

Abstract
Motor neuron diseases such as amyotrophic lateral sclerosis (ALS) are now recognized as multi‐system disorders also involving various non‐motor neuronal cell types. The precise extent and mechanistic basis of non‐motor neuron damage in human ALS and ALS animal models remain however unclear. To address this, we here studied progressive motor neuronopathy (pmn) mice carrying a missense loss‐of‐function mutation in tubulin binding cofactor E (TBCE). These mice manifest a particularly aggressive form of motor axon dying back and display a microtubule loss, similar to that induced by human ALS‐linked TUBA4A mutations. Using whole nerve confocal imaging of pmn × thy1.2‐YFP16 fluorescent reporter mice and electron microscopy, we demonstrate axonal discontinuities, bead‐like spheroids and ovoids in pmn suralis nerves indicating prominent sensory neuropathy. The axonal alterations qualitatively resemble those in phrenic motor nerves but do not culminate in the loss of myelinated fibers. We further show that the pmn mutation decreases the level of TBCE, impedes microtubule polymerization in dorsal root ganglion (DRG) neurons and causes progressive loss of microtubules in large and small caliber suralis axons. Live imaging of axonal transport using GFP‐tagged tetanus toxin C‐fragment (GFP‐TTC) demonstrates defects in microtubule‐based transport in pmn DRG neurons, providing a potential explanation for the axonal alterations in sensory nerves. This study unravels sensory neuropathy as a pathological feature of mouse pmn, and discusses the potential contribution of cytoskeletal defects to sensory neuropathy in human motor neuron disease.
Keywords: axon degeneration, microtubule, motor neuron disease, sensory neuropathy, spheroid
Introduction
The neurodegenerative disease amyotrophic lateral sclerosis (ALS) is characterized by progressive degeneration of motor neurons in spinal cord, brainstem and cerebral cortex 5. Increasing evidence indicates however that ALS affects not only motor neurons but also additional types of neurons involved in cognitive, sensory, autonomous and other functions 8, 22, 75, 76 leading to the notion that ALS is a multisystem disorder 42, 63, 68, 75.
Sensory impairments in human ALS have been well documented by clinical, electrophysiologial and histological studies. ALS patients complain about pain 10 or abnormal laryngeal sensation 1 more frequently than controls. They further present elevated thresholds for vibration 23, 45 or altered touch‐pressure sensation 31. Electrophysiological examinations have detected reduced nerve conduction velocities 50, 62, altered sensory nerve action potentials (SNAP) 23, 26 and delayed somatosensory evoked potentials (SEPs) 21, 28, 41 even in the absence of sensory signs. Furthermore, histopathological analyses have documented loss of dorsal root ganglion (DRG) neurons or dorsal root myelinated fibers 35, axonal degeneration/regeneration in suralis nerves 26 or reduced densities of intraepidermal nerve fibers 11, 81 in a high proportion of ALS cases. The molecular mechanisms of sensory nerve degeneration in ALS remain however incompletely understood. This is at least partly caused by our insufficient knowledge on the precise pathogenic mechanisms in ALS which can be triggered by mutations in at least 25 different genes 39.
Recently, rare forms of human ALS have been found associated with mutations in the TUBA4A gene which encodes the major α‐tubulin isoform in adult brain 65. Overexpression of human ALS‐associated TUBA4A mutants in cultured motor neurons causes severe alterations of the microtubule network suggesting microtubule destruction as a pathogenic mechanism 65. Microtubule destruction is also an essential feature of mutant progressive motor neuronopathy (pmn) mice which carry a missense mutation in the tubulin binding cofactor E (TBCE) gene 6, 25, 40, a chaperone that assists in tubulin folding, microtubule polymerization 48, 71 and microtubule maintenance 6, 40. We previously showed that TBCE is strongly expressed in motor neurons where it localizes to the Golgi apparatus 4. We further demonstrated that TBCE function is critical for axonal tubulin routing 59 while destabilization of TBCE through the pmn missense mutation (p.Trp524Gly) causes retrograde loss of axonal microtubules and dying back of motor axons 40, 59.
Mutant pmn mice suffer from a severe and rapidly progressive form of motor neuron degeneration which starts in the hindlimbs and leads to death of the animals before 7 weeks of age 60. In addition, pmn mice manifest progressive hearing loss 51, 79, indicating that the sensory system is not spared. Using YFP imaging and electron microscopy, we here demonstrate prominent signs of peripheral sensory neuropathy in pmn mice. We show that reduced TBCE expression and reduced microtubule polymerization in pmn DRG neurons are associated with defective microtubule‐dependent transport and progressive microtubule loss in DRG axons, providing a mechanistic basis for the peripheral sensory neuropathy.
Methods
Mouse lines
Thy1.2‐YFP‐16 mice (B6.CgTg (Thy1‐YFP)16Jrs/J) were obtained from the Jackson laboratory. Mutant pmn, Xt pmn 40 and pmn thy1.2‐YFP‐16 mice were maintained on a mixed genetic background (C57/BL6 × 129/SvJ) using intercrosses (>F5) and genotyped by PCR 59. All experiments with animals were performed in strict compliance with institutional, national and European legislation. In particular, authorization to conduct research on animals had been delivered by Préfecture des Bouches du Rhône on 12.12.2006 (licence n° 13.301); research with genetically modified animals had been approved by French Ministry of Research and Education on 10.7.2013 (decision n°6545) and ethical aspects of animal research had been approved by the Ethics Committee on Animal Experimentation n°71 on 7.4.2014 and were registered by French Ministry of Research and Education.
Antibodies and reagents
Primary antibodies were as follows [supplier, dilution in immunofluorescence (IF), and western blot (WB)]: rabbit anti‐TBCE (SA53, IF 1:300, WB 1:500, custom made), mouse anti‐p115, (BD Transduction Laboratories; IF 1:300), mouse anti βIII‐tubulin (TuJ1, Babco, IF 1:2000), rabbit anti‐γ‐tubulin, rabbit anti Δ‐Tyr (Detyr) tubulin (Dr. A. Andrieux, IF 1:5000). Fluorochrome‐conjugated secondary antibodies were from Molecular Probes (Carlsbad, CA).
Additional reagents were from the following suppliers: PBS, Hbss, trypsin, culture media and supplements (Invitrogen, Carlsbad, CA), polyornithin, taxol, nocodazole (Sigma), Vectashield (Vector laboratories, Burlingame, CA), Complete protease inhibitors (Roche, Basel, Switzerland), Ketamine (Bayer, Leverkusen, Germany), Xylazine (Mérial, Lyon, France), Superfrost Plus glass slides and coverslips (Menzel, Schwerte, Germany).
Conventional and confocal fluorescence microscopy
Mice deeply anesthetized by intraperitoneal injection of Ketamine and Xylazine were perfused with Sorensen buffer phosphate buffer pH 7.4 containing 3000 I.U. Heparin per liter followed by paraformaldehyde (PFA) 4% (w/v) in Sorensen buffer. Lumbar spinal cords were dissