

Abstract
Autophagy is an intracellular lysosomal degradation process, which plays an important role in cell growth and development, and keeping cellular homeostasis in all eukaryotes. Autophagy has multiple physiological functions, including protein degradation, organelle turnover and response to stress. Emerging evidences support the notion that dysregulation of autophagy might be critical for pathogenesis of amyotrophic lateral sclerosis (ALS). The autophagy dysregulation in motor neurons of ALS may occur in different steps of the autophagic process. Recent studies have shown that two ALS associated proteins, TDP‐43 and superoxide dismutase 1 (SOD1), are involved in the abnormal autophagy regulation. Furthermore, it is reported that several genetic mutations in ALS disturb the autophagic process in the motor neurons. This review will provide new evidence of autophagy dysregulation as a critical pathogenic process leading to ALS, and will discuss the prospect of future therapeutic targets using autophagic regulation to treat this disease.
Keywords: amyotrophic lateral sclerosis, autophagy, Cu/Zn superoxide dismutase 1, dynein, lysosome, dysregulation
INTRODUCTION
Amyotrophic lateral sclerosis (ALS) is an adult‐onset devastating neurodegenerative disease. The pathological hallmark of ALS is the selective death of motor neurons in the brain and spinal cord (6). Although the causes of most cases of ALS are not yet undefined, it is believed that a toxic gain of function resulting from abnormal protein aggregation is probably one of the mechanisms contributing to this disease (52). In eukaryotic cells, there are two main systems for degradation of cytoplasmic proteins: the ubiquitin‐proteasome system (UPS) and autophagy 34, 54. The autophagic pathway has been implicated in several neurodegenerative diseases such as Alzheimer's disease (AD), Huntington's disease (HD), Parkinson's disease (PD), frontotemporal dementia and ALS (67). Post‐mortem and experimental neuropathological studies have shown increased number of autophagosomes in the motor neurons of the spinal cord of sporadic and familial ALS patients 42, 46, 58.
Several studies suggest indirectly that activation of autophagy may be protective in some neurodegenerative diseases by enhancing the removal of toxic protein aggregates (10). Targeting autophagy with rapamycin, which induces autophagy through the inhibition of the mammalian target of rapamycin (mTOR) pathway, decreases the abnormal protein aggregates and alleviates disease progression in animal models of HD and other neurodegenerative disorders 55, 60. However, rapamycin could also affect aggregate levels through its well‐known effects on protein translation. It should be pointed out that autophagy may not only operate as a protein degradation mechanism, but also its impairment may lead to detrimental effects in neuronal function and survival 3, 67. Actually, emerging evidence supports the point of view that dysregulation of autophagy (defects in autophagic flux or in specific autophagy‐regulatory processes) contributes to neurodegeneration. In this review article, we provide insight into autophagy dysregulation mechanisms in ALS, which may not only enhance our understanding of the disease pathogenesis, but also help in the design of future therapies to treat this devastating disease.
THE AUTOPHAGIC PROCESS
Autophagy is an evolutionally conserved pathway involving the lysosome that is induced by trophic factor deprivation or starvation (33). According to the different pathways by which cargo is delivered to the lysosome, autophagy is divided into three forms: chaperone‐mediated autophagy (CMA); microautophagy; and macroautophagy (45). In CMA, cytoplasmic proteins containing a KFERQ motif is specifically recognized by a cytosolic chaperone, heat‐shock cognate 70 (HSC70), which targets the cargo protein to the CMA receptor lysosome‐associated membrane protein type‐2A (LAMP2A) 37, 43. A number of proteins of neuropathological significance, including amyloid precursor protein (APP) and α‐synuclein, contain this targeting sequence, supporting a possible role of CMA in neurodegenerative diseases (37). Microautophagy is a poorly understood non‐selective process, in which small quantities of cytoplasm enter the lysosome for degradation through invagination, protrusion or septation of the lysosomal membrane (61). Finally, macroautophagy is characterized by the formation of a double‐membrane delivery vesicle, the autophagosome. Among these three forms of autophagy, macroautophagy is the most studied form, hereafter referred to as autophagy.
Autophagy is a bulk degradation pathway, beginning with the formation of a phagophore (or isolation membrane), which expands to encase portions of cytoplasm containing proteins and organelles into an autophagosome (45). At present, microtubule‐associated light chain 3‐II (LC3‐II) is the major protein marker specifically associated with the autophagosome in eukaryotes (27). Autophagosomes are trafficked along microtubules in a dynein‐dependent manner to lysosomes (45). They mature through fusion with endosomal vesicles or multivesicular bodies (MVBs; amphisomes), then fusing with lysosomes to form autolysomes (5). Finally, the cytoplasmic contents are degraded by lysosomal acid hydrolases. Because the morphology of autophagosomes, amphisomes and autolysosomes form a continuum, the term “autophagic vacuoles” is used frequently to refer to all these structures.
