

Abstract
Amyotrophic lateral sclerosis (ALS) may be accompanied by frontotemporal dementia (FTD). We report a case of glial mixed tau and TDP‐43 proteinopathies in a Japanese patient diagnosed clinically as having ALS‐D. Autopsy revealed loss of lower motor neurons and degeneration of the pyramidal tracts in the spinal cord and brain stem. The brain showed frontotemporal lobar degeneration (FTLD), the most severe neuronal loss and gliosis being evident in the precentral gyrus. Although less severe, such changes were also observed in other brain regions, including the basal ganglia and substantia nigra. AT8 immunostaining revealed that predominant occurrence of astrocytic tau lesions termed globular astrocytic inclusions (GAIs) was a feature of the affected regions. These GAIs were Gallyas‐Braak negative. Neuronal and oligodendrocytic tau lesions were comparatively scarce. pS409/410 immunostaining also revealed similar neuronal and glial TDP‐43 lesions. Interestingly, occasional co‐localization of tau and TDP‐43 was evident in the GAIs. Immunoblot analyses revealed band patterns characteristic of a 4‐repeat (4R) tauopathy, corticobasal degeneration and a TDP‐43 proteinopathy, ALS/FTLD‐TDP Type B. No mutations were found in the MAPT or TDP ‐43 genes. We consider that this patient harbored a distinct, sporadic globular glial mixed 4R tau and TDP‐43 proteinopathy associated with motor neuron disease and FTD.
Keywords: amyotrophic lateral sclerosis, astrocyte, frontotemporal lobar degeneration, motor neuron disease, tau, TDP‐43
Introduction
The association of amyotrophic lateral sclerosis (ALS), the most common motor neuron disease (MND), with frontotemporal dementia (FTD) has been described 31, 34, 40. Since the discovery of a nuclear protein, TDP‐43, as the pathological protein of frontotemporal lobar degeneration (FTLD) and ALS 5, 32, it has now been widely recognized that ALS (MND) and FTLD‐TDP (dementia) are part of a spectrum of TDP‐43 proteinopathy encompassing ALS at one end and FTLD‐TDP at the other 16, 17. Some patients with ALS may have accompanying dementia (ALS‐D) characterized by FTLD with TDP‐43 pathology 33. On the other hand, some patients with FTLD‐TDP may develop ALS during the disease course (FTLD‐MND); TDP‐43 pathology has been described in lower motor neurons in FTLD‐TDP patients with, and even without, ALS 36.
The clinical picture of MND with FTD, or FTLD, may also be a feature in some tauopathies. We previously reported a sporadic 4‐repeat (4R) tauopathy with FTLD, parkinsonism and MND in three Japanese patients, one with evident dementia; the most striking feature was the occurrence of unique astrocytic tau lesions 14, 35 different in morphology from tufted astrocytes (TAs) in progressive supranuclear palsy (PSP), or astrocytic plaques (APs) in corticobasal degeneration (CBD) 12. Recently, based on a compilation of cases of atypical tauopathies, including our above three cases 14, 35, a new category of 4R tauopathy designated globular glial tauopathies (GGT) has been proposed. These are characterized by tau‐positive “globular” glial inclusions in oligodendrocytes (GOIs) and astrocytes (GAIs), and exhibit the clinical features of MND and/or FTD 2. Retrospectively, clinical diagnoses of PSP and CBD (or corticobasal syndrome) are common in patients with GGT 2.
Here we report the neuropathological, biochemical and genetic findings in a case characterized by the occurrence of globular astrocytic tau and TDP‐43 inclusions in a Japanese patient diagnosed clinically as having ALS‐D.
Patient and Methods
The present study was conducted within the frame of a project, “Neuropathological and Molecular Genetic Investigation of CNS Degenerative Diseases,” approved by the Institutional Review Board of the University of Niigata. Informed consent was obtained from the patient's family prior to genetic analyses.
Case report
A 76‐year‐old Japanese woman became aware of gait disturbance and subsequently developed dysarthria. On examination, she showed atrophy and fasciculation in the tongue, a hypoactive gag reflex and muscle weakness in the upper extremities. Increased deep tendon reflexes were also present in the upper and lower extremities, with positive Babinski sign in both legs. About 10 months after onset, at the age of 77, she was diagnosed as having ALS.
Over the following year, she developed difficulty in swallowing and underwent gastrostomy for tube feeding. Her mental performance deteriorated rapidly and a state of apathy ensued; at this stage, the clinical diagnosis of ALS‐D was made. She also suffered from progressive respiratory distress and underwent tracheotomy for artificial respiratory support. At the age of 78 years, she eventually became bedridden in a totally locked‐in state. Brain computed tomography (CT) scan performed at the age of 81 years revealed frontotemporal atrophy (Figure 1). At the age of 85 years, the patient died of septic acute cholecystitis, about 9 years after the onset of the disease. There were no parkinsonian features during the disease course. There had been no family history of ALS (MND), dementia or other neurological disease.
Figure 1.
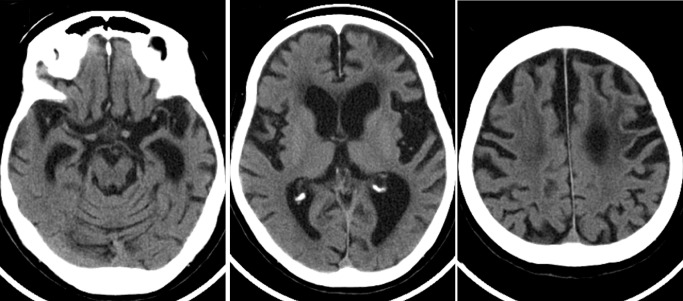
The computed tomography scan obtained 5 years after disease onset shows asymmetric atrophy (left > right) of the frontal and temporal lobes with dilatation of the lateral ventricle and enlargement of the Sylvian fissure. Decreased density of the fronto‐parietal white matter is also evident.
A general autopsy was performed about 4 h after death, at which time the brain weighed 910 g (brainstem and cerebellum, 140 g).
Neuropathological examination
The brain and spinal cord were fixed with 20% buffered formalin, and multiple tissue blocks were embedded in paraffin. Histological examinations were performed on 4‐μm thick sections using several stains: hematoxylin‐eosin, Klüver‐Barrera and Gallyas‐Braak. In addition, selected sections were immunostained after optimal pretreatment with mouse monoclonal antibodies against hyperphosphorylated tau (AT8; Innogenetics, Ghent, Belgium; 1:200), β‐amyloid (Dako, Glostrup, Denmark; 1:100; formic acid for 5 minutes), p62 (BD Biosciences, San Jose, CA, USA; 1:1000; microwave for 20 minutes), phosphorylated TDP‐43 (pS409/410; Cosmo Bio, Tokyo, Japan; 1:5000; autoclave at 120°C in 0.05 M citrate buffer for 10 minutes), FUS (Sigma‐Aldrich, St. Louis, MO, USA; 1:50), and 3R and 4R tau (RD3 and RD4 11; Upstate, Charlottesville, VA, USA; 1:3000 and 1:100, respectively; serial pretreatment with formic acid for 5 and 20 minutes, and microwave for 20 and 20 s, respectively), and rabbit polyclonal antibody against ubiquitin (Dako; 1:800), 4R tau (anti‐4R 10, 20; Cosmo Bio 1:500; serial pretreatment with formic acid for 5 minutes and microwave for 20 s) and phospho‐PHF‐tau pThr212/pSer214 (AT100; Thermo Scientific, Cergy Pontoise, France; 1:200). Bound antibodies were visualized by the peroxidase–polymer‐based method using a Histofine Simple Stain MAX‐PO kit (Nichirei, Tokyo, Japan) with diaminobenzidine as the chromogen. Immunostained sections were lightly counterstained with hematoxylin.
A double‐labeling immunofluorescence study was performed on sections obtained from the precentral (motor) cortex using rabbit polyclonal anti‐phosphorylated TDP‐43 (pS409/410; Cosmo Bio; 1:2000) and mouse monoclonal anti‐hyperphosphorylated tau (AT8; 1:2000), and with rabbit polyclonal anti‐glial fibrillary acidic protein (GFAP) (Dako; 1:800) and mouse monoclonal anti‐phosphorylated TDP‐43 (pS409/410; Cosmo Bio; 1:2000). The second antibodies used were Alexa Fluor 488 goat anti‐mouse IgG (Molecular Probes Eugene, OR, USA; 1:1000) and Alexa Fluor 555 goat anti‐rabbit IgG (Molecular Probes Eugene, OR, USA; 1:1000), and Alexa Fluor 488 goat anti‐rabbit IgG (1:1000) and Alexa Fluor 555 goat anti‐mouse IgG (1:1000), respectively. The sections were treated with an Autofluorescence Eliminator Reagent (Millipore, Billerica, MA, USA), mounted under glass coverslips using VectaShield mounting medium with 4,6‐diamidino‐2‐phenylindole nuclear stain (Vector Laboratories, Burlingame, CA, USA) and analyzed using a Carl Zeiss confocal laser scanning microscope (LSM510‐V4.0).
A double‐immunolabeling electron microscopy study was also carried out. Small tissue blocks of motor cortex fixed in 20% buffered formalin were embedded in LR white resin (London Resin, Reading, UK). Ultrathin sections were then cut, placed on Formvar‐coated nickel grids, incubated in a mixture of GFAP (1:200) and AT8 (1:10) and reacted with a mixture of 40‐nm gold colloidal particle‐conjugated anti‐rabbit IgG (British BioCell, Cardiff, UK; 1:30) and 20 nm gold colloidal particle‐conjugated anti‐mouse IgG (British BioCell; 1:30). They were then stained with uranyl acetate and lead citrate, and examined with a Hitachi H‐7100 electron microscope at 75 kV.
Biochemical analyses of tau and TDP‐43
Biochemical analysis of tau was performed using postmortem frozen tissues from the frontal and motor cortices of the present case as well as from the frontal cortex of typical PSP and CBD cases (one each) as described previously 14. The samples before and after dephosphorylation and a mixture of recombinant human tau containing the six isoforms were subjected to sodium dodecyl sulfate polyacrylamide gel electrophoresis. Sarkosyl‐insoluble tau was visualized using a phosphorylation‐independent anti‐tau antibody, T46 (Zymed, South San Francisco, CA, USA; 1:1000), which recognizes the C‐terminal regions (amino acid residues 404–441) of human tau.
In addition, biochemical analysis of TDP‐43 was performed using postmortem frozen tissues from the frontal and motor cortex of the present case and a GGT case (case 1 14), as well as from the frontal cortex of FTLD‐TDP type A, B and C cases (one each), as described previously 19, 25 with minor modifications. Briefly, to distinguish the 20–25‐kDa band pattern clearly, we used a large polyacrylamide gel (184 × 185 mm) and electrophoresed the samples at 200 V for 16 h at 4°C. The separated samples were analyzed by immunoblotting with a mouse monoclonal anti‐pS409/410 antibody (1:2000).
Sequencing of MAPT and some other genes
Genomic DNAs were extracted from frozen samples of cerebellar cortex. Mutation analysis for MAPT and some other genes, including APP, PSEN1, PSEN2, PGRN, TDP‐43 and C9ORF72, was carried out as described previously 15, 21, 24. The APOE genotype was determined by direct cycle sequencing as well as the TaqMan method.
Results
Neuropathological findings
Atrophy of the frontal and temporal lobes was a prominent feature (Figure 2). In the spinal cord, atrophy of the anterior nerve roots was evident. The cerebellum was unremarkable.
Figure 2.
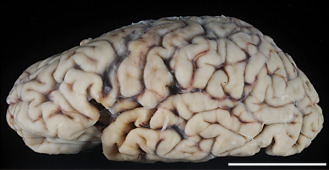
Atrophy of the frontal and temporal lobes is evident with perisylvian accentuation. Bar = 5 cm.
Histologically, loss of myelin and axons was observed in the spinal white matter except for the posterior columns, being most severe in the corticospinal tracts (Figure 3A). Severe neuronal loss and gliosis were evident in the spinal anterior horns (Figure 3B) and the brainstem lower motor neuron nuclei (Figure 3F). No Bunina bodies were found. Immunostaining with AT8 and pS409/410 revealed a small number of tau‐ (Figure 3C,E,G) and TDP‐43‐positive inclusions (Figure 3D,H), respectively, in the lower motor neurons and glial cells. Morphologically unique astrocytic tau and TDP‐43 lesions were a characteristic feature; the astrocytic lesions were recognized to be GAIs (Figure 3E) 2, 14, which were occasionally immunopositive for ubiquitin and p62. These tau and TDP‐43 lesions were completely immunonegative for FUS (data not shown).
Figure 3.
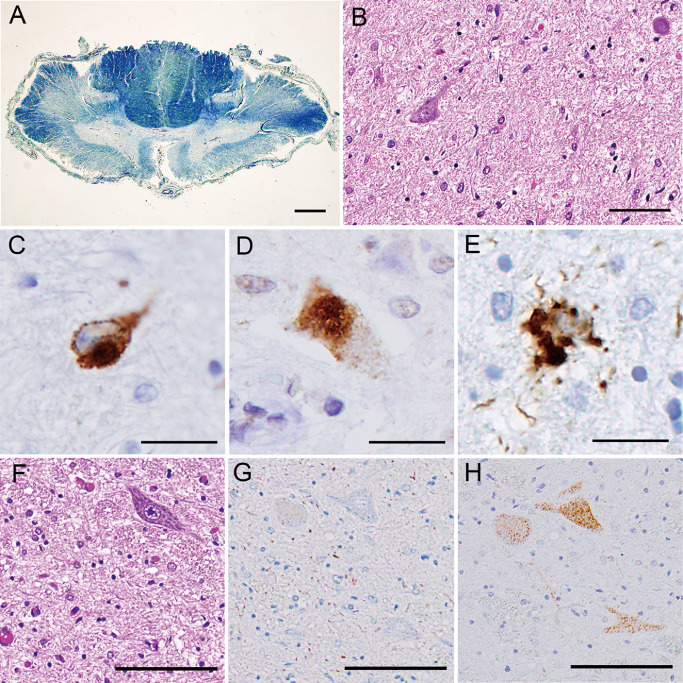
A. The spinal cord at the cervical level showing myelin pallor in the anterolateral columns, especially the lateral corticospinal tracts (Klüver‐Barrera). B–E. The anterior horns at the lumbar level showing severe neuronal loss and gliosis (B), tau‐positive (C) and TDP‐43‐positive (D) motor neurons, and tau‐positive astrocyte (E); note cytoplasmic multiple, globular deposits of tau (globular astrocytic inclusions) (E). F–H. The facial nucleus showing severe neuronal loss and gliosis (F). Two serial sections showing tau‐negative (G) and TDP‐43‐positive (H) motor neurons; note scattered tau‐positive neuropil threads (G). Bars = 1 mm in A; 100 μm in B; 20 μm in C–E; 100 μm in F–H.
