

Abstract
The unexpectedly high frequency and universality of alterations to the chromatin machinery is one of the most striking themes emerging from the current deluge of cancer genomics data. Medulloblastoma (MB), a malignant pediatric brain tumor, is no exception to this trend, with a wealth of recent studies indicating multiple alterations at all levels of chromatin processing. MB is typically now regarded as being composed of four major molecular entities (WNT, SHH, Group 3 and Group 4), which vary in their clinical and biological characteristics. Similarities and differences across these subgroups are also reflected in the specific chromatin modifiers that are found to be altered in each group, and each new cancer genome sequence or microarray profile is adding to this important knowledge base. These data are fundamentally changing our understanding of tumor developmental pathways, not just for MB but also for cancer as a whole. They also provide a new class of targets for the development of rational, personalized therapeutic approaches. The mechanisms by which these chromatin remodelers are dysregulated in MB, and the consequences both for future basic research and for translation to the clinic, will be examined here.
Keywords: chromatin, epigenetics, histone, medulloblastoma.
Introduction
Brain tumors currently account for more than a quarter of all deaths from pediatric cancer 3. Medulloblastoma (MB), the most common embryonal brain tumor, is a major contributor to this unfortunate figure. Improvements in patient outcomes over the past few decades have largely been achieved through advances in the application of chemotherapy and craniospinal radiation, as opposed to the biology‐driven risk stratification that has been so successful in treating leukemia, for example 58. A significant fraction of patients will still die from their disease, and those who survive often experience long‐term side effects 19, 42, 63. Thus, there is clearly scope for a shift toward more rational patient stratification and for the application of personalized targeted therapy based on sound tumor‐biological data 55.
In the recent era of high‐throughput genomics, several major advances have been made in terms of our understanding of the biology behind MB tumorigenesis 47. Arguably, the most important paradigm shift has been the recognition that MB is not a single entity, but rather is composed of multiple molecular subgroups (WNT, SHH, Group 3 and Group 4) that differ in their age and gender distribution, clinical outcome and molecular genetic features 10, 34, 35, 48, 75, 76. Despite these differences, some common threads have also emerged. One such feature that is found across subgroups is the recurrent alteration of the chromatin remodeling machinery, as discussed in the following paragraphs. Even here, however, the importance of molecular classification is highlighted by the fact that certain types of chromatin modifier alterations are restricted to specific subgroups.
The packaging and organization of chromatin is the principal mechanism by which cell‐specific transcriptional programs are established and maintained during organismal development 8, 39, 41, including the specification of neural cell types 28, 73. This organization is mediated in large part by an array of post‐translational modifications (PTMs) at key residues on histone tails—the so‐called histone code 29, 36. Genes involved in this process of chromatin regulation are typically grouped into four main categories. First, there are “writers” of covalent histone marks such as histone methyltransferases (HMTs) and histone acetyltransferases (HATs) that establish patterns of PTMs on histone tails. Next, there are “erasers,” histone demethylases (HDMs) and histone deacetylases (HDACs), which can remove these PTMs. An additional class of genes, the “readers” of PTMs, binds to specific histone tail motifs in order to coordinate the downstream consequences of these marks. Finally, there are also large‐scale remodelers of nucleosome structure and chromatin architecture such as the SWI/SNF, INO80 or CHD/NuRD complexes 11, 25, 38, 61, 78, or CTCF 52.
Disruptions of these normal regulatory processes, including in nervous system formation, have been linked to multiple disorders 23, 37, 64. Indeed, it has become increasingly clear in recent years that alterations in how chromatin structure controls cellular processes are one of the most important features of human cancer, irrespective of tumor site or histology 9, 12, 84. Intriguingly, mutations at key regulatory residues in histone proteins themselves (histone 3.1 and the variant 3.3), as well as the histone chaperones ATRX and DAXX, have recently been identified in a subset of another malignant pediatric brain tumor—glioblastoma 33, 68, 81 (see also Fontebasso et al in this issue). Importantly, these alterations in chromatin modifying genes also provide new classes of candidates for targeted therapeutic intervention, with several compounds already showing promise in preclinical or clinical settings 13, 56.
The purpose of this article is to highlight what is currently known about the role of dysregulation of chromatin remodeling in MB and to provide an outlook as to how this knowledge may be expanded upon in the future, as focus shifts toward translating these findings into benefit for patients.
Copy‐Number Changes and Altered Chromatin in MB
The first strong evidence for a role of altered chromatin in MB development emerged from genome‐wide investigation of DNA copy‐number alterations 49. A study by Northcott et al identified multiple changes affecting chromatin modifying genes, including focal gain or amplification of the histone acetyltransferase KAT6A (formerly MYST3) and the lysine demethylases JMJD2B and JMJD2C, together with deletions of polycomb group genes (L3MBTL2, L3MBTL3 and SCML2) and histone methyltransferases (EHMT1, SMYD4). Many of these genes converge on regulation of H3K9 methylation status, and hypomethylation at H3K9 was revealed by immunohistochemical analysis to occur in 41% of tumor samples investigated 49. The same study demonstrated that re‐expression of L3MBTL3 in DAOY cells, in which the gene is deleted, could restore H3K9 methylation and block cellular proliferation.