Most of the steps in the autophagic process require the cytoskeletal network. Efficient autophagosome trafficking is essential for autophagic degradation in neurons or other cells 35, 68. Several proteins associated with microtubule transport, such as dynein, histone deacetylase 6 (HDAC6) and p62/SQSTM1 (p62), play crucial roles in the autophagic process 40, 51. Chemical interference with dynein function leads to an accumulation of autophagosomes, suggesting impaired autophagosome/lysosome fusion (68). HDAC6, which interacts with dynein complex, appears to be important for trafficking ubiquitined proteins and lysosomes 24, 29. These data indicate that the impairment of microtubule transport retards the autophagosome/lysosome fusion.
As a complementary pathway for the UPS, which serves as the primary route of degradation for short‐lived proteins, autophagy is responsible for degrading long‐lived proteins and intact organelles or protein complexes (45). Selective forms of autophagy is classified into several subtypes: mitophagy (for mitochondria), nucleophagy (for nucleus), ribophagy (for ribosomes), pexophagy (for peroxisomes), reticulophagy (for endoplasmic reticulum) and xenophagy (for microorganisms) (22).
AUTOPHAGY AND NEURODEGENERATION
Now, at least 35 autophagy‐related (Atg) proteins are considered to be involved in mammalian autophagy initiation and execution (49). It is known that autophagy is essential to neuronal homeostasis, and may in some settings be neuroprotective (41). Mice with conditional knockout of Atg genes die prematurely with extensive neurodegeneration and ubiquitin‐positive pathology, which illustrate the importance of autophagy in neurodegeneration (36). However, when autophagy is excessively induced or defective, it can result in autophagic cell death (type II cell death), which is defined by the massive accumulation of autophagosomes without nuclear condensation (59). Growing evidence supports a role of autophagic dysfunction as a contributing factor in various neurodegenerative diseases associated with abnormal aggregation of disease‐related mutant proteins 3, 67. For example, mutations in presenilin 1, huntingtin, α‐synuclein, parkin, LRRK2 and dynein have been shown to dysregulate autophagic process either at the initiation or maturation steps, or at the substrates recognition (67).
Autophagic vacuoles form at a rate in parallel with protein degradation. Under pathological conditions the rate of autophagic vacuoles formation may exceed the rate of degradation, and such state is called “autophagic stress.” Autophagic stress has been recognized as harmful for cell survival in lysosomal storage disease and neurodegenerative diseases 9, 12. In other words, defects in initiation and maturation stages of autophagic process and imbalances between induction and clearance of autophagosomes can collectively develop autophagic stress 9, 12. Under such condition, neurons are more vulnerable to genetic and environmental disturbance‐induced degeneration.
MOLECULES INVOLVED IN AUTOPHAGY DYSREGULATION IN ALS
Superoxide dismutase 1 (SOD1)
Altered autophagy in SOD1‐G93A mice
Most cases of ALS are sporadic; only ∼5–10% are familial. Mutations in Cu/Zn SOD1 account for approximately 20% of classic familial ALS (57). Transgenic mice overexpressing mutant forms of the SOD1 protein, especially SOD1‐G93A, have been widely used to model ALS. Studies report the altered autophagy in G93A mice that starts from the presymptomatic stage of the disease (42). The number of LC3‐labeled autophagic vacuoles is significant increased in the motor neurons of the spinal cords of G93A mice compared with the age‐matched controls (42). However, it is not known whether the increased autophagic vacuoles in motor neurons are the result of autophagy induction or autophagy flux impairment. Furthermore, while autophagosomes are frequently observed in dying neurons, it is unclear whether autophagy actively participates in the process of motor neuron death in ALS. Accumulating evidence suggests that defects in autophagic flux or in specific autophagy‐regulatory processes, rather than simply induction, may contribute to the motor neuron degeneration (4). A recent report indicates that treatment of G93A mice with rapamycin significantly inactivates the mTOR signaling pathway, causes further accumulation of autophagic vacuoles, but fails to reduce the level of mutant SOD1 aggregates in the spinal cord of transgenic G93A mice (71). This study indicates the possibility of abnormal autophagic flux in ALS. Increased autophagic activity may augment the motor neuron degeneration with the extra accumulation of autophagosomes, causing a state of autophagic stress 9, 12.
SOD1 protein and autophagy
Motor neuron degeneration in ALS is characterized by mitochondrial damage and abnormal protein aggregates or inclusions, which show different morphological features, such as Lewy bodies, skein inclusions and Bunina inclusions (52). Several important proteins including SOD1 and TAR DNA‐binding protein of 43 kDa (TDP‐43) have been identified within these inclusions in both sporadic and familial ALS 1, 50. SOD1 protein is considered as one of the most important components of ALS‐associated inclusions in motor neurons. Accumulating evidence indicates that a gain of toxic function of mutant SOD1 proteins is the cause of ALS (6). Mutant SOD1 can be degraded by autophagy and by the UPS in neuronal and non‐neuronal cells (28). Furthermore, an in vitro study shows that rapamycin can decrease SOD1 aggregates and reduce mutant SOD1‐mediated toxicity (28). Growing evidence supports the assumption that ubiquitin‐interacting proteins such as p62 and HDAC6 are involved in the selective autophagy pathway that targets protein aggregates for clearance (69). Recently, it has been reported that mutant SOD1 can be recognized by p62 and targeted for the autophagy‐lysosome degradation pathway in motor neurons (18). Although autophagy induction may have benefit effects in SOD1 degradation, mutant SOD1 can inhibit the autophagic process and lead to defective clearance (70). Dynein is reported to play an important role in autophagosome trafficking and/or fusion with lysosomes 31, 65. Mutant SOD1 can alter the cellular localization of dynein and inhibit the dynein/dynactin function in neurons (70). It is worth noting that mutant SOD1 impairs the axonal transport of mitochondria and membrane‐bound organelles, leading to their accumulation in the perinuclear location (70). These findings suggest the possibility that mutant SOD1 protein may induce dysfunction of autophagy in ALS.
TDP‐43
TDP‐43 is a nuclear protein functioning in the regulation of transcription and mRNA splicing. Mutations in TDP‐43 are identified in patients with familial and sporadic ALS (50). The neuropathology associated with TDP‐43 mutations consists of inclusions in neurons and glial cells in the spinal cords and brains 50, 62. Furthermore, TDP‐43 is a major component of ubiquitinated protein aggregates found in ALS patients (2). Thus, it is important to explore the degradation of TDP‐43 in ALS patients and experimental models. Under normal conditions, TDP‐43 proteins locate in the nucleus and are degraded through the proteasome pathway. However, in ALS, aberrant modifications and cleavage of TDP‐43 generate the 25 kDa fragments. Because the 25 kDa fragments lack the nuclear localization signal, they aggregate in the cytoplasm (72). The accumulation of abnormally modified TDP‐43 in ALS patients indicates that dysfunction in the intracellular protein degradation systems may be involved in the disease pathogenesis. Recent studies show that inhibition of either the UPS or autophagy dramatically increases TDP‐43 aggregation, suggesting that TDP‐43 can be degraded by both degradative systems (63). Autophagy induction by rapamycin is associated with reduced cytoplasmic TDP‐43 accumulation in neuronal cell lines (8). In addition, an autophagy enhancer, trehalose, can reduce TDP‐43 aggregates by an mTOR‐independent pathway way (20). TDP‐43 aggregates colocalize with the marker of autophagy LC3 and the adaptor protein p62 7, 63. Over‐expression of p62 reduces TDP‐43 aggregation in an autophagy and proteasome‐dependent manner (7). A recent report demonstrates that mice overexpressing mutant TDP‐43 develop a progressive and fatal neurodegenerative disease reminiscent of ALS (66). In this mice model, aggregates of ubiquitinated proteins selectively accumulated in spinal motor neurons and frontal cortex neurons (66). Because the ubiquitinated proteins are the substrates for both UPS and autophagy pathways, it is likely that mutant TDP‐43 may impair protein clearance pathways in motor neurons.
Dynein/dynactin complex
DCTN1 encodes the p150 subunit of dynactin and mutations in this gene can cause familial ALS (47). Transgenic mice that express mutant DCTN1 show increased LC3‐II proteins, and accumulated ubiquitin‐positive aggregates and autophagic vacuoles in affected motor neurons (38). In fact, microtubule‐based vesicular trafficking is essential for delivery of autophagosomes to lysosomes and subsequent fusion, and impaired dynein‐mediated trafficking is associated with impaired autophagosome/lysosome fusion and reduced protein clearance (70). Dynactin is a hereomultimeric protein complex, which binds to both microtubules and cytoplasmic dynein. Dynein promotes the movement of the autophagosome to regions rich in lysosomes (56). Furthermore, it is reported that the mutations in dynein link motor neuron degeneration to defects in axonal transport in SOD1 mutant mice model (21). Another protein that interacts with dynactin, is dynamitin, which inhibits dynein‐based cellular trafficking acts by causing disassembly of dynactin (44). Overexpressing dynamitin can result in motor neuron degeneration in an experimental model (39). These findings raise the possible roles of the dynein/dynactin complex in autophagosome abnormalities observed in ALS.
Charged multivesicular body protein‐2B (CHMP2B)
The endosomal sorting complexes required for transport (ESCRT) are necessary for sorting of integral membrane proteins into MVBs, and their subsequent degradation in the lysosome (16). Mutations in the ESCRT‐III subunit CHMP2B, the mammalian orthologue of Vps2, are associated with frontotemporal dementia and ALS 12, 13. Studies report that autophagic degradation is inhibited in cells depleted of ESCRT subunits and in cells expressing CHMP2B mutants, leading to accumulation of protein aggregates containing ubiquitinated proteins and the cargo adapter p62 (16). Moreover, it is reported that either ESCRT depletion or CHMP2B mutations cause aggregation of TDP‐43 and other proteins (16). These results indicate that efficient autophagic degradation requires functional MVBs, and provide a possible explanation to the autophagic dysregulation in patients with CHMP2B mutations.
Valosin‐containing protein (VCP)
Recently, mutations in VCP have been identified in familial ALS patients, which have previously been considered as causative factors for inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia (IBMPFD) (64). VCP is a member of the AAA‐ATPase family, which interacts with various ubiquitin‐binding partners to regulate multiple ubiquitin‐dependent processes (26). VCP also participates in regulating the autophagic pathway. Loss of VCP activity results in autophagosome accumulation. After autophagy induction, autophagosomes fail to mature into autolysosomes in VCP mutant models (26). Interestingly, VCP works with HDAC6 to control autophagosome maturation and aggresome formation in a ubiquitin‐dependent mechanism (25). Thus, autophagosome maturation defects may contribute to the pathogenesis of ALS in VCP mutant patients.
Ubiquilin 2
UBQLIN 2 encodes the ubiquitin‐like protein ubiquilin 2; mutations in this gene cause dominantly inherited, chromosome‐X‐linked ALS and ALS/dementia (14). Ubiquilin 2, a member of the ubiquilin family, regulates the degradation of ubiquitinated misfolded proteins. Deng et al. reported that the mutations in UBQLIN 2 lead to an impairment of protein degradation in ALS patients, especially the ubiquitin‐mediated proteasomal degradation (14). Because ubiquitin modification of protein aggregates also provides a specific recognition signal for autophagic degradation through p62, it is believed that ubiquilin 2 may influence the autophagic process in ALS.
MECHANISMS OF AUTOPHAGIC DYSREGULATION IN ALS
Because the autophagy process involves multiple steps, it will be interesting to know when and where the autophagic dysfunction may occur in ALS. Figure 1 summarizes the possible mechanisms implicated for how the proteins above can affect the autophagic process in ALS.
Figure 1.
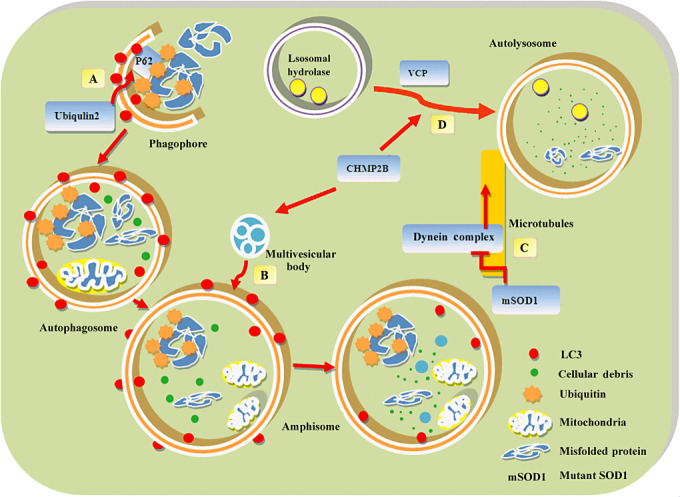
The tentative mechanisms of autophagic dysregulation in ALS. First, the autophagosome formation phase (A). p62, linking ubiquitinated protein aggregates and LC3, brings the ubiquitinated cargo to autophagosome through its interaction with LC3. Ubiquilin 2 may influence substrate recognition through its interaction with p62 or LC3. Second, the autophagosome maturation phase (B). Autophagosomes fusion with multivesicular bodies (MVB) to form an amphisome. Charged multivesicular body protein‐2B (CHMP2B) is a component of MVB, which sorts integral membrane proteins into MVB. Mutations of CHMP2B may impair the fusion of autophagosomes to lysosomes. Third, the autophagosome trafficking phase (C). Autophagosomes move to lysosomes in a microtubule‐ and dynein complex‐dependent manner. Mutations in dynein or dynactin cause defective autophagosome movement. Furthermore, mutant superoxide dismutase 1 (SOD1) proteins may impair autophagosomes clearance through inhibition of dynein/dynactin function. Fourth, the lysosomal fusion phase (D). The fusion of autophagosomes to lysosomes is an important step to complete the autophagic process. Mutation of valosin‐containing protein (VCP) or CHMP2B can lead to impairment in autophagosome/lysosome fusion.
Substrates sequestration
It is now well accepted that selective autophagy plays a major role in the clearance of various cellular structures, including protein aggregates and abnormal mitochondria (32). The clearance of protein aggregates requires the ubiquitination of autophagic substrates 15, 23. Molecules that bind both polyubiquitinated protein and membrane components of the autophagosome mediate the selective autophagic process. P62, a receptor for ubiquitinated protein aggregates is one such adapter, bringing ubiquitinated cargo to the autophagosome through its interaction with LC3 (51). Furthermore, substrate recognition by p62 is not limited to misfolded protein aggregates, but also includes organelles such as mitochondria (30). It is envisaged that mutations in Ubiquilin 2 may change the ubiquitin modification on substrates, influence recognition by p62 and reduce the clearance of ubiquitinated misfolded proteins.
Autophagosome clearance
Recent studies indicate that increased number of autophagic vacuoles is not necessarily associated with increased autophagic flux 3, 4, 67. In SOD1‐G93A mice, rapamycin treatment increases autophagic vacuoles in the motor neurons of spinal cord. However, it causes accumulation of p62, a marker of autophagic flux, which is degraded by autophagy (71). The increased autophagic vacuoles and accumulated p62 raise the possibility of impaired autophagosomes clearance in SOD1‐G93A transgenic mice, which may result from the inhibition of the fusion between autophagosomes and lysosomes and/or impaired protein degradation in the autolysosome.
As a membrane trafficking pathway, autophagy carries cytoplasmic components to the lysosome for degradation. During this process, autophagosomes move to the lysosome in a microtubule‐ and dynein/dynactin complex‐dependent manner (31). The dynein complex plays an important role in the process of autophagosome/lysosome fusion (65). Mutations in dynein or dynactin affecting the dynein machinery cause defective autophagosome movement and autophagosome/lysosome fusion in the motor neurons (38). It is speculated that mutant SOD1 protein may impair autophagosomes clearance through inhibition of dynein/dynactin function in SOD1‐G93A transgenic mice (70).
Similar autophagosome/lysosome fusion defects have also been observed in the mouse models of CHMP2B or VCP mutations. CHMP2B is a component of the MVB, which sorts and delivers membrane proteins to lysosomes through a ubiquitin‐dependent mechanism (16). Mutation of VCP can lead to impairment in autophagosome/lysosome fusion (26). These observations confirm the autophagic dysregulation in ALS, with potential impact on developing effective therapies.
TARGETING AUTOPHAGY IN ALS THERAPY
The recent assumption that autophagy induction might have therapeutic potential for ALS has led to clinical trials to evaluate the effects of several autophagic inducers such as lithium and rapamycin for ALS patients. Lithium has been reported to reduce motor neuron loss in SOD1‐G93A mice, probably through the activation of autophagy and increase of the mitochondriogenesis (17). However, using the same strain of ALS mice and similar treatment protocols, two other investigators did not observed therapeutic or neuroprotective effects of lithium; instead it caused an earlier onset of the disease and a reduction of survival time 19, 53. Moreover, a recent clinical trial reported that therapeutic or subtherapeutic doses of lithium did not differ in the primary outcome of efficacy (survival or loss of autonomy) in ALS patients (11). In addition, concerns about the safety of lithium carbonate as a therapy for ALS patients have been raised after the halting of a trial that found serious adverse effects in its participants. Although rapamycin shows neuroprotective effects in HD and PD models through autophagy‐mediated protein clearance 55, 60, the impact of rapamycin treatment is controversial in ALS 28, 71. In vitro treatment with rapamycin induces the degradation of SOD1 proteins and exerts neuroprotective effects in neurons expressing mutant SOD1 (28). However, an in vivo study reported that rapamycin treatment exacerbates motor neuron loss and disease progression, accompanied by autophagy induction in SOD1‐G93A mice (71). Importantly, no alteration of mutant SOD1 levels was detected after rapamycin treatment, even though it is a known substrate of the autophagy pathway (71). These studies suggest the possibility that autophagy activity or flux may be impaired in SOD1 mutant ALS mice. In‐depth studies are required to directly manipulate the possible impact of autophagy in disease progression and pathogenesis of ALS. The use of rapmaycin analogs (CCI‐779) or molecules that induce mTOR‐independent autophagy, such as trehalose disaccharide, or combinatorial strategies to repair the autophagy deficit and also enhance the activation of the pathway, may be useful to solve this question (48). However, there is a need to explore new autophagic regulators with higher specificity and lower side effects.
CONCLUSIONS
The protein degradation system plays an important role in neurodegenerative diseases. The contribution of autophagy in the pathology of ALS has not yet been fully elucidated. Although autophagic alteration has been confirmed in the ALS patients and experimental models, it remains controversial whether activating autophagy is beneficial or detrimental for the motor neuron degeneration. Accumulating evidence suggests an impaired autophagic process in ALS, caused by the ALS‐associated protein aggregation or genetic mutations. Manipulating the autophagy process is thus a complicated dilemma. It is anticipated that more specific autophagic regulators will be discovered and deeper understanding of the autophagy biology will be advanced in the near future, which will help decode the mystery of autophagy in ALS pathogenesis, and evaluate the therapeutic value of autophagy modulators for this devastating disease.
ACKNOWLEDGMENTS
Work in our laboratory was supported by grant from the National Natural Science Foundation (81171201, 30970925 and 81000541), the National Basic Research Program of China from Science and Technology Commission (2010CB945200 and 2011CB510003).