Neuronal loss and gliosis were also observed in many brain regions, being particularly severe in the motor and premotor cortices (motor > premotor) (Figure 4A), and the subcortical white matter showed severe myelin pallor with loss of axons (Figure 4B). AT8 immunostaining revealed numerous tau‐positive structures mainly in the cortices (Figure 4C); pS409/410 immunostaining also revealed neuronal and astrocytic TDP‐43 lesions in the affected cortices (Figure 4D). Many swollen, achromatic neurons (ballooned neurons) were observed; they often showed granulovacuolar degeneration (Figure 4E) and were immunopositive for tau (Figure 4F). In the affected cortices, predominant occurrence of astrocytic tau lesions (Figure 4G), together with numerous neuropil threads (randomly arranged short cell processes), was a characteristic feature; again, most of the astrocytic lesions were recognized to be GAIs (Figure 4H). Scattered neuronal tau lesions (pretangles/tangles) were evident. A comparatively much smaller number of oligodendrocytic tau lesions were found in the cortex and white matter; they mostly appeared as coiled bodies and a few were recognized to be GOIs (Figure 4I). The GAIs and most of the tau‐positive neuropil threads were hardly visible by Gallyas‐Braak staining (Figure 4J), suggesting that the neuropil threads were mainly of astrocytic origin. Moreover, predominant occurrence of astrocytic TDP‐43 lesions with similar morphology was a feature (Figure 4K,L). The distribution and severity of neuronal loss, and tau and TDP‐43 lesions, are shown in Table 1.
Figure 4.
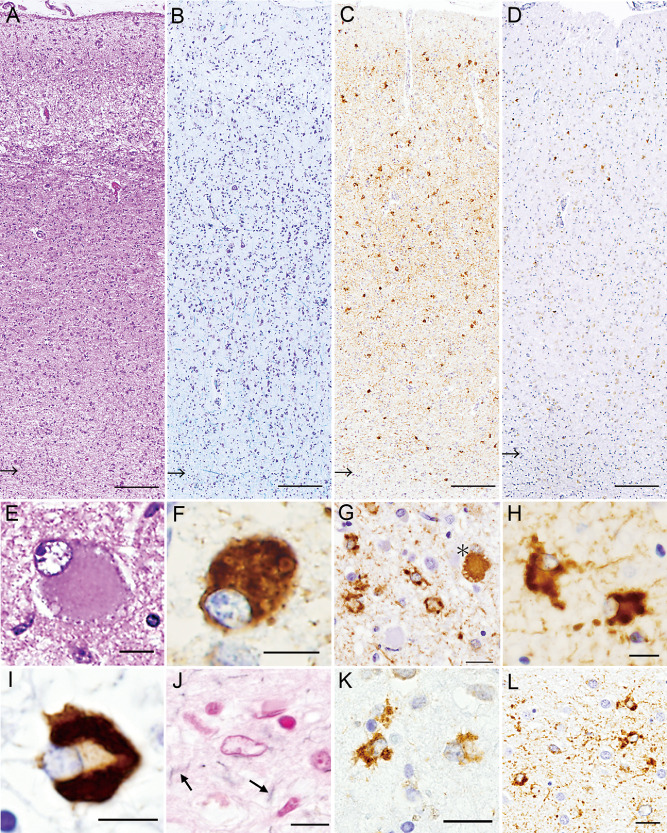
A–D. The precentral (motor) cortex and subcortical white matter (arrows indicate the gray–white matter junctions). Marked neuronal loss and gliosis is evident with microvacuolation in layer II (A). The white matter shows severe loss of myelin; note blurred gray–white matter junction (Klüver‐Barrera) (B). Numerous tau‐positive structures, including neuropil threads, are evident mainly in the cortical area (C). TDP‐43‐positive neuronal and glial cells are scattered mainly in layers II–III (D). E–J. The motor cortex. Ballooned neurons often show granulovacuolar degeneration (E) and are immunopositive for tau (F). Many unique astrocytic tau lesions are evident; note a comparatively large neuronal tau lesion (a ballooned neuron with vacuolar degeneration) indicated by an asterisk (G). The astrocytic tau lesions are those of globular astrocytic inclusions (GAIs) (H). In a unique oligodendrocytic tau lesion, somewhat spherical inclusions are evident in the cytoplasm (globular oligodendrocytic inclusions) (I). Only faintly argyrophilic processes are seen (arrows); note a pale astrocytic nucleus (center) (J). K, L. Astrocytic TDP‐43 lesions (GAIs) in the motor cortex (K) and caudate nucleus (L). Bars = 200 μm in A–D; 10 μm in E, F; 20 μm in G; 10 μm in H–J; 20 μm in K, L.
Table 1.
