

Abstract
The administration of reserpine to rodents was one of the first models used to investigate the pathophysiology and screening for potential treatments of Parkinson's disease (PD). The reserpine model was critical to the understanding of the role of monoamine system in the regulation of motor and affective disorders, as well as the efficacy of current PD treatments, such as L‐DOPA and dopamine agonists. Nevertheless, with the introduction of toxin‐induced and genetic models of PD, reserpine became underused. The main rationale to this drawback was the supposed absence of reserpine construct validity with PD. Here, we highlight classical and recent experimental findings that support the face, pharmacological, and construct validity of reserpine PD model and reason against the current rationale for its underuse. We also aim to shed a new perspective upon the model by discussing the main challenges and potentials for the reserpine model of PD.
Keywords: animal model, dopamine, Parkinson's disease, reserpine, rodent
Introduction
Parkinson's disease (PD) is the second most common neurodegenerative disorder after Alzheimer's disease. Its onset is rarely before the age of 50 years and a sharp increase of the incidence occurs after the age of 60 years 19. PD affects approximately 1%–2% of the population over the age of 60 63, with a higher prevalence in men than in women 19, 62. Most importantly, it is a disorder with progressive onset and escalating deterioration of quality of life 28. Therefore, PD is a social and economic burden to countries with increasing life expectancy, and for this reason, the scientific interest in the disorder is continuously emphasized.
PD diagnosis is based on its cardinal motor symptoms, which include bradykinesia, rigidity, resting tremor, and postural instability 108. However, even though PD is essentially a motor disorder, patients present equally incapacitating nonmotor symptoms. Furthermore, those symptoms may appear previously or concomitantly to motor symptoms 126 and include sleep disorders 83, 134, 152, anxiety 154, depression 15, 97, neuropathic pain and nociceptive sensitization 27, 72, 196, impulsivity 160, 203, 204, dementia and executive function impairment 1, 7, 49, 123, olfactory dysfunction 7, 60, and constipation 48, 152.
The motor alterations are a consequence of dopaminergic neuronal loss in the substantia nigra (SN) 92, 108, where the main dopaminergic projection to the motor‐regulating nucleus in the basal ganglia originates 52, 120. Nonetheless, loss of dopaminergic neurons in the ventral tegmental area (VTA)—projecting to limbic areas and to prefrontal cortex—is also reported in PD 192, 197. This loss results in emotional and cognitive deficits 154, 165. Furthermore, other neurotransmission disturbances are described, as revealed by histopathological markers in serotonergic 101, 194, noradrenergic 28, 211, 213, and cholinergic 197, 211 neurons.
Studies have also characterized the neurochemical alterations in PD at the cellular and genetic levels. Five to 10% of PD cases are traced to familial heritage and studies have identified some genes that underlie rare familial forms of the disease 206. This approach highlighted genes involved in cellular pathways implicated in synaptic function (SNCA: α‐synuclein), ubiquitin‐proteasome protein degradation (Parkin and UCHL1), respiratory chain (PINK1), protein phosphorylation (LRRK2), and oxidative stress response (DJ‐1) 59, 163, 202, 206. Hence, impairment of these pathways leads to oxidative stress and defective protein folding, signaling, and degradation 47, 104, 114, 184. Finally, the accumulation of defective protein aggregates—mainly constituted by α‐synuclein, parkin, and ubiquitin, known as Lewy's bodies 200—is followed by cell death. Thus, the pathogenesis of PD primarily relates to the generation of oxidative stress and accumulation of defective proteins.
The genetic alterations are in accordance with epidemiological associations to PD. These associations comprise exposure to environmental toxins that act on the respiratory chain 42, 143, 195—such as pesticides, heavy metals, and carbon monoxide—and neuroinflammation 88, 200. Both events result in the generation of toxic reactive oxygen (ROS) and reactive nitrogen species, giving rise to cell damage and eventually cell death. In brief, PD harbors the oxidative imbalance as a common molecular pathway to cellular stress and neurodegeneration. Thus, animal models of PD aim to reproduce the aforementioned cellular and molecular damages 44, 61, 129, while clinical and preclinical therapeutic strategies target different candidate steps of these pathways to slow PD progression 34, 91.
Animal Models of PD
Current studies use genetic and neurotoxic approaches to reproduce pathophysiological hallmarks in animal models of PD. In genetic studies, some strategies focus on the overexpression of normal or truncated autosomal dominant genes, such as SNCA 23, 105, 137, 205 and LRRK2 117, 118, and knockout or knockdown of autosomal recessive genes, as Parkin, PINK1, or DJ‐1 106, 107, 157, 191. Nevertheless, none of these strategies recapitulates the key clinical and neuropathological features of PD and they only account for 5%–10% of PD cases 206. As a result, the most frequently used strategy is to induce oxidative imbalance and dopamine (DA) depletion by the administration of toxins or drugs that act upon dopaminergic neurons 37, 44, 61, 71, 129, 136, 167, 177, 210.
1‐Methyl‐4‐phenyl‐1,2,3,6‐tetrahydropyridine (MPTP) and 6‐hydroxydopamine (6‐OHDA) are the most used toxins in animal models of PD because of their rather selective actions upon dopaminergic neurons 9, 18, 61, 129. Both enter the dopaminergic neuron by the DA transporter (DAT) and inhibit the complex I in the respiratory chain, causing adenosine triphosphate (ATP) reduction, oxidative damage, protein aggregation, cell death, and DA depletion 61, 94, 129, 181. MPTP is a highly lipophilic protoxin that readily crosses the blood–brain barrier when peripherally administered 161. Once in the brain, MPTP is converted by glial monoamine oxidase (MAO)‐B into its intermediate 1‐methyl‐4‐phenyl‐2,3,dihydropyridinium, which is rapidly oxidized into 1‐methyl‐4‐phenylpyridinium and then reabsorbed by the dopaminergic neuron through the DAT 45. A disadvantage of this model is that rodents are more resilient to cell damage induced by MPTP compared with primates. This results in the need for higher dosages and increased variability in neurodegeneration within treated animals 43, 61, 170. In addition, there is a high risk of contamination to researchers because of the handling of large doses of MPTP and the respective biological waste 155.
6‐OHDA, on the other hand, does not cross the blood–brain barrier and is directly administered into the brain 18, 26, 61, 170. Contrastingly from MPTP, 6‐OHDA enters noradrenergic neurons as well, through the noradrenaline (NA) transporter (NAT) 29. This lack of specificity is usually resolved by the coadministration of inhibitors of NA and serotonin (5‐HT) reuptake, such as nortriptyline or desipramine 27, 56, 188. Although safer regarding contamination risk compared to MPTP, bilateral administration of 6‐OHDA results in extensive neuronal loss and severe motor impairment followed by death. After administration, animals need tube‐feeding because of aphagia and adipsia 55, 198. In order to avoid these issues, most studies perform the unilateral lesion with 6‐OHDA and assess motor deficit by inducing unilateral rotating behavior with dopaminergic agonists 171, 188. Although rotational behavior lacks face validity with PD 55, some studies evaluate forelimb akinesia (evaluated by adjusted stepping and limb‐use asymmetry tests) after unilateral 6‐OHDA administration 145, 169, 183. Nevertheless, even though the forelimb akinesia provides face validity, the unilateral lesion is still a weak approach to mimic PD pathology and symptomatology.
Alternatively, studies have employed environmental toxins such as rotenone, paraquat and maneb to model PD in rodents 9. Of those, rotenone is the most used because of its lipophilic structure, easiness to cross biological membranes, ability to inhibit complex I, and generate ROS 16, 93, 172. However, despite its close relationship to epidemiological risk factors of PD, rotenone's lack of selective action results in systemic and peripheral toxicity 74, 151, 158 and highly variable dopaminergic lesions 22, 43, 172, 212.
Finally, the administration of reserpine—an inhibitor of the vesicular transporter of monoamines in the central nervous system (VMAT2)—was one of the earliest animal models of PD. Reserpine is an alkaloid extracted from Rauwolfia serpentine and was first used as a potent antihypertensive drug because of its capacity to deplete cellular monoamine content 76, 125, 150. The clinical use of reserpine led to the observation that patients chronically treated with reserpine developed lethargy, depression, and motor dyskinesia, implicating the monoamine system in the pathophysiology of affective and motor disorders 76, 102. Readily after, reserpine was used in rodents to mimic parkinsonian motor and nonmotor impairments 17, 38, 39, 51, 69, 164, 175. Although considered outdated in comparison with the aforementioned models, the reserpine model mimics key features of PD symptomatology, neurochemistry, and pharmacology. For this reason, the model was useful to elucidate the relevance of dopaminergic neurotransmission to motor control as well as to screen for candidate drugs for treatment of PD. This review will highlight a new perspective upon the model and reason against the current rationale for the undervaluation of the reserpine‐induced parkinsonism model.
Motor and Nonmotor Behavioral Impairment in the Reserpine Model
The relationship between reserpine and PD was first reported by Carlsson et al, who observed that the akinetic state induced by reserpine in rodents was alleviated by L‐DOPA 38, 39. At doses varying from 1 to 10 mg/kg, reserpine induces a wide range of motor impairments that resemble PD, mainly akinesia, hypokinesia, catalepsy, limb rigidity, and oral tremor 17, 51, 164. These motor features are a consequence of the blockage of VMAT2 201, leading to total monoamine depletion, including DA, NA, and 5‐HT.
Besides the typical motor impairment, reserpine is also able to produce aversive 70, 174 and recognition 167 memory deficits, anxiety‐like behavior 25, 112, depressive and anhedonic‐like behaviors 10, 11, 175, and nociceptive sensitization 10, 11, 119, 144. Moreover, the memory impairment and the anxiety‐like behavior were described in a dose range (0.1–0.5 mg/kg) that did not produce motor impairment 25, 70, 167, 174. This outcome allowed the dissociation of an important confounding factor in behavioral analyses.
More recently, the repeated treatment with low doses of reserpine (0.1 mg/kg) has been suggested as a progressive model of PD 71, 167. Under this treatment regimen, animals progressively developed motor impairment in the open field, catalepsy bar, and oral movement tests after repeated injections of a low dose (0.1 mg/kg) of reserpine. Deficits in these motor tests recapitulate main motor symptoms of PD, such as hypokinesia and bradykinesia, in the open field and catalepsy bar test (ie, slowness and difficulty to initiate movements) and resting tremor in the oral movement test.
In the aforementioned study 167, the motor impairments were preceded by cognitive impairment in the novel object recognition task. This impairment was also accompanied by neuronal alterations compatible with the pathophysiology of PD such as reduction in tyrosine hydroxylase (TH) immunostaining 167 and increased lipid peroxidation in the striatum 71. Furthermore, the object recognition index positively correlated with VTA immunostaining for TH, suggesting neuronal pathways disruption other than the nigrostriatal pathway playing an important role in nonmotor symptoms of PD. In addition, the object recognition deficit occurred after a 1‐h interval between training and test sessions 167, but not when the two sessions were 24‐h apart 71. In other words, reserpine‐treated rats presented short‐term, but not for long‐term, memory deficit previously to motor deficits. Thus, performance in the task requires recognition and executive functions. These findings are in accordance with early PD symptomatic description, as executive function, attention deficit and episodic and procedural memory impairment have been described 20, 65, 115, 160, 162, 204. Furthermore, acute administration of low dose of reserpine resulted in emotional processing deficits in aversive memory tasks, such as context conditioning 70 and discriminative avoidance 40 task, but not motor impairment. In parallel, immobility in the forced swim test correlated with pain indexes, indicating a comorbid relationship between different reserpine‐induced nonmotor symptoms 10. Similarly, PD nonmotor impairments comprise anxiety 154, depression 15, 97, and nociceptive sensitization 30, 72, 196. Thus, nonmotor findings induced by reserpine resemble nonmotor PD symptoms, reinforcing reserpine's face validity as a PD model.
Pharmacological and Predictive Quality of the Reserpine Model
The use of reserpine was critical to the first demonstration of the therapeutic efficacy of L‐DOPA 38, 178. This effect was shortly after observed in humans 54 and the reserpine model was established for screening of potential symptomatic treatment efficacy of new drugs for PD. Indeed, besides L‐DOPA, the reserpine model predicted other current symptomatic anti‐Parkinson treatments: apomorphine 85, pramipexole 68, 122, ropinirole 77, rotigotine 199, pergolide 51, 98, bromocriptine 98, 99, and cabergoline 133. Likewise, reserpine‐induced motor impairment is also reversed by agents that are used in association with L‐DOPA, for example: muscarinic antagonists, such as benztropine and trihexyphenidyl 85; MAO‐B or catechol‐O‐methyltransferase (COMT) inhibitors, such as selegiline 51, 176, rasagiline 73, and tolcapone 121; and amantadine 51, 53, 85, 100, 176. Table 1 summarizes different types of motor impairment induced by reserpine that are reversed by these drugs. In fact, reserpine is still currently used to assess anti‐parkinsonian efficacy of novel agents, such as D3 receptor agonists 80, inhibitors of glutamate release 103, group III metabotropic glutamate receptor agonists or positive allosteric modulators 14, 32, 142, group I muscarinic metabotropic receptor antagonists or allosteric modulator 207, and mixed adenosine A2A/A1 antagonists 13, 173.
Table 1.
Predictive validity of reserpine Parkinson's disease (PD) model effectiveness for symptomatic treatment of different motor disturbances in PD. The table was constructed and updated according to the table presented by Duty and Jenner (61). The drug list was compiled from the Parkinson's UK website: parkinsons.org.uk/content/drug‐treatments‐parkinsons (accessed 6 October 2014). Abbreviations: COMT = catechol‐O‐methyltransferase; DA = dopamine; MAO = monoamine oxidase
| Treatment | Rigidity | Hypokinesia | Catalepsy | Tremor | Oral dyskinesia | References |
|---|---|---|---|---|---|---|
| L‐DOPA ± Carbidopa | + | + | + | + | − | 51, 85, 99, 133, 176 |
| DA agonists | ||||||
| Bromocriptine | + | + | + | − | − | 98, 99, 133, 176 |
| Cabergoline | + | + | + | − | − | 133 |
| Pergoline | + | + | + | + | − | 51, 98, 122 |
| Pramipexole | − | + | + | − | − | 68, 122 |
| Ropinirole | − | − | + | − | − | 77 |
| Apomorphine | + | + | + | − | − | 85, 98, 99 |
| Glutamate antagonists | ||||||
| Amantadine | + | + | − | + | − | 51, 85, 176 |
| Anticholinergics | − | − | − | − | − | − |
| Orphenadrine | − | − | − | − | − | − |
| Procyclidine | − | − | − | − | − | − |
| Trihexyphenidyl | + | − | − | − | − | 85 |
| Benztropine | + | − | − | − | − | 85 |
| COMT inhibitors | ||||||
| Entacapone | − | − | − | − | − | − |
| Tolcapone | − | − | − | − | − | − |
| MAO‐B inhibitors | ||||||
| Rasagiline | − | + | − | − | − | 73 |
| Selegiline | + | + | − | − | + | 51, 176 |
| Antioxidative and Dietary therapy | ||||||
| Vitamin E | − | − | − | − | + | 3, 66 |
| Co‐enzyme Q10 | − | − | − | − | − | − |
| Miscellaneous | − | − | − | − | + | 5, 24, 139, 147, 148 |
Reserpine is also employed in the screening for antioxidant and anti‐inflammatory treatments to prevent motor impairments such as dyskinesia 5, 10, 24, 66, 139, 147, 148. Current literature on oral dyskinesia implicates oxidative stress on the pathophysiology of the disorder 3, 4, 136, 186, 187. Accordingly, monoamine depletion in reserpine‐treated rats is followed by increase of reactive oxygen and nitrogen species and cell damage 179. The metabolism of catecholamine (CA) intrinsically results in ROS formation, which is increased as a consequence of free CA in the cytoplasm of reserpine‐treated rats 127, 156. Thus, oxidative stress and cell damage sums up to the monoamine depletion to impair motor performance. For this reason, treatment with antioxidants is able to revert reserpine‐induced oxidative stress and oral dyskinesia 3, 147. Finally, the treatment with 40 mg/kg vitamin E concomitant to the repeated treatment with 0.1 mg/kg reserpine 71, 167 prevented cognitive and motor impairments 168, as well as the reduction of TH immunostaining in rats (unpublished data).
These neurochemical imbalances resemble features of PD, as oxidative stress and DA depletion, which are keystones of the pathophysiology of the disease 33, 79. Thus, the pharmacological mechanism of reserpine comprises important qualities of PD pathophysiology and constitutes a good model for screening for candidate drugs to both symptomatic treatment and possible slowing of PD symptom progression. This advantage is reinforced by its low toxicity to researchers, low cost, and reproducibility among laboratories, which points out the reserpine model of PD as a suitable model for drug screening.
Molecular and Neurochemical Features of the Reserpine Model
Despite the robust face and pharmacological validities, the current literature does not recognize reserpine as a useful PD model, arguing the lack of construct validity 61. This drawback is due to the experimental observations that (i) reserpine do not induce neurodegeneration and protein aggregation 61, 208; (ii) motor performance, monoamine content, and TH staining are partially restored after treatment interruption 144, 167; and (iii) reserpine lacks specificity regarding dopaminergic neurotransmission 10, 11, 119, 141, 144.
Nevertheless, the behavioral and neurochemical features of reserpine administration are highly reproducible with little variance across studies. Reserpine peripherally administered in the dose range of 1–10 mg/kg is known to produce a robust (70%–95%) depletion of monoamine content in several brain areas 10, 11, 58, 63, 86, 90, 119, 141, 144, 189; for a summary, see Table 2). This monoamine depletion starts 30 minutes after reserpine injection and may endure up to 14 days, finally returning to normal levels after 21 days of retrieval 90, 144. At first, the absence of specificity was considered a disadvantage regarding accurate modeling of PD neurochemistry. However, there is evidence of relevant alterations in 5‐HT and NA imbalances in PD as well 28, 101, 194, 211, 213. This argues in favor of the resemblance of the neurochemical disruptions in the reserpine model with those in PD. Moreover, this characteristic is especially important to the aforementioned nonmotor deficits of PD. For instance, NA and 5‐HT transmissions are related to cognitive and emotional function 130, 175. Accordingly, reserpine treatment results in monoamine depletion in areas involved in emotional processing—as the amygdala 119—and cognition—as the hippocampus, cortex 9, 10, and prefrontal cortex 144. Furthermore, repeated reserpine treatment reduces TH staining in the hippocampus, prefrontal cortex, dorsal striatum, VTA, SN pars compacta (SNpc), and locus coeruleus 167.
Table 2.
Monoamine content depletion induced by different reserpine treatment regimens in rodents. Abbreviations: 5‐HT = serotonin; BLA = basolateral amygdala; CTX = cortex; DA = dopamine; HPC = hippocampus; NA = noradrenaline; N/A = not applicable; PFC = prefrontal cortex; SN = substantia nigra; STR = striatum; THA = thalamus
| Dose (mg/kg) | Structure | Time window | DA | NA | 5‐HT | References |
|---|---|---|---|---|---|---|
| (50×) 0.01 | STR | 24 h | 0% | ∼45% | 0% | 141 |
| (50×) 0.1 | STR | 24 h | ∼90% | ∼90% | ∼65% | |
| (50×) 1.0 | STR | 24 h | ∼95% | ∼90% | ∼90% | |
| 5.0 | SN | 2 h | ∼85% | N/A | N/A | 90 |
| 24 h | ∼70% | |||||
| STR | 2 h | >95% | ||||
| 24 h | >95% | |||||
| 1.0 | STR | 6 h | ∼80% | N/A | ∼50% | Unpublished data |
| 24 h | ∼90% | ∼80% | ||||
| 96 h | ∼75% | ∼80% | ||||
| 5.0 | STR | 24 h | ∼95% | N/A | N/A | 65 |
| 5.0 | STR | 24 h | ∼70% | N/A | N/A | 189 |
| 10.0 | STR | 18 h | ∼95% | N/A | N/A | 86 |
| STR* | 18 h | >95% | ||||
| 1.0 | STR | 24 h | ∼55% | N/A | N/A | 58 |
| (3×) 1.0 | BLA | 24 h | ∼75% | ∼80% | ∼70% | 119 |
| (3×) 1.0 | CTX | 48 h | ∼75% | ∼60% | ∼70% | 10 |
| (3×) 1.0 | CTX | 48 h | ∼80% | ∼70% | ∼80% | 11 |
| HPC | 48 h | ∼70% | ∼60% | ∼85% | ||
| 3.0 | THA* | 24 h | ∼75% | >95% | >95% | 144 |
| PFC* | 24 h | ∼90% | >95% | ∼90% |
*Microdialysis studies.
Time window refers to time after last reserpine injection.
Finally, acute or short‐term DA depletion by reserpine treatment results in upregulation of D1, but not D2 46, 132, 189. Nevertheless, long‐term treatment also leads to D2 upregulation 140, 193. These neurochemical modifications also occur because of dopaminergic denervation in untreated PD patients. Functional imaging techniques report upregulation of D2 receptor, whereas upregulation of D1 is not yet clearly defined 87, 95.
Another highly reproducible biochemical alteration in the reserpine model is the induction of oxidative stress. Reserpine, in the dose range of 1–10 mg/kg, is able to induce decreases in catalase, superoxide dismutase, total content of reduced glutathione, and ATP. Similarly, it increases glutathione peroxidase activity, oxidized glutathione, lipid peroxidation, nitric oxide (NO), and iron 2, 3, 4, 10, 11, 24, 35, 36, 63, 66, 71, 119, 138, 139, 147, 149, 159, 166, 174, 179, 186, 187; for a summary, see Table 3). Overall, there is an increase in oxidative damage. Nevertheless, some studies report contradicting results. Those differences seem to emerge from different dosage, treatment regimen, and brain area studied. For example, repeated treatment with low doses of reserpine (0.1 mg/kg) produced cumulative effects upon lipid peroxidation in the striatum, but not hippocampus, of rats 71. As well, catalase activity is generally reduced in all brain areas—except for the striatum in which some studies found increased activity 186, 187 or no significant differences 4, 66. This opposite outcome may be due to a differential fine‐tuning of catalase activity regulation in the striatum, as catecholaminergic metabolism intrinsically leads to oxidative stress 127, 156. In fact, hydrogen peroxide (H2O2) is one of the main products of CA metabolism by MAO‐A 127, 156, and naturally one may speculate that catalase in catecholaminergic neurotransmission is differentially modulated by increases in H2O2 in order to provide antioxidant protection. Indeed, this is endorsed by the observation that catecholaminergic neurons are relatively abundant in populations of catalase‐positive microperoxisomes 124. Thus, it seems that treatment duration and brain area studied define the extent of oxidative damage induced by reserpine.
Table 3.
Molecular changes related to oxidative stress induced by different reserpine treatment regimens in rodents. Abbreviations: CAT = catalase; GPX = glutathione peroxidase; GSH = reduced glutathione; GSSG = oxidized glutathione; GST = glutathione‐S‐transferase; LPO = lipid peroxide; NO = nitric oxide; NS = not significant; SOD = superoxide dismutase
| Structure | Dose (mg/kg) | Time window | CAT | SOD | GPX | GST | GSH | GSSG | GSSG/GSH | LPO | NO | References |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Total brain | 5.0 | 24 h | ↓ | ↓ | ↑ | 65 | ||||||
| (3×) 1.0 | 3 h | ↓ | ↓ | ↓ | ↑ | 147 | ||||||
| (3×) 1.0 | 24 h | ↓ | ↓ | ↓ | ↑ | 138 | ||||||
| (3×) 1.0 | 24 h | ↓ | ↓ | ↓ | ↑ | 139 | ||||||
| (3×) 1.0 | 17 days | ↓ | ↓ | ↓ | ↑ | 166 | ||||||
| Cortex | (2×) 1.0 | 24 h | NS | 149 | ||||||||
| (3×) 1.0 | 24 h | NS | 35 | |||||||||
| (3×) 1.0 | 48 h | ↓ | ↓ | ↓ | ↑ | ↑ | 11 | |||||
| (3×) 1.0 | 48 h | ↓ | ↓ | ↑ | ↑ | 10 | ||||||
| (3×) 1.0 | 96 h | NS | 159 | |||||||||
| 10 | 2 h | NS | 180 | |||||||||
| Striatum | (10×) 0.1 | 24 h | ↑ | 2 | ||||||||
| (10×) 0.1 | 48 h | ↑ | 71 | |||||||||
| (2×) 0.5 | 24 h | NS | 66 | |||||||||
| (2×) 1.0 | 24 h | NS | NS | 4 | ||||||||
| (2×) 1.0 | 24 h | ↑ | ↑ | 187 | ||||||||
| (2×) 1.0 | 24 h | ↑ | 3 | |||||||||
| (2×) 1.0 | 24 h | ↑ | 36 | |||||||||
| (2×) 1.0 | 24 h | ↑ | ↓ | 186 | ||||||||
| (2×) 1.0 | 24 h | NS | 149 | |||||||||
| (3×) 1.0 | 24 h | ↑ | 35 | |||||||||
| (3×) 1.0 | 96 h | NS | 159 | |||||||||
| 5.0 | 90 minutes | ↑ | NS | ↑ | ↑ | 24 | ||||||
| 10 | 2 h | ↑ | 180 | |||||||||
| Hippocampus | (10×) 0.1 | 48 h | NS | 71 | ||||||||
| (2×) 1.0 | 24 h | NS | 149 | |||||||||
| (3×) 1.0 | 48 h | ↓ | ↓ | ↓ | ↑ | ↑ | 11 | |||||
| (3×) 1.0 | 48 h | ↓ | ↓ | ↑ | ↑ | 10 | ||||||
| 5.0 | 90 minutes | NS | NS | ↑ | ↑ | 24 | ||||||
| Substantia nigra | (2×) 1.0 | 24 h | NS | 149 | ||||||||
| Basolateral amygdala | (3×) 1.0 | 24 h | ↓ | ↑ | 119 |
Time window refers to time after last reserpine injection.
The oxidative stress induced by reserpine is related to increased DA metabolism as a result of the reduction on the number of DA molecules in the vesicle 146 and increased DA turnover 67, 141, 179. Accordingly, MAO‐A inhibitor reverts L‐DOPA and reserpine induced increase in oxidized glutathione 179, 180. In addition, free DA and metabolites in the cytoplasm results in auto‐oxidation of DA and DOPAC to their corresponding reactive quinones—DA‐Q and DOPAC‐Q, respectively—12, 127, 156, which contributes to cell apoptosis and synuclein dimerization 84.
The generation of highly reactive molecules results in early cell damage—as consistently evidenced by lipid peroxidation (Table 3)—initiating proinflammatory signaling by tumor necrosis factor (TNF)‐α and interleukin (IL)‐1β 10, 11. Subsequently, the increase in proinflammatory cytokines activates microglia, which leads to a vicious circle of adhesion, inflammation, and release of more cytokines. Activated microglia upon dopaminergic neurons also results in increased NO 10, 11, 24. Afterwards, NO—in the presence of superoxide (O2 −)—produces peroxynitrite (NO3 −) 127, 156, which is highly reactive and has been shown to inactivate TH via S‐thiolation on cysteine residues 8, 96, 110, 111. In this context, repeated treatment with a low dose of reserpine (0.1 mg/kg) resulted in reduced TH immunostaining in several brain areas—that is hippocampus, prefrontal cortex, dorsal striatum, SNpc, and VTA 167.
Ultimately, these events may terminate in the commitment with apoptotic pathways. In other words, there is a reduction in anti‐apoptotic molecules, as Bcl‐2 63, 119, and an increase in proapoptotic molecules, as caspase‐3 10, 11, 119.
Nevertheless, whether reserpine leads to permanent cell damage or neurodegeneration is not clear yet. In this respect, repeated treatment with 0.1 mg/kg of reserpine every other day for 20 days resulted in a reduction of TH immunostaining that was partially reversed after 30 days of treatment withdrawal 167. Likewise, the same protocol increased α‐synuclein immunostaining in SN and dorsal striatum and these effects were reversed after treatment interruption (data not published). Of notice, such increase did not result in protein inclusions and studies addressing if actual neuronal loss occurs are currently being held. Thus, in light of the current evidence (extent of TH reduction and α‐synuclein increase, restauration of motor performance, and reversion of reduction in TH and α‐synuclein immunostaining after interruption of treatment), data regarding the repeated low‐dose reserpine treatment should be interpreted in terms of TH expression reduction rather than neurodegeneration.
On the other hand, some evidence support long‐lasting or permanent cellular and behavioral changes within a high dose chronic reserpine treatment. Treatment with 1 mg/kg of reserpine every other day for 6 weeks resulted in persistent behavioral and neurochemical changes (oral dyskinesia, DA depletion and D1 and D2 receptor upregulation) up to 60 days after treatment withdrawal 140. Thus, we do not discard the possibility of some extent of permanent cell damage or cell death after reserpine treatment, depending on dose and/or length of treatment.
In this context, untreated VMAT2 genetically deficient mice—which express only 5% of functional VMAT2—presents age‐associated neurodegeneration in SNpc, locus coeruleus, and dorsal raphe, followed by α‐synuclein accumulation and TH and tyramine transporter immunostaining reduction 41, 185. This VMAT2‐deficient mice also presents L‐DOPA responsive motor impairment, twofold increase in DA concentration in cytosol, reduction in TH phosphorylation associated with catechol feedback, 95% of DA depletion, and increased DA turnover 50, 135, 185. Moreover, these alterations are accompanied by nonmotor impairments, such as deficit in olfactory discrimination, delayed gastric emptying, altered sleep latency, anxiety‐like behavior, and age‐dependent depressive behavior 185. In short, all behavioral and neurochemical alterations in VMAT2‐deficient mice resemble the effects of reserpine treatment. As both reserpine and VMAT2‐deficient mice models are similar in terms of functional construct, we speculate that neurodegeneration is a plausible outcome in long‐term VMAT2 functional blockade by reserpine treatment. As mentioned earlier, this issue is currently under investigation.
In conclusion, reserpine treatment is able to induce (i) monoamine depletion, (ii) oxidative stress, (iii) inflammation, (iv) proapoptotic commitment, (v) reduction in tyrosine hydroxylase and increase in α‐synuclein immunostaining, and (vi) DA receptors upregulation (for summary of neurochemical events after reserpine administration, see Figure 1). Despite that there is still no evidence of some important pathological features of PD—such as protein aggregation, permanent cellular damage, and neurodegeneration—most of the reserpine‐induced neurochemical alterations are clearly reminiscent of PD pathophysiology and thus holds a satisfactory resemblance to PD phenomenology. Therefore, the lack of construct validity should not be an argument against the use of the reserpine model to study PD.
Figure 1.
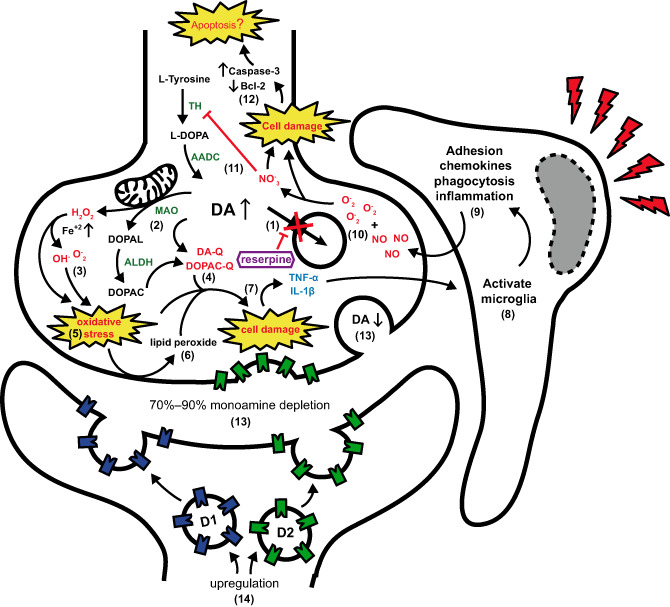
Neurochemical and molecular events after reserpine treatment. (1) Reserpine precludes dopamine (DA) storage. (2) Increased DA is metabolized in the cytoplasm (3) generating reactive oxygen species (ROS) and (4) highly reactive quinones (DA‐Q and DOPAC‐Q) (5) resulting in oxidative stress and (6) lipid peroxidation.(7) Accumulation of ROS and reactive quinones leads to cell damage and proinflammatory signalization. (8) Activation of microglia by tumor necrosis factor (TNF)‐α and interleukin (IL)‐1β (9) amplify proinflammatory signalization resulting in (10) nitric oxide (NO) increase and peroxynitrite (NO3 ‐) formation with free superoxide (O2 ‐). (11) NO3 ‐ inhibits tyrosine hydroxylase (TH) activity and (12) reinforces cell damage committing cell fate in proapoptotic signalization. At the same time, (13) monoamine depletion in synaptic cleft results in (14) upregulation of D1 and D2 receptors on the postsynaptic and presynaptic membrane. AADC, aromatic L‐amino acid decarboxylase; ALDH, aldehyde dehydrogenases; MAO, monoamine oxidase.
It should be noted that the aforementioned toxin‐based animal models do not account for all pathophysiological features of PD as well. 6‐OHDA leads to neurodegeneration and motor impairment, but studies have not shown protein inclusions, while MPTP administration resulted in Lewy's body‐like inclusions specifically in particular mice lineages. Likewise, rotenone treatment induces Lewy's body‐like inclusions and neurodegeneration in rats, but the extent of neurodegeneration is highly variable 78, 81, 109, 113, 128, 190.
Final Considerations
In addition to the aforementioned features, one might question if the reserpine model mimics risk factors of PD, such as age and sex, for example. Neurochemical studies regarding age‐related effects of reserpine treatment found that older rats presents reduced DA turnover 6 and a tendency to reduced DA recovery 153 compared with younger animals. Furthermore, oral dyskinesia is increased in older rats 2, 4, 35 and reserpine treatment results in cumulative 182 and persistent 21 oral dyskinesia in older animals. However, current literatures have not directly addressed the influence of age on other reserpine‐induced motor deficits. Up to date, the low‐dose repeated reserpine treatment has been conducted with 6‐month‐old rats (unlike studies with other parkinsonism‐inducing drugs, which are usually conducted with 3‐month‐old animals), but the studies did not include other age groups 71, 167.
Moreover, regarding sex differences, we have recently conducted the low‐dose repeated reserpine treatment (0.1 mg/kg) in male and female Swiss mice and found that female mice took longer to develop motor impairment in the catalepsy (Figure 2A,B) and oral dyskinesia (Figure 2C) tests (refer to Figure 2 legend for methods and statistical analysis). Conversely, other study reported increased oral dyskinesia in female mice that was inconsistent at different time points 174. Contradicting results regarding oral dyskinesia might be explained by differences in protocol—that is length of treatment, dosage, and type of motor parameter (vacuous chewing vs. jaw twitching). Nevertheless, studies with CD‐1 mice have suggested that female animals present a more efficient VMAT2 function 57, 58, which could explain the need of a longer treatment for female mice to develop the motor alterations (data displayed in Figure 2). Importantly, this result is in accordance with the lower incidence of PD in women 19, 62 and adds to the similarities between the reserpine model and the clinical condition.
Figure 2.