This work was recently expanded upon in a comprehensive copy‐number profiling analysis of more than 1000 MBs by an international consortium led by the same Toronto group 50, which confirmed the importance of copy‐number alterations at chromatin modifying genes in a subset of cases. These alterations were particularly enriched in Group 4 MBs, and novel homozygous deletions on chromosome X affecting the H3K27 demethylase KDM6A (also called UTX) were identified in this subgroup. Interestingly, this gene was also found to be recurrently mutated in the same subgroup, as described in the following paragraphs.
It is also interesting to note that alterations of chromatin architecture may be a cause of certain structural changes in a cell's DNA rather than simply a consequence. For example, a process of catastrophic rearrangement occurring at a single time point [termed chromothripsis 70] has recently been linked to TP53‐mutated SHH‐subgroup MB (SHH‐MB) 60. One hypothesis as to how these dramatic rearrangements might arise is through a critical loss of specialized chromatin structures at the telomeres, resulting in chromosome end‐to‐end fusions and subsequent mechanical shearing during mitosis 77. A link between germline TP53 mutation [as was frequently observed in SHH‐MB with chromothripsis 60], accelerated telomere shortening and age of tumor onset has previously been established 72. It will therefore be of interest to further investigate the role of telomeric chromatin alterations in the generation of these complex DNA copy‐number changes.
Mutations of Chromatin Modifying Genes—Insights from Large‐Scale Sequencing Studies
The first large‐scale, unbiased sequencing effort in MB, published by Parsons et al in 2011, provided a number of further insights into the role of chromatin modifying genes in this tumor type 53. Several truncating mutations in the histone methyltransferases MLL2 and MLL3 were reported, suggesting a tumor suppressor function of these two genes in MB. Alterations in chromatin modifiers in general were clearly a recurring theme, with rarer mutations being found in the histone demethylase KDM6B, as well as the SWI/SNF chromatin remodeling complex members SMARCA4 and ARID1A, among others. The subgroup specificity of these changes, however, was not clear in this study.
The theme of chromatin modification was also an obvious feature of three next‐generation sequencing studies published in the summer of 2012 30, 57, 62, and was summarized in a recent review and meta‐analysis 47. One‐third of all tumors, across all subgroups, were reported as having a mutation in a gene mapping to the Gene Ontology (GO) term “Chromatin modification” (GO:0015168). A summary of recurrently mutated chromatin modifiers, and their frequency of mutation, is shown in Figure 1.
Figure 1.
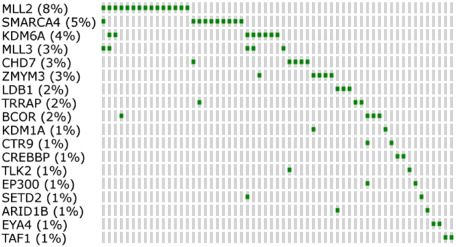
Chromatin modifier genes recurrently altered in medulloblastoma (MB). This figure indicates those genes related to the GO term “Chromatin Modification” (GO:0015168), which are mutated at least twice in the discovery cohorts (whole‐exome or whole‐genome sequencing, n = 189) of the three next‐generation sequencing studies described herein 30, 57, 62. Percentages by gene names indicate the overall frequency of mutation in this gene across the three studies. NB—cases with no mutation in the listed genes are not shown.
Mutations of SMARCA4, as identified in the Parsons et al study, were found to be largely restricted to WNT and Group 3 MB, where they were identified in 25% and 11%, respectively, of tumors in these subgroups 47. Rarer mutations in ARID1B and ARID2 were also observed, again implicating the SWI/SNF complex as an important component of MB development. The question of whether there may be a link between hyperactivation of c‐Myc oncogene signaling (common to WNT and Group 3 tumors) and alterations in this complex will be an interesting avenue for further investigation.
SHH‐MBs showed a strong enrichment for alterations in several members of the nuclear receptor co‐repressor (N‐CoR) complex, which is associated with histone deacetylation and is thought to repress target genes by inducing chromatin condensation 51. Genes of this complex mutated in SHH‐MB include BCL6 co‐repressor (BCOR), LIM‐domain binding 1 (LDB1) and G‐protein pathway suppressor 2 (GPS2). In addition, LIM‐domain only 4 (LMO4), a binding partner of LDB1 14, has been identified as a recurrently amplified gene in SHH‐MB 50, further supporting a key role for this complex in the pathogenesis of this subgroup.
One of the most striking single gene changes was the recurrent inactivating mutation of KDM6A in 12% of Group 4 tumors 47. This gene acts in concert with the most commonly mutated chromatin modifier in MB as a whole, MLL2, to elevate H3K4me3 and remove H3K27me3 marks in order to activate target genes 65, 66. Robinson et al further speculated that this loss of KDM6A function, as well as additional mutations in ZMYM3 and CHD7 (regulators of H3K4me3‐mediated patterns) and overexpression of the H3K27 methyltransferase EZH2, may lead to an imbalance between trimethylated H3K4 and H3K27 and altered differentiation signaling in this subgroup 62 (see Figure 2).
Figure 2.
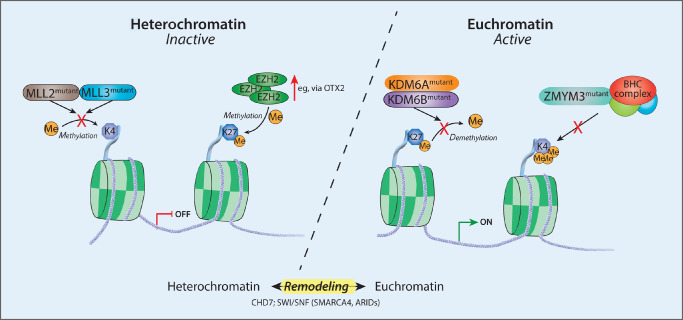
Schematic of key chromatin‐related mutations in medulloblastoma (MB). This schematic view indicates some of the recurrently mutated chromatin modifier genes in medulloblastoma, and their possible effects on disrupting chromatin marks, with a particular focus on the disrupted H3K4/H3K27 methylation frequently observed in Group 3 and Group 4 tumors.
In a follow‐up study of these sequencing efforts, Dubuc et al screened a large series of MBs specifically for mutations in MLL2 and KDM6A 17. MLL2 mutations were seen in approximately 8% of MBs, with roughly half of those predicted to be truncating alterations and no clear enrichment in a particular MB subgroup. Alterations of KDM6A, in contrast, were largely restricted to Group 4 tumors (in keeping with the above sequencing and copy‐number studies) and were mostly truncating 17. The two alterations were found to be mutually exclusive, as might be expected from the above‐noted similarities in the predicted outcome of these changes. When further considering copy‐number and transcriptional changes of EZH2, KDM6A and KDM6B, the authors found further support for the concept postulated by Robinson et al 62 that a subset of Group 3 and Group 4 MBs may show an enrichment of H3K27me3 marks (“K27+”). This was further supported by immunohistochemical staining for various histone marks, as well as an overrepresentation of differential expression of polycomb repressive complex 2 (PRC2) targets (which are typically linked to H3K27me3 levels) in Group 3 and Group 4 tumors with or without this K27+ phenotype 17.
As with copy‐number changes, evidence has recently emerged that the relationship between these DNA mutations and chromatin architecture may not be solely a one‐way process. For example, it seems that certain marks of heterochromatin, particularly H3K9 trimethylation, are highly enriched in regions of the genome that show a higher rate of mutation 67, indicating perhaps that DNA repair processes are not as efficient in these repressive chromatin domains. It is unclear as yet whether a similar phenomenon may be shaping the mutational landscape of somatic changes in MB.
Other Links Between Chromatin Alterations and MB
As noted earlier, several of the reported alterations in chromatin modifying genes seem to converge on methylation of lysine 27 of histone 3, particularly elevated H3K27me3 in Group 3 and Group 4 tumors 17, 62. In a similar vein, one recent report investigated the link between polycomb group genes (required for induction of H3K27 methylation) and the transcription factor OTX2 7, which is occasionally amplified and almost universally overexpressed in Group 3 and Group 4 tumors 1, 6, 50. The authors found upregulation of multiple genes belonging to PRC1 and PRC2, including EZH2, EED and SUZ12, across MB subgroups, but particularly in Group 3 and Group 4 MBs 7. Upon silencing of OTX2 in the MB cell line D425, Bunt et al saw downregulation of these polycomb genes in conjunction with induction of H3K27 demethylases (e.g. KDM6A, KDM6B and KDM7A), resulting in reduced levels of H3K27me3 7. Thus, further investigation of the role of OTX2 as a regulator of key chromatin modifiers in certain MB subgroups may be warranted.
In addition to these OTX2‐mediated changes, other examples of polycomb deregulation have also been reported. In particular, the PRC1 regulator and stem cell self‐renewal factor BMI1 has been linked to SHH‐MB [the only MB subgroup in which OTX2 is essentially not expressed 1]. Using Bmi1‐null mice, Leung et al first showed in 2004 that Bmi1 is essential for normal cerebellar development 40. The same study identified a subset of human MB characterized by concordant overexpression of PTCH1 and BMI1. This was built upon by Michael et al in 2008, in a study which used a SmoA1 mouse model to demonstrate that Bmi1 is essential for SHH‐MB tumorigenesis 43. A recent report also identified a feedback loop between hedgehog pathway activation and BMI1, whereby Shh ligand can induce expression of BMI1 in MB tumor‐initiating cells, which then further upregulates hedgehog target genes 79.
Outlook
The current era of MB research is an exciting one. Significant advances have been made over the past few years, with the development of important subgroup classification schema and the identification of multiple prognostic markers. As large‐scale genomics projects have come to fruition, chromatin remodeling defects have taken center stage in multiple studies. There is still much to be done, however, in translating this increased knowledge into true clinical benefit. While the genetic alterations affecting chromatin remodelers are becoming better understood, the downstream epigenetic consequences of these alterations remain relatively understudied. In addition, it is not clear whether these changes in chromatin architecture are really seen across the whole genome, or whether they affect only a subset of specific targets. The transcriptional consequences of mutations in chromatin modifiers are also still somewhat unclear. More detailed characterization of the MB epigenome will therefore be an important area of focus in the coming years. Several such studies are under way, for example, under the auspices of International Cancer Genome Consortium (ICGC) projects in Germany and Canada or the Pediatric Cancer Genome Project in the USA, to look at global DNA methylation (Illumina Infinium 450k arrays and whole‐genome bisulfite sequencing), non‐coding RNAs (RNA‐seq, miRNA‐seq) and patterns of critical histone modifications (ChIP‐seq).
The repertoire of potential therapeutic targets has also expanded rapidly in the genomics era. Many novel therapeutic agents targeting chromatin modifiers are currently in development or in early stage trials, although not many have achieved Food and Drug Administration (FDA) approval as yet 5. The best known class of approved drugs are histone deacetylase inhibitors such as vorinostat 31 and romidepsin 26, which are both in phase I/II clinical trials for pediatric brain tumors (see http://www.clinicaltrials.gov). Vorinostat has shown some promising results in preclinical MB models 44, 46, 69, 74, and knockdown of specific HDACs in MB cells results in decreased proliferation and cell viability 45. More recently, inhibition of bromodomain‐containing proteins, which interact with acetylated histones and recruit transcriptional regulators 59, has been suggested as one way of indirectly targeting Myc‐driven tumorigenesis 15. This is of particular interest given the strong transcriptional upregulation and/or amplification of c‐Myc in WNT and Group 3 MBs. Targeting the aberrant regulation of H3K27 methylation observed in MB has also been suggested as a therapeutic avenue 17, 62. As further support for this concept, DZNep (3‐deazaneplanocin A), an inhibitor of the H3K27 methyltransferase EZH2, has recently been shown to suppress MB cell growth in vitro 2. Phase II clinical trials of EZH2 inhibitors are now part of the Children's Oncology Group (COG) blueprint for MB research 20. Particularly for those tumors with elevated OTX2 expression, the combination of polycomb inhibition with a differentiating agent such as all‐trans retinoic acid (ATRA) has also been suggested 4, 7, 16. As it seems, however, that the global process of histone methylation is affected, rather than a single gene or pathway, therapeutic targeting of these alterations may prove challenging.
Finally, the development of tumor models that faithfully recapitulate these epigenetic alterations will also be crucial, both for further characterizing the biological changes resulting from epigenetic dysregulation and for the development and testing of novel therapeutics. The majority of the established MB cell lines are thought to most closely resemble Group 3 MB, with no WNT models and only poor representation of the SHH and Group 4 subgroups. As with any long‐term culture model, there are also concerns as to how well these lines truly reflect the primary tumor. As such, a lot of effort is currently being put into developing suitable in vivo models. There are already genetically engineered mouse models (GEMMs) of three of the four molecular subgroups of MB 18: WNT 21, SHH 22, 24, 27, 80, 82, 83 and Group 3 32, 54. A MYCN‐driven model that may resemble Group 4 tumors in certain contexts has also been established 71. None of these GEMMs, however, are driven by alterations in chromatin modifiers, and it will therefore be necessary to build a larger repertoire to model the variety of changes identified through high‐throughput genomics. A number of research groups are also building collections of xenografted tumor material from the various molecular subgroups, which will help recapitulate the wide spectrum of alterations observed in primary tumors and provide a useful additional tool for preclinical testing of targeted agents 85.
Thus, while it is right to acknowledge the multiple advances that have recently been made in the field, building on these insights into the role of chromatin remodeling in MB in order to transfer this knowledge to the bedside will be one of the major challenges in the emerging post‐genomics era.
Acknowledgments
The authors are supported by the PedBrain Tumor Project contributing to the International Cancer Genome Consortium, funded by German Cancer Aid (109252) and by the German Federal Ministry of Education and Research (BMBF, Grant No. 01KU1201A); the Dutch Cancer Foundations KWF (2010‐4713) and KIKA (MK), and the Roman Herzog Postdoctoral Fellowship funded by the Hertie Foundation and the German Cancer Research Center (PAN). Dr Christian Smith is acknowledged for assistance with artwork.
References
- 1. Adamson DC, Shi Q, Wortham M, Northcott PA, Di C, Duncan CG et al (2010) OTX2 is critical for the maintenance and progression of Shh‐independent medulloblastomas. Cancer Res 70:181–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Alimova I, Venkataraman S, Harris P, Marquez VE, Northcott PA, Dubuc A et al (2012) Targeting the enhancer of zeste homologue 2 in medulloblastoma. Int J Cancer 131:1800–1809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Altekruse SF, Kosary CL, Krapcho M, Neyman N, Aminou R, Waldron W et al (2010) SEER Cancer Statistics Review, 1975–2007. National Cancer Institute: Bethesda, MD. [Google Scholar]
- 4. Bai R, Siu IM, Tyler BM, Staedtke V, Gallia GL, Riggins GJ (2010) Evaluation of retinoic acid therapy for OTX2‐positive medulloblastomas. Neuro Oncol 12:655–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Baylin SB, Jones PA (2011) A decade of exploring the cancer epigenome—biological and translational implications. Nat Rev Cancer 11:726–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Boon K, Eberhart CG, Riggins GJ (2005) Genomic amplification of orthodenticle homologue 2 in medulloblastomas. Cancer Res 65:703–707. [PubMed] [Google Scholar]
- 7. Bunt J, Hasselt NA, Zwijnenburg DA, Koster J, Versteeg R, Kool M (2012) OTX2 sustains a bivalent‐like state of OTX2‐bound promoters in medulloblastoma by maintaining their H3K27me3 levels. Acta Neuropathol. [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 8. Butler JS, Koutelou E, Schibler AC, Dent SY (2012) Histone‐modifying enzymes: regulators of developmental decisions and drivers of human disease. Epigenomics 4:163–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chi P, Allis CD, Wang GG (2010) Covalent histone modifications—miswritten, misinterpreted and mis‐erased in human cancers. Nat Rev Cancer 10:457–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cho YJ, Tsherniak A, Tamayo P, Santagata S, Ligon A, Greulich H et al (2010) Integrative genomic analysis of medulloblastoma identifies a molecular subgroup that drives poor clinical outcome. J Clin Oncol 29:1424–1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Conaway RC, Conaway JW (2009) The INO80 chromatin remodeling complex in transcription, replication and repair. Trends Biochem Sci 34:71–77. [DOI] [PubMed] [Google Scholar]
- 12. Conte M, Altucci L (2012) Molecular pathways: the complexity of the epigenome in cancer and recent clinical advances. Clin Cancer Res 18:5526–5534. [DOI] [PubMed] [Google Scholar]
- 13. Dawson MA, Kouzarides T (2012) Cancer epigenetics: from mechanism to therapy. Cell 150:12–27. [DOI] [PubMed] [Google Scholar]
- 14. Deane JE, Ryan DP, Sunde M, Maher MJ, Guss JM, Visvader JE, Matthews JM (2004) Tandem LIM domains provide synergistic binding in the LMO4:Ldb1 complex. Embo J 23:3589–3598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Delmore JE, Issa GC, Lemieux ME, Rahl PB, Shi J, Jacobs HM et al (2011) BET bromodomain inhibition as a therapeutic strategy to target c‐Myc. Cell 146:904–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Di C, Liao S, Adamson DC, Parrett TJ, Broderick DK, Shi Q et al (2005) Identification of OTX2 as a medulloblastoma oncogene whose product can be targeted by all‐trans retinoic acid. Cancer Res 65:919–924. [PubMed] [Google Scholar]
- 17. Dubuc AM, Remke M, Korshunov A, Northcott PA, Zhan SH, Mendez‐Lago M et al (2012) Aberrant patterns of H3K4 and H3K27 histone lysine methylation occur across subgroups in medulloblastoma. Acta Neuropathol. [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Eberhart CG (2012) Three down and one to go: modeling medulloblastoma subgroups. Cancer Cell 21:137–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gajjar A, Chintagumpala M, Ashley D, Kellie S, Kun LE, Merchant TE et al (2006) Risk‐adapted craniospinal radiotherapy followed by high‐dose chemotherapy and stem‐cell rescue in children with newly diagnosed medulloblastoma (St Jude Medulloblastoma‐96): long‐term results from a prospective, multicentre trial. Lancet Oncol 7:813–820. [DOI] [PubMed] [Google Scholar]
- 20. Gajjar A, Packer RJ, Foreman NK, Cohen K, Haas‐Kogan D, Merchant TE on behalf of the COG Brain Tumor Committee (2012) Children's Oncology Group's 2013 blueprint for research: central nervous system tumors. Pediatr Blood Cancer. [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gibson P, Tong Y, Robinson G, Thompson MC, Currle DS, Eden C et al (2010) Subtypes of medulloblastoma have distinct developmental origins. Nature 468:1095–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Goodrich LV, Milenkovic L, Higgins KM, Scott MP (1997) Altered neural cell fates and medulloblastoma in mouse patched mutants. Science 277:1109–1113. [DOI] [PubMed] [Google Scholar]