Distribution and severity of major histological findings in the studied patient
| Neuronal loss and gliosis | Tau‐positive | pTDP‐43‐positive | |||||
|---|---|---|---|---|---|---|---|
| Neurons | Astrocytes/oligodendrocytes | Threads | Neurons | Astrocytes/oligodendrocytes | Threads | ||
| Cerebral cortex | |||||||
| Frontal | 1 | 1 | 2/1 | 1 | 0 | 1/1 | 0 |
| Premotor/motor | 2 | 2 | 2/1 | 2 | 1 | 2/0 | 1 |
| Cerebral white matter | 2 | 1/1 | 1 | 0/1 | 0 | ||
| Parietal | 1 | 2 | 2/1 | 1 | 1 | 1/0 | 0 |
| Temporal | 1 | 2 | 2/1 | 1 | 1 | 2/1 | 1 |
| Transentorhinal | 2 | 2 | 2/1 | 2 | 2 | 2/0 | 2 |
| Occipital | 1 | 2 | 2/1 | 1 | 1 | 1/1 | 0 |
| Subcortical areas | |||||||
| Hippocampus (CA1‐subiculum) | 1(2) | 2(2) | 2/1(2/2) | 2 | 0(0) | 1/0(1/0) | 0 |
| Dentate gyrus | 1 | 1 | 1/0 | 1 | 1 | 0/0 | 0 |
| Amygdala | 2 | 1 | 1/1 | 1 | 1 | 1/1 | 1 |
| Basal nucleus of Meynert | 1 | 2 | 1/1 | 2 | 0 | 0/0 | 0 |
| Caudate/putamen | 1/1 | 1/1 | 2/1/2/1 | 2/2 | 1/1 | 1/1/1/1 | 0/0 |
| Globus pallidus | 1 | 1 | 1/2 | 2 | 1 | 1/1 | 0 |
| Internal capsule | 1/1 | 2 | 0/1 | 0 | |||
| Thalamus | 1 | 2 | 1/1 | 1 | 1 | 1/1 | 0 |
| Subthalamic nucleus | 2 | 1 | 1/1 | 1 | 1 | 0/0 | 0 |
| Brain stem | |||||||
| Midbrain tectum | 1 | 2 | 2/1 | 2 | 1 | 1/1 | 0 |
| Oculomotor nucleus | 1 | 2 | 2/2 | 2 | 0 | 1/1 | 1 |
| Red nucleus | 1 | 2 | 2/2 | 2 | 1 | 0/1 | 0 |
| Substantia nigra | 1 | 2 | 1/2 | 2 | 1 | 0/1 | 0 |
| Cerebral peduncle | 1 | 0/1 | 2 | 0/1 | 0 | ||
| Locus ceruleus | 2 | 2 | 0/1 | 1 | 0 | 0/1 | 0 |
| Trigeminal motor nucleus | 2 | 1 | 0/1 | 1 | 0 | 1/0 | 0 |
| Facial nucleus | 2 | 1 | 0/1 | 1 | 2 | 0/1 | 0 |
| Pontine nucleus | 0 | 1 | 1/1 | 1 | 0 | 0/0 | 0 |
| Fibers in pontine base | 0/1 | 2 | 0/0 | 0 | |||
| Hypoglossal nucleus | 2 | 0 | 0/0 | 0 | 0 | 0/0 | 0 |
| Inferior olivary nucleus | 0 | 1 | 1/1 | 1 | 1 | 1/1 | 1 |
| Cerebellum | |||||||
| Cerebellar cortex | 0 | 0 | 0/0 | 0 | 0 | 0/0 | 0 |
| Dentate nucleus | 1 | 1 | 0/0 | 1 | 0 | 0/0 | 0 |
| Cerebellar white matter | 0/0 | 1 | 0 | 0/0 | 0 | ||
| Spinal cord | |||||||
| Anterior horn | 2 | 1 | 1/0 | 1 | 1 | 1/0 | 0 |
| Intermediate area | 2 | 0 | 0/0 | 0 | 1 | 1/1 | 0 |
| Clarke's column | 1 | 0 | 0/0 | 0 | 0 | 0/0 | 0 |
| Posterior horn | 1 | 0 | 0/0 | 0 | 0 | 1/0 | 0 |
| Spinal white matter | 1/0 | 0 | 1/1 | 0 | |||
The presence and severity of the histopathological features are represented as 0 = none, 1 = minimal/mild and 2 = moderate/severe. Tau‐ and TDP‐43‐positive astrocytes and oligodendrocytes observed appeared mostly as globular astrocytic inclusions and coiled bodies, respectively.
Immunostaining with AT100 also clearly depicted neuronal and glial tau lesions in the spinal cord and brain, the pathological tau burden and distribution pattern being almost the same as those recognized by immunostaining with AT8 (data not shown).
Double‐labeling immunofluorescence indicated that occasional co‐localization of tau and TDP‐43 was a feature in the GAIs (Figure 5A–C) and neuropil threads (Figure 5D–F), but this phenomenon was not detected in the neuronal tau or TDP‐43 inclusions (Figure 5G–I). No co‐localization of tau and TDP‐43 in the neuronal inclusions was also evident in serial sections (Figure 3G,H). Double‐labeling immunofluorescence with antibodies against GFAP and TDP‐43 also revealed the frequent presence of TDP‐43‐positive globular cytoplasmic inclusions in GFAP‐positive astrocytes (Figure 5J–L).
Figure 5.
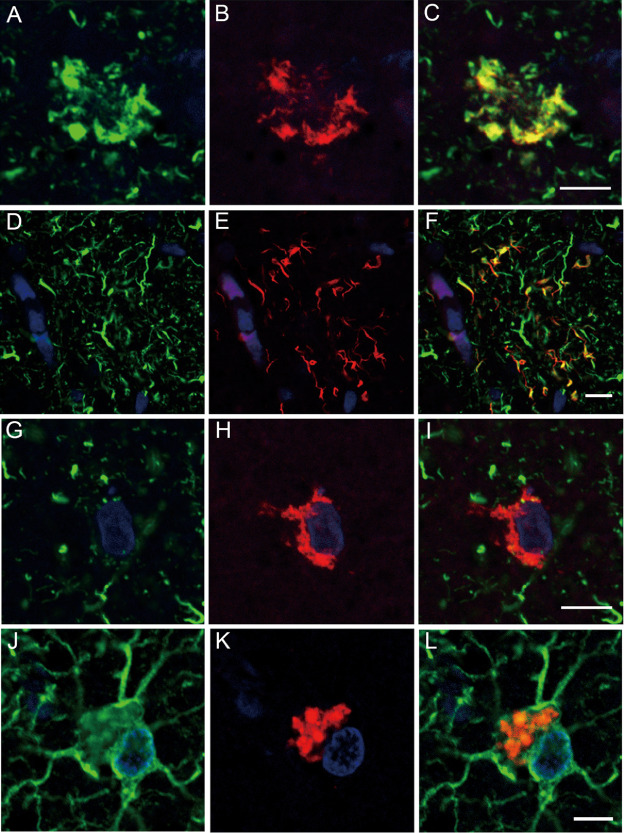
A–L. Double‐labeling immunofluorescence in the motor cortex. A–C. Tau (green, A) and TDP‐43 (red, B) are occasionally co‐localized (merge, C) in the affected astrocytes forming globular astrocytic inclusions (GAIs). D–F. Tau (green, D) and TDP‐43 (red, E) are rarely found co‐localized (merge, F) in the cell processes probably of the affected astrocytes. G–I. Tau (green, G) and TDP‐43 (red, H) are not co‐localized (merge, I) in the affected neurons. J–L. Glial fibrillary acidic protein (green, J) and TDP‐43 (red, K) are sometimes co‐localized (merge, L) in the affected astrocytes forming GAIs. Bars = 5 μm.
With regard to 4R tau, RD4 immunostaining (Figure 6A) revealed far fewer tau lesions than AT8 immunostaining (Figure 4C). However, as was the case for AT8 immunostaining, anti‐4R immunostaining demonstrated numerous tau‐positive structures, including GAIs and neuropil threads (Figure 6B,C). Again, these anti‐4R‐positive astrocytic tau lesions and most of the neuropil threads were basically Gallyas‐Braak negative (Figure 6D). In addition to the GAIs (Figure 6E), many astrocytic tau lesions indistinguishable from PSP‐type TAs (Figure 6F) and CBD‐type APs (Figure 6G) became recognizable upon immunostaining with anti‐4R. Neuronal tau lesions were also more strongly stained and more frequently observed by immunostaining with anti‐4R (Figure 6H) than with RD4. Immunostaining with RD3 revealed positive pretangle/tangles that were mostly limited to the hippocampus.