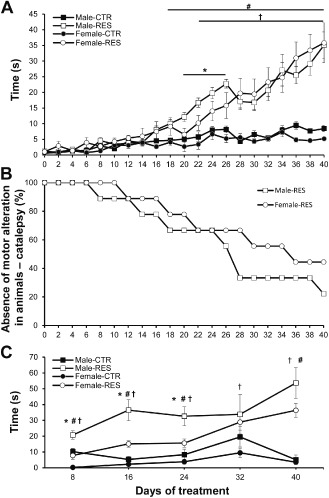
Motor deficits of repeated low‐dose reserpine treatment in male and female mice. Male and female Swiss mice (6 months old; n = 9 per group) were repeatedly treated every other day with reserpine (0.1 mg/kg) (RES) or vehicle (CTR) for 40 days according to the protocol previously described for rats 71, 167. (A) Latency to step down in the catalepsy bar test. Mice were gently positioned with both forepaws in an elevated bar (6 cm). Catalepsy score was the mean of three measures of the latency to step down. Two‐way analysis of variance (ANOVA) with repeated measures revealed effect of time [f(20, 160) = 39.53, P < 0.001], treatment [f(3, 24) = 12.97, P < 0.001], and time vs. treatment interaction [f(60, 480) = 7.93, P < 0.001]. (B) Percentage of male and female reserpine‐treated mice without motor impairment (catalepsy test) across treatment. Animals were considered to present motor impairment when the catalepsy score was above the mean plus two standard errors of the mean of the respective CTR group. Gehan‐Breslow‐Wilcoxon test revealed that more female rats did not present motor deficit in the catalepsy bar test compared with male rats (chi‐square = 4.065, P = 0.043). (C) Oral movement test. Mice were positioned in a small cage (20 × 25 × 20 cm) surrounded by mirrors and jaw‐twitching time (s) was quantified within a 10‐minute session by two blind observers. Two‐way ANOVA with repeated measures revealed effect of time [f(5, 40) = 15.86, P < 0.001], treatment [f(3, 24) = 25.58, P < 0.001], and sex [f(1, 8) = 42.07, P < 0.001], as well as interactions for time vs. treatment [f(15, 120) = 7.55, P < 0.001], sex vs. time [f(5, 40) = 18.35, P = 0.002], and sex vs. treatment [f(1, 24) = 37.93, P = 0.003]. For all graphs, †P < 0.05 Female‐RES vs. Female‐CTR; #P < 0.05 male‐RES vs. male‐CTR; and *P < 0.05 male‐RES vs. female‐RES (Tukey's post hoc test for each day).
The exposed prospect of reserpine‐induced behavioral, pharmacological, and neurochemical effects restates the use of reserpine as a valuable and promising model for PD study. Thus, the current underuse of reserpine to investigate PD features should be reconsidered. Of notice, the use of reserpine could be important to the relevance of VMAT2 functionality to PD in humans. Indeed, polymorphisms in promoter regions that increases transcription of VMAT2 are protective against PD 31, 82 and reduction in VMAT2 and its mRNA in nigrostriatal neurons have been reported in PD patients 89, 131. Furthermore, VMAT2 is present in Lewy's bodies in the SN of PD patients 209 and VTA dopaminergic neurons that are spared in PD harbors higher levels of VMAT2 131. Finally, increased cytoplasmic DA influences the conformational state of α‐synuclein, promoting stabilization of its pathogenic form 75, 116. Thus, because functional VMAT2 expression is protective against dopaminergic neurodegeneration, its long‐term blockage might represent an interesting approach to model PD.
In conclusion, we believe that the scientific effort on reserpine PD model validation should focus in answering whether neurodegeneration and cell death occur after chronic reserpine treatment, as well as the exploitation of the model to investigate progression of symptoms and neurochemical features of PD pathophysiology. We recently presented a low‐dose reserpine‐induced progressive model of PD that could be useful to investigate such inquiry 71, 167. Therefore, in view of the presented experimental evidence, the reserpine‐induced PD model in rodents reaches robust face and pharmacological validity criteria, besides presenting a significant number of neurochemical and molecular features that closely resemble the pathophysiology of the disease. Taken together, these characteristics render the reserpine model a useful tool for PD basic research.
References
- 1. Aarsland D, Andersen K, Larsen JP, Lolk A (2003) Prevalence and characteristics of dementia in Parkinson disease: an 8‐year prospective study. Arch Neurol 60:387–392. [DOI] [PubMed] [Google Scholar]
- 2. Abílio V, Vera J, Ferreira L, Duarte C, Carvalho R, Grassl C et al (2002) Effects of melatonin on orofacial movements in rats. Psychopharmacology (Berl) 161:340–347. [DOI] [PubMed] [Google Scholar]
- 3. Abílio VC, Araujo CCS, Bergamo M, Calvente PRV, D'Almeida V, Ribeiro RA, Frussa‐Filho R (2003) Vitamin E attenuates reserpine‐induced oral dyskinesia and striatal oxidized glutathione/reduced glutathione ratio (GSSG/GSH) enhancement in rats. Prog Neuropsychopharmacol Biol Psychiatry 27:109–114. [DOI] [PubMed] [Google Scholar]
- 4. Abílio VC, Silva RH, Carvalho RC, Grassl C, Calzavara MB, Registro S et al (2004) Important role of striatal catalase in aging‐ and reserpine‐induced oral dyskinesia. Neuropharmacology 47:263–272. [DOI] [PubMed] [Google Scholar]
- 5. Al‐Bloushi S, Safer A‐M, Afzal M, Mousa SA (2009) Green tea modulates reserpine toxicity in animal models. J Toxicol Sci 34:77–87. [DOI] [PubMed] [Google Scholar]
- 6. Algeri S, Achilli G, Calderini G, Perego C, Ponzio F, Toffano G (1987) Age‐related changes in metabolic responses to chronic monoamine depletion in central dopaminergic and serotonergic systems of rats treated with reserpine. Neurobiol Aging 8:61–66. [DOI] [PubMed] [Google Scholar]
- 7. Anang JBM, Gagnon J‐F, Bertrand J‐A, Romenets SR, Latreille V, Panisset M et al (2014) Predictors of dementia in Parkinson disease: a prospective cohort study. Neurology 83:1253–1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ara J, Przedborski S, Naini AB, Jackson‐Lewis V, Trifiletti RR, Horwitz J, Ischiropoulos H (1998) Inactivation of tyrosine hydroxylase by nitration following exposure to peroxynitrite and 1‐methyl‐4‐phenyl‐1,2,3,6‐tetrahydropyridine (MPTP). Proc Natl Acad Sci U S A 95:7659–7663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Arif IA, Khan HA (2010) Environmental toxins and Parkinson's disease: putative roles of impaired electron transport chain and oxidative stress. Toxicol Ind Health 26:121–128. [DOI] [PubMed] [Google Scholar]
- 10. Arora V, Chopra K (2013) Possible involvement of oxido‐nitrosative stress induced neuro‐inflammatory cascade and monoaminergic pathway: underpinning the correlation between nociceptive and depressive behaviour in a rodent model. J Affect Disord 151:1041–1052. [DOI] [PubMed] [Google Scholar]
- 11. Arora V, Kuhad A, Tiwari V, Chopra K (2011) Curcumin ameliorates reserpine‐induced pain‐depression dyad: behavioural, biochemical, neurochemical and molecular evidences. Psychoneuroendocrinology 36:1570–1581. [DOI] [PubMed] [Google Scholar]
- 12. Asanuma M, Miyazaki I, Ogawa N (2003) Dopamine‐ or L‐DOPA‐induced neurotoxicity: the role of dopamine quinone formation and tyrosinase in a model of Parkinson's disease. Neurotox Res 5:165–176. [DOI] [PubMed] [Google Scholar]
- 13. Atack JR, Shook BC, Rassnick S, Jackson PF, Rhodes K, Drinkenburg WH et al (2014) JNJ‐40255293, a novel adenosine A2A/A1 antagonist with efficacy in preclinical models of Parkinson's disease. ACS Chem Neurosci 5:1005–1019. [DOI] [PubMed] [Google Scholar]
- 14. Austin PJ, Betts MJ, Broadstock M, O'Neill MJ, Mitchell SN, Duty S (2010) Symptomatic and neuroprotective effects following activation of nigral group III metabotropic glutamate receptors in rodent models of Parkinson's disease. Br J Pharmacol 160:1741–1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Barone P (2011) Treatment of depressive symptoms in Parkinson's disease. Eur J Neurol 18:11–15. [DOI] [PubMed] [Google Scholar]
- 16. Bashkatova V, Alam M, Vanin A, Schmidt WJ (2004) Chronic administration of rotenone increases levels of nitric oxide and lipid peroxidation products in rat brain. Exp Neurol 186:235–241. [DOI] [PubMed] [Google Scholar]
- 17. Baskin P, Salamone J (1993) Vacuous jaw movements in rats induced by acute reserpine administration: interactions with different doses of apomorphine. Pharmacol Biochem Behav 46:793–797. [DOI] [PubMed] [Google Scholar]
- 18. Beal MF (2001) Experimental models of Parkinson's disease. Nat Rev Neurosci 2:325–332. [DOI] [PubMed] [Google Scholar]
- 19. Benito‐León J, Bermejo‐Pareja F, Morales‐González JM, Porta‐Etessam J, Trincado R, Vega S, Louis ED (2004) Incidence of Parkinson disease and parkinsonism in three elderly populations of central Spain (NEDICES). Neurology 62:734–741. [DOI] [PubMed] [Google Scholar]
- 20. Benito‐León J, Louis ED, Posada IJ, Sánchez‐Ferro Á, Trincado R, Villarejo A et al (2011) Population‐based case‐control study of cognitive function in early Parkinson's disease (NEDICES). J Neurol Sci 310:176–182. [DOI] [PubMed] [Google Scholar]
- 21. Bergamo M, Abílio VC, Queiroz CMT, Barbosa‐Júnior HN, Abdanur LRA, Frussa‐Filho R (1997) Effects of age on a new animal model of tardive dyskinesia. Neurobiol Aging 18:623–629. [DOI] [PubMed] [Google Scholar]
- 22. Betarbet R, Sherer TB, Mackenzie G, Garcia‐Osuna M, Panov AV, Greenamyre JT (2000) Chronic systemic pesticide exposure reproduces features of Parkinson's disease. Nat Neurosci 3:1301–1306. [DOI] [PubMed] [Google Scholar]
- 23. Lo Bianco C, Ridet J‐L, Schneider BL, Déglon N, Aebischer P (2002) Alpha‐synucleinopathy and selective dopaminergic neuron loss in a rat lentiviral‐based model of Parkinson's disease. Proc Natl Acad Sci U S A 99:10813–10818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bilska A, Dubiel M, Sokołowska‐Jezewicz M, Lorenc‐Kocib E, Włodek L (2007) Alpha‐lipoic acid differently affects the reserpine‐induced oxidative stress in the striatum and prefrontal cortex of rat brain. Neuroscience 146:1758–1771. [DOI] [PubMed] [Google Scholar]
- 25. Bisong SA, Brown R, Osim EE (2010) Comparative effects of Rauwolfia vomitoria and chlorpromazine on locomotor behaviour and anxiety in mice. J Ethnopharmacol 132:334–339. [DOI] [PubMed] [Google Scholar]
- 26. Blesa J, Phani S, Jackson‐Lewis V, Przedborski S (2012) Classic and new animal models of Parkinson's disease. J Biomed Biotechnol 2012:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bonito‐Oliva A, Masini D, Fisone G (2014) A mouse model of non‐motor symptoms in Parkinson's disease: focus on pharmacological interventions targeting affective dysfunctions. Front Behav Neurosci 8:290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Braak H, Del K, Rüb U, de Vos RAI, Jansen ENH, Braak E (2003) Staging of brain pathology related to sporadic Parkinson's disease. Neurobiol Aging 24:197–211. [DOI] [PubMed] [Google Scholar]
