

Abstract
Pediatric high‐grade astrocytomas (HGAs) account for 15–20% of all pediatric central nervous system tumors. These neoplasms predominantly involve the supratentorial hemispheres or the pons—diffuse intrinsic pontine gliomas (DIPG). Assumptions that pediatric HGAs are biologically similar to adult HGAs have recently been challenged, and the development of effective therapeutic modalities for DIPG and supratentorial HGA hinges on a better understanding of their biological properties. Here, 20 pediatric HGAs (9 DIPGs and 11 supratentorial HGAs) were subject to gene expression profiling following approval by the research ethics board at our institution. Many of these tumors showed expression signatures composed of genes that promote G1/S and G2/M cell cycle progression. In particular, Aurora kinase B (AURKB) was consistently and highly overexpressed in 6/9 DIPGs and 8/11 HGAs. Array data were validated using quantitative real‐time PCR and immunohistochemistry, as well as cross‐validation of our data set with previously published series. Inhibition of Aurora B activity in DIPG and in pediatric HGA cell lines resulted in growth arrest accompanied by morphological changes, cell cycle aberrations, nuclear fractionation and polyploidy as well as a reduction in colony formation. Our data highlight Aurora B as a potential therapeutic target in DIPG.
Keywords: astrocytoma, Aurora kinase, DIPG, glioma, pediatric
Introduction
Pediatric high‐grade astrocytomas (HGAs) account for 15%–20% of all pediatric central nervous system tumors 6. These neoplasms predominantly involve the supratentorial hemispheres or the pons—diffuse intrinsic pontine gliomas (DIPG). For both entities, survival is poor. For supratentorial HGA, 5‐year survival is 15%–30% 6. For DIPG, the outcome is even worse with a median survival of approximately 10 months and <10% 2‐year survival 13, 14. Furthermore, for DIPG surgical resection is not an option and tissue biopsies are not usually undertaken, with diagnosis based on typical clinical presentation and MRI findings. For this reason, surgical material is rarely available for study 2. Clinical investigations into the effects of adjuvant chemotherapy on patient survival have not thus far shown any benefit, and radiotherapy is predominantly palliative, offering some symptom control for a limited period of time 12. A major contributing factor to the inefficacy of previous clinical trials was the assumption that pediatric DIPGs would show similar biologic properties and chemosensitivity to adult HGAs. However, this notion has recently been challenged with both our group and others demonstrating genetic differences between HGAs arising in the pediatric and adult age groups 22, 29. In order to further our understanding of the biology of DIPG, we undertook expression array analysis of a group of these tumors and analyzed the data with a focus on finding potential therapeutic targets.
Materials and Methods
Patients and samples
Snap frozen tumor tissue was obtained after research ethics board approval and parental consent from 20 patients (9 DIPG and 11 HGA) who were treated at the Hospital for Sick Children between 2003 and 2007. Patient details are given in Table S1. Tumor tissue was snap frozen as soon as possible after surgery or death. The median post‐mortem interval was 16 h (range 9–40 h). Percent tumor content was assessed by light microscopy examination of tissue sections by a neuropathologist and estimated to range from 70% to 100% across all the samples with the remaining tissue comprising infiltrated brain or necrosis. RNA from the samples was extracted using the QIAshredder™ and RNeasy® kit (Qiagen, Mississauga, ON, Canada) according to the manufacturer's protocol. RNA quality was determined using the Agilent 2100 Bioanalyzer (Agilent Technologies, Böblingen, Germany). RNA Integrity Number (RIN) ranged from 2.3 to 9.2 with those autopsy cases with longer post‐mortem intervals generally having lower RINs. Three DIPG autopsy samples with RIN equal to or greater than 6 were submitted for hybridization to the HumanWG‐6 v3.0 array (Illumina®, San Diego, CA, USA). The remainder of the samples was hybridized to the HumanRef‐8 v3.0 WG‐DASL array from Illumina. Microarray data were analyzed using Partek® Genomics Suite™ v6.5 (Partek Incorporated, St. Louis, MO, USA). Common probes from both beadchip platforms, numbering 18 404 probes were selected for data analysis. Samples were normalized and batch effect was accounted for using median‐centered quantile normalization. Data analysis was conducted on log2 transformed data. Differentially expressed genes were identified using one‐way ANOVA and significance was corrected for FDR (false discovery rate). Biologically relevant processes of the top 50 most highly over‐ and underexpressed genes were analyzed using GOstat (Fiat Lux, Melbourne, VIC, Australia).
Quantitative real‐time PCR
RNA was synthesized to cDNA using Omniscript® RT kit (Qiagen) as per manufacturer's instructions. Quantitative real‐time PCR was conducted on an ABI Step One Plus™ RT‐PCR (Applied Biosystems, Foster City, CA, USA) system using the SYBR Green PCR Master Mix (Applied Biosystems). The primers used were as follows: AURKB ACATCTTAACGCGGCACTTC and ATGAAATGGCTTTTCTTCTCC, HPRT1 (internal control/housekeeping gene) TTATGGACAGGACTGAACGTC and GATGTAATCCAGCAGGTCAG.
Immunohistochemistry
High‐grade astrocytoma tissue microarrays (TMA) were constructed in our laboratory. Tissue adequacy and diagnostic accuracy (anaplastic astrocytoma or glioblastoma multiforme) was reviewed by neuropathology for each patients' pathological block. Areas representative of tumor histology were identified on each block, and at least three cores were obtained for each sample. A variety of control tissues were lined along the periphery of the array including cerebrum, cerebellum, liver, placenta, lung, large bowel, spinal cord, heart and kidney.
