant cells presented with a strong cytoplasmic binding. The box indicates the area of higher magnification shown in (B). Magnification: (A) 25×, (B) 200×.
Table S1. Clinical, histological, immunohistochemical and molecular characteristics of the studied Pleomorphic Xanthoastrocytomas. ID = internal patient identifier; Loc = location; Seq = BRAF codon 600 sequencing; wt = wild type; V600E = substitution of valine (V) by a glutamic acid (E); 599insT = 3‐bp insertion at codon position 599 resulting in an additional threonine (T); y = yes; n = no; + = positive; − = negative; VE1 = BRAF V600E mutation‐specific antibody clone VE1; Diag = diagnosis; PXA II = pleomorphic xanthoastrocytoma, WHO grade II; PXA waf = pleomorphic xanthoastrocytoma with anaplastic features; reti = reticulin deposits; EGB = eosinophilic granular bodies; PC/MC = pleomorphic cells/multinucleated cells; XC = xanthomatous cells; TM = tumor matrix; Ro = Rosenthal fibers; Ca = calcium deposits; Hm = hemosiderin; Mi = mitoses; DN = dysplastic neurons; LC = lymphocytic cuffs; PCN = prominent capillary network; EP = endothelial proliferation; Nec = necrosis; GC = giant cells; NI = nuclear inclusions; AG = angiocentric growth; IRS = semiquantitative immunoreactive score; NA = information/data not available.
Table S2. Clinical, histological, immunohistochemical and molecular characteristics of the studied Giant Cell Glioblastomas. ID = internal patient identifier; Loc = location; Seq = BRAF codon 600 sequencing; wt = wild type; y = yes; n = no; + = positive; − = negative; VE1 = BRAF V600E mutation‐specific antibody clone VE1; Diag = diagnosis; gcGBM = giant‐cell glioblastoma, WHO grade IV; Reti = reticulin.
Table S3. p16, VE1 and CD34 staining status (+ = positive; − = negative) according to temporal/nontemporal location in pleomorphic xanthoastrocytoma (A), giant‐cell glioblastoma (B) and ganglioglioma (C).