- 23. Graff J, Mansuy IM (2009) Epigenetic dysregulation in cognitive disorders. Eur J Neurosci 30:1–8. [DOI] [PubMed] [Google Scholar]
- 24. Hallahan AR, Pritchard JI, Hansen S, Benson M, Stoeck J, Hatton BA et al (2004) The SmoA1 mouse model reveals that notch signaling is critical for the growth and survival of sonic hedgehog‐induced medulloblastomas. Cancer Res 64:7794–7800. [DOI] [PubMed] [Google Scholar]
- 25. Hargreaves DC, Crabtree GR (2011) ATP‐dependent chromatin remodeling: genetics, genomics and mechanisms. Cell Res 21:396–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Harrison SJ, Bishton M, Bates SE, Grant S, Piekarz RL, Johnstone RW et al (2012) A focus on the preclinical development and clinical status of the histone deacetylase inhibitor, romidepsin (depsipeptide, Istodax(®)). Epigenomics 4:571–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hatton BA, Villavicencio EH, Tsuchiya KD, Pritchard JI, Ditzler S, Pullar B et al (2008) The Smo/Smo model: hedgehog‐induced medulloblastoma with 90% incidence and leptomeningeal spread. Cancer Res 68:1768–1776. [DOI] [PubMed] [Google Scholar]
- 28. Hirabayashi Y, Gotoh Y (2010) Epigenetic control of neural precursor cell fate during development. Nat Rev Neurosci 11:377–388. [DOI] [PubMed] [Google Scholar]
- 29. Jenuwein T, Allis CD (2001) Translating the histone code. Science 293:1074–1080. [DOI] [PubMed] [Google Scholar]
- 30. Jones DTW, Jager N, Kool M, Zichner T, Hutter B, Sultan M et al (2012) Dissecting the genomic complexity underlying medulloblastoma. Nature 488:100–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kavanaugh SM, White LA, Kolesar JM (2010) Vorinostat: a novel therapy for the treatment of cutaneous T‐cell lymphoma. Am J Health Syst Pharm 67:793–797. [DOI] [PubMed] [Google Scholar]
- 32. Kawauchi D, Robinson G, Uziel T, Gibson P, Rehg J, Gao C et al (2012) A mouse model of the most aggressive subgroup of human medulloblastoma. Cancer Cell 21:168–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Khuong‐Quang DA, Buczkowicz P, Rakopoulos P, Liu XY, Fontebasso AM, Bouffet E et al (2012) K27M mutation in histone H3.3 defines clinically and biologically distinct subgroups of pediatric diffuse intrinsic pontine gliomas. Acta Neuropathol 124:439–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kool M, Korshunov A, Remke M, Jones DTW, Schlanstein M, Northcott PA et al (2012) Molecular subgroups of medulloblastoma: an international meta‐analysis of transcriptome, genetic aberrations, and clinical data of WNT, SHH, Group 3, and Group 4 medulloblastomas. Acta Neuropathol 123:473–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kool M, Koster J, Bunt J, Hasselt NE, van Lakeman A, Sluis P et al (2008) Integrated genomics identifies five medulloblastoma subtypes with distinct genetic profiles, pathway signatures and clinicopathological features. PLoS ONE 3:e3088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kouzarides T (2007) Chromatin modifications and their function. Cell 128:693–705. [DOI] [PubMed] [Google Scholar]
- 37. Lagali PS, Corcoran CP, Picketts DJ (2010) Hippocampus development and function: role of epigenetic factors and implications for cognitive disease. Clin Genet 78:321–333. [DOI] [PubMed] [Google Scholar]
- 38. Lai AY, Wade PA (2011) Cancer biology and NuRD: a multifaceted chromatin remodelling complex. Nat Rev Cancer 11:588–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lee TI, Jenner RG, Boyer LA, Guenther MG, Levine SS, Kumar RM et al (2006) Control of developmental regulators by polycomb in human embryonic stem cells. Cell 125:301–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Leung C, Lingbeek M, Shakhova O, Liu J, Tanger E, Saremaslani P et al (2004) Bmi1 is essential for cerebellar development and is overexpressed in human medulloblastomas. Nature 428:337–341. [DOI] [PubMed] [Google Scholar]
- 41. Li B, Carey M, Workman JL (2007) The role of chromatin during transcription. Cell 128:707–719. [DOI] [PubMed] [Google Scholar]
- 42. Mabbott DJ, Penkman L, Witol A, Strother D, Bouffet E (2008) Core neurocognitive functions in children treated for posterior fossa tumors. Neuropsychology 22:159–168. [DOI] [PubMed] [Google Scholar]