Five micrometer sections were cut from the TMA, mounted on positively charged slides and placed in xylene to remove paraffin wax and hydrated by immersion in decreasing concentrations of ethanol in distilled water. Immunohistochemical (IHC) staining on the TMAs was performed using anti‐Aurora kinase B (anti‐AURKB) antibody (1:250, Abcam, Cambridge, MA, USA) and the Vector‐Elite® avidin‐biotin complex detection system (Vector Laboratories, Burlingame, CA, USA), which utilizes the avidin‐biotin complex method and 3,3'diaminobenzidine (DAB) for immunodetection. All sections were heat treated to facilitate antigen retrieval and blocked for endogenous biotin and peroxidase. Staining of positive and negative controls was run in tandem with each TMA. The sections were counterstained with hematoxylin. The sections were scored for percent nuclear positivity by two independent observers blinded to the clinical data. The distribution of nuclear positivity was not linear. Cases showed either <1% or 10%–20% strong nuclear staining. The latter were considered positive for the purposes of this study when strength and distribution of staining was averaged over three cores. For this antibody, there was general concordance among the cores for a particular case. For the samples that had cores on the TMA and expression array data, there was correlation between AURKB overexpression and IHC positivity.
Cell lines and functional studies
Functional studies were performed in a mouse derived brainstem glioma cell line 4, a primary DIPG line derived from a diffuse intrinsic pontine glioma with glioblastoma multiforme pathology from a 7‐year‐old boy who had undergone treatment at our institution; DIPG27 and two pediatric high‐grade astrocytoma cell lines, SJ‐G2 and SF‐188 18, 28. SJ‐G2 cells were cultured in DMEM (Wisent, Quebec City, QC, Canada), 10% FBS, 4 mM L‐glutamine, 10 mM sodium bicarbonate and 1% penicillin/streptomycin. These cells were established from a glioblastoma multiforme from a 5‐year‐old girl 18. SF‐188 cells were derived from a glioblastoma multiforme from an 8‐year‐old boy 3. SF‐188 cells were cultured in DMEM/F12 (Wisent) supplemented with 10% FBS, 1% L‐glutamine and 1% antibiotic/antimycotic (Wisent). The mouse derived brainstem glioma cell line was from a nestin Ntv‐a;Ink4a‐ARF −/− mouse induced with injection of RCAS‐PDGF‐B‐expressing DF1 cells into the pons as previously described 4. This cell line was cultured in DMEM (Wisent) supplemented with 10% FBS, 1% penicillin/streptomycin. DIPG27 cells were cultured in suspension in NeuroCult NS‐Basal medium (human) supplemented with 200 mM L‐glutamine, 1% antibiotic/antimycotic, 1% N2, supplement, 1% B27 supplement, 75ng/mL BSA, 200 μg/mL hEGF, 4 μg/mL hFGF and 1 mg/mL Heparin. All cell lines were incubated at 37°C and 5% CO2. AURKB was inhibited in the cells using reversine [N'‐cyclohexyl‐N‐(4‐morpholonophenyl)‐7H‐purine‐2,6‐diamine] (Sigma, Oakville, ON, Canada) or VX‐680 [N‐[4‐[4‐(4‐methylpiperazin‐1‐yl)‐6‐[(5‐methyl‐1H‐pyrazol‐3‐yl)amino]pyrimidin‐2‐yl]sylfanylphenyl)cyclopropanecarboxamide] (Selleck Chemicals, Burlington, ON, Canada). Reversine was added to cell media in concentrations ranging from 0.01 to 10 μM 9. Cells were treated with VX‐680 in concentrations ranging from 0.3 to 100 nM. The DMSO equivalent of reversine or VX‐680 concentrations was used in the vehicle control samples. Cell proliferation and viability was assessed through cell counting using trypan blue as well as cytotoxic MTT assay using Cell Proliferation Kit 1 (Roche, Basel, Switzerland) as recommended by the manufacturer. Relative cell viability was determined by taking absorbance readings of treated and control cells with an ELISA plate reader at 560 nm. Effects of reversine and VX‐680 on colony formation of SJ‐G2, SF‐188 and mouse‐derived brainstem glioma cells were assessed by clonogenic assay as described previously 25.
Cell cycle analysis of reversine, VX‐680 and vehicle treated cells using propidium iodide staining (BD Biosciences, Mississauga, ON, Canada) was conducted using FACScan™ system (BD Biosciences). Samples were analyzed using the CellQuest™ Pro software (BD Biosciences). Sub‐G0 and S‐phase subfractions were calculated as averages of duplicate experiments.
Western blot
Western blotting of total cell lysates from reversine‐ and vehicle‐treated cells was conducted using antibodies against AURKB (1:10000, Cell Signaling, Danvers, MA, USA), Histone H3 (1:1000, Abcam), phospho‐Histone H3 (Ser10) (1:1000, Millipore, Billerica, MA, USA) and actin (1:1000, Sigma, St. Louis, MO, USA).
Immunofluorescence staining
DIPG27, SF‐188, SJ‐G2 and mouse derived brainstem glioma cells were cultured on cover slips. The cells were treated with reversine, VX‐680 or vehicle and incubated for 72 h post‐treatment. Immunofluorescence staining was performed using anti‐GFAP antibody in a 1:1000 dilution (Sigma, Oakville).
Statistical analysis
Statistical significance for cell line MTT assays and growth curve data was conducted using either analysis of variance (ANOVA) for multiple comparisons or Student's t‐test. All experiments were repeated in triplicate, and error bars represent the standard deviation.
Results
Expression profiling of pediatric DIPG reveals a proliferative profile
mRNA expression profiles of DIPGs, HGAs and non‐neoplastic control brain tissue were performed on the Illumina HumanWG‐6 or HumanRef‐8 beadchip arrays. Unsupervised hierarchical clustering of the most variably expressed genes among all samples segregated tumors and non‐neoplastic controls into distinct classes. The number of cases was too small to reliably segregate them into further subclasses. Classification of the pediatric tumors with respect to the proneural, mesenchymal and proliferative signature gene classification scheme of adult HGA 24 did not segregate the pediatric tumors into any particular class.