- 29. Breese GR, Traylor TD (1971) Depletion of brain noradrenaline and dopamine by 6‐hydroxydopamine. Br J Pharmacol 42:88–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Brefel‐Courbon C, Ory‐Magne F, Thalamas C, Payoux P, Rascol O (2013) Nociceptive brain activation in patients with neuropathic pain related to Parkinson's disease. Parkinsonism Relat Disord 19:548–552. [DOI] [PubMed] [Google Scholar]
- 31. Brighina L, Riva C, Bertola F, Saracchi E, Fermi S, Goldwurm S, Ferrarese C (2013) Analysis of vesicular monoamine transporter 2 polymorphisms in Parkinson's disease. Neurobiol Aging 34:1712.e9–1712.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Broadstock M, Austin PJ, Betts MJ, Duty S (2012) Antiparkinsonian potential of targeting group III metabotropic glutamate receptor subtypes in the rodent substantia nigra pars reticulata. Br J Pharmacol 165 (4b):1034–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Brundin P, Li J‐Y, Holton JL, Lindvall O, Revesz T (2008) Research in motion: the enigma of Parkinson's disease pathology spread. Nat Rev Neurosci 9:741–745. [DOI] [PubMed] [Google Scholar]
- 34. Brundin P, Barker RA, Conn PJ, Dawson TM, Kieburtz K, Lees AJ et al (2013) Linked clinical trials—the development of new clinical learning studies in Parkinson's disease using screening of multiple prospective new treatments. J Parkinsons Dis 3:231–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Burger M, Fachinetto R, Calegari L, Paixão MW, Braga AL, Rocha JBT (2004) Effects of age on reserpine‐induced orofacial dyskinesia and possible protection of diphenyl diselenide. Brain Res Bull 64:339–345. [DOI] [PubMed] [Google Scholar]
- 36. Burger ME, Alves A, Callegari L, Athayde FR, Nogueira CW, Zeni G, Rocha JBT (2003) Ebselen attenuates reserpine‐induced orofacial dyskinesia and oxidative stress in rat striatum. Prog Neuropsychopharmacol Biol Psychiatry 27:135–140. [DOI] [PubMed] [Google Scholar]
- 37. Cannon JR, Tapias VM, Na HM, Honick AS, Drolet RE, Greenamyre JT (2009) A highly reproducible rotenone model of Parkinson's disease. Neurobiol Dis 34:279–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Carlsson A, Lindqvist M, Magnusson T (1957) 3,4‐dihydroxyphenylalanine and 5‐hydroxytryptophan as reserpine antagonists. Nature 180:1200. [DOI] [PubMed] [Google Scholar]
- 39. Carlsson M, Carlsson A (1989) Marked locomotor stimulation in monoamine‐depleted mice following treatment with atropine in combination with clonidine. J Neural Transm Park Dis Dement Sect 1:317–322. [DOI] [PubMed] [Google Scholar]
- 40. Carvalho RC, Patti CC, Takatsu‐Coleman AL, Kameda SR, Souza CF, Garcez‐do‐Carmo L et al (2006) Effects of reserpine on the plus‐maze discriminative avoidance task: dissociation between memory and motor impairments. Brain Res 1122:179–183. [DOI] [PubMed] [Google Scholar]
- 41. Caudle WM, Richardson JR, Wang MZ, Taylor TN, Guillot TS, McCormack AL et al (2007) Reduced vesicular storage of dopamine causes progressive nigrostriatal neurodegeneration. J Neurosci 27:8138–8148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Caudle WM, Guillot TS, Lazo CR, Miller GW (2012) Industrial toxicants and Parkinson's disease. Neurotoxicology 33:178–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Cenci MA, Whishaw IQ, Schallert T (2002) Animal models of neurological deficits: how relevant is the rat? Nat Rev Neurosci 3:574–579. [DOI] [PubMed] [Google Scholar]
- 44. Chesselet M‐F, Richter F (2011) Modelling of Parkinson's disease in mice. Lancet Neurol 10:1108–1118. [DOI] [PubMed] [Google Scholar]
- 45. Chiba K, Trevor A, Castagnoli N (1984) Metabolism of the neurotoxic tertiary amine, MPTP, by brain monoamine oxidase. Biochem Biophys Res Commun 120:574–578. [DOI] [PubMed] [Google Scholar]
- 46. Chipkin RE, McQuade RD, Iorio LC (1987) D1 and D2 dopamine binding site up‐regulation and apomorphine‐induced stereotypy. Pharmacol Biochem Behav 28:477–482. [DOI] [PubMed] [Google Scholar]
- 47. Chuang RS, Gitler AD (2013) Parallel PARKing: Parkinson's genes function in common pathway. Neuron 77:377–379. [DOI] [PubMed] [Google Scholar]
- 48. Clairembault T,Leclair‐Visonneau L,Neunlist M,Derkinderen P (2014) Enteric glial cells: New players in Parkinson's disease? Mov Disord doi: 10.1002/mds.25979 [DOI] [PubMed] [Google Scholar]
- 49. Coelho M, Marti MJ, Sampaio C, Ferreira JJ, Valldeoriola F, Rosa MM, Tolosa E (2015) Dementia and severity of parkinsonism determines the handicap of patients in late‐stage Parkinson's disease: the Barcelona‐Lisbon cohort. Eur J Neurol 22(2):305–312. [DOI] [PubMed] [Google Scholar]
- 50. Colebrooke RE, Humby T, Lynch PJ, McGowan DP, Xia J, Emson PC (2006) Age‐related decline in striatal dopamine content and motor performance occurs in the absence of nigral cell loss in a genetic mouse model of Parkinson's disease. Eur J Neurosci 24:2622–2630. [DOI] [PubMed] [Google Scholar]
- 51. Colpaert FC (1987) Pharmacological characteristics of tremor, rigidity and hypokinesia induced by reserpine in rat. Neuropharmacology 26:1431–1440. [DOI] [PubMed] [Google Scholar]
- 52. Da Cunha C, Wietzikoski EC, Dombrowski P, Bortolanza M, Santos LM, Boschen SL, Miyoshi E (2009) Learning processing in the basal ganglia: a mosaic of broken mirrors. Behav Brain Res 199:157–170. [DOI] [PubMed] [Google Scholar]
- 53. Danysz W, Gossel M, Zajaczkowski W (1994) Are NMDA antagonistic properties relevant for antiparkinsonian‐like activity in rats? Case of amantadine and memantine. J Neural Transm 7:155–166. [DOI] [PubMed] [Google Scholar]
- 54. Degkwitz R, Frowein R, Kulenkampff C, Mohs U (1960) On the effects of L‐dopa in man and their modification by reserpine, chlorpromazine, iproniazid and vitamin B6. Klin Wochenschr 38:120–123. [DOI] [PubMed] [Google Scholar]
- 55. Deumens R, Blokland A, Prickaerts J (2002) Modeling Parkinson's disease in rats: an evaluation of 6‐OHDA lesions of the nigrostriatal pathway. Exp Neurol 175:303–317. [DOI] [PubMed] [Google Scholar]
- 56. Didonet JJ, Cavalcante JC, Souza LDS, Costa MSMO, André E, Soares‐Rachetti VDP et al (2014) Neuropeptide S counteracts 6‐OHDA‐induced motor deficits in mice. Behav Brain Res 266:29–36. [DOI] [PubMed] [Google Scholar]
- 57. Dluzen DE, McDermott JL (2008) Sex differences in dopamine‐ and vesicular monoamine‐transporter functions: Implications for methamphetamine use and neurotoxicity. Ann N Y Acad Sci 1139:140–150. [DOI] [PubMed] [Google Scholar]
- 58. Dluzen DE, Bhatt S, McDermott JL (2008) Differences in reserpine‐induced striatal dopamine output and content between female and male mice: implications for sex differences in vesicular monoamine transporter 2 function. Neuroscience 154:1488–1496. [DOI] [PubMed] [Google Scholar]
- 59. Dodson MW, Guo M (2007) Pink1, Parkin, DJ‐1 and mitochondrial dysfunction in Parkinson's disease. Curr Opin Neurobiol 17:331–337. [DOI] [PubMed] [Google Scholar]
- 60. Driver‐Dunckley E, Adler CH, Hentz JG, Dugger BN, Shill HA, Caviness JN et al (2014) Olfactory dysfunction in incidental Lewy body disease and Parkinson's disease. Parkinsonism Relat Disord 20:1260–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Duty S, Jenner P (2011) Animal models of Parkinson's disease: a source of novel treatments and clues to the cause of the disease. Br J Pharmacol 164:1357–1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Eeden SKVD, Tanner CM, Bernstein AL, Fross RD, Leimpeter A, Bloch DA, Nelson LM (2003) Incidence of Parkinson's disease: variation by age, gender, and race/ethnicity. Am J Epidemiol 157:1015–1022. [DOI] [PubMed] [Google Scholar]
- 63. Elbaz A, Bower JH, Maraganore DM, McDonnell SK, Peterson BJ, Ahlskog JE et al (2002) Risk tables for parkinsonism and Parkinson's disease. J Clin Epidemiol 55:25–31. [DOI] [PubMed] [Google Scholar]
- 64. Elgh E, Domellöf M, Linder J, Edström M, Stenlund H, Forsgren L (2009) Cognitive function in early Parkinson's disease: a population‐based study. Eur J Neurol 16:1278–1284. [DOI] [PubMed] [Google Scholar]
- 65. El‐Ghazaly MA, Sadik NAH, Rashed ER, Abd‐El‐Fattah AA (2013) Neuroprotective effect of EGb761(R) and low‐dose whole‐body γ‐irradiationin a rat model of Parkinson's disease. Toxicol Ind Health 21:1–17. [DOI] [PubMed] [Google Scholar]
- 66. Faria RR, Abílio VC, Grassl C, Chinen CC, Negrão LTR, de Castro JPMV et al (2005) Beneficial effects of vitamin C and vitamin E on reserpine‐induced oral dyskinesia in rats: critical role of striatal catalase activity. Neuropharmacology 48:993–1001. [DOI] [PubMed] [Google Scholar]
- 67. Fekete MI, Szentendrei T, Herman JP, Kanyicska B (1980) Effects of reserpine and antidepressants on dopamine and DOPAC (3,4‐dihydroxyphenylacetic acid) concentrations in the striatum, olfactory tubercle and median eminence of rats. Eur J Pharmacol 64:231–238. [DOI] [PubMed] [Google Scholar]
- 68. Ferger B, Buck K, Shimasaki M, Koros E, Voehringer P, Buerger E (2010) Continuous dopaminergic stimulation by pramipexole is effective to treat early morning akinesia in animal models of Parkinson's disease: a pharmacokinetic‐pharmacodynamic study using in vivo microdialysis in rats. Synapse 64:533–541. [DOI] [PubMed] [Google Scholar]
- 69. Fernagut PO, Diguet E, Labattu B, Tison F (2002) A simple method to measure stride length as an index of nigrostriatal dysfunction in mice. J Neurosci Methods 113:123–130. [DOI] [PubMed] [Google Scholar]
- 70. Fernandes VS, Ribeiro AM, Melo TG, Godinho M, Barbosa FF, Medeiros DS et al (2008) Memory impairment induced by low doses of reserpine in rats: possible relationship with emotional processing deficits in Parkinson disease. Prog Neuropsychopharmacol Biol Psychiatry 32:1479–1483. [DOI] [PubMed] [Google Scholar]
- 71. Fernandes VS, Santos JR, Leão AHFF, Medeiros AM, Melo TG, Izídio GS et al (2012) Repeated treatment with a low dose of reserpine as a progressive model of Parkinson's disease. Behav Brain Res 231:154–163. [DOI] [PubMed] [Google Scholar]
- 72. Fil A, Cano‐de‐la‐Cuerda R, Muñoz‐Hellín E, Vela L, Ramiro‐González M, Fernández‐de‐Las‐Peñas C (2013) Pain in Parkinson disease: a review of the literature. Parkinsonism Relat Disord 19:285–294. [DOI] [PubMed] [Google Scholar]
