

Abstract
Cerebral amyloid angiopathy (CAA) is one of the main causes of intracerebral hemorrhage (ICH) in the elderly. Matrix metalloproteinases (MMPs) have been implicated in blood–brain barrier disruption and ICH pathogenesis. In this study, we determined the levels MMP‐2 and MMP‐9 in plasma and their brain expression in CAA‐associated hemorrhagic stroke. Although MMP‐2 and MMP‐9 plasma levels did not differ among patients and controls, their brain expression was increased in perihematoma areas of CAA‐related hemorrhagic strokes compared with contralateral areas and nonhemorrhagic brains. In addition, MMP‐2 reactivity was found in β‐amyloid (Aβ)‐damaged vessels located far from the acute ICH and in chronic microbleeds. MMP‐2 expression was associated to endothelial cells, histiocytes and reactive astrocytes, whereas MMP‐9 expression was restricted to inflammatory cells. In summary, MMP‐2 expression within and around Aβ‐compromised vessels might contribute to the vasculature fatal fate, triggering an eventual bleeding.
Keywords: cerebral amyloid angiopathy, hemorrhagic stroke, intracerebral hemorrhage, matrix metalloproteinase, MMP‐2, MMP‐9
INTRODUCTION
Cerebral amyloid angiopathy (CAA) is characterized by the deposition of amyloid protein, of which β‐amyloid (Aβ) is the most common form, in the media and adventitia of small‐ and mid‐sized arteries, and, less frequently, in veins, of the cerebral cortex and the leptomeninges (31). In addition to the high incidence of CAA in Alzheimer's disease (AD) (11), this disorder is clinically significant for being one of the main causes of symptomatic lobar intracerebral hemorrhage (ICH) in the elderly. Clinical manifestations of CAA include white matter changes associated with cortical microbleeds and progressive dementia (16). Microbleeds appear as small hypointense lesions on gradient‐echo T2*‐weighted magnetic resonance imaging (MRI) sequences and clusters of hemosiderin‐containing macrophages. Small and asymptomatic microbleeds commonly accompany the larger symptomatic macrobleeds (9).
The mechanisms of vessel rupture triggering ICH because of Aβ deposition have not been yet elucidated. Neuropathological studies have demonstrated that Aβ deposits within brain vessel walls are related to abnormalities in the vasculature, including microaneurysms and fibrinoid necrosis, which are closely correlated with the occurrence of ICH 15, 29. A mechanical stress produced by compact and fibrillar Aβ in the vessel wall has been proposed as a possible cause of its rupture. In this regard, proteolytic systems might also form part of the molecular pathway involved in the alteration of the blood–brain barrier (BBB).
Matrix metalloproteinases (MMPs) are a family of zinc‐dependent endopeptidases responsible for the degradation and remodeling of extracellular matrices. The members of the MMP family with gelatinase activity, MMP‐2 and MMP‐9 (gelatinases), have been widely associated with BBB disruption and ICH 17, 19, 21, 22.
Despite the ability to degrade a range of protein substrates, as certain components of the basal lamina, MMP‐2 and MMP‐9 can proteolyse different Aβ peptides in vitro 4, 7, 10, 20, 32. In fact, increased expression of MMP‐9 has been found around Aβ deposits in post‐mortem AD brains (4) and Baig et al (5) demonstrated that the percentage of active MMP‐9 is greater in the frontal cortex of AD brains than in controls.
Here, we aimed to investigate the possible alteration of MMP‐2 and MMP‐9 in plasma of CAA patients and their expression and localization in different brain areas and damaged vasculature of CAA‐associated hemorrhagic stroke patients.
METHODS
Study patients
Consecutive patients aged ≥55 years who presented to the Vall d'Hebron Hospital and were diagnosed as primary lobar ICH were recruited. For this purpose, all patients underwent a set of diagnostic tests, including routine blood biochemistry, coagulation tests, computed tomography (CT) scan and brain MRI with either CT angiography or magnetic resonance angiography when appropriate, as well as conventional angiography in selected patients. We excluded patients with a lobar ICH related to vascular malformation, impaired coagulation or oral anticoagulant intake, traumatic brain injury, tumoral bleedings and those who underwent a surgical procedure. Patients with a baseline MRI study showing multiple lobar and deep microbleeds, suggesting the existence of a mixed pathology were excluded as well. Baseline clinical and demographic characteristics were collected and patients were classified as having a probable or possible CAA, according to the Boston criteria at baseline (12). Briefly, the presence of multiple and exclusively lobar, cortical, or corticosubcortical hemorrhages, detected by gradient‐echo MRI sequences, was defined as probable CAA‐related ICH, whereas the diagnosis of possible CAA was defined by a single lobar hemorrhagic lesion, without any other finding explaining the ICH. Patients were followed up every 6 months at the outpatient clinic, as part of their clinical evaluation. Follow‐up duration was variable, ranging from 20 to 36 months. Control participants were healthy volunteers, elder than 65 years, classified free of neurovascular and cardiovascular history, and familiar history of stroke by direct interview before recruitment. Informed written consent was obtained from all subjects, and the local Ethics Committee approved the study. All subjects were of Caucasian ancestry.
MRI protocol
A baseline MRI was performed in all patients during the first week. All MRI examinations were obtained using a 1.5‐T whole body scanner. Images obtained included axial T2‐weighted turbo spin‐echo [(3700/90/2) (TR/TE/excitations)], axial T1‐weighted spin‐echo [(550/14/2)], turbo fluid‐attenuated inversion recovery [(9000/110/2)] and axial T2‐weighted susceptibility‐based echo‐planar gradient‐echo sequence [(0.8/29/1)]. All images were evaluated by a neurologist and a neuroradiologist blinded to the clinical information to detect hemorrhages (microhemorrhages and macrohemorrhages), and the presence and degree of leukoaraiosis. Hemorrhages were defined as hypointense foci on gradient‐echo sequences and recorded according to size (microhemorrhages ≤5 mm in diameter, macrohemorrhages >5 mm in diameter). Leukoaraiosis was defined as irregular hyperintensities extending into the deep white matter in fluid‐attenuated inversion recovery or T2‐weighted MRI scans. The degree of cerebral leukoaraiosis was estimated using a modification of the age‐related white matter change rating scale, according to our institutional protocol (30). Leukoaraiosis score was recorded in the hemisphere not affected by the hemorrhage, except in cases of both hemisphere involvements.
MMP‐2/MMP‐9 determination in plasma samples
Blood samples were collected from acute possible/probable CAA patients (within the first 24 h after the symptoms onset; n = 33), chronic possible/probable CAA patients (during the patient's follow‐up, at least 6 months after the event; n = 15) and controls (n = 21). The distribution and number of samples are represented in the Supporting Information Figure S1. Peripheral blood was collected in ethylenediaminetetraacetic acid tubes and plasma was immediately separated by centrifugation at 3500 rpm for 15 minutes and stored at −80°C until analysis was performed.
MMP‐2 and MMP‐9 levels were assayed by enzyme‐linked immunosorbent assay (ELISA) in 96‐well microtiter plates (Amersham Matrix Metalloproteinase‐2 or ‐9, Human, Biotrak ELISA System, GE Healthcare, Barcelona, Spain). The ELISA kits recognize the free and complexed precursors of MMP‐2 and MMP‐9 (proMMP‐2/proMMP‐9), respectively. Each sample was tested in duplicate, and the coefficient of variation was <20%.
CAA‐related ICH brain tissue samples for MMP‐9/MMP‐2 immunofluorescence and immunoblotting analysis
For the immunoblotting and immunofluorescence studies, four deceased patients who had a CAA‐associated hemorrhagic stroke within the previous 3 days were included in the study. On autopsy, hemorrhagic area was delimited by an experienced neuropathologist, who obtained brain tissue from perihematoma (PH) and contralateral (CL) areas. A morphological diagnosis of CAA was made in all cases. Three control subjects who had died from other, noninflammatory, non‐neurologic diseases were included. Patients' characteristics and the causes of death are presented in Table 1. All patients or their representatives had consented to participate in this research protocol before the tissue donation, and all samples were obtained from 2 to 20 h after decease.
Table 1.
Cause of death and patients' characteristics. matrix metalloproteinase (MMP)‐2 and MMP‐9 expression in brain samples was evaluated using different methodologies. Abbreviations: IF = immunofluorescence; IB = immunoblotting; IH = immunohistochemistry. AD = Alzheimer's disease; CAA = cerebral amyloid angiopathy; HCV = hepatitis c virus; HTN = hypertension; DM = diabetes mellitus; DL = dyslipidemia; COPD = chronic obstructive pulmonary disease.
| Case number | Methodology | Age | Sex | AD | CAA | Cause of death | Other observations |
|---|---|---|---|---|---|---|---|
| 1 | IF, IB, IH | 84 | Male | IV | + | Intracerebral hemorrhage in the left frontal/parietal lobes | HTN |
| 2 | IF, IB, IH | 88 | Female | V–VI | + | Intracerebral hemorrhage in the right frontal/parietal lobes | — |
| 3 | IF, IB, IH | 90 | Female | V–VI | + | Intracerebral hemorrhage in the left frontal/parietal/ occipital lobes | COPD, HTN |
| 4 | IF, IB, IH | 79 | Female | IV | + | Intracerebral hemorrhage in the left parietal lobe (ventricular invasion) | — |
| 5 | IH | 66 | Male | II | + | Intracerebral hemorrhage in the right frontal/parietal lobes (ventricular invasion) | HTN |
| 6 | IH | 65 | Male | — | + | Heart failure | Hepatic transplantation, HCV‐positive |
| 10 | IF, IB | 83 | Male | — | — | Legionellosis | — |
| 11 | IF, IB | 64 | Female | — | — | Digestive hemorrhage | — |
| 12 | IF, IB | 80 | Male | — | — | Heart and kidney failure | — |
| 7 | IH | 49 | Female | — | — | Heart failure/pulmonary hypertension | — |
| 8 | IH | 63 | Male | — | — | Large B‐cell lymphoma/pulmonary edema | DM, DL, ischemic cardiomyopathy |
| 9 | IH | 72 | Male | — | — | Septic shock | HTN, DL, COPD |
For immunofluorescence analysis, frozen sections (12 µm) were fixed in ice‐cold acetone for 15 inutes and then washed in phosphate‐buffered saline (PBS) for 5 minutes. The sections were blocked with 10% bovine serum albumin (w/v) per 0.3% Triton X‐100 (v/v) in PBS (blocking buffer) for 30 minutes at room temperature (RT), and then incubated overnight (ON) at 4°C with mouse antihuman MMP‐9 or MMP‐2 1:100 (Chemicon, Millipore, Hertfordshire, UK) in blocking buffer. After washing in PBS, the sections were incubated with goat Alexa‐Fluor 568 antirabbit immunoglobulin G antibodies (Invitrogen, Carlsbad, CA, USA) diluted 1:500 in blocking buffer for 1 h at RT. Finally, the sections were thoroughly washed and then mounted on coverslips using Vectashield with 46‐diamidino‐2‐phenyl indole (DAPI, Vector Laboratories, Burlingame, CA, USA) for nuclear staining.
For immunoblotting of brain homogenates, tissue samples frozen in liquid nitrogen and then stored at −80°C were weighed out and then mixed in cold lysis buffer (50 mM Tris‐HCl, pH 7.6, 150 mM NaCl, 5 mM CaCl2, 0.05% Brij‐35, 0.02% NaN3 and 1% Triton X‐100) containing protease inhibitors (1 mM phenylmethanesulfonylfluoride and 7 µg/mL aprotinin). MMP‐9 and MMP‐2 protein content were detected by Western blot in brain homogenates. Briefly, an equal protein amount (30 µg) was separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis and transferred onto nitrocellulose membrane. For MMP‐9 Western blot, samples were diluted in nonreducing sample buffer, whereas for MMP‐2 Western blot, samples were diluted in 10% 2‐mercaptoethanol and boiled for 5 minutes. Nonspecific bindings were blocked for 1 h with 10% (w/v) of nonfatty milk in PBS, 0.1% Tween 20, before membranes were incubated ON with human anti‐MMP‐9 or MMP‐2 (1:500; Chemicon, El Segundo, CA, USA) at 4°C. Then, membranes were incubated with secondary antibody antimouse‐horseradish peroxidase (HRP) (DAKO, Glostrup, Denmark; 1:1000) for 1 h at RT. Membranes were developed using ECL® detection reagents from Millipore. Immunodetection of glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH) (Ambion, Austin, TX, USA) was performed to verify that equal amounts of total protein were loaded for each sample.
MMP‐2/MMP‐9 and amyloid load in CAA vessels/lesions
With the purpose of studying the implication of MMP‐2 and MMP‐9 in CAA patient's brain vasculature, nine cases were used to carry out a immunohistochemistry study. From them, six deceased cases were classified as severe CAA whereas the rest did not present any sign of CAA at autopsy and were included as controls. Patients' characteristics and the causes of death are presented in Table 1. The distribution and number of samples are represented in the Supporting Information Figure S1.
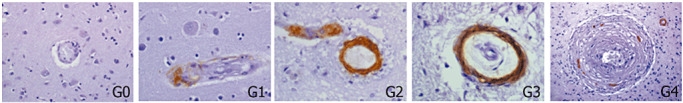
For the immunohistochemistry, 10 hematoxylin‐eosin‐stained slides representative of different cerebral areas were reviewed from each case, selecting all vascular lesions in those sections. Only the parenchymal CAA‐related lesions were studied, not the meningeal amyloid affected vessels. These lesions were graded following the criteria described by Vonsattel in 1991 and modified by Greenberg and Vonsattel in 1997. Grade 0 CAA is given when no amyloid can be found in the vessel wall. Grade 1 CAA lesions show some amyloid deposit in otherwise normal vessels. The thickening of the vessel wall with a stiff appearance is characteristic of a grade 2 CAA lesion, where the media is replaced by amyloid. Grade 3 CAA shows cracking of the vessel wall, creating a “vessel‐within‐vessel” appearance, affecting at least 50% of the circumference of the vessel. In grade 4 CAA lesions, scarring of the wall is observed, with fibrinoid necrosis and traces of intermingled amyloid deposits. Moreover, chronic bleeds, defined as hemosiderin or hematoidin deposits with or without the presence of macrophages, were also selected from all CAA or control sections. AD pathology was evaluated and staged according to Braak and Braak's criteria (6) .
Consecutive sections of the selected blocks were stained with anti‐Aβ, anti‐MMP‐2 and anti‐MMP‐9 antibodies, and evaluated for their positivity within the affected vessels. In sections from CAA patients, a total of 30 morphologically healthy vessels negative for Aβ staining (grade 0 CAA) and 60 positive for Aβ staining (grade 1 and 2 CAA) were randomly selected. In sections from control cases, 30 vessels were also randomly selected, and defined lesions were also determined. The positivity of MMP‐9, MMP‐2 and Aβ staining in damaged or healthy brain vessels was systematically defined by two different expert neuropathologists. The procedure was carried out with brain samples fixed for 48 h with formalin 10%, embedded in paraffin and cut into 8‐µm sections using a microtome. Paraffin was removed, endogenous peroxidases were blocked with methanol‐H2O2 and unspecific binding sites were blocked with 10% normal goat serum for 2 h. Sections were incubated with mouse antihuman MMP‐9 1:100 (Chemicon) and mouse antihuman MMP‐2 1:25 (Chemicon), mouse antihuman β‐Amyloid Clone 6F/3D 1:500 (DAKO) for 1 h at RT. Secondary antibody (goat antimouse horseradish peroxidase, 1:500) was applied for 30 minutes. Immunoreactive sites were developed with 3, 3′‐diaminobenzidine (DAB) solution and serial sections were stained with hematoxylin‐eosin for assessing morphology. Cell‐specific markers were used to identify different MMP‐2/‐9‐positive cell types: anti‐CD68 for macrophages and histiocytes, anti‐CD31 for endothelial cells and anti‐GFAP for reactive astrocytes. Fibrinoid necrosis was detected by phosphotungstic acid hematoxylin stain (Mallory's protocol).
Statistical analysis
Statistical analyses were performed using SPSS software, 15.0 (SPSS Inc., Chicago, IL, USA). Intergroup differences were calculated by Fisher's exact test for categorical variables. MMP‐2 and MMP‐9 were normally distributed (Kolmogorov–Smirnov). Values are expressed as mean ± standard deviation, Student's t‐test and ANOVA, and Bonferroni correction; tests were performed to assess intergroup differences. Pearson's correlations were assessed between variables.
RESULTS
MMP‐2 /MMP‐9 level in possible/probable CAA patients
Demographic characteristics and relevant clinical and radiological variables related to CAA of the patients are shown in Table 2. MMP‐2/MMP‐9 and Aβ plasma level were measured in CAA patients and controls. We found that plasma MMP‐2 levels were lower during the acute phase after ICH (1280.53 ± 529.54 ng/mL) compared with chronic CAA–ICH patients or controls, although the difference did not reach statistical significance. Chronic CAA–hemorrhagic stroke patients showed a median plasma MMP‐2 concentration of 1459.29 ± 330.58 ng/mL, similar to controls (1458.99 ± 355.25 ng/mL). Plasma levels of MMP‐9 were slightly higher, but not significant when measured within the first 24 h after the CAA‐associated hemorrhagic event (87.86 ± 66.99 ng/mL), than in the chronic phase (77.95 ± 53.56 ng/mL) or in elderly control subjects (66.54 ± 38.10 ng/mL; data not shown). None of the demographic parameters or clinical variables was associated with MMP‐2 or MMP‐9 plasma level.
Table 2.
Demographic characteristics and cerebral amyloid angiopathy (CAA) neuroimaging features of acute (24 h after the hemorrhagic stroke) and chronic (more than 6 months after the last hemorrhagic stroke) CAA patients.
| CAA Acute patients | CAA Chronic patients | |
|---|---|---|
| n = 15 | n = 33 | |
| Age at first hemorrhagic stroke | 71.5 ± 4.9 | 74.5 ± 6.9 |
| Females | 5 (33.3%) | 13 (39.4%) |
| Hypertension | 6 (42.9%) | 14 (51.9%) |
| Diabetes mellitus | 0 (0%) | 2 (20%) |
| Prestroke cognitive decline | 3 (33.3%) | 7 (29.2%) |
| Previous stroke | 1 (7.7%) | 3 (11.1%) |
| Presence of multiple hemorrhages | 3 (20%) | 6 (20.6%) |
| Meningeal siderosis | 8 (72.7%) | 3 (33.3%) |
| Severe leukoaraiosis | 5 (41.7%) | 5 (35.7%) |
| Probable CAA | 12 (80%) | 21 (67.7%) |
High content of MMP‐9 and MMP‐2 in the CAA‐associated ICH
Immunoblotting studies of MMP‐9 and MMP‐2 in brains of CAA patients who died because of the fatal hemorrhage showed that pro‐MMP‐9 (95 kDa) and pro‐MMP‐2 (72 kDa) content was significantly higher in hemorrhagic areas (PH) than in CL hemisphere or nonhemorraghic brains (pro‐MMP‐2, P = 0.029 and pro‐MMP‐9, P = 0.039) (Figure 1A). The cellular localization of MMP‐2 and MMP‐9 was assayed in PH and the corresponding CL areas by immunofluorescence. We found MMP‐9 mainly located in brain vessels and some neurons in both areas. MMP‐2 expression, which was mainly located in medium‐sized brain vessels and capillaries, was more pronounced in the PH area, being especially well‐defined in the inner layer of the vessel wall (Figure 1B, see inserts). Aβ‐deposition in blood vessels was analysed using thioflavin S staining and allowed us to conclude that not all MMP‐2‐positive vessels were affected by CAA (data not shown).
Figure 1.
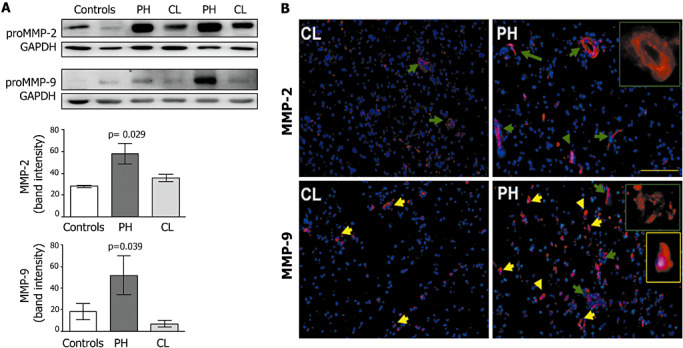
A. Representative pro‐matrix metalloproteinase (MMP)‐2 and pro‐MMP‐9 Western blots of perihematoma (PH) and contralateral (CL) areas of cerebral amyloid angiopathy (CAA) hemorrhagic stroke patients and control brains and densitometric quantification of the corresponding bands. B. Representative MMP‐2 and MMP‐9 inmunofluorescence of PH and CL areas of CAA hemorrhagic stroke patients. Green arrows show positive staining in brain vessels and yellow arrow mark positive neurons. Bars = 50 µm.
MMP‐2 and MMP‐9 expression in CAA‐related vascular damage
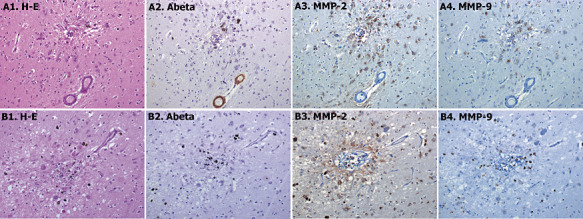
Aβ and hematoxylin‐eosin staining allowed the recognition of CAA‐affected vessels from sections of CAA patient's brains. Following previously described morphological criteria (see Methods section), 30 CAA‐grade 0, 60 CAA‐grade 1 and 60 grade 2 vessels were randomly selected. By definition, all of grade 1 and 2 CAA lesions were positive for Aβ staining (Figure 2A2). Whereas MMP‐9 expression was practically absent in those cases (Figure 2A4), MMP‐2 was expressed in reactive astrocytes surrounding Aβ‐affected vessels (2, 3). Its expression was clearly associated with the Aβ load in the vessel, being more pronounced in grade 2 CAA (60.1%) than grade 1 (26.6%) (Table 3). Furthermore, MMP‐2 expression was higher around those vessels that contain Aβ in the vascular wall and in the surrounding cerebral parenchyma. Indeed, MMP‐2 was expressed in reactive astrocytes associated with both vascular Aβ deposition and neuritic plaques.
Figure 2.
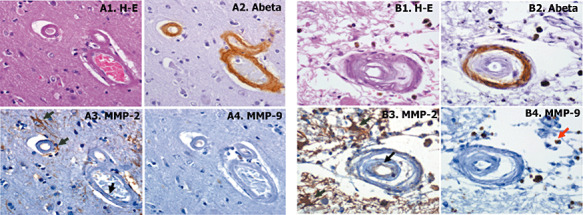
A. Representative example of cerebral amyloid angiopathy (CAA) grade 2; brain vessels that present the media muscle layer replaced by amyloid. B. Representative example of CAA grade 3; vessel with double‐barrel appearance. Serial section were stained with hematoxylin‐eosin (H‐E) staining (A1 and B1), anti‐β‐amyloid (A2 and B2), anti‐matrix metalloproteinase (MMP)‐2 (A3 and B3) and anti‐MMP‐9 (A4 and B4) antibodies. Cellular positive staining is indicated with black arrows for endothelial cells and green arrows for reactive astrocytes 400× magnification.