To further investigate the expression signature in pediatric DIPG and HGA we compared the average expression of all Illumina probe sets between the pediatric tumors and non‐neoplastic control brain samples. The top fifty most significantly over‐ and underexpressed genes (Figure 1A, P < 0.01, adjusted for multiple comparisons) were overexpressed at least twofold on average or underexpressed by 0.6‐fold on average in the tumors. To determine the biological processes associated with these genes, we assessed their GO (gene ontology) classification using GOstat. This analysis revealed GO clusters that were significantly overrepresented in the tumors with highly significant enrichment for genes associated with regulation of cell cycle process (P = 0.00267), cell cycle phase/mitosis (P = 0.0103), and chromosome organization/cell division (P = 0.0262), including AURKB, CCNB1, CCNB2, CDK2, CDC2, BIRC5 and FOXM1. Interestingly, these GO biological processes closely resembled the mitosis and cell‐cycle module (MCM) gene expression signature previously identified in a subset of adult GBM 17.
Figure 1.
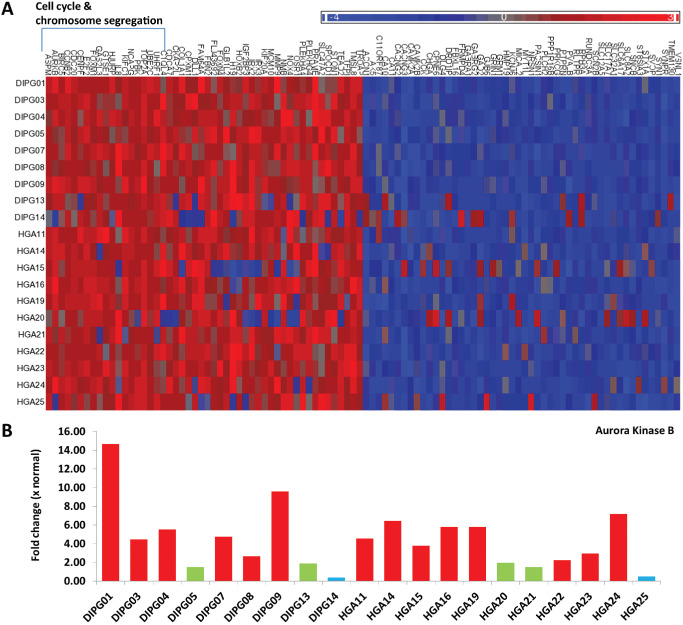
A. Top 50 over‐ and underexpressed genes in pediatric supratentorial high‐grade astrocytoma (HGA) and diffuse intrinsic pontine glioma (DIPG). Red: overexpressed, green: normal expression level compared with non‐neoplastic brain and blue: underexpressed compared with non‐neoplastic brain. 18/50 (36%) of the overexpressed genes were associated with cell cycle and chromosome segregation. B. Analysis of microarray data reveals Aurora kinase B (AURKB) is overexpressed in 6/9 DIPGs and 8/11 HGAs with mRNA levels 2.23 to 14.65 times that of the normal.
Analysis of the 10 most recurrently, highly overexpressed genes again revealed eight of these to be associated with mitosis and cell cycle. Of these, TOP2A (Figure S1) and AURKB were the two most highly overexpressed genes on average across all of the samples. AURKB was overexpressed in 6/9 (66%) of DIPGs and 8/11 (73%) of HGAs with a mean fold change of 5.73 (range 2.23–14.65) overexpression compared with normal brain (Figure 1B). Overexpression of AURKB was confirmed in 14/22 (64%) tumors by qPCR (Figure 2A) and in 30/46 (65%) tumors by immunohistochemistry (Figure 2B and C). Interestingly, expression profiling of three untreated mouse‐derived brainstem glioma tumors (PDGF; Ink4a‐ARF‐/‐) revealed AURKB to be significantly upregulated 5.69‐fold (P = 0.004) compared with three age‐matched and background‐matched normal mouse brainstems (data not shown). Examination of published data sets revealed AURKB to be overexpressed in 16/35 (46%) pediatric HGAs (Figure S2) and 21/26 (81%) DIPG (Figure S3) 22, 23. Combined, our data suggest that AURKB overexpression is a frequent event, occurring in about 65% of both pediatric DIPGs and supratentorial HGAs.
Figure 2.
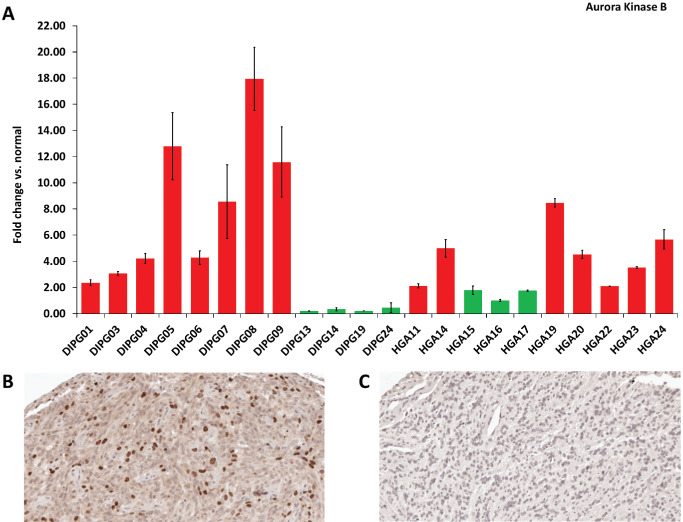
(A) Quantitative polymerase chain reaction validation of Aurora kinase B (AURKB) expression levels in pediatric DIPG and supratentorial HGA. The y‐axis represents the fold change in expression of the tumor sample compared to non‐neoplastic brain. 14 of 22 samples (64%) over‐expressed AURKB (P < 0.05, fold change >2). Error bars represent the standard error of the mean (SEM) of triplicates. Red: overexpression of AURKB and Green: normal expression of AURKB. Immunohistochemical staining of HGA tissue microarrays for Aurora kinase B revealed both positive (B) and negative (C) tumors. Strong nuclear Aurora kinase B positivity found in 10%–20% of cells was present in 65% of samples. Abbreviations: DIPG = diffuse intrinsic pontine glioma; HGA = high‐grade astrocytoma.