Figure 6.
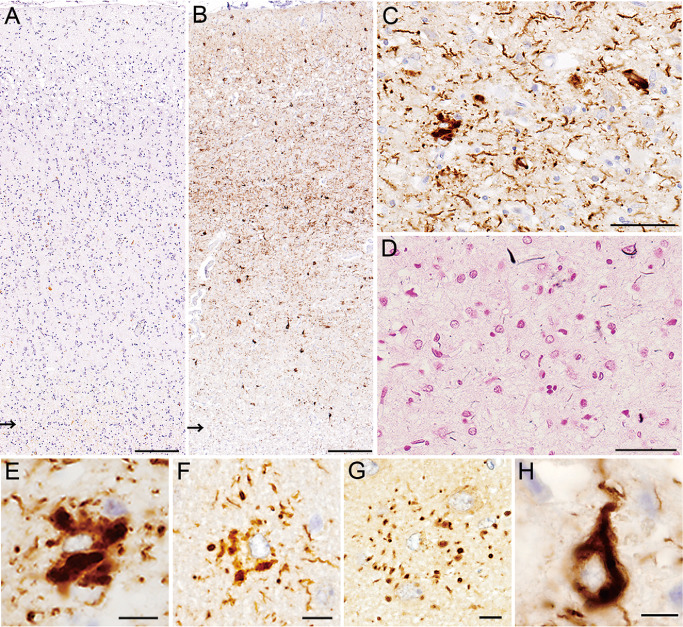
A, B. Only a small number of positive structures are visible with RD4 (A), whereas numerous positive structures are depicted with anti‐4R (B) (arrows indicate the gray–white matter junctions). C, D. Most of the structures recognized by anti‐4R (C) hardly show argyrophilia (D, Gallyas‐Braak). E–G. A variety of astrocytic tau lesions recognized by anti‐4R, including globular astrocytic inclusions (E), and those reminiscent of tufted astrocytes (F) and astrocytic plaques (G). H. Neuronal tau lesions are also clearly visible with anti‐4R. A–E, H. Motor cortex. F, G. Frontal cortex. Bars = 200 μm in A, B; 50 μm in C, D; 10 μm in E–H.
At the ultrastructural level, the presence of bundles of tubular structures (∼20 nm in diameter) was confirmed in the affected astrocytic cytoplasm (Figure 7).
Figure 7.
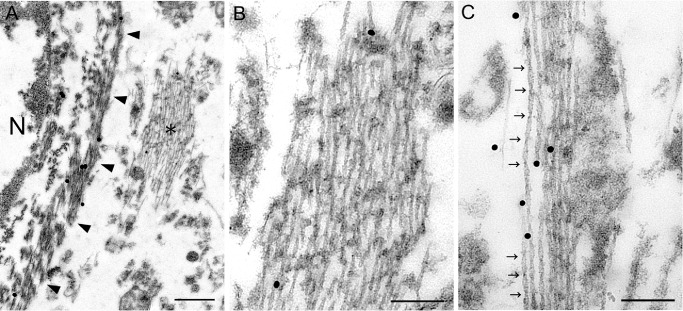
A–C. Glial fibrillary acidic protein (GFAP) and AT8 double‐immunolabeling electron microscopy; GFAP is probed by large (40 nm) particles, and AT8 by small (20 nm) particles. This affected astrocyte (N, nucleus) shows GFAP‐labeled filaments (arrowheads: glial filaments) and AT8‐labeled tubules (asterisk) in the cytoplasm (A). Higher magnification view of the area indicated by asterisk in (A) demonstrates that these longitudinally cut tubules are mostly straight (B). In the cytoplasm of another affected astrocyte at higher magnification, the presence of twisted tubules with periodic constrictions (arrows) is evident (C). Bars = 500 nm in A; 200 nm in B, C.
In the present case, the significant presence of AT8‐positive pretangles/tangles was mostly limited to the hippocampus, but many Aβ‐positive senile plaques were widely scattered in the cerebral cortices, including the motor cortex (neurofibrillary tangles : senile plaques = II : C 7, 8; Aβ deposition, Thal phase 3 39).
Sarkosyl‐insoluble tau and TDP‐43
Immunoblot analysis using dephosphorylated samples from the frontal cortex revealed two major bands that were aligned with the recombinant 4R tau isoforms of 412 (4R, 1N) and 383 (4R, 1N) amino acids in the present case (data not shown). Immunoblot analysis using non‐dephosphorylated samples was performed in the frontal and motor cortices. The sample from the frontal cortex revealed a band pattern of low‐molecular mass tau fragments compatible with that obtained in a case of CBD; however, the sample from the motor cortex revealed no apparent bands of low‐molecular mass tau fragments (Figure 8A).
Figure 8.
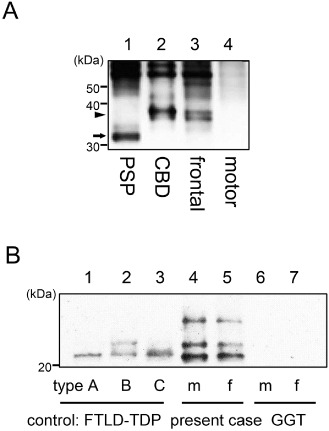
A. Immunoblot analysis of the sarkosyl‐insoluble fraction before dephosphorylation. The band pattern of low‐molecular mass tau fragments in a progressive supranuclear palsy case shows a prominent band of 33 kDa (lane 1, arrow), while that in a corticobasal degeneration (CBD) case shows a prominent band of 37 kDa (lane 2, arrowhead). In the present case, the frontal cortex clearly shows a prominent band of 37 kDa characteristic of CBD. However, the motor cortex shows no such band patterns of low‐molecular mass tau fragments. B. Immunoblot analysis of the sarkosyl‐insoluble fraction from the motor cortex (m; lanes 4, 6) and frontal cortex (f; lane 5, 7) of the present case and a globular glial tauopathies (GGT) case, respectively, as well as from the frontal cortex of frontotemporal lobar degeneration (FTLD)‐TDP type A (lane 1), type B (lane 2) and type C (lane 3) (one each) as controls. The band pattern of TDP‐43 C‐terminal fragments differs between the three FTLD‐TDP subtypes (lanes 1–3). The present case shows two prominent bands of 23 and 24 kDa (lanes 4, 5); this finding is also a feature in a case of FTLD‐TDP type B (lane 2). A GGT case has no visible bands (lane 6, 7).
With regard to TDP‐43, immunoblot analysis of the sarkosyl‐insoluble samples from the present case revealed a band pattern of TDP‐43 C‐terminal fragments compatible with that obtained in a case of FTLD‐TDP type B; no bands of TDP‐43 were evident in a case of GGT (Figure 8B).
Detection of gene mutations
There were no mutations in the coding regions of the MAPT, TDP‐43, APP, PSEN1, PSEN2 and PGRN genes. No C9ORF72 repeat expansion was observed. The genotype of APOE was ε3/ε4.