Figure 3.
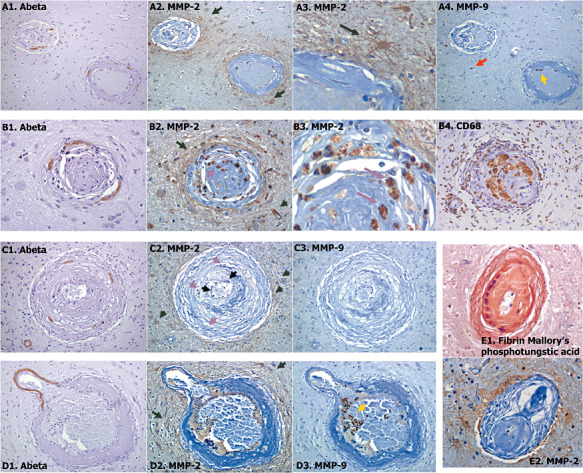
Different representative examples of cerebral amyloid angiopathy grade 4 (A–E). Serial sections were stained with anti‐β‐amyloid (Aβ) (A1, B1, C1 and D1), anti‐matrix metalloproteinase (MMP)‐2 (A2, A3, B2, B3, C2, D2 and E2), anti‐MMP‐9 (A4, C3 and D3) or anti‐CD68 (B4) antibodies. Fibrinoid necrosis in some sections was detected through Mallory phosphotungstic acid staining (E1). Cellular positive staining is indicated with black arrows for endothelial cells, green arrows for reactive astrocytes, pink arrows for histiocytes, red arrows for macrophages and yellow arrows for neutrophils. A: 100× magnification, B–E: 400× magnification. A3 and B3 images correspond to zoomed images from A2 and B2, respectively.
Table 3.
Number of positive vessels for β‐amyloid (Aβ), matrix metalloproteinase (MMP)‐2 or MMP‐9 immunostaining from the total number of vessels of each group, according to cerebral amyloid angiopathy (CAA) severity following the pathological criteria described in the Methods section. Representative Aβ immunohistochemistry of CAA different grades.
| Aβ | MMP‐9 | MMP‐2 | |
|---|---|---|---|
| Positivity within the vessel wall | Cellular positivity within or around the vessel | Cellular positivity (a) with or (b) around the vessel | |
| CAA (n = 6) | |||
| Grade 0 | 0/30 (0%) | 0/30 (0%) | (a) 3/30 (10%) |
| (b) 0/30 (0%) | |||
| Grade 1 | 60/60 (100%) | 0/60 (0%) | (a) 0/60 (0%) |
| (b) 16/60 (26.7%) | |||
| Grade 2 | 60/60 (100%) | 2/60 (3.3%) | (a) 12/60 (20%) |
| (b) 37/60 (61.7%) | |||
| Grade 3 | 10/11 (90.1%) | 0/11 (0%) | (a) 7/11 (63.6%) |
| (b) 11/11 (100%) | |||
| Grade 4 | 12/24 (50%) | 1/24 (4.2%) | (a) 12/24 (50%) |
| (b) 24/24 (100%) | |||
| Controls (n = 6) | 0/30 (0%) | 0/30 (0%) | (a) 6/30 (20%) |
| (b) 0/30 (0%) | |||
| |||
Elements shown in bold indicate that the positive staining is ≥50%.
In grade 3 CAA lesions, Aβ reactivity was very frequent (10 of 11 lesions, 91%) and restricted to the external membrane of the vessel (Figure 2B2). We also observed a very consistent MMP‐2 expression in astrocytes surrounding the lesions (100% of the cases; Figure 2B3 shows one example of grade 3 CAA), although some vessels (7 of 11 lesions, 63.6%) also expressed MMP‐2 in the endothelium (Figure 2B3). MMP‐9 staining was only observed in some isolated macrophages around the grade 3 CAA lesions (Figure 2B4).
On the contrary, Aβ within the vessel wall was only found in 50% of grade 4 CAA lesions, showing a very focal and segmented staining (different examples in Figure 3A1,B1,C1,D1). Gliosis was prominent around these fibrous thickened vessels, and reactive astrocytes were strongly positive for MMP‐2 (100% of the cases; Figure 3A2,A3,B2,C2,D2). Interestingly, MMP‐2 was also expressed within the compromised vessel in 50% of grade 4 CAA lesions, mainly located in infiltrated histiocytes (tissue macrophages; Figure 3B2,B3,C2) within the vessel wall. The presence of histiocytes in some of these lesions was confirmed by CD68 reactivity, which is a cellular marker of macrophages, as shown in Figure 3B4. Occasional well‐defined microaneurysms were found in CAA cases (Figure 3D), and fibrinoid necrosis was also confirmed in some of these lesions, as shown by the positivity of fibrin Mallory's phosphotungstic acid staining in the example in Figure 3E1. In this case, MMP‐2 was highly expressed in astrocytes around the damaged vessel (Figure 3E2). Regarding MMP‐9 expression in grade 4 CAA, it was again mainly found in some macrophages and neutrophils (Figure 3A4,C3,D3). None of the control cases showed Aβ or MMP‐9 reactivity within the vessels, whereas 20% of morphologically healthy vessels showed a very mild expression of MMP‐2. Number of lesions and percentage of Aβ, MMP‐2 and MMP‐9 positivity are shown in Table 3.