Inhibition of AURKB leads to cell cycle arrest in pediatric HGA cell lines
Because of the frequency and degree of overexpression of AURKB in pediatric HGA, we next tested the effects of AURKB inhibitors on a mouse‐derived brainstem glioma cell line and two pediatric HGA cell lines, SF‐188 and SJ‐G2 18, 28. Treatment of the cell lines with reversine caused a dose‐dependent decrease in cell number compared with vehicle‐treated cells, with maximum effect observed at 5 μM for mouse‐derived brainstem glioma (Figure 3A), 2 μM for SJ‐G2 (Figure S4A) and 5 μM for SF‐188 (Figure S5A). VX‐680 treatment of these cell lines over 72 h resulted in significant decrease in overall cell viability by 100nM for the mouse derived brainstem glioma (Figure 3B) and for SJ‐G2 (Figure S4B) and 18nM for SF‐188 (Figure S5B). On Western blot, treatment with the AURKB inhibitors resulted in a decrease in phosphorylated histone H3 (ser10) as compared with vehicle‐treated controls with total histone H3 levels unaffected (data not shown), indicating effective inhibition of AURKB activity 9. The specificity of reversine and VX‐680 as Aurora kinase inhibitors has previously been tested against panels of other kinases 9, 15.
Figure 3.
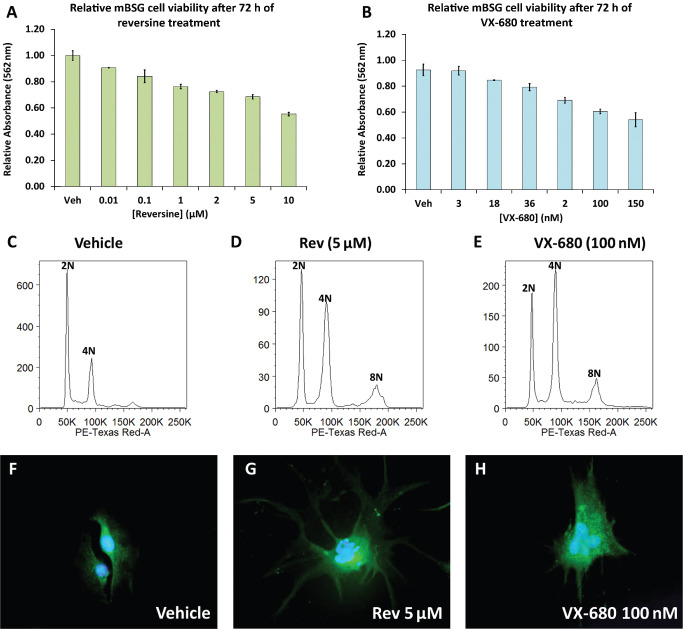
MTT assay reveals a dose‐dependent decrease in cell viability in mouse derived brainstem glioma cells treated with reversine (P < 0.001) (A) and VX‐680 (P < 0.001) (B) after 72 h of treatment. The error bars represent the standard deviation. Propidium iodide based cell sorting of mouse derived brainstem glioma cells (C–E) after 72 h treatment with 5 μM reversine or 100 nM VX‐680 respectively reveals increased cell populations with 4N and 8N DNA content as compared to vehicle control. Treatment of mouse derived brainstem glioma cells for 72 h with 5 μM reversine or 100 nM VX‐680 increases cell size compared with vehicle‐treated control and leads to irregular‐shaped nuclei and micronuclei (F–H). Images F–H represent immunofluorescent staining for GFAP (green) with DAPI counter‐stain (blue) and were taken at 400× magnification.
Flow cytometry revealed that after 48 h of reversine treatment the population of G2/M phase cells was markedly increased as compared with the DMSO‐treated controls for the three cell lines. Similar results were observed for VX‐680 treatment. The two peaks represented cell populations with 2N (G1) and 4N (G2/M). Cell cycle analysis after 72 h resulted in a third peak, most likely representing a polyploidy state of (8N) (mouse‐derived brainstem glioma Figure 3C–E, SJ‐G2 Figure S4C–E, and SF‐188 Figure S5C–E). Immunofluorescence staining of reversine and VX‐680‐treated cells for GFAP counterstained with DAPI revealed morphological changes including irregular, large multinucleated cells (mouse derived brainstem glioma Figure 3F–H, SJ‐G2 Figure S4F–H & SF‐188 Figure S5F–H). Phase contrast microscopy images also highlighted changes in size and shape of reversine‐ and VX‐680‐treated cells (data not shown).
In the presence of reversine or VX‐680 over 96 h, there was minimal growth of SF‐188 (Figure 4A) and no growth of SJ‐G2 or mouse‐derived BSG cells (Figure 4B and C), with a statistical difference in cell number apparent by 48 h (P < 0.005). Importantly, following removal of the drug, there was no further growth rate increase of SF‐188 and mouse‐derived brainstem glioma cells for at least 96 h (Figure 4A and C), and SJ‐G2 began to undergo cell death with a 75% decrease in cell number by 96 h (Figure 4B). Growth rate and percent viability were significantly decreased in all cell lines treated with reversine or VX‐680 when compared with vehicle‐treated controls. In addition, reversine and VX‐680 treatment significantly inhibited colony formation of SJ‐G2, SF‐188 and mouse‐derived brainstem glioma cells compared with vehicle treated controls as determined by clonogenic assay (Figure 4D–F).