- 43. Michael LE, Westerman BA, Ermilov AN, Wang A, Ferris J, Liu J et al (2008) Bmi1 is required for hedgehog pathway‐driven medulloblastoma expansion. Neoplasia 10:1343–1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Milde T, Lodrini M, Savelyeva L, Korshunov A, Kool M, Brueckner LM et al (2012) HD‐MB03 is a novel Group 3 medulloblastoma model demonstrating sensitivity to histone deacetylase inhibitor treatment. J Neurooncol 110:335–348. [DOI] [PubMed] [Google Scholar]
- 45. Milde T, Oehme I, Korshunov A, Kopp‐Schneider A, Remke M, Northcott P et al (2010) HDAC5 and HDAC9 in medulloblastoma: novel markers for risk stratification and role in tumor cell growth. Clin Cancer Res 16:3240–3252. [DOI] [PubMed] [Google Scholar]
- 46. Muscal JA, Scorsone KA, Zhang L, Ecsedy JA, Berg SL (2012) Additive effects of vorinostat and MLN8237 in pediatric leukemia, medulloblastoma, and neuroblastoma cell lines. Invest New Drugs. [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Northcott PA, Jones DTW, Kool M, Robinson GW, Gilbertson RJ, Cho YJ et al (2012) Medulloblastomics: the end of the beginning. Nat Rev Cancer 12:818–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Northcott PA, Korshunov A, Witt H, Hielscher T, Eberhart CG, Mack S et al (2011) Medulloblastoma comprises four distinct molecular variants. J Clin Oncol 29:1408–1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Northcott PA, Nakahara Y, Wu X, Feuk L, Ellison DW, Croul S et al (2009) Multiple recurrent genetic events converge on control of histone lysine methylation in medulloblastoma. Nat Genet 41:465–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Northcott PA, Shih DJ, Peacock J, Garzia L, Morrissy AS, Zichner T et al (2012) Subgroup‐specific structural variation across 1,000 medulloblastoma genomes. Nature 488:49–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Oberoi J, Fairall L, Watson PJ, Yang JC, Czimmerer Z, Kampmann T et al (2011) Structural basis for the assembly of the SMRT/NCoR core transcriptional repression machinery. Nat Struct Mol Biol 18:177–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ohlsson R, Renkawitz R, Lobanenkov V (2001) CTCF is a uniquely versatile transcription regulator linked to epigenetics and disease. Trends Genet 17:520–527. [DOI] [PubMed] [Google Scholar]
- 53. Parsons DW, Li M, Zhang X, Jones S, Leary RJ, Lin JC et al (2011) The genetic landscape of the childhood cancer medulloblastoma. Science 331:435–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Pei Y, Moore CE, Wang J, Tewari AK, Eroshkin A, Cho YJ et al (2012) An animal model of MYC‐driven medulloblastoma. Cancer Cell 21:155–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Pizer BL, Clifford SC (2009) The potential impact of tumour biology on improved clinical practice for medulloblastoma: progress towards biologically driven clinical trials. Br J Neurosurg 23:364–375. [DOI] [PubMed] [Google Scholar]
- 56. Popovic R, Licht JD (2012) Emerging epigenetic targets and therapies in cancer medicine. Cancer Discov 2:405–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Pugh TJ, Weeraratne SD, Archer TC, Pomeranz Krummel DA, Auclair D, Bochicchio J et al (2012) Medulloblastoma exome sequencing uncovers subtype‐specific somatic mutations. Nature 488:106–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Pui CH, Carroll WL, Meshinchi S, Arceci RJ (2011) Biology, risk stratification, and therapy of pediatric acute leukemias: an update. J Clin Oncol 29:551–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Rahman S, Sowa ME, Ottinger M, Smith JA, Shi Y, Harper JW, Howley PM (2011) The Brd4 extraterminal domain confers transcription activation independent of pTEFb by recruiting multiple proteins, including NSD3. Mol Cell Biol 31:2641–2652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Rausch T, Jones DTW, Zapatka M, Stutz AM, Zichner T, Weischenfeldt J et al (2012) Genome sequencing of pediatric medulloblastoma links catastrophic DNA rearrangements with TP53 mutations. Cell 148:59–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Reisman D, Glaros S, Thompson EA (2009) The SWI/SNF complex and cancer. Oncogene 28:1653–1668. [DOI] [PubMed] [Google Scholar]
- 62. Robinson G, Parker M, Kranenburg TA, Lu C, Chen X, Ding L et al (2012) Novel mutations target distinct subgroups of medulloblastoma. Nature 488:43–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Rutkowski S, von Hoff K, Emser A, Zwiener I, Pietsch T, Figarella‐Branger D et al (2010) Survival and prognostic factors of early childhood medulloblastoma: an international meta‐analysis. J Clin Oncol 28:4961–4968. [DOI] [PubMed] [Google Scholar]