MMP‐2 and MMP‐9 expression in chronic bleedings
Fifteen chronic bleedings were found through the frontal, temporoparietal and occipital lobes only among the six CAA cases, identified by the presence of hemosiderin or other iron storage complexes in brain scar tissue (Figure 4A1,B1). Around 50% (7/15) of these lesions showed some remaining Aβ‐positivity (Figure 4A2,B2). MMP‐2 was mainly expressed in astrocytes surrounding the scar (Figure 4A3,B3) and MMP‐9 was only found in some iron‐containing macrophages (Figure 4A4,B4).
Figure 4.
Representative chronic bleeding found in cerebral amyloid angiopathy patients' brains. Serial sections were stained with hematoxylin‐eosin (H‐E) staining (A), anti‐β‐amyloid (Aβ) (B), anti‐matrix metalloproteinase (MMP)‐2 (C) and anti‐MMP‐9 (D) antibodies. 100× magnification.
DISCUSSION
The objective of the present study was to determine the possible contribution of MMP‐9 and MMP‐2 to hemorrhagic events associated with Aβ deposition in the wall of cerebral vessels. We aimed to find an association between gelatinase expression and Aβ level among CAA patients. Our results provided the following evidences: (i) plasma MMP‐2 and MMP‐9 levels did not discriminate between CAA patients and controls; (ii) acute MMP‐9 and MMP‐2 levels were higher in hemorrhagic areas of CAA brains; (iii) MMP‐2 was strongly expressed in reactive astrocytes around affected vessels and associated with Aβ load, (iv) the most damaged and fragile vessels affected in CAA also expressed MMP‐2 in infiltrated histiocytes within the vessel wall; and (v) MMP‐9 was only present in some inflammatory blood cells around CAA‐affected vessels.
First, we determined plasma MMP levels in CAA hemorrhagic stroke patients. We only found a slight increase in soluble MMP‐9 levels during the acute phase of ICH, although it did not differ significantly from controls. We previously described higher MMP‐9 plasma levels after ICH (1), but those results already showed differences between MMP‐9 levels regarding deep and lobar ICH. Thus, the contribution of soluble MMP‐9 in amyloid‐related ICH might be more complex than in deeper hemorrhages and would need further investigation. Our results regarding the slight decrease in MMP‐2 levels during the first 24 h after the stroke are in accordance with previous determinations in global hemorrhagic stroke patients (2) and do not show important alterations of this protease in plasma of CAA cases compared with controls. Nevertheless, although the total concentration of plasma proMMP‐2 and proMMP‐9 do not seem to be highly altered in CAA‐associated ICH, changes in protease activity cannot be ruled out and should be determined in the future.
Concerning the presence of MMP‐2 and MMP‐9 in CAA‐associated hemorrhagic stroke brains, we observed an overexpression of both gelatinases in the PH compared with the CL area or to healthy brains. Although it would be extremely interesting to determine their expression in the precise site where the hemorrhage started, the amyloid‐infiltrated microvessel responsible for the brain macrobleeding is very hard to identify, especially after the tissue destruction that occurs during brain hemorrhage. Nevertheless, this increase in brain gelatinase expression in the tissue surrounding the hematoma is likely a consequence, rather than a cause, of the massive inflammatory reaction that occurs following ICH, as it has been shown after different types of acute brain injury in humans 21, 26, 27. In fact, we suggest that MMP‐2 and MMP‐9 activity might contribute to the hematoma extent and brain edema after the initial hemorrhagic event, as shown in different ICH in vivo models 19, 22.
To further study the possible contributions of MMP‐2 and MMP‐9 to brain bleeding, we determined their expression in damaged vessels from CAA patients' brains, which were the most susceptible and fragile sites where the initial rupture of the vessel may occur. We based our study on the classification of Greenberg and Vonsattel pathological criteria (8) because this method accurately scores the severity of CAA in single blood vessels. Other pathological CAA classifications have been described 3, 18, 25, which give a regional score and the overall severity of CAA across the brain. Although these ranking criteria have been very valuable to evaluate CAA in AD and dementia cases, the aim of the present work was to correlate the expression of MMP‐2 and MMP‐9 in particular lesions associated to Aβ deposition.
An important finding of our study was the presence of MMP‐2‐enriched reactive astrocytes surrounding vessels with CAA and old bleeding wounds. As expected, the gliosis rate around the affected vasculature was associated with the lesion's severity, being especially important in the most highly developed lesions. Previous studies by Maeda et al (15) already described the presence of many reactive astrocytes around the dilated vascular segments in human CAA brains. In fact, MMP‐2 was also found in reactive astrocytes around Aβ plaques in an AD transgenic animal model (33) and near cerebral microvascular fibrillar amyloid deposits in the triple‐transgenic amyloid precursor protein (APP) mouse model (Tg‐SwDI) brains as well (14). The presence of reactive astrocytes surrounding the fragile vessels might be understood as a protective barrier to maintain the vessel integrity. Indeed, the fact that glial MMP‐2 might digest Aβ material located in the damaged vessels is an hypothesis to consider, although MMP‐2 does not seem to be able to degrade fibrillar Aβ material, as it easily cleaves soluble peptides in vitro 7, 10.
Immunohistochemically, and in contrast with what we found for MMP‐2, MMP‐9 expression in leaked vessels or old bleeding lesions was only found in blood inflammatory cells, such as macrophages and neutrophils. We did not observe MMP‐9 positivity in astrocytes surrounding vascular amyloid deposits, as described for Aβ plaques in AD transgenic mice models 32, 33. In our human samples, we obtained an acute increased expression of MMP‐9 after ICH, although we could not demonstrate an alteration of this protease within or around fragile CAA vessels before their rupture. A possible MMP‐9 activity deleterious effect could take place in the secondary injury after initial bleeding, being part of the inflammatory cascade, as previously proposed for other ICH etiologies (21). Indeed, MMP‐9 expression in CAA vessels showing evidence of microbleedings was observed in APPsw transgenic mice (13). However, the presence of MMP‐9 surrounding acute bleeding in CAA patients still needs to be demonstrated.