Figure 4.
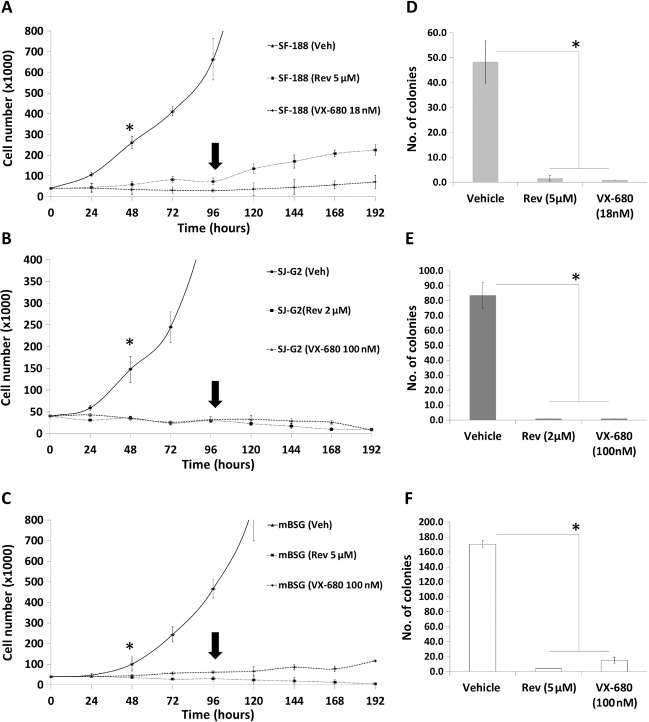
Cell viability of SF‐188, SJ‐G2 and mouse‐derived brainstem glioma cells measured over a period of 8 days (192 h) by trypan blue exclusion assay. Cells were cultured in the presence of reversine (5 μM for SF‐188, 2 μM for SJ‐G2 and 5 μM for mouse‐derived brainstem glioma) or VX‐680 (18 nM for SF‐188, and 100 nM for SJ‐G2 and mouse derived brainstem glioma) from days 1–4. On day 4 (arrow), media was replaced with fresh media not containing reversine. Time course reveals very slow SF‐188 (A) or no SJ‐G2 (B) and mouse‐derived brainstem glioma (C) cell growth following reversine or VX‐680 treatment when compared to vehicle‐treated controls with a statistically significant difference in cell number by 44 h (asterisk: SF‐188, P = 0.0037; SJ‐G2, P = 0.0008; mouse‐derived brainstem glioma, P = 0.0126). Following removal of the drug, there was no further growth rate increase of SF‐188 or mouse derived brainstem glioma for 96 h (cell viability of SF‐188 cells was reduced by ∼20%; P > 0.1, cell viability of mouse derived brainstem glioma was reduced by 37%; P < 0.05), and SJ‐G2 cell numbers began to decrease (cell viability began steadily decreasing, reaching 75% decrease in cell viability by day 8; P < 0.001). Colony formation assay following treatment with reversine or VX‐680 for 72 h resulted in a significant decrease or complete lack of colony formation in all treated cells (P < 0.0001); SF‐188 (D), SJ‐G2 (E) and mouse‐derived brainstem glioma (F).
DIPG27 cells were treated with 5 μM reversine or 18nM VX‐680 for 72 h. Cells treated with reversine or VX‐680 showed significant reduction in cell number (Figure 5B) and perturbed morphology under phase contrast microscopy (data not shown). Immunofluorescence staining of treated DIPG27 cells for GFAP and counterstained with DAPI showed similar changes to reversine‐ or VX‐680‐treated cell lines, including multinucleated cells, increased cell size and decreased cell number (Figure 5C–E). Untreated and vehicle‐treated DIPG27 cells had bipolar processes, whereas treatment of these cells with reversine or VX‐680 resulted in larger cells with multiple processes.
Figure 5.
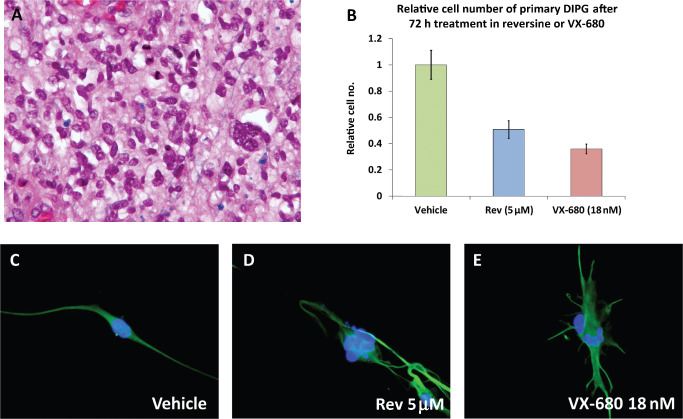
H&E staining of DIPG 27 (200×) (A). Treatment of DIPG27 cells with reversine or VX‐680 for 72 h reduced cell numbers (B) and increased cell size as compared to vehicle treated control and lead to irregular shaped nuclei and micronuclei (C–E). Images C–E represent immunofluorescent staining for GFAP (green) with DAPI counter‐stain (blue) and were taken at 400× magnification. AAbreviations: H&E = Hematoxylin and eosin; DIPG = diffuse intrinsic pontine glioma.