REFERENCES
- 1. Andersen PM (2006) Amyotrophic lateral sclerosis associated with mutations in the CuZn superoxide dismutase gene. Curr Neurol Neurosci Rep 6:37–46. [DOI] [PubMed] [Google Scholar]
- 2. Arai T, Hasegawa M, Akiyama H, Ikeda K, Nonaka T, Mori H et al (2006) TDP‐43 is a component of ubiquitin‐positive tau‐negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun 351:602–611. [DOI] [PubMed] [Google Scholar]
- 3. Baehrecke EH (2005) Autophagy: dual roles in life and death? Nat Rev Mol Cell Biol 6:505–510. [DOI] [PubMed] [Google Scholar]
- 4. Banerjee R, Beal MF, Thomas B (2010) Autophagy in neurodegenerative disorders: pathogenic roles and therapeutic implications. Trends Neurosci 33:541–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Berg TO, Fengsrud M, Stromhaug PE, Berg T, Seglen PO (1998) Isolation and characterization of rat liver amphisomes. Evidence for fusion of autophagosomes with both early and late endosomes. J Biol Chem 273:21883–21892. [DOI] [PubMed] [Google Scholar]
- 6. Bhatt JM, Gordon PH (2007) Current clinical trials in amyotrophic lateral sclerosis. Expert Opin Investig Drugs 16:1197–1207. [DOI] [PubMed] [Google Scholar]
- 7. Brady OA, Meng P, Zheng Y, Mao Y, Hu F (2010) Regulation of TDP‐43 aggregation by phosphorylation and p62/SQSTM1. J Neurochem 116:248–259. [DOI] [PubMed] [Google Scholar]
- 8. Caccamo A, Majumder S, Deng JJ, Bai Y, Thornton FB, Oddo S (2009) Rapamycin rescues TDP‐43 mislocalization and the associated low molecular mass neurofilament instability. J Biol Chem 284:27416–27424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cherra SJ 3rd, Chu CT (2008) Autophagy in neuroprotection and neurodegeneration: a question of balance. Future Neurol 3:309–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cheung ZH, Ip NY (2011) Autophagy deregulation in neurodegenerative diseases—recent advances and future perspectives. J Neurochem 118:317–325. [DOI] [PubMed] [Google Scholar]
- 11. Chio A, Borghero G, Calvo A, Capasso M, Caponnetto C, Corbo M et al (2010) Lithium carbonate in amyotrophic lateral sclerosis: lack of efficacy in a dose‐finding trial. Neurology 75:619–625. [DOI] [PubMed] [Google Scholar]
- 12. Chu CT (2006) Autophagic stress in neuronal injury and disease. J Neuropathol Exp Neurol 65:423–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cox LE, Ferraiuolo L, Goodall EF, Heath PR, Higginbottom A, Mortiboys H et al (2010) Mutations in CHMP2B in lower motor neuron predominant amyotrophic lateral sclerosis (ALS). PLoS ONE 5:e9872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Deng HX, Chen W, Hong ST, Boycott KM, Gorrie GH, Siddique N et al (2011) Mutations in UBQLN2 cause dominant X‐linked juvenile and adult‐onset ALS and ALS/dementia. Nature 477:211–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Elsasser S, Finley D (2005) Delivery of ubiquitinated substrates to protein‐unfolding machines. Nat Cell Biol 7:742–749. [DOI] [PubMed] [Google Scholar]
- 16. Filimonenko M, Stuffers S, Raiborg C, Yamamoto A, Malerod L, Fisher EM et al (2007) Functional multivesicular bodies are required for autophagic clearance of protein aggregates associated with neurodegenerative disease. J Cell Biol 179:485–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Fornai F, Longone P, Cafaro L, Kastsiuchenka O, Ferrucci M, Manca ML et al (2008) Lithium delays progression of amyotrophic lateral sclerosis. Proc Natl Acad Sci USA 105:2052–2057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gal J, Strom AL, Kwinter DM, Kilty R, Zhang J, Shi P et al (2009) Sequestosome 1/p62 links familial ALS mutant SOD1 to LC3 via an ubiquitin‐independent mechanism. J Neurochem 111:1062–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gill A, Kidd J, Vieira F, Thompson K, Perrin S (2009) No benefit from chronic lithium dosing in a sibling‐matched, gender balanced, investigator‐blinded trial using a standard mouse model of familial ALS. PLoS ONE 4:e6489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gomes C, Escrevente C, Costa J (2010) Mutant superoxide dismutase 1 overexpression in NSC‐34 cells: effect of trehalose on aggregation, TDP‐43 localization and levels of co‐expressed glycoproteins. Neurosci Lett 475:145–149. [DOI] [PubMed] [Google Scholar]
- 21. Hafezparast M, Klocke R, Ruhrberg C, Marquardt A, Ahmad‐Annuar A, Bowen S et al (2003) Mutations in dynein link motor neuron degeneration to defects in retrograde transport. Science 300:808–812. [DOI] [PubMed] [Google Scholar]
- 22. He C, Klionsky DJ (2009) Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet 43:67–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Husnjak K, Elsasser S, Zhang N, Chen X, Randles L, Shi Y et al (2008) Proteasome subunit Rpn13 is a novel ubiquitin receptor. Nature 453:481–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Iwata A, Riley BE, Johnston JA, Kopito RR (2005) HDAC6 and microtubules are required for autophagic degradation of aggregated huntingtin. J Biol Chem 280:40282–40292. [DOI] [PubMed] [Google Scholar]