Discussion
In the present case, ALS symptoms/signs were the presenting manifestations, that is, fasciculation and muscle weakness, increased deep tendon reflexes and positive Babinski sign as a pathological reflex. In addition, subsequent development of rapidly progressive mental deterioration and apparent frontotemporal atrophy on brain CT scan were characteristic. No extrapyramidal symptoms/signs were evident throughout the entire disease course.
Pathologically, severe loss of motor neurons was observed in the spinal cord and brain stem. In addition, FTLD was evident; the motor cortex showed most severe neuronal loss and gliosis and the subcortical white matter and descending pyramidal tracts showed most severe loss of myelin and axons. The degeneration was characterized by the occurrence of tau and TDP‐43 lesions affecting both the neuronal and glial cells; predominant occurrence of morphologically unique astrocytic tau and TDP‐43 lesions termed GAIs was a striking feature. These GAIs were not visible by Gallyas‐Braak staining. Genetically, no mutations were found in the MAPT, TDP‐43 or other genes associated with dementia and/or FTLD, suggesting that the patient's disease had been sporadic.
We previously reported three cases of sporadic 4R tauopathy with FTLD, parkinsonism and MND. Clinically, upper motor neuron signs (spasticity, hyperreflexia and pathological reflexes) and extrapyramidal signs (tremor, rigidity and bradykinesia) were a feature, the clinical picture being somewhat different from that observed in the present case. Neuropathologically, predominant occurrence of astrocytic tau lesions very similar, if not identical, to those seen in the present case was a characteristic feature; such astrocytic tau lesions showed no evident argyrophilia by Gallyas‐Braak silver impregnation 14, 35. These three cases have now been classified into GGT type III 2 (discussed below). With regard to the neuropathology, the regional severity and distribution pattern were basically similar to those observed in the present case (Table 1; also see table 2 14). Thus, it may be possible to consider that in the present case, extrapyramidal symptoms/signs must have been masked by the preceding, more severely progressive degeneration involving the lower and upper motor neuron systems.
It was noteworthy that numerous 4R tau lesions were visible using a new antibody, anti‐4R. Surprisingly, astrocytic 4R tau lesions indistinguishable from TAs and APs were also easily recognizable with this antibody. We confirmed in this study that neuronal and glial tau lesions observed in the previous three cases were also clearly reproduced with anti‐4R (they were also visible with RD4 14, 35). Dan et al 10 reported that post‐translational deamidation took place at asparagine residue 279 (N279) in the RD4 epitope of 4R tau, and that RD4 was not able to recognize N279D‐4R tau. They also mentioned that the deamidation was observed in the RD4 epitope of tau in Alzheimer's disease (AD), but not in CBD or PSP 10, 20. Although the widespread occurrence of many senile plaques was a feature of the present patient with an APOE ε4 allele 23, 42, the clinical course and entire pathological picture were not those of AD. In the present case, the pathomechanism by which RD4‐negative, anti‐4R‐positive deamidated tau had accumulated in the neuronal and glial cells remains uncertain.
In addition to the aforementioned three cases reported by our group 14, 35, a number of cases have so far been described as novel 4R tauopathies, showing multiple system tauopathy (MST) 37, MST with dementia 6, atypical PSP with corticospinal tract (CST) degeneration 22, atypical tauopathy with massive involvement of the white matter 18, white matter tauopathy with globular glial inclusions 26 and GGT presenting with MND or FTD 1. These 4R tauopathies appeared to be heterogeneous in terms of clinical and pathological features. Recently, however, Ahmed et al reviewed these reported cases and proposed a new category of 4R tauopathy designated GGT 2. They divided these cases into three subtypes according to their clinical features and predominant areas of degeneration: type I, FTD with a FT distribution of pathology; type II, pyramidal features reflecting motor cortex involvement and CST degeneration; and type III, a combination of FTD and MND with the FT cortex, motor cortex and CST being severely affected 2. They also stated that extrapyramidal features can be present in type II and III cases, and that significant degeneration of the white matter is a feature of all GGT subtypes 2.
The distinctive feature of GGT is occurrence of glial globular tau inclusions, that is, GOIs and GAIs, the latter being consistently Gallyas‐Braak negative. Thus, the morphological and staining properties of GAIs are different from those shown in TAs and APs, which are characteristic of PSP and CBD, respectively (see Figure 6 14). The present case was reasonably considered to be an additional example of GGT type III 2. In cases of GGT type I, much heavier accumulation of tau‐positive fibrils and GOIs is observed in the subcortical white matter 2, 26. In such cases, the white matter degeneration appears to be caused primarily by certain metabolic alterations and subsequent death of oligodendrocytes with abnormal tau accumulation (GOIs, neuropil threads and coiled bodies). In the present case, tau accumulation was inconspicuous in the affected white matter. Severe myelin pallor with axonal loss in the motor and premotor subcortical white matter appeared to parallel the severe neuronal loss in the covering cerebral cortices, suggesting that the former was caused secondarily by the latter.
GGT share some neuropathological and biochemical features with PSP: accumulation of 4R tau in glial cells and the presence of low‐molecular mass tau fragments at ∼35 kDa 2, 5. Of great importance was that the present case showed a prominent band of low‐molecular mass tau fragments at ∼40 kDa characteristic of CBD 5. It was also noteworthy that occurrence of astrocytic tau lesions indistinguishable from CBD‐type APs, which were visible with anti‐4R, as well as ballooned neurons 12, was evident in the affected cerebral cortex.
It is unclear whether or not immunoblot band patterns would be identical in different brain regions in affected individuals with GGT. One case of GGT was reported to have a shared immunoblot feature of PSP and CBD 4, showing a more prominent band at ∼40 kDa in the frontal cortex and a more prominent band at ∼35 kDa in the motor cortex 14; this finding seems to be very important when considering the propagation hypothesis for pathological tau protein 3, 9. In the present case, in comparison with the recovery of sarkosyl‐insoluble tau in the frontal cortex, the motor cortex was apparently inefficient, with no evident band patterns suggestive of the PSP or CBD type. The reason for these biochemical results remains unexplained.
Of great interest was that in the present case, neuronal and glial TDP‐43 lesions were also observed; occasional co‐localization of tau and TDP‐43 was a feature of the GAIs. Accumulation of TDP‐43 can occur in glial cells in ALS, but it is a phenomenon involving almost exclusively oligodendrocytes 33, 38. In the affected cerebral cortex, TDP‐43‐positive neuronal inclusions were found mainly in layers II–III with a few TDP‐43‐positive neurites, being classified as FTLD‐TDP pathology Type B 28, and immunoblot analysis also revealed a band pattern of ALS and FTLD‐TDP type B 41. We have similarly studied three cases of GGT type III 14, 35, showing no neuronal or glial TDP‐43 lesions (data not shown). It was also noteworthy that TDP‐43 inclusions in astrocytes have been reported in cases of FTLD and Lewy body disease, with mutations in the PGRN and SNCA genes, respectively 27.