- 64. Santos‐Reboucas CB, Pimentel MM (2007) Implication of abnormal epigenetic patterns for human diseases. Eur J Hum Genet 15:10–17. [DOI] [PubMed] [Google Scholar]
- 65. Schuettengruber B, Chourrout D, Vervoort M, Leblanc B, Cavalli G (2007) Genome regulation by polycomb and trithorax proteins. Cell 128:735–745. [DOI] [PubMed] [Google Scholar]
- 66. Schuettengruber B, Martinez AM, Iovino N, Cavalli G (2011) Trithorax group proteins: switching genes on and keeping them active. Nat Rev Mol Cell Biol 12:799–814. [DOI] [PubMed] [Google Scholar]
- 67. Schuster‐Bockler B, Lehner B (2012) Chromatin organization is a major influence on regional mutation rates in human cancer cells. Nature 488:504–507. [DOI] [PubMed] [Google Scholar]
- 68. Schwartzentruber J, Korshunov A, Liu XY, Jones DTW, Pfaff E, Jacob K et al (2012) Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 482:226–231. [DOI] [PubMed] [Google Scholar]
- 69. Spiller SE, Ravanpay AC, Hahn AW, Olson JM (2006) Suberoylanilide hydroxamic acid is effective in preclinical studies of medulloblastoma. J Neurooncol 79:259–270. [DOI] [PubMed] [Google Scholar]
- 70. Stephens PJ, Greenman CD, Fu B, Yang F, Bignell GR, Mudie LJ et al (2011) Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell 144:27–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Swartling FJ, Grimmer MR, Hackett CS, Northcott PA, Fan QW, Goldenberg DD et al (2010) Pleiotropic role for MYCN in medulloblastoma. Genes Dev 24:1059–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Tabori U, Nanda S, Druker H, Lees J, Malkin D (2007) Younger age of cancer initiation is associated with shorter telomere length in Li‐Fraumeni syndrome. Cancer Res 67:1415–1418. [DOI] [PubMed] [Google Scholar]
- 73. Takizawa T, Meshorer E (2008) Chromatin and nuclear architecture in the nervous system. Trends Neurosci 31:343–352. [DOI] [PubMed] [Google Scholar]
- 74. Tang Y, Yacoub A, Hamed HA, Poklepovic A, Tye G, Grant S, Dent P (2012) Sorafenib and HDAC inhibitors synergize to kill CNS tumor cells. Cancer Biol Ther 13:567–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Taylor MD, Northcott PA, Korshunov A, Remke M, Cho YJ, Clifford SC et al (2012) Molecular subgroups of medulloblastoma: the current consensus. Acta Neuropathol 123:465–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Thompson MC, Fuller C, Hogg TL, Dalton J, Finkelstein D, Lau CC et al (2006) Genomics identifies medulloblastoma subgroups that are enriched for specific genetic alterations. J Clin Oncol 24:1924–1931. [DOI] [PubMed] [Google Scholar]
- 77. Tusell L, Pampalona J, Soler D, Frias C, Genesca A (2010) Different outcomes of telomere‐dependent anaphase bridges. Biochem Soc Trans 38:1698–1703. [DOI] [PubMed] [Google Scholar]
- 78. Wang GG, Allis CD, Chi P (2007) Chromatin remodeling and cancer, Part II: ATP‐dependent chromatin remodeling. Trends Mol Med 13:373–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Wang X, Venugopal C, Manoranjan B, McFarlane N, O'Farrell E, Nolte S et al (2012) Sonic hedgehog regulates Bmi1 in human medulloblastoma brain tumor‐initiating cells. Oncogene 31:187–199. [DOI] [PubMed] [Google Scholar]
- 80. Wetmore C, Eberhart DE, Curran T (2000) The normal patched allele is expressed in medulloblastomas from mice with heterozygous germ‐line mutation of patched. Cancer Res 60:2239–2246. [PubMed] [Google Scholar]
- 81. Wu G, Broniscer A, McEachron TA, Lu C, Paugh BS, Becksfort J et al (2012) Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non‐brainstem glioblastomas. Nat Genet 44:251–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Wu X, Northcott PA, Dubuc A, Dupuy AJ, Shih DJ, Witt H et al (2012) Clonal selection drives genetic divergence of metastatic medulloblastoma. Nature 482:529–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Yang ZJ, Ellis T, Markant SL, Read TA, Kessler JD, Bourboulas M et al (2008) Medulloblastoma can be initiated by deletion of Patched in lineage‐restricted progenitors or stem cells. Cancer Cell 14:135–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. You JS, Jones PA (2012) Cancer genetics and epigenetics: two sides of the same coin? Cancer Cell 22:9–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Zhao X, Liu Z, Yu L, Zhang Y, Baxter P, Voicu H et al (2012) Global gene expression profiling confirms the molecular fidelity of primary tumor‐based orthotopic xenograft mouse models of medulloblastoma. Neuro Oncol 14:574–583. [DOI] [PMC free article] [PubMed] [Google Scholar]