Despite CAA being associated with several entities (28), the presence of fibrinoid necrosis and microaneurysms in the affected tissue has been shown to closely correlate with the occurrence of spontaneous ICH (29). Thus, our study was particularly focused in the study of MMPs expression in severe pathological CAA lesions. In this regard, we observed that MMP‐2 was frequently present in the endothelial layers of vessels with “vessel‐within‐vessel” appearance. It was also often expressed in the endothelium and infiltrated histiocytes of fibrous dilated vessels, even though these vessels presented only some patchy amyloid staining. Therefore, the cellular expression of MMP‐2 within the brain vessels seems to be associated with more damaged vasculature, indicating its possible contribution to the delayed degeneration of the vessel wall. The presence of proteases in infiltrated histiocytes might be important for the fibrotic material compression process, enhancing cell necrosis of the compromised vessel.
An unavoidable limitation of our study is the possible bias in determining MMP level in affected vessels because the immunohistochemical staining allowed simultaneous rating of the vascular lesion. Our data agree with a recent report that shows how MMP‐2 and MMP‐9 are not highly expressed in vessels with Aβ deposition per se. The authors found a mild MMP‐2 staining and a strong MMP‐19 and ‐26 reactivity in positive Aβ vessels, although those vessels that presented severe lesions were not specifically analyzed (24). On the other hand, it is important to mention that aneurysm formation and fibrinoid necrosis are also found in deep intracerebral, hypertension‐related hemorrhages (23). Then, the specificity of MMP‐2 and MMP‐9 in CAA‐related ICH still needs to be determined.
In conclusion, cellular expression of MMP‐2 in degenerated and compromised brain vessels might contribute to the fracture of the vessel wall and weaken the area around bleeding lesions, triggering the CAA‐associated hemorrhagic stroke.
Supporting information
Figure S1. A diagram indicating the number of patients included in the different parts of the study.
Supporting info item
ACKNOWLEDGMENTS
E. M.‐S. is supported by the Rio Hortega program (CM09/00143) and P. D. is supported by the Miguel Servet program, both from the Spanish Ministry of Science and Innovation (Instituto de Salud Carlos III). The Neurovascular Research Laboratory belongs to the Spanish stroke research network RENEVAS (RD06/0026/0010) and has received a grant from the Spanish Fondo de Investigaciones Sanitarias PI070737. We thank Dr Joaquín Serena and Susanna Camós for their help and kind offer to use the Luminex® 100™ in the Parc Científic i tecnològic de la Universitat de Girona. We also thank the neuroradiologists, residents and nurses of the emergency department at Vall d'Hebron Hospital for their invaluable help in performing this study.
Disclosure Statement: None.
REFERENCES
- 1. Abilleira S, Montaner J, Molina CA, Monasterio J, Castill J, Alvarez‐Sabín J (2003) Matrix metalloproteinase‐9 concentration after spontaneous intracerebral hemorrhage. J Neurosurg 99:65–70. [DOI] [PubMed] [Google Scholar]
- 2. Alvarez‐Sabín J, Delgado P, Abilleira S, Molina CA, Arenillas J, Ribó M et al (2004) Temporal profile of matrix metalloproteinases and their inhibitors after spontaneous intracerebral hemorrhage: relationship to clinical and radiological outcome. Stroke 35:1316–1322. [DOI] [PubMed] [Google Scholar]
- 3. Attems J (2005) Sporadic cerebral amyloid angiopathy: pathology, clinical implications, and possible pathomechanisms. Acta Neuropathol 110:345–359. [DOI] [PubMed] [Google Scholar]
- 4. Backstrom JR, Lim GP, Cullen MJ, Tökés ZA (1996) Matrix metalloproteinase‐9 (MMP‐9) is synthesized in neurons of the human hippocampus and is capable of degrading the amyloid‐beta peptide (1‐40). J Neurosci 16:7910–7919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Baig S, Kehoe PG, Love S (2008) MMP‐2, ‐3 and ‐9 levels and activity are not related to Abeta load in the frontal cortex in Alzheimer's disease. Neuropathol Appl Neurobiol 34:205–215. [DOI] [PubMed] [Google Scholar]
- 6. Braak H, Braak E (1991) Neuropathological stageing of Alzheimer‐related changes. Acta Neuropathol (Berl) 82:239–259. [DOI] [PubMed] [Google Scholar]
- 7. Crouch PJ, Tew DJ, Du T, Nguyen DN, Caragounis A, Filiz G et al (2009) Restored degradation of the Alzheimer's amyloid‐beta peptide by targeting amyloid formation. J Neurochem 108:1198–1207. [DOI] [PubMed] [Google Scholar]
- 8. Greenberg SM, Vonsattel JP (1997) Diagnosis of cerebral amyloid angiopathy. Sensitivity and specificity of cortical biopsy. Stroke 28:1418–1422. [DOI] [PubMed] [Google Scholar]
- 9. Greenberg SM, Nandigam RN, Delgado P, Betensky RA, Rosand J, Viswanathan A et al (2009) Microbleeds versus macrobleeds: evidence for distinct entities. Stroke 40:2382–2386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hernandez‐Guillamon M, Mawhirt S, Fossati S, Blais S, Pares M, Penalba A et al (2010) Matrix metalloproteinase 2 (MMP‐2) degrades soluble vasculotropic amyloid‐beta E22Q and L34V mutants, delaying their toxicity for human brain microvascular endothelial cells. J Biol Chem 285:27144–27158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Jellinger KA (2002) Alzheimer disease and cerebrovascular pathology: an update. J Neural Transm 109:813–836. [DOI] [PubMed] [Google Scholar]