- 25. Ju JS, Miller SE, Hanson PI, Weihl CC (2008) Impaired protein aggregate handling and clearance underlie the pathogenesis of p97/VCP‐associated disease. J Biol Chem 283:30289–30299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ju JS, Fuentealba RA, Miller SE, Jackson E, Piwnica‐Worms D, Baloh RH et al (2009) Valosin‐containing protein (VCP) is required for autophagy and is disrupted in VCP disease. J Cell Biol 187:875–888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T et al (2000) LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J 19:5720–5728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kabuta T, Suzuki Y, Wada K (2006) Degradation of amyotrophic lateral sclerosis‐linked mutant Cu,Zn‐superoxide dismutase proteins by macroautophagy and the proteasome. J Biol Chem 281:30524–30533. [DOI] [PubMed] [Google Scholar]
- 29. Kawaguchi Y, Kovacs JJ, McLaurin A, Vance JM, Ito A, Yao TP (2003) The deacetylase HDAC6 regulates aggresome formation and cell viability in response to misfolded protein stress. Cell 115:727–738. [DOI] [PubMed] [Google Scholar]
- 30. Kim PK, Hailey DW, Mullen RT, Lippincott‐Schwartz J (2008) Ubiquitin signals autophagic degradation of cytosolic proteins and peroxisomes. Proc Natl Acad Sci USA 105:20567–20574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kimura S, Noda T, Yoshimori T (2008) Dynein‐dependent movement of autophagosomes mediates efficient encounters with lysosomes. Cell Struct Funct 33:109–122. [DOI] [PubMed] [Google Scholar]
- 32. Kirkin V, McEwan DG, Novak I, Dikic I (2009) A role for ubiquitin in selective autophagy. Mol Cell 34:259–269. [DOI] [PubMed] [Google Scholar]
- 33. Klionsky DJ (2007) Autophagy: from phenomenology to molecular understanding in less than a decade. Nat Rev Mol Cell Biol 8:931–937. [DOI] [PubMed] [Google Scholar]
- 34. Klionsky DJ, Emr SD (2000) Autophagy as a regulated pathway of cellular degradation. Science 290:1717–1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kochl R, Hu XW, Chan EY, Tooze SA (2006) Microtubules facilitate autophagosome formation and fusion of autophagosomes with endosomes. Traffic 7:129–145. [DOI] [PubMed] [Google Scholar]
- 36. Komatsu M, Waguri S, Ueno T, Iwata J, Murata S, Tanida I et al (2005) Impairment of starvation‐induced and constitutive autophagy in Atg7‐deficient mice. J Cell Biol 169:425–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kon M, Cuervo AM (2010) Chaperone‐mediated autophagy in health and disease. FEBS Lett 584:1399–1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Laird FM, Farah MH, Ackerley S, Hoke A, Maragakis N, Rothstein JD et al (2008) Motor neuron disease occurring in a mutant dynactin mouse model is characterized by defects in vesicular trafficking. J Neurosci 28:1997–2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. LaMonte BH, Wallace KE, Holloway BA, Shelly SS, Ascano J, Tokito M et al (2002) Disruption of dynein/dynactin inhibits axonal transport in motor neurons causing late‐onset progressive degeneration. Neuron 34:715–727. [DOI] [PubMed] [Google Scholar]
- 40. Lee JY, Koga H, Kawaguchi Y, Tang W, Wong E, Gao YS et al (2010) HDAC6 controls autophagosome maturation essential for ubiquitin‐selective quality‐control autophagy. EMBO J 29:969–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Levine B, Kroemer G (2008) Autophagy in the pathogenesis of disease. Cell 132:27–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Li L, Zhang X, Le W (2008) Altered macroautophagy in the spinal cord of SOD1 mutant mice. Autophagy 4:290–293. [DOI] [PubMed] [Google Scholar]
- 43. Massey AC, Zhang C, Cuervo AM (2006) Chaperone‐mediated autophagy in aging and disease. Curr Top Dev Biol 73:205–235. [DOI] [PubMed] [Google Scholar]
- 44. Melkonian KA, Maier KC, Godfrey JE, Rodgers M, Schroer TA (2007) Mechanism of dynamitin‐mediated disruption of dynactin. J Biol Chem 282:19355–19364. [DOI] [PubMed] [Google Scholar]
- 45. Mizushima N (2007) Autophagy: process and function. Genes Dev 21:2861–2873. [DOI] [PubMed] [Google Scholar]
- 46. Morimoto N, Nagai M, Ohta Y, Miyazaki K, Kurata T, Morimoto M et al (2007) Increased autophagy in transgenic mice with a G93A mutant SOD1 gene. Brain Res 1167:112–117. [DOI] [PubMed] [Google Scholar]
- 47. Munch C, Sedlmeier R, Meyer T, Homberg V, Sperfeld AD, Kurt A et al (2004) Point mutations of the p150 subunit of dynactin (DCTN1) gene in ALS. Neurology 63:724–726. [DOI] [PubMed] [Google Scholar]
- 48. Nassif M, Hetz C (2011) Targeting autophagy in ALS: a complex mission. Autophagy 7:450–453. [DOI] [PubMed] [Google Scholar]