Discussion
DIPGs are one of the main causes of brain tumor death in children. After decades of clinical trials, largely based on protocols and biological data for adult brain tumors, no effective treatment has yet been found. Here we have undertaken gene expression profiling of a series of pediatric DIPGs and HGAs, uncovering a large subset of tumors demonstrating a proliferative phenotype with overexpression of M phase, DNA replication and chromosome organization (MRC) genes, especially AURKB, CCNB1, CCNB2, CDK2, CDC2, BIRC5 and FOXM1. The gene signature that segregates adult GBM into three subtypes (proneural, mesenchymal and proliferative) described by Phillips et al did not segregate our pediatric HGA data set into the any subtype. Nevertheless, genes that also play roles in cell proliferation according to their GOstat annotation were identified to be frequently overexpressed in our pediatric HGAs including AURKB.
Aurora kinases are a family of three highly homologous serine/threonine kinases that play a critical role in regulating many of the processes that are pivotal to mitosis 10. AURKB is the catalytic component of the chromosomal passenger complex (CPC) that is critical for the correct progression through and completion of mitosis 11, 16. The CPC initially forms along chromosome arms and then concentrates at the centromere before finally localizing to the spindle midzone during cytokinesis. Functions of the CPC include condensation of chromosomes, formation of the bipolar spindle, attachment of the chromosomes to the mitotic spindle, regulation of the spindle checkpoint and completion of cytokinesis. Alterations in Aurora kinase signaling are associated with mitotic errors and have been closely linked to chromosomal aneuploidy and genomic instability in cancer 10. Since their discovery in the 1990s, Aurora kinases have been strongly linked to the progression of human cancers. Aurora kinase A maps to human chromosome 20q13 and AURKB to human chromosome 17q13.1, which are loci altered frequently in human cancers. Overexpression of Aurora kinases A and B is seen in many cancers, including hepatocellular carcinoma, colorectal cancer, ovarian carcinoma and adult GBM 1, 5, 7, 30. Aurora kinase inhibitors are an intense area of research for the development of anticancer therapies with 10 inhibitors currently in phase I and II clinical trials for adult cancers 8, 10.
AURKB inhibition has been reported to cause perturbed spindle biorientation, chromosome misalignment, and induce polyploidy and micronuclei formation 19, 20. Similar morphologic findings were identified in our pediatric HGA cell lines and our DIPG line. Sorting cells based on cell cycle after 48 h of reversine or VX‐680 treatment suggested cells were accumulating in the G2/M phase of the cell cycle, while after 72 h, additional peaks were seen, suggesting 4N, 8N or aneuploid high DNA content. Similar effects of AURKB inhibition were seen in a previous studies by D'Alise et al 9 and Rao et al 26, where treatment of HCT‐116 cells with reversine and VX‐680, respectively led to doubling of their DNA content, exit from mitosis and impaired cytokinesis. In vivo mouse xenograft experiments of renal cell carcinoma using AURKB inhibition with VX‐680 showed inhibited tumor growth and Aurora kinase signaling 21. The same group reported G2/M cell cycle arrest and apoptosis in renal cell carcinoma cell lines 21. Importantly, the effect of reversine and VX‐680 on SF‐188, SJ‐G2 and mouse‐derived brainstem glioma cells lasted at least 96 h after removal of the drug from the culture medium suggesting the cells were unable to recover from the blockade. Further, the removal of the inhibitor led to cell death with a modest 15% decrease in cell number seen in SF‐188 but a 75% reduction in SJ‐G2. Others have shown that inhibition of AURKB in cell culture eventually causes apoptotic cell death 27.
In this study, we uncovered, using expression profiling, a significant subset of pediatric HGAs and DIPGs demonstrate a proliferative profile including overexpression of AURKB. We have shown that targeting AURKB results in cell cycle arrest and polyploidy followed by cell death upon removal of the blockade in pediatric high‐grade astrocytoma cell lines. We have also shown that AURKB inhibition in primary DIPG cells has the same effect as on established cell lines. This important preclinical data supports the potential of Aurora kinase inhibitors for use in clinical trials for this devastating disease.
Supporting information
Table S1. Patient information. Abbreviations: Dx = diagnosis; GBM = glioblastoma multiforme; AA = anaplastic astrocytoma.
Figure S1. Analysis of microarray data reveals TOP2A is overexpressed in 7/9 DIPGs and 11/11 HGAs with mRNA levels 2.40–24.04 times that of the normal. Red bars represent samples with significant overexpression of TOP2A (P < 0.05, fold change >2). Red: overexpression of TOP2A and green: normal expression of TOP2A.
Figure S2. Analysis of a previously published expression microarray data set 22 revealed AURKB overexpression in 16/35 (46%) pediatric high‐grade astrocytomas. Red bars represent samples with significant over‐expression of AURKB (P < 0.05, fold change >2).
Figure S3. Analysis of a previously published expression microarray data set 22 revealed AURKB overexpression in 21/26 (81%) pediatric high‐grade brainstem gliomas. Red bars represent samples with significant overexpression of AURKB (P < 0.05, fold change >2).
Figure S4. MTT assay reveals a dose‐dependent decrease in cell viability in SJ‐G2 cells treated with reversine (P < 0.001) (A) and VX‐680 (P < 0.001) (B) after 72 h of treatment. The error bars represent the standard deviation. Propidium iodide‐based cell sorting of SJ‐G2 cells (C–E) after 72 h treatment with 2 μM reversine or 100nM VX‐680, respectively reveals increased cell populations with 4N and 8N DNA content as compared to vehicle control. Treatment of SJ‐G2 cells for 72 h with 2 μM reversine or 100nM VX‐680 increases cell size compared with vehicle treated control and leads to irregular shaped nuclei and micronuclei (F–H). Images F–H represent immunofluorescent staining for GFAP (green) with DAPI counter‐stain (blue) and were taken at 400× magnification.