- 73. Finberg JPM, Youdim MBH (2002) Pharmacological properties of the anti‐Parkinson drug rasagiline; modification of endogenous brain amines, reserpine reversal, serotonergic and dopaminergic behaviours. Neuropharmacology 43:1110–1118. [DOI] [PubMed] [Google Scholar]
- 74. Fleming SM, Zhu C, Fernagut PO, Mehta A, DiCarlo CD, Seaman RL, Chesselet M (2004) Behavioral and immunohistochemical effects of chronic intravenous and subcutaneous infusions of varying doses of rotenone. Exp Neurol 187:418–429. [DOI] [PubMed] [Google Scholar]
- 75. Follmer C, Roma L, Einsiedler CM, Lara A, Moncores M, Weissmu G et al (2007) Dopamine affects the stability, hydration, and packing of protofibrils and fibrils of the wild type and variants of α‐synuclein. Biochemistry 46:472–482. [DOI] [PubMed] [Google Scholar]
- 76. Freis ED (1954) Mental depression in hypertensive patients treated for long periods with large doses of reserpine. N Engl J Med 251:1006–1008. [DOI] [PubMed] [Google Scholar]
- 77. Fukuzaki K, Kamenosono T, Nagata R (2000) Effects of ropinirole on various parkinsonian models in mice, rats, and cynomolgus monkeys. Pharmacol Biochem Behav 65:503–508. [DOI] [PubMed] [Google Scholar]
- 78. Galte D, Terzioglu M (2008) Parkinson's disease: genetic vs. toxin‐induced rodent models. FEBS J 275:1384–1391. [DOI] [PubMed] [Google Scholar]
- 79. Gao HM, Hong JS (2011) Gene‐environment interactions: key to unraveling the mystery of Parkinson's disease. Prog Neurobiol 94:1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Ghosh B, Antonio T, Reith M, Dutta A (2010) (4‐(2‐((5‐Hydroxy‐1, 2, 3, 4‐tetrahydronaphthalen‐2‐yl)(propyl) amino) ethyl) piperazin‐1‐yl) quinolin‐8‐ol and its analogues as highly potent dopamine D2/D3 agonists. J Med Chem 53:2114–2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Giráldez‐Pérez RM, Antolín‐Vallespín M, Muñoz MD, Sánchez‐Capelo A (2014) Models of α‐synuclein aggregation in Parkinson's disease. Acta Neuropathol Commun 2:176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Glatt CE, Wahner AD, White DJ, Ruiz‐Linares A, Ritz B (2006) Gain‐of‐function haplotypes in the vesicular monoamine transporter promoter are protective for Parkinson disease in women. Hum Mol Genet 15:299–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Gómez‐Esteban JC, Tijero B, Somme J, Ciordia R, Berganzo K, Rouco I et al (2011) Impact of psychiatric symptoms and sleep disorders on the quality of life of patients with Parkinson's disease. J Neurol 258:494–499. [DOI] [PubMed] [Google Scholar]
- 84. Goldstein DS, Sullivan P, Cooney A, Jinsmaa Y, Sullivan R, Gross DJ et al (2012) Vesicular uptake blockade generates the toxic dopamine metabolite 3,4‐dihydroxyphenylacetaldehyde in PC12 cells: relevance to the pathogenesis of Parkinson's disease. J Neurochem 123:932–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Goldstein J, Barnett A, Malick J (1975) The evaluation of anti‐parkinson drugs on reserpine‐induced rigidity in rats. Eur J Pharmacol 33:183–188. [DOI] [PubMed] [Google Scholar]
- 86. Gołembiowska K, Dziubina A (2012) The effect of adenosine A(2A) receptor antagonists on hydroxyl radical, dopamine, and glutamate in the striatum of rats with altered function of VMAT2. Neurotox Res 22:150–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Guttman M (1992) Dopamine receptors in Parkinson's disease. Neurol Clin 10:377–386. [PubMed] [Google Scholar]
- 88. Hald A, Lotharius J (2005) Oxidative stress and inflammation in Parkinson's disease: is there a causal link? Exp Neurol 193:279–290. [DOI] [PubMed] [Google Scholar]
- 89. Harrington KA, Augood SJ, Kingsbury AE, Foster OJF, Emson PC (1996) Dopamine transporter (DAT) and synaptic vesicle amine transporter (VMAT2) gene expression in the substantia nigra of control and Parkinson's disease. Brain Res Mol Brain Res 36:157–162. [DOI] [PubMed] [Google Scholar]
- 90. Heeringa MJ, Abercrombie ED (1995) Biochemistry of somatodendritic dopamine release in substantia nigra: an in vivo comparison with striatal dopamine release. J Neurochem 65:192–200. [DOI] [PubMed] [Google Scholar]
- 91. Henchcliffe C, Severt WL (2011) Disease modification in Parkinson's disease. Drugs Aging 28:605–615. [DOI] [PubMed] [Google Scholar]
- 92. Henderson JM, Stanic D, Tomas D, Patch J, Horne MK, Bourke D, Finkelstein DI (2005) Postural changes after lesions of the substantia nigra pars reticulata in hemiparkinsonian monkeys. Behav Brain Res 160:267–276. [DOI] [PubMed] [Google Scholar]
- 93. Höglinger GU, Féger J, Prigent A, Michel PP, Parain K, Champy P et al (2003) Chronic systemic complex I inhibition induces a hypokinetic multisystem degeneration in rats. J Neurochem 84:491–502. [DOI] [PubMed] [Google Scholar]
- 94. Hsieh Y, Mounsey RB, Teismann P (2011) MPP+‐induced toxicity in the presence of dopamine is mediated by COX‐2 through oxidative stress. Naunyn Schmiedebergs Arch Pharmacol 384:157–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Hurley MJ, Jenner P (2006) What has been learnt from study of dopamine receptors in Parkinson's disease? Pharmacol Ther 111:715–728. [DOI] [PubMed] [Google Scholar]
- 96. Ischiropoulos H (2003) Biological selectivity and functional aspects of protein tyrosine nitration. Biochem Biophys Res Commun 305:776–783. [DOI] [PubMed] [Google Scholar]
- 97. Ishihara L, Brayne C (2006) A systematic review of depression and mental illness preceding Parkinson's disease. Acta Neurol Scand 113:211–220. [DOI] [PubMed] [Google Scholar]
- 98. Johnels B (1982) Locomotor hypokinesia in the reserpine‐treated rat: drug effects from the corpus striatum and nucleus accumbens. Pharmacol Biochem Behav 17:283–289. [DOI] [PubMed] [Google Scholar]
- 99. Johnson A, Loew D, Vigouret J (1976) Stimulant properties of bromocriptine on central dopamine receptors in comparison to apomorphine, (+)‐amphetamine and L‐dopa. Br J Pharmacol 56:59–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Jurna I, Grossmann W, Nell T (1973) Depression by amantadine of tremor induced by reserpine and oxotremorine in the rat. Naunyn Schmiedebergs Arch Pharmacol 152:141–152. [Google Scholar]
- 101. Kalaitzakis ME, Gentleman SM, Pearce RKB (2013) Disturbed sleep in Parkinson's disease: anatomical and pathological correlates. Neuropathol Appl Neurobiol 39:644–653. [DOI] [PubMed] [Google Scholar]
- 102. Kane JM, Smith JM (1982) Tardive dyskinesia; prevalence and risk factors, 1959 to 1979. Arch Gen Psychiatry 39:473–481. [DOI] [PubMed] [Google Scholar]
- 103. Kaur S, Starr M (1996) Motor effects of lamotrigine in naive and dopamine‐depleted mice. Eur J Pharmacol 304:1–6. [DOI] [PubMed] [Google Scholar]
- 104. Kim SW, Ko HS, Dawson VL, Dawson TM (2005) Recent advances in our understanding of Parkinson's disease. Drug Discov Today Dis Mech 2:427–433. [Google Scholar]
- 105. Kirik D, Annett LE, Burger C, Muzyczka N, Mandel RJ, Bjo A (2003) Nigrostriatal alpha‐synucleinopathy induced by viral vector‐mediated overexpression of human alpha‐synuclein: a new primate model of Parkinson's disease. Proc Natl Acad Sci U S A 100:2884–2889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S et al (1998) Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 392:605–608. [DOI] [PubMed] [Google Scholar]
- 107. Kitada T, Pisani A, Porter DR, Yamaguchi H, Tscherter A, Martella G et al (2007) Impaired dopamine release and synaptic plasticity in the striatum of PINK1‐deficient mice. Proc Natl Acad Sci U S A 104:11441–11446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Klockgether T (2004) Parkinson's disease: clinical aspects. Cell Tissue Res 318:115–120. [DOI] [PubMed] [Google Scholar]
- 109. Korecka JA, Eggers R, Swaab DF, Bossers K, Verhaagen J (2013) Modeling early Parkinson's disease pathology with chronic low dose MPTP treatment. Restor Neurol Neurosci 31:155–167. [DOI] [PubMed] [Google Scholar]
- 110. Kuhn DM, Aretha CW, Geddes TJ (1999) Peroxynitrite inactivation of tyrosine hydroxylase: mediation by sulfhydryl oxidation, not tyrosine nitration. J Neurosci 19:10289–10294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Kuhn DM, Sakowski SA, Sadidi M, Geddes TJ (2004) Nitrotyrosine as a marker for peroxynitrite‐induced neurotoxicity: the beginning or the end of the end of dopamine neurons? J Neurochem 89:529–536. [DOI] [PubMed] [Google Scholar]
- 112. LaBuda CJ, Fuchs PN (2002) Catecholamine depletion by reserpine blocks the anxiolytic actions of ethanol in the rat. Alcohol 26:55–59. [DOI] [PubMed] [Google Scholar]
- 113. Lane E, Dunnett S (2008) Animal models of Parkinson's disease and L‐dopa induced dyskinesia: how close are we to the clinic? Psychopharmacology (Berl) 199:303–312. [DOI] [PubMed] [Google Scholar]
- 114. Lee FJS, Liu F (2008) Genetic factors involved in the pathogenesis of Parkinson's disease. Brain Res Rev 58:354–364. [DOI] [PubMed] [Google Scholar]
- 115. Lees AJ, Smith E (1983) Cognitive deficits in the early stages of Parkinson's disease. Brain 106:257–270. [DOI] [PubMed] [Google Scholar]
- 116. Li HT, Lin DH, Luo XY, Zhang F, Ji LN, Du HN et al (2005) Inhibition of alpha‐synuclein fibrillization by dopamine analogs via reaction with the amino groups of alpha‐synuclein. Implication for dopaminergic neurodegeneration. FEBS J 272:3661–3672. [DOI] [PubMed] [Google Scholar]
- 117. Li X, Patel JC, Wang J, Avshalumov M V, Nicholson C, Buxbaum JD et al (2010) Enhanced striatal dopamine transmission and motor performance with LRRK2 overexpression in mice is eliminated by familial Parkinson's disease mutation G2019S. J Neurosci 30:1788–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Lin X, Parisiadou L, Gu X‐L, Wang L, Shim H, Sun L et al (2009) Leucine‐rich repeat kinase 2 regulates the progression of neuropathology induced by Parkinson's‐disease‐related mutant alpha‐synuclein. Neuron 64:807–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Liu S, Zhao R, Li X, Guo H, Tian Z (2014) Attenuation of reserpine‐induced pain/depression dyad by gentiopicroside through downregulation of GluN2B receptors in the amygdala of mice. Neuromolecular Med 16:350–359. [DOI] [PubMed] [Google Scholar]
- 120. Lotharius J, Brundin P (2002) Pathogenesis of Parkinson's disease: dopamine, vesicles and alpha‐synuclein. Nat Rev Neurosci 3:932–942. [DOI] [PubMed] [Google Scholar]
- 121. Maj J, Rog Z, Skuza G, Sowifiska H, Superata J (1990) Behavioural and neurochemical effects of Ro 40–7592, a new COMT inhibitor with a potential therapeutic activity in Parkinson's disease. J Neural Transm 2:101–112. [DOI] [PubMed] [Google Scholar]