Dobson‐Stone et al reported a form of familial FTD‐ALS linked to 16p12.1‐q12.2 in two autopsy cases where neuronal tau and TDP‐43 lesions were observed, but no mention was made of glial TDP‐43 lesions 13; they considered the family to have both FTLD‐tau (CBD‐type) 12 and FTLD‐TDP Type B pathologies 28. Recently, Mckee et al reported patients with chronic traumatic encephalopathy (CTE) showing TDP‐43 proteinopathy and MND 29, 30. A clinicopathological entity, CTE, is triggered by repetitive mild traumatic brain injury, and is pathologically characterized by widespread deposition of hyperphosphorylated tau appearing as neurofibrillary tangles. Of great interest is that in CTE, neuronal and glial TDP‐43 lesions were also observed, and some cases may develop progressive MND 29. In the present case, occurrence of both tau and TDP‐43 lesions was a feature, but there was no history of brain injury.
In conclusion, we have described an unusual combination of 4R tauopathy and TDP‐43 proteinopathy in an 85‐year‐old Japanese woman diagnosed clinically as having ALS‐D (MND with FTD). The co‐occurrence of tau‐ and TDP‐43‐positive GAIs, with occasional co‐localization of both proteins, as well as the co‐presence of immunoblot features of CBD 5 and ALS/FTLD‐TDP type B 41 appeared to be more than a mere coincidence. Finally, we consider that the present patient represents a distinct, sporadic mixed 4R tau and TDP‐43 proteinopathy associated MND and FTD (GGT type III 2/FTLD‐TDP Type B 28). With regard to the 4R tauopathy, it was very interesting to note that most of the tau lesions were composed of RD4‐negative (undetectable), but anti‐4R‐positive (detectable) deamidated tau, and that recovery of sarkosyl‐insoluble tau was apparently inefficient in the motor cortex, which was the area most severely affected by tau pathology. These findings strongly suggest that the tau disease process in the present patient was more complicated than in the two representative 4R tauopathies, PSP and CBD.
Conflict of Interest
The authors declare no conflict of interest.
Acknowledgments
We are indebted to Dr. O. Yokota and Dr. S. Kuroda, Department of Psychiatry, Okayama University, for providing specimens of a GGT‐type III case. We also thank C. Tanda, S. Nigorikawa, J. Takasaki, H. Saito, T. Fujita and S. Egawa for their technical assistance, and M. Machida and Y. Ueda for secretarial assistance. This work was supported by Grants‐in‐Aid, 26430052 (to Y. T.), 25253065 (to M. N.) and 26250017 (to H. T.), from Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology, Japan.
References
- 1. Ahmed Z, Doherty KM, Silveira‐Moriyama L, Bandopadhyay R, Lashley T, Mamais A et al (2011) Globular glial tauopathies (GGT) presenting with motor neuron disease or frontotemporal dementia: an emerging group of 4‐repeat tauopathies. Acta Neuropathol 122:415–428. [DOI] [PubMed] [Google Scholar]
- 2. Ahmed Z, Bigio EH, Budka H, Dickson DW, Ferrer I, Ghetti B et al (2013) Globular glial tauopathies (GGT): consensus recommendations. Acta Neuropathol 126:537–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ahmed Z, Cooper J, Murray TK, Garn K, McNaughton E, Clarke H et al (2014) A novel in vivo model of tau propagation with rapid and progressive neurofibrillary tangle pathology: the pattern of spread is determined by connectivity, not proximity. Acta Neuropathol 127:667–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Arai T, Ikeda K, Akiyama H, Nonaka T, Hasegawa M, Ishiguro K et al (2004) Identification of amino‐terminally cleaved tau fragments that distinguish progressive supranuclear palsy from corticobasal degeneration. Ann Neurol 55:72–79.14705114 [Google Scholar]
- 5. Arai T, Hasegawa M, Akiyama H, Ikeda K, Nonaka T, Mori H et al (2006) TDP‐43 is a component of ubiquitin‐positive tau‐negative inclusion in frontotemopral lobar degeneration and amyotrophic lateral clerosis. Biochem Biophys Res Commun 351:602–611. [DOI] [PubMed] [Google Scholar]
- 6. Bigio EH, Lipton AM, Yen SH, Hutton ML, Baker M, Nacharaju P et al (2001) Frontal lobe dementia with novel tauopathy: sporadic multiple system tauopathy with dementia. J Neuropathol Exp Neurol 60:328–341. [DOI] [PubMed] [Google Scholar]
- 7. Braak H, Alafuzoff I, Arzberger T, Kretzschmar H, Del Tredici K (2006) Staging of Alzheimer disease‐associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol 112:389–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Braak H, Braak E (1991) Neuropathological staging of Alzheimer‐related changes. Acta Neuropathol 82:239–259. [DOI] [PubMed] [Google Scholar]
- 9. Clavaguera F, Akatsu H, Fraser G, Crowther RA, Frank S, Hench J et al (2013) Brain homogenates from human tauopathies induce tau inclusions in mouse brain. Proc Natl Acad Sci U S A 110:9535–9540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dan A, Takahashi M, Masuda‐Suzukake M, Kametani F, Nonaka T, Kondo H et al (2013) Extensive deamidation at asparagine residue 279 accounts for weak immunoreactivity of tau with RD4 antibody in Alzheimer's disease brain. Acta Neuropathol Commun 1:54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. de Silva R, Lashley T, Gibb G, Hanger D, Hope A, Reid A et al (2003) Pathological inclusion bodies in tauopathies contain distinct complements of tau with three or four microtubule‐binding repeat domains as demonstrated by new specific monoclonal antibodies. Neuropathol Appl Neurobiol 29:288–302. [DOI] [PubMed] [Google Scholar]
- 12. Dickson DW (1999) Neuropathologic differentiation of progressive supranuclear palsy and corticobasal degeneration. J Neurol 246 (Suppl. 2):II6–II15. [DOI] [PubMed] [Google Scholar]
- 13. Dobson‐Stone C, Luty AA, Thompson EM, Blumbergs P, Brooks WS, Short CL et al (2013) Frontotemporal dementia‐amyotrophic lateral sclerosis syndrome locus on chromosome 16p12.1‐q12.2: genetic, clinical and neuropathological analysis. Acta Neuropathol 125:523–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Fu YJ, Nishihira Y, Kuroda S, Toyoshima Y, Ishihara T, Shinozaki M et al (2010) Sporadic four‐repeat tauopathy with frontotemporal lobar degeneration, Parkinsonism, and motor neuron disease: a distinct clinicopathological and biochemical disease entity. Acta Neuropathol 120:21–32. [DOI] [PubMed] [Google Scholar]
- 15. Gass J, Cannon A, Mackenzie IR, Boeve B, Baker M, Adamson J et al (2006) Mutations in progranulin are a major cause of ubiquitin‐positive frontotemporal lpbar degeneration. Hum Mol Genet 15:2988–3001. [DOI] [PubMed] [Google Scholar]