Figure S5. MTT assay reveals a dose‐dependent decrease in cell viability in SF‐188 cells treated with reversine (P < 0.001) (A) and VX‐680 (P < 0.001) (B) after 72 h of treatment. The error bars represent the standard deviation. Propidium iodide‐based cell sorting of SF‐188 cells (C–E) after 72 h treatment with 5 μM reversine or 18 nM VX‐680, respectively, reveals increased cell populations with 4N and 8N DNA content as compared with vehicle control. Treatment of SF‐188 cells for 72 h with 5 μM reversine or 18 nM VX‐680 increases cell size compared with vehicle‐treated control and leads to irregular‐shaped nuclei and micronuclei (F–H). Images F–H represent immunofluorescent staining for GFAP (green) with DAPI counter‐stain (blue) and were taken at 400× magnification.
Acknowledgments
We would like to thank the National Brain Tumor Society for providing funding for this research.
Research support: This work was supported by an operating grant from the National Brain Tumor Society and the Canadian Institutes of Health Research [MOP 115004].
References
- 1. Aihara A, Tanaka S, Yasen M, Matsumura S, Mitsunori Y, Murakata A et al (2010) The selective Aurora B kinase inhibitor AZD1152 as a novel treatment for hepatocellular carcinoma. J Hepatol 52:63–71. [DOI] [PubMed] [Google Scholar]
- 2. Angelini P, Hawkins C, Laperriere N, Bouffet E, Bartels U (2011) Post mortem examinations in diffuse intrinsic pontine glioma: challenges and chances. J Neurooncol 101:75–81. [DOI] [PubMed] [Google Scholar]
- 3. Bax DA, Little SE, Gaspar N, Perryman L, Marshall L, Viana‐Pereira M et al (2009) Molecular and phenotypic characterisation of paediatric glioma cell lines as models for preclinical drug development. Plos ONE 4:e5209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Becher OJ, Hambardzumyan D, Walker TR, Helmy K, Nazarian J, Albrecht S et al (2010) Preclinical evaluation of radiation and perifosine in a genetically and histologically accurate model of brainstem glioma. Cancer Res 70:2548–2557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bischoff JR, Anderson L, Zhu Y, Mossie K, Ng L, Souza B et al (1998) A homologue of Drosophila aurora kinase is oncogenic and amplified in human colorectal cancers. EMBO J 17:3052–3065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Broniscer A, Gajjar A (2004) Supratentorial high‐grade astrocytoma and diffuse brainstem glioma: two challenges for the pediatric oncologist. Oncologist 9:197–206. [DOI] [PubMed] [Google Scholar]
- 7. Chen YJ, Chen CM, Twu NF, Yen MS, Lai CR, Wu HH et al (2009) Overexpression of Aurora B is associated with poor prognosis in epithelial ovarian cancer patients. Virchows Arch 455:431–440. [DOI] [PubMed] [Google Scholar]
- 8. Cheung CH, Coumar MS, Hsieh HP, Chang JY (2009) Aurora kinase inhibitors in preclinical and clinical testing. Expert Opin Investig Drugs 18:379–398. [DOI] [PubMed] [Google Scholar]
- 9. D'Alise AM, Amabile G, Iovino M, Di Giorgio FP, Bartiromo M, Sessa F et al (2008) Reversine, a novel Aurora kinases inhibitor, inhibits colony formation of human acute myeloid leukemia cells. Mol Cancer Ther 7:1140–1149. [DOI] [PubMed] [Google Scholar]
- 10. Dar AA, Goff LW, Majid S, Berlin J, El‐Rifai W (2010) Aurora kinase inhibitors—rising stars in cancer therapeutics? Mol Cancer Ther 9:268–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ditchfield C, Johnson VL, Tighe A, Ellston R, Haworth C, Johnson T et al (2003) Aurora B couples chromosome alignment with anaphase by targeting BubR1, Mad2, and Cenp‐E to kinetochores. J Cell Biol 161:267–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Donaldson SS, Laningham F, Fisher PG (2006) Advances toward an understanding of brainstem gliomas. J Clin Oncol 24:1266–1272. [DOI] [PubMed] [Google Scholar]
- 13. Freeman CR, Perilongo G (1999) Chemotherapy for brain stem gliomas. Childs Nerv Syst 15:545–553. [DOI] [PubMed] [Google Scholar]
- 14. Hargrave D, Bartels U, Bouffet E (2006) Diffuse brainstem glioma in children: critical review of clinical trials. Lancet Oncol 7:241–248. [DOI] [PubMed] [Google Scholar]
- 15. Harrington EA, Bebbington D, Moore J, Rasmussen RK, Ajose‐Adeogun AO, Nakayama T et al (2004) VX‐680, a potent and selective small‐molecule inhibitor of the Aurora kinases, suppresses tumor growth in vivo. Nat Med 10:262–267. [DOI] [PubMed] [Google Scholar]
- 16. Hauf S, Cole RW, LaTerra S, Zimmer C, Schnapp G, Walter R et al (2003) The small molecule Hesperadin reveals a role for Aurora B in correcting kinetochore‐microtubule attachment and in maintaining the spindle assembly checkpoint. J Cell Biol 161:281–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hodgson JG, Yeh RF, Ray A, Wang NJ, Smirnov I, Yu M et al (2009) Comparative analyses of gene copy number and mRNA expression in glioblastoma multiforme tumors and xenografts. Neuro Oncol 11:477–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hosoi H, Dilling MB, Liu LN, Danks MK, Shikata T, Sekulic A et al (1998) Studies on the mechanism of resistance to rapamycin in human cancer cells. Mol Pharmacol 54:815–824. [DOI] [PubMed] [Google Scholar]
- 19. Kitzen JJ, de Jonge MJ, Verweij J (2010) Aurora kinase inhibitors. Crit Rev Oncol Hematol 73:99–110. [DOI] [PubMed] [Google Scholar]