- 122. Maj J, Rogóz Z, Skuza G, Kołodziejczyk K (1997) The behavioural effects of pramipexole, a novel dopamine receptor agonist. Eur J Pharmacol 324:31–37. [DOI] [PubMed] [Google Scholar]
- 123. Mata IF, Leverenz JB, Weintraub D, Trojanowski JQ, Hurtig HI, Van Deerlin VM et al (2014) APOE, MAPT, and SNCA genes and cognitive performance in Parkinson disease. JAMA Neurol 71:1405–1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. McKenna O, Arnold G, Holtzman E (1976) Microperoxisome distribution in the central nervous system of the rat. Brain Res 117:181–194. [DOI] [PubMed] [Google Scholar]
- 125. McQueen EG, Doyle AE, Smirk FH (1954) Mechanism of hypotensive action of reserpine, an alkaloid of Rauwolfia serpentina. Nature 174:1015. [DOI] [PubMed] [Google Scholar]
- 126. Mehndiratta M, Garg RK, Pandey S (2011) Nonmotor symptom complex of Parkinson's disease—an under‐recognized entity. J Assoc Physicians India 59:302–308. [PubMed] [Google Scholar]
- 127. Meiser J, Weindl D, Hiller K (2013) Complexity of dopamine metabolism. Cell Commun Signal 11:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Meredith GE, Halliday GM, Totterdell S (2004) A critical review of the development and importance of proteinaceous aggregates in animal models of Parkinson's disease: new insights into Lewy body formation. Parkinsonism Relat Disord 10:191–202. [DOI] [PubMed] [Google Scholar]
- 129. Meredith GE, Sonsalla PK, Chesselet MF (2008) Animal models of Parkinson's disease progression. Acta Neuropathol 115:385–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Millan MJ, Agid Y, Brüne M, Bullmore ET, Carter CS, Clayton NS et al (2012) Cognitive dysfunction in psychiatric disorders: characteristics, causes and the quest for improved therapy. Nat Rev Drug Discov 11:141–168. [DOI] [PubMed] [Google Scholar]
- 131. Miller GW, Erickson JD, Perez JT, Penland SN, Mash DC, Rye DB, Levey AI (1999) Immunochemical analysis of vesicular monoamine transporter (VMAT2) protein in Parkinson's disease. Exp Neurol 148:138–148. [DOI] [PubMed] [Google Scholar]
- 132. Missale C, Nisoli E, Liberini P, Rizzonelli P, Memo M, Buonamici M et al (1989) Repeated reserpine administration up‐regulates the transduction mechanisms of D1 receptors without changing the density of [3H]SCH 23390 binding. Brain Res 483:117–122. [DOI] [PubMed] [Google Scholar]
- 133. Miyagi M, Arai N, Taya F, Itoh F, Komatsu Y, Kojima M, Isaji M (1996) Effect of cabergoline, a long‐acting dopamine D2 agonist, on reserpine‐treated rodents. Biol Pharm Bull 11:1499–1502. [DOI] [PubMed] [Google Scholar]
- 134. Monderer R, Thorpy M (2009) Sleep disorders and daytime sleepiness in Parkinson's disease. Curr Neurol Neurosci Rep 9:173–180. [DOI] [PubMed] [Google Scholar]
- 135. Mooslehner KA, Chan PM, Xu W, Liu L, Smadja C, Humby T et al (2001) Mice with very low expression of the vesicular monoamine transporter 2 gene survive into adulthood: potential mouse model for parkinsonism. Mol Cell Biol 21:5321–5331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Morin N, Jourdain VA, Di Paolo T (2014) Modeling dyskinesia in animal models of Parkinson disease. Exp Neurol 256:105–116. [DOI] [PubMed] [Google Scholar]
- 137. Mulcahy P, O'Doherty A, Paucard A, O'Brien T, Kirik D, Dowd E (2013) The behavioural and neuropathological impact of intranigral AAV‐α‐synuclein is exacerbated by systemic infusion of the Parkinson's disease‐associated pesticide, rotenone, in rats. Behav Brain Res 243:6–15. [DOI] [PubMed] [Google Scholar]
- 138. Nade VS, Shendye NV, Kawale LA, Patil NR, Khatri ML (2013) Protective effect of nebivolol on reserpine‐induced neurobehavioral and biochemical alterations in rats. Neurochem Int 63:316–321. [DOI] [PubMed] [Google Scholar]
- 139. Naidu PS, Singh A, Kulkarni SK (2006) Effect of Withania somnifera root extract on reserpine‐induced orofacial dyskinesia and cognitive dysfunction. Phytother Res 20:140–146. [DOI] [PubMed] [Google Scholar]
- 140. Neisewander JL, Lucki I, McGonigle P (1991) Neurochemical changes associated with the persistence of spontaneous oral dyskinesia in rats following chronic reserpine treatment. Brain Res 558:27–35. [DOI] [PubMed] [Google Scholar]
- 141. Neisewander JL, Castañeda E, Davis DA (1994) Dose‐dependent differences in the development of reserpine‐induced oral dyskinesia in rats: support for a model of tardive dyskinesia. Psychopharmacology (Berl) 116:79–84. [DOI] [PubMed] [Google Scholar]
- 142. Niswender C, Johnson K, Weaver C, Jones C, Xiang Z, Luo Q et al (2008) Discovery, characterization, and antiparkinsonian effect of novel positive allosteric modulators of metabotropic glutamate receptor 4. Mol Pharmacol 74:1345–1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Noyce AJ, Bestwick JP, Silveira‐Moriyama L, Hawkes CH, Giovannoni G, Lees AJ, Schrag A (2012) Meta‐analysis of early nonmotor features and risk factors for Parkinson disease. Ann Neurol 72:893–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Oe T, Tsukamoto M, Nagakura Y (2010) Reserpine causes biphasic nociceptive sensitivity alteration in conjunction with brain biogenic amine tones in rats. Neuroscience 169:1860–1871. [DOI] [PubMed] [Google Scholar]
- 145. Olsson M, Nikkhah G (1995) Forelimb akinesia in the rat Parkinson model: differential effects of dopamine agonists and nigral transplants as assessed by a new stepping test. J Neurosci 15:3863–3875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Omiatek DM, Bressler AJ, Cans A‐S, Andrews AM, Heien ML, Ewing AG (2013) The real catecholamine content of secretory vesicles in the CNS revealed by electrochemical cytometry. Sci Rep 3:1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Patil R, Kasture S (2012) Protective effect of Rubia cordifolia on reserpine‐induced orofacial dyskinesia. Nat Prod Res 26:2159–2161. [DOI] [PubMed] [Google Scholar]
- 148. Patil R, Dhawale K, Gound H, Gadakh R (2012) Protective effect of leaves of Murraya koenigii on reserpine‐induced orofacial dyskinesia. Iran J Pharm Res 11:635–641. [PMC free article] [PubMed] [Google Scholar]
- 149. Pereira R, Fachinetto R, Prestes AS, Wagner C, Sudati JH, Boligon AA et al (2011) Valeriana officinalis ameliorates vacuous chewing movements induced by reserpine in rats. J Neural Transm 118:1547–1557. [DOI] [PubMed] [Google Scholar]
- 150. Peter D, Jimenez J, Liu Y, Kim J, Edwards RH (1994) The chromaffin granule and synaptic vesicle amine transporters differ in substrate recognition and sensitivity to inhibitors. J Biol Chem 269:7231–7237. [PubMed] [Google Scholar]
- 151. Phinney AL, Andringa G, Bol JGJM, Wolters EC, van Muiswinkel FL, van Dam A‐MW, Drukarch B (2006) Enhanced sensitivity of dopaminergic neurons to rotenone‐induced toxicity with aging. Parkinsonism Relat Disord 12:228–238. [DOI] [PubMed] [Google Scholar]
- 152. Plouvier AOA, Hameleers RJMG, van den Heuvel EAJ, Bor HH, Olde Hartman TC, Bloem BR et al (2014) Prodromal symptoms and early detection of Parkinson's disease in general practice: a nested case‐control study. Fam Pract 31:373–378. [DOI] [PubMed] [Google Scholar]
- 153. Ponzio F, Achilli G, Calderini G, Ferretti P, Perego C, Toffano G, Algeri S (1984) Depletion and recovery of neuronal monoamine storage in rats of different ages treated with reserpine. Neurobiol Aging 5:101–104. [DOI] [PubMed] [Google Scholar]
- 154. Prediger RDS, Matheus FC, Schwarzbold ML, Lima MMS, Vital MABF (2012) Anxiety in Parkinson's disease: a critical review of experimental and clinical studies. Neuropharmacology 62:115–124. [DOI] [PubMed] [Google Scholar]
- 155. Przedborski S, Jackson‐Lewis V, Naini AB, Petzinger G, Miller R, Akram M (2001) The parkinsonian toxin (MPTP): a technical review of its utility and safety. J Neurochem 76:1265–1274. [DOI] [PubMed] [Google Scholar]
- 156. Qi Z, Miller GW, Voit EO (2008) Computational systems analysis of dopamine metabolism. PLoS ONE 3:e2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Ramsey CP, Tsika E, Ischiropoulos H, Giasson BI (2010) DJ‐1 deficient mice demonstrate similar vulnerability to pathogenic Ala53Thr human alpha‐syn toxicity. Hum Mol Genet 19:1425–1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Ravenstijn PGM, Merlini M, Hameetman M, Murray TK, Ward MA, Lewis H et al (2008) The exploration of rotenone as a toxin for inducing Parkinson's disease in rats, for application in BBB transport and PK‐PD experiments. J Pharmacol Toxicol Methods 57:114–130. [DOI] [PubMed] [Google Scholar]
- 159. Reckziegel P, Peroza LR, Schaffer LF, Ferrari MC, de Freitas CM, Bürger ME, Fachinetto R (2013) Gallic acid decreases vacuous chewing movements induced by reserpine in rats. Pharmacol Biochem Behav 104:132–137. [DOI] [PubMed] [Google Scholar]
- 160. Redgrave P, Rodriguez M, Smith Y, Rodriguez‐Oroz MC, Lehericy S, Bergman H et al (2010) Goal‐directed and habitual control in the basal ganglia: implications for Parkinson's disease. Nat Rev Neurosci 11:760–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Riachi NJ, Lamanna C, Harik I (1989) Entry of 1‐methyl‐4‐phenyl‐1,2,3,6‐tetrahydropyridine into rat brain. J Pharmacol Exp Ther 249:744–748. [PubMed] [Google Scholar]
- 162. Rodriguez‐Oroz MC, Jahanshahi M, Krack P, Litvan I, Macias R, Bezard E, Obeso JA (2009) Initial clinical manifestations of Parkinson's disease: features and pathophysiological mechanisms. Lancet Neurol 8:1128–1139. [DOI] [PubMed] [Google Scholar]
- 163. Sai Y, Zou Z, Peng K, Dong Z (2012) The Parkinson's disease‐related genes act in mitochondrial homeostasis. Neurosci Biobehav Rev 36:2034–2043. [DOI] [PubMed] [Google Scholar]
- 164. Salamone J, Baskin P (1996) Vacuous jaw movements induced by acute reserpine and low‐dose apomorphine: possible model of parkinsonian tremor. Pharmacol Biochem Behav 53:179–183. [DOI] [PubMed] [Google Scholar]
- 165. Salgado‐Pineda P, Delaveau P, Blin O, Nieoullon A (2005) Dopaminergic contribution to the regulation of emotional perception. Clin Neuropharmacol 28:228–237. [DOI] [PubMed] [Google Scholar]
- 166. Sanghavi CR, Barhate SA, Mahajan MS, Mohan M, Kasture SB (2011) Korean ginseng extract attenuates reserpine‐induced orofacial dyskinesia and improves cognitive dysfunction in rats. Nat Prod Res 25:704–715. [DOI] [PubMed] [Google Scholar]
- 167. Santos JR, Cunha JAS, Dierschnabel AL, Campêlo CLC, Leão AHFF, Silva AF et al (2013) Cognitive, motor and tyrosine hydroxylase temporal impairment in a model of parkinsonism induced by reserpine. Behav Brain Res 253:68–77. [DOI] [PubMed] [Google Scholar]
- 168. Sarmento‐Silva AJ, Lima RH, Cabral A, Meurer Y, Ribeiro AM, Silva RH (2014) Alpha‐tocopherol counteracts cognitive and motor deficits induced by repeated treatment with reserpine. Biochem Pharmacol 4:153. doi: 10.4172/2167-0501.1000153 [DOI] [Google Scholar]
- 169. Schallert T, Fleming SM, Leasure JL, Tillerson JL, Bland ST (2000) CNS plasticity and assessment of forelimb sensorimotor outcome in unilateral rat models of stroke, cortical ablation, parkinsonism and spinal cord injury. Neuropharmacology 39:777–787. [DOI] [PubMed] [Google Scholar]
- 170. Schober A (2004) Classic toxin‐induced animal models of Parkinson's disease: 6‐OHDA and MPTP. Cell Tissue Res 318:215–224. [DOI] [PubMed] [Google Scholar]
- 171. Schwarting RKW, Huston JP (1996) The unilateral 6‐hydroxydopamine lesion model in behavioral brain research: analysis of functional deficits, recovery and treatments. Prog Neurobiol 50:275–331. [DOI] [PubMed] [Google Scholar]
- 172. Sherer TB, Kim JH, Betarbet R, Greenamyre JT (2003) Subcutaneous rotenone exposure causes highly selective dopaminergic degeneration and alpha‐synuclein aggregation. Exp Neurol 179:9–16. [DOI] [PubMed] [Google Scholar]
- 173. Shook BC, Rassnick S, Osborne MC, Davis S, Westover L, Boulet J et al (2010) In vivo characterization of a dual adenosine A2A/A1 receptor antagonist in animal models of Parkinson's disease. J Med Chem 53:8104–8115. [DOI] [PubMed] [Google Scholar]
- 174. Silva RH, Abílio VC, Torres‐Leite D, Bergamo M, Chinen CC, Claro FT et al (2002) Concomitant development of oral dyskinesia and memory deficits in reserpine‐treated male and female mice. Behav Brain Res 132:171–177. [DOI] [PubMed] [Google Scholar]
- 175. Skalisz LL, Beijamini V, Joca SL, Vital MABF, Da Cunha C, Andreatini R (2002) Evaluation of the face validity of reserpine administration as an animal model of depression—Parkinson's disease association. Prog Neuropsychopharmacol Biol Psychiatry 26:879–883. [DOI] [PubMed] [Google Scholar]
- 176. Skuza G, Rogoz Z, Quack G, Danysz W (1994) Memantine, amantadine, and L‐deprenyl potentiate the action of L‐dopa in monoamine‐depleted rats. J Neural Transm 98:57–67. [DOI] [PubMed] [Google Scholar]
- 177. Sonsalla PK, Zeevalk GD, German DC (2008) Chronic intraventricular administration of 1‐methyl‐4‐phenylpyridinium as a progressive model of Parkinson's disease. Parkinsonism Relat Disord 14 (Suppl. 2):S116–S118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Sourkes TL (1971) Actions of levodopa and dopamine in the central nervous system. JAMA 218:1909–1911. [PubMed] [Google Scholar]
- 179. Spina MB, Cohen G (1989) Dopamine turnover and glutathione oxidation: implications for Parkinson disease. Proc Natl Acad Sci U S A 86:1398–1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Spina N, Cohen GS, York B, York N, July NY (1988) Exposure of school synaptosomes of oxidized glutathione1 to L‐Dopa increases levels. J Pharmacol Exp Ther 247:502–507. [PubMed] [Google Scholar]
- 181. Sriram K, Pai KS, Boyd MR, Ravindranath V (1997) Evidence for generation of oxidative stress in brain by MPTP: in vitro and in vivo studies in mice. Brain Res 749:44–52. [DOI] [PubMed] [Google Scholar]
- 182. Steinpreis RE, Salamone JD (1993) Effects of acute haloperidol and reserpine administration on vacuous jaw movements in three different age groups of rats. Pharmacol Biochem Behav 46:405–409. [DOI] [PubMed] [Google Scholar]
- 183. Sutton AC, Yu W, Calos ME, Smith AB, Ramirez‐Zamora A, Molho ES et al (2013) Deep brain stimulation of the substantia nigra pars reticulata improves forelimb akinesia in the hemiparkinsonian rat. J Neurophysiol 109:363–374. [DOI] [PubMed] [Google Scholar]
- 184. Takahashi H, Wakabayashi K (2001) The cellular pathology of Parkinson's disease. Neuropathology 21:315–322. [DOI] [PubMed] [Google Scholar]
- 185. Taylor TN, Caudle WM, Shepherd KR, Noorian A, Jackson CR, Iuvone PM et al (2009) Nonmotor symptoms of Parkinson's disease revealed in an animal model with reduced monoamine storage capacity. J Neurosci 29:8103–8113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Teixeira AM, Trevizol F, Colpo G, Garcia SC, Charão M, Pereira RP et al (2008) Influence of chronic exercise on reserpine‐induced oxidative stress in rats: behavioral and antioxidant evaluations. Pharmacol Biochem Behav 88:465–472. [DOI] [PubMed] [Google Scholar]
- 187. Teixeira AM, Reckziegel P, Müller L, Pereira RP, Roos DH, Rocha JBT, Bürger ME (2009) Intense exercise potentiates oxidative stress in striatum of reserpine‐treated animals. Pharmacol Biochem Behav 92:231–235. [DOI] [PubMed] [Google Scholar]
- 188. Thiele SL, Warre R, Nash JE (2012) Development of a unilaterally‐lesioned 6‐OHDA mouse model of Parkinson's disease. J Vis Exp 60:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Thomas KL, Rose S, Jenner P, Marsden CD (1992) Acute reserpine treatment induces down regulation of D1 dopamine receptor associated adenylyl cyclase activity in rat striatum. Biochem Pharmacol 44:83–91. [DOI] [PubMed] [Google Scholar]
- 190. Tieu K (2011) A guide to neurotoxic animal models of Parkinson's disease. Cold Spring Harb Perspect Med 1:a009316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. Tohru K, Youren T, Gautier CA, Jie S (2009) Absence of nigral degeneration in aged parkin/DJ‐1/PINK1 triple knockout mice. J Neurochem 111:696–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Torack RM, Morris JC (1992) Tyrosine hydroxylase‐like (TH) immunoreactivity in Parkinson's disease and Alzheimer's disease. J Neural Transm Park Dis Dement Sect 4:165–171. [DOI] [PubMed] [Google Scholar]
- 193. Traub M, Reches A, Wagner HR, Fahn S (1986) Reserpine‐induced up‐regulation of dopamine D2 receptors in the rat striatum is enhanced by denervation but not by chronic receptor blockade. Neurosci Lett 70:245–249. [DOI] [PubMed] [Google Scholar]
- 194. Tredici K, Braak H (2012) Spinal cord lesions in sporadic Parkinson's disease. Acta Neuropathol 124:643–664. [DOI] [PubMed] [Google Scholar]
- 195. Tsuboi Y (2012) Environmental‐genetic interactions in the pathogenesis of Parkinson's disease. Exp Neurobiol 21:123–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196. Tykocki T, Kornakiewicz A, Mandat T, Nauman P (2013) Pain perception in patients with Parkinson's disease. J Clin Neurosci 20:663–666. [DOI] [PubMed] [Google Scholar]
- 197. Uhl GR, Hedreen JC, Price DL (1985) Parkinson's disease: loss of neurons from the ventral tegmental area contralateral to therapeutic surgical lesions. Neurology 35:1215–1218. [DOI] [PubMed] [Google Scholar]
- 198. Ungerstedt U (1971) Adipsia and Aphagia after 6‐Hydroxydopamine induced degeneration of the nigro‐striatal dopamine system. Acta Physiol Scand 82 (S367):95–122. [DOI] [PubMed] [Google Scholar]
- 199. van der Weide J, de Vries J, Tepper PG, Horn AS (1986) Pharmacological profiles of three new, potent and selective dopamine receptor agonists: N‐0434, N‐0437 and N‐0734. Eur J Pharmacol 125:273–282. [DOI] [PubMed] [Google Scholar]
- 200. Venderova K, Park DS (2012) Programmed cell death in Parkinson's disease. Cold Spring Harb Perspect Med 2:a009365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. Vergo S, Johansen JL, Leist M, Lotharius J (2007) Vesicular monoamine transporter 2 regulates the sensitivity of rat dopaminergic neurons to disturbed cytosolic dopamine levels. Brain Res 1185:18–32. [DOI] [PubMed] [Google Scholar]
- 202. Vives‐Bauza C, de Vries RL, Tocilescu MA, Przedborski S (2009) Is there a pathogenic role for mitochondria in Parkinson's disease? Parkinsonism Relat Disord 15 (Suppl. 3):S241–S244. [DOI] [PubMed] [Google Scholar]
- 203. Voon V, Dalley JW (2011) Parkinson disease: impulsive choice‐Parkinson disease and dopaminergic therapy. Nat Rev Neurol 7:541–542. [DOI] [PubMed] [Google Scholar]
- 204. Voon V, Mehta AR, Hallett M (2011) Impulse control disorders in Parkinson's disease: recent advances. Curr Opin Neurol 24:324–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205. Wakamatsu M, Ishii A, Iwata S, Sakagami J, Ukai Y, Ono M et al (2008) Selective loss of nigral dopamine neurons induced by overexpression of truncated human alpha‐synuclein in mice. Neurobiol Aging 29:574–585. [DOI] [PubMed] [Google Scholar]
- 206. Wood‐Kaczmar A, Gandhi S, Wood NW (2006) Understanding the molecular causes of Parkinson's disease. Trends Mol Med 12:521–528. [DOI] [PubMed] [Google Scholar]
- 207. Xiang Z, Thompson AD, Jones CK, Lindsley CW, Conn PJ (2012) Roles of the M1 muscarinic acetylcholine receptor subtype in the regulation of basal ganglia function and implications for the treatment of Parkinson's disease. J Pharmacol Exp Ther 340:595–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208. Yadav S, Dixit A, Agrawal S, Singh A, Srivastava G, Singh AK et al (2012) Rodent models and contemporary molecular techniques: notable feats yet incomplete explanations of Parkinson's disease pathogenesis. Mol Neurobiol 46:495–512. [DOI] [PubMed] [Google Scholar]
- 209. Yamamoto S, Fukae J, Mori H, Mizuno Y, Hattori N (2006) Positive immunoreactivity for vesicular monoamine transporter 2 in Lewy bodies and Lewy neurites in substantia nigra. Neurosci Lett 396:187–191. [DOI] [PubMed] [Google Scholar]
- 210. Yazdani U, German DC, Liang C‐L, Manzino L, Sonsalla PK, Zeevalk GD (2006) Rat model of Parkinson's disease: chronic central delivery of 1‐methyl‐4‐phenylpyridinium (MPP). Exp Neurol 200:172–183. [DOI] [PubMed] [Google Scholar]
- 211. Zarow C, Lyness SA, Mortimer JA, Chui HC (2003) Neuronal loss is greater in the locus coeruleus than nucleus basalis and substantia nigra in Alzheimer and Parkinson diseases. Arch Neurol 60:337–341. [DOI] [PubMed] [Google Scholar]
- 212. Zhu C, Vourc'h P, Fernagut P‐O, Fleming SM, Lacan S, Dicarlo CD et al (2004) Variable effects of chronic subcutaneous administration of rotenone on striatal histology. J Comp Neurol 78:418–426. [DOI] [PubMed] [Google Scholar]
- 213. Zweig RM, Cardillo JE, Cohen M, Giere S, Hedreen JC (1993) The locus coeruleus and dementia in Parkinson's disease. Neurology 43:986–991. [DOI] [PubMed] [Google Scholar]