- 16. Geser F, Martinez‐Lage M, Kwong LK, Lee VM, Trojanowski JQ (2009) Amyotrophic lateral sclerosis, frontotemporal dementia and beyond: the TDP‐43 diseases. J Neurol 256:1205–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Geser F, Lee VM, Trojanowski JQ (2010) Amyotrophic lateral sclerosis and frontotemporal lobar degeneration: a spectrum of TDP‐43 proteinopathies. Neuropathology 30:103–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Giaccone G, Marcon G, Mangieri M, Morbin M, Rossi G, Fetoni V et al (2008) Atypical tauopathy with massive involvement of the white matter. Neuropathol Appl Neurobiol 34:468–472. [DOI] [PubMed] [Google Scholar]
- 19. Hasegawa M, Arai T, Nonaka T, Kametani F, Yoshida M, Hashizume Y et al (2008) Phosphorylated TDP‐43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Ann Neurol 64:60–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hasegawa M, Watanabe S, Kondo H, Akiyama H, Mann DM, Saito Y, Murayama S (2014) 3R and 4R tau isoforms in paired helical filaments in Alzheimer's disease. Acta Neuropathol 127:303–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ikeuchi T, Kaneko H, Miyashita A, Nozaki H, Kasuga K, Tsukie T et al (2008) Mutational analysis in early‐onset familial dementia in the Japanese population. The role of PSEN1 and MAPT R406W mutations. Dement Geriatr Cogn Disord 26:43–49. [DOI] [PubMed] [Google Scholar]
- 22. Josephs KA, Katsuse O, Beccano‐Kelly DA, Lin WL, Uitti RJ, Fujino Y et al (2006) Atypical progressive supranuclear palsy with corticospinal tract degeneration. J Neuropathol Exp Neurol 65:396–405. [DOI] [PubMed] [Google Scholar]
- 23. Kamboh MI, Sanghera DK, Ferrell RE, DeKosky ST (1995) APOE*4‐associated Alzheimer's disease risk is modified by alpha 1‐antichymotrypsin polymorphism. Nat Genet 10:486–488. [DOI] [PubMed] [Google Scholar]
- 24. Konno T, Shiga A, Tsujino A, Sugai A, Kato T, Kanai K et al (2012) Japanese amyotrophic lateral sclerosis with GGGCC hexanucleotide repeat expansion in C9ORF72. J Neurol Neurosurg Psychiatry 84:398–401. [DOI] [PubMed] [Google Scholar]
- 25. Kosaka T, Fu YJ, Shiga A, Ishidaira H, Tan CF, Tani T et al (2012) Primary lateral sclerosis: upper‐motor‐predominant amyotrophic lateral sclerosi with frontotemporal lobar degeneration—immunohistochemical and biochemical analyses of TDP‐43. Neuropathology 32:373–384. [DOI] [PubMed] [Google Scholar]
- 26. Kovacs GG, Majtenyi K, Spina S, Murrell JR, Gelpi E, Hoftberger R et al (2008) White matter tauopathy with globular glial inclusions: a distinct sporadic frontotemporal lobar degeneration. J Neuropathol Exp Neurol 67:963–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lin W‐L, Castanedes‐Casey M, Dickson DW (2009) Transactivation response DNA‐binding protein 43 microvasculopathy in frontotemporal degeneration and familial Lewy body disease. J Neuropathol Exp Neurol 68:1167–1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mackenzie IR, Neumann M, Baborie A, Sampathu DM, Du Plessis D, Jaros E et al (2011) A harmonized classification system for FTLD‐TDP pathology. Acta Neuropathol 122:111–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. McKee AC, Gavett BE, Stern RA, Nowinski CJ, Cantu RC, Kowall NW et al (2010) TDP‐43 proteinopathy and motor neuron disease in chronic traumatic encephalopathy. J Neuropathol Exp Neurol 69:918–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. McKee AC1, Stern RA, Nowinski CJ, Stein TD, Alvarez VE, Daneshvar DH et al (2013) The spectrum of disease in chronic traumatic encephalopathy. Brain 136:43–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Nakano I (2000) Frontotemporal dementia with motor neuron disease (amyotrophic lateral sclerosis with dementia). Neuropathology 20:68–75. [DOI] [PubMed] [Google Scholar]
- 32. Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT et al (2006) Uniquitinated TDP‐43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314:130–133. [DOI] [PubMed] [Google Scholar]
- 33. Nishihira Y, Tan CF, Onodera O, Toyoshima Y, Yamada M, Morita T et al (2008) Sporadic amyotrophic lateral sclerosis: two pathological patterns shown by analysis of distribution of TDP‐43 immunoreactive neuronal and glial cytoplasmic inclusions. Acta Neuropathol 116:169–182. [DOI] [PubMed] [Google Scholar]
- 34. Piao YS, Wakabayashi K, Kakita A, Yamada M, Hayashi S, Morita T et al (2003) Neuropathology with clinical correlations of amyotrophic lateral sclerosis: 102 autopsy cases examined between 1962 and 2000. Brain Pathol 13:10–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Piao YS, Tan CF, Iwanaga K, Kakita A, Takano H, Nishizawa M et al (2005) Sporadic four‐repeat tauopathy with frontotemporal degeneration, parkinsonism and motor neuron disease. Acta Neuropathol 110:600–609. [DOI] [PubMed] [Google Scholar]
- 36. Riku Y, Watanabe H, Yoshida M, Tatsumi S, Mimuro M, Iwasaki Y et al (2014) Lower motor neuron involvement in TAR DNA‐binding protein of 43 kDa‐related frontotemporal lobar degeneration and amyotrophic lateral sclerosis. JAMA Neurol 71:172–179. [DOI] [PubMed] [Google Scholar]
- 37. Spina S, Farlow MR, Unverzagt FW, Kareken DA, Murrell JR, Fraser G et al (2008) The tauopathy associated with mutation +3 in intron 10 of tau: characterization of the MSTD family. Brain 131:72–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Tan CF, Eguchi H, Tagawa A, Onodera O, Iwasaki T, Tsujino A et al (2007) TDP‐43 immunoreactivity in neuronal inclusions in familial amyotrophic lateral sclerosis with or without SOD1 gene mutation. Acta Neuropathol 113:535–542. [DOI] [PubMed] [Google Scholar]
- 39. Thal DR, Rüb U, Orantes M, Braak H (2002) Phases of A beta‐deposition in the human brain and its relevance for the development of AD. Neurology 58:1791–1800. [DOI] [PubMed] [Google Scholar]
- 40. Toyoshima Y, Piao YS, Tan CF, Morita M, Tanaka M, Oyanagi K et al (2003) Pathological involvement of the motor neuron system and hippocampal formation in motor neuron disease‐inclusion dementia. Acta Neuropathol 106:50–56. [DOI] [PubMed] [Google Scholar]
- 41. Tsuji H, Arai T, Kametani F, Nonaka T, Yamashita M, Suzukake M et al (2012) Molecular analysis and biochemical classification of TDP‐43 proteinopathy. Brain 135:3380–3391. [DOI] [PubMed] [Google Scholar]
- 42. Yamaguchi H, Sugihara S, Ogawa A, Oshima N, Ihara Y (2001) Alzheimer beta amyloid deposition enhanced by apoE epsilon4 gene precedes neurofibrillary pathology in the frontal association cortex of nondemented senior subjects. J Neuropathol Exp Neurol 60:731–739. [DOI] [PubMed] [Google Scholar]