- 49. Nazarko VY, Nazarko TY, Farre JC, Stasyk OV, Warnecke D, Ulaszewski S et al (2011) Atg35, a micropexophagy‐specific protein that regulates micropexophagic apparatus formation in Pichia pastoris. Autophagy 7:375–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT et al (2006) Ubiquitinated TDP‐43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314:130–133. [DOI] [PubMed] [Google Scholar]
- 51. Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H et al (2007) p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem 282:24131–24145. [DOI] [PubMed] [Google Scholar]
- 52. Pasinelli P, Brown RH (2006) Molecular biology of amyotrophic lateral sclerosis: insights from genetics. Nat Rev Neurosci 7:710–723. [DOI] [PubMed] [Google Scholar]
- 53. Pizzasegola C, Caron I, Daleno C, Ronchi A, Minoia C, Carri MT et al (2009) Treatment with lithium carbonate does not improve disease progression in two different strains of SOD1 mutant mice. Amyotroph Lateral Scler 10:221–228. [DOI] [PubMed] [Google Scholar]
- 54. Ravid T, Hochstrasser M (2008) Diversity of degradation signals in the ubiquitin‐proteasome system. Nat Rev Mol Cell Biol 9:679–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Ravikumar B, Vacher C, Berger Z, Davies JE, Luo S, Oroz LG et al (2004) Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet 36:585–595. [DOI] [PubMed] [Google Scholar]
- 56. Ravikumar B, Acevedo‐Arozena A, Imarisio S, Berger Z, Vacher C, O'Kane CJ et al (2005) Dynein mutations impair autophagic clearance of aggregate‐prone proteins. Nat Genet 37:771–776. [DOI] [PubMed] [Google Scholar]
- 57. Rosen DR (1993) Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 364:362. [DOI] [PubMed] [Google Scholar]
- 58. Sasaki S (2011) Autophagy in spinal cord motor neurons in sporadic amyotrophic lateral sclerosis. J Neuropathol Exp Neurol 70:349–359. [DOI] [PubMed] [Google Scholar]
- 59. Scarlatti F, Granata R, Meijer AJ, Codogno P (2009) Does autophagy have a license to kill mammalian cells? Cell Death Differ 16:12–20. [DOI] [PubMed] [Google Scholar]
- 60. Tain LS, Mortiboys H, Tao RN, Ziviani E, Bandmann O, Whitworth AJ (2009) Rapamycin activation of 4E‐BP prevents parkinsonian dopaminergic neuron loss. Nat Neurosci 12:1129–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Todde V, Veenhuis M, van der Klei IJ (2009) Autophagy: principles and significance in health and disease. Biochim Biophys Acta 1792:3–13. [DOI] [PubMed] [Google Scholar]
- 62. Van Deerlin VM, Leverenz JB, Bekris LM, Bird TD, Yuan W, Elman LB et al (2008) TARDBP mutations in amyotrophic lateral sclerosis with TDP‐43 neuropathology: a genetic and histopathological analysis. Lancet Neurol 7:409–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Wang X, Fan H, Ying Z, Li B, Wang H, Wang G (2010) Degradation of TDP‐43 and its pathogenic form by autophagy and the ubiquitin‐proteasome system. Neurosci Lett 469:112–116. [DOI] [PubMed] [Google Scholar]
- 64. Watts GD, Wymer J, Kovach MJ, Mehta SG, Mumm S, Darvish D et al (2004) Inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia is caused by mutant valosin‐containing protein. Nat Genet 36:377–381. [DOI] [PubMed] [Google Scholar]
- 65. Webb JL, Ravikumar B, Rubinsztein DC (2004) Microtubule disruption inhibits autophagosome‐lysosome fusion: implications for studying the roles of aggresomes in polyglutamine diseases. Int J Biochem Cell Biol 36:2541–2550. [DOI] [PubMed] [Google Scholar]
- 66. Wegorzewska I, Bell S, Cairns NJ, Miller TM, Baloh RH (2009) TDP‐43 mutant transgenic mice develop features of ALS and frontotemporal lobar degeneration. Proc Natl Acad Sci USA 106:18809–18814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Wong E, Cuervo AM (2010) Autophagy gone awry in neurodegenerative diseases. Nat Neurosci 13:805–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Yamamoto M, Suzuki SO, Himeno M (2010) The effects of dynein inhibition on the autophagic pathway in glioma cells. Neuropathology 30:1–6. [DOI] [PubMed] [Google Scholar]
- 69. Yao TP (2010) The role of ubiquitin in autophagy‐dependent protein aggregate processing. Genes Cancer 1:779–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. hang F, Strom AL, Fukada K, Lee S, Hayward LJ, Zhu H (2007) Interaction between familial amyotrophic lateral sclerosis (ALS)‐linked SOD1 mutants and the dynein complex. J Biol Chem 282:16691–16699. [DOI] [PubMed] [Google Scholar]
- 71. Zhang X, Li L, Chen S, Yang D, Wang Y, Wang Z, Le W (2011) Rapamycin treatment augments motor neuron degeneration in SOD1(G93A) mouse model of amyotrophic lateral sclerosis. Autophagy 7:412–425. [DOI] [PubMed] [Google Scholar]
- 72. Zhang YJ, Xu YF, Cook C, Gendron TF, Roettges P, Link CD et al (2009) Aberrant cleavage of TDP‐43 enhances aggregation and cellular toxicity. Proc Natl Acad Sci USA 106:7607–7612. [DOI] [PMC free article] [PubMed] [Google Scholar]