- 20. Lens SM, Voest EE, Medema RH (2010) Shared and separate functions of polo‐like kinases and aurora kinases in cancer. Nat Rev Cancer 10:825–841. [DOI] [PubMed] [Google Scholar]
- 21. Li Y, Zhang ZF, Chen J, Huang D, Ding Y, Tan MH et al (2010) VX680/MK‐0457, a potent and selective Aurora kinase inhibitor, targets both tumor and endothelial cells in clear cell renal cell carcinoma. Am J Transl Res 2:296–308. [PMC free article] [PubMed] [Google Scholar]
- 22. Paugh BS, Qu C, Jones C, Liu Z, Adamowicz‐Brice M, Zhang J et al (2010) Integrated molecular genetic profiling of pediatric high‐grade gliomas reveals key differences with the adult disease. J Clin Oncol 28:3061–3068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Paugh BS, Broniscer A, Qu C, Miller CP, Zhang J, Tatevossian RG et al (2011) Genome‐wide analyses identify recurrent amplifications of receptor tyrosine kinases and cell‐cycle regulatory genes in diffuse intrinsic pontine glioma. J Clin Oncol 29:3999–4006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Phillips HS, Kharbanda S, Chen R, Forrest WF, Soriano RH, Wu TD et al (2006) Molecular subclasses of high‐grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell 9:157–173. [DOI] [PubMed] [Google Scholar]
- 25. Rafehi H, Orlowski C, Georgiadis GT, Ververis K, El‐Osta A, Karagiannis TC (2011) Clonogenic assay: adherent cells. J Vis Exp 13:25731–25734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Rao B, van Leeuwen IM, Higgins M, Campbel J, Thompson AM, Lane DP, Lain S (2010) Evaluation of an Actinomycin D/VX‐680 aurora kinase inhibitor combination in p53‐based cyclotherapy. Oncotarget 1:639–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Stolz A, Vogel C, Schneider V, Ertych N, Kienitz A, Yu H, Bastians H (2009) Pharmacologic abrogation of the mitotic spindle checkpoint by an indolocarbazole discovered by cellular screening efficiently kills cancer cells. Cancer Res 69:3874–3883. [DOI] [PubMed] [Google Scholar]
- 28. Valera ET, de Freitas Cortez MA, de Paula Queiroz RG, de Oliveira FM, Brassesco MS, Jabado N et al (2009) Pediatric glioblastoma cell line shows different patterns of expression of transmembrane ABC transporters after in vitro exposure to vinblastine. Childs Nerv Syst 25:39–45. [DOI] [PubMed] [Google Scholar]
- 29. Zarghooni M, Bartels U, Lee E, Buczkowicz P, Morrison A, Huang A et al (2010) Whole‐genome profiling of pediatric diffuse intrinsic pontine gliomas highlights platelet‐derived growth factor receptor alpha and poly (ADP‐ribose) polymerase as potential therapeutic targets. J Clin Oncol 28:1337–1344. [DOI] [PubMed] [Google Scholar]
- 30. Zeng WF, Navaratne K, Prayson RA, Weil RJ (2007) Aurora B expression correlates with aggressive behaviour in glioblastoma multiforme. J Clin Pathol 60:218–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Patient information. Abbreviations: Dx = diagnosis; GBM = glioblastoma multiforme; AA = anaplastic astrocytoma.
Figure S1. Analysis of microarray data reveals TOP2A is overexpressed in 7/9 DIPGs and 11/11 HGAs with mRNA levels 2.40–24.04 times that of the normal. Red bars represent samples with significant overexpression of TOP2A (P < 0.05, fold change >2). Red: overexpression of TOP2A and green: normal expression of TOP2A.
Figure S2. Analysis of a previously published expression microarray data set 22 revealed AURKB overexpression in 16/35 (46%) pediatric high‐grade astrocytomas. Red bars represent samples with significant over‐expression of AURKB (P < 0.05, fold change >2).
Figure S3. Analysis of a previously published expression microarray data set 22 revealed AURKB overexpression in 21/26 (81%) pediatric high‐grade brainstem gliomas. Red bars represent samples with significant overexpression of AURKB (P < 0.05, fold change >2).
Figure S4. MTT assay reveals a dose‐dependent decrease in cell viability in SJ‐G2 cells treated with reversine (P < 0.001) (A) and VX‐680 (P < 0.001) (B) after 72 h of treatment. The error bars represent the standard deviation. Propidium iodide‐based cell sorting of SJ‐G2 cells (C–E) after 72 h treatment with 2 μM reversine or 100nM VX‐680, respectively reveals increased cell populations with 4N and 8N DNA content as compared to vehicle control. Treatment of SJ‐G2 cells for 72 h with 2 μM reversine or 100nM VX‐680 increases cell size compared with vehicle treated control and leads to irregular shaped nuclei and micronuclei (F–H). Images F–H represent immunofluorescent staining for GFAP (green) with DAPI counter‐stain (blue) and were taken at 400× magnification.
Figure S5. MTT assay reveals a dose‐dependent decrease in cell viability in SF‐188 cells treated with reversine (P < 0.001) (A) and VX‐680 (P < 0.001) (B) after 72 h of treatment. The error bars represent the standard deviation. Propidium iodide‐based cell sorting of SF‐188 cells (C–E) after 72 h treatment with 5 μM reversine or 18 nM VX‐680, respectively, reveals increased cell populations with 4N and 8N DNA content as compared with vehicle control. Treatment of SF‐188 cells for 72 h with 5 μM reversine or 18 nM VX‐680 increases cell size compared with vehicle‐treated control and leads to irregular‐shaped nuclei and micronuclei (F–H). Images F–H represent immunofluorescent staining for GFAP (green) with DAPI counter‐stain (blue) and were taken at 400× magnification.