- 12. Knudsen KA, Rosand J, Karluk D, Greenberg SM (2001) Clinical diagnosis of cerebral amyloid angiopathy: validation of the Boston criteria. Neurology 56:537–539. [DOI] [PubMed] [Google Scholar]
- 13. Lee JM, Yin KJ, Hsin I, Chen S, Fryer JD, Holtzman DM et al (2003) Matrix metalloproteinase‐9 and spontaneous hemorrhage in an animal model of cerebral amyloid angiopathy. Ann Neurol 54:379–382. [DOI] [PubMed] [Google Scholar]
- 14. Liao MC, Van Nostrand WE (2010) Degradation of soluble and fibrillar amyloid beta‐protein by matrix metalloproteinase (MT1‐MMP) in vitro . Biochemistry 49:1127–1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Maeda A, Yamada M, Itoh Y, Otomo E, Hayakawa M, Miyatake T (1993) Computer‐assisted three‐dimensional image analysis of cerebral amyloid angiopathy. Stroke 24:1857–1864. [DOI] [PubMed] [Google Scholar]
- 16. Maia LF, Vasconcelos C, Seixas S, Magalhães R, Correia M (2006) Lobar brain hemorrhages and white matter changes: clinical, radiological and laboratorial profiles. Cerebrovasc Dis 22:155–161. [DOI] [PubMed] [Google Scholar]
- 17. Montaner J, Molina CA, Monasterio J, Abilleira S, Arenillas JF, Ribó M et al (2003) Matrix metalloproteinase‐9 pretreatment level predicts intracranial hemorrhagic complications after thrombolysis in human stroke. Circulation 107:598–603. [DOI] [PubMed] [Google Scholar]
- 18. Olichney JM, Hansen LA, Hofstetter CR, Grundman M, Katzman R, Thal LJ (1995) Cerebral infarction in Alzheimer's disease is associated with severe amyloid angiopathy and hypertension. Arch Neurol 52:702–708. [DOI] [PubMed] [Google Scholar]
- 19. Power C, Henry S, Del Bigio MR, Larsen PH, Corbett D, Ima Y et al (2003) Intracerebral hemorrhage induces macrophage activation and matrix metalloproteinases. Ann Neurol 53:731–742. [DOI] [PubMed] [Google Scholar]
- 20. Roher AE, Kasunic TC, Woods AS, Cotter RJ, Ball MJ, Fridman R (1994) Proteolysis of A beta peptide from Alzheimer disease brain by gelatinase A. Biochem Biophys Res Commun 205:1755–1761. [DOI] [PubMed] [Google Scholar]
- 21. Rosell A, Ortega‐Aznar A, Alvarez‐Sabín J, Fernández‐Cadenas I, Ribó M, Molina CA et al (2006) Increased brain expression of matrix metalloproteinase‐9 after ischemic and hemorrhagic human stroke. Stroke 37:1399–1406. [DOI] [PubMed] [Google Scholar]
- 22. Rosenberg GA, Navratil M (1997) Metalloproteinase inhibition blocks edema in intracerebral hemorrhage in the rat. Neurology 48:921–926. [DOI] [PubMed] [Google Scholar]
- 23. Rosenblum WI (2003) Cerebral hemorrhage produced by ruptured dissecting aneurysm in miliary aneurysm. Ann Neurol 54:376–378. [DOI] [PubMed] [Google Scholar]
- 24. Tanskanen M, Myllykangas L, Saarialho‐Kere U, Paetau A (2011) Matrix metalloproteinase‐19 expressed in cerebral amyloid angiopathy. Amyloid 18:3–9. [DOI] [PubMed] [Google Scholar]
- 25. Thal DR, Ghebremedhin E, Orantes M, Wiestler OD (2003) Vascular pathology in Alzheimer disease: correlation of cerebral amyloid angiopathy and arteriosclerosis/lipohyalinosis with cognitive decline. J Neuropathol Exp Neurol 62:1287–1301. [DOI] [PubMed] [Google Scholar]
- 26. Vilalta A, Sahuquillo J, Rosell A, Poca MA, Riveiro M, Montaner J (2008a) Moderate and severe traumatic brain injury induce early overexpression of systemic and brain gelatinases. Intensive Care Med 34:1384–1392. [DOI] [PubMed] [Google Scholar]
- 27. Vilalta A, Sahuquillo J, Poca MA, De Los Rios J, Cuadrado E, Ortega‐Aznar A et al (2008b) Brain contusions induce a strong local overexpression of MMP‐9. Results of a pilot study. Acta Neurochir Suppl 102:415–419. [DOI] [PubMed] [Google Scholar]
- 28. Vinters HV (1987) Cerebral amyloid angiopathy. A critical review. Stroke 18:311–324. [DOI] [PubMed] [Google Scholar]
- 29. Vonsattel JP, Myers RH, Hedley‐Whyte ET, Ropper AH, Bird ED, Richardson EP Jr (1991) Cerebral amyloid angiopathy without and with cerebral hemorrhages: a comparative histological study. Ann Neurol 30:637–649. [DOI] [PubMed] [Google Scholar]
- 30. Wahlund LO, Barkhof F, Fazekas F, Bronge L, Augustin M, Sjogren M et al (2001) A new rating scale for age‐related white matter changes applicable to MRI and CT. Stroke 32:1318–1322. [DOI] [PubMed] [Google Scholar]
- 31. Yamada M (2000) Cerebral amyloid angiopathy: an overview. Neuropathology 20:8–22. [DOI] [PubMed] [Google Scholar]
- 32. Yan P, Hu X, Song H, Yin K, Bateman RJ, Cirrito JR et al (2006) Matrix metalloproteinase‐9 degrades amyloid‐beta fibrils in vitro and compact plaques in situ . J Biol Chem 281:24566–24574. [DOI] [PubMed] [Google Scholar]
- 33. Yin KJ, Cirrito JR, Yan P, Hu X, Xiao Q, Pan X et al (2006) Matrix metalloproteinases expressed by astrocytes mediate extracellular amyloid‐beta peptide catabolism. J Neurosci 26:10939–10948. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. A diagram indicating the number of patients included in the different parts of the study.
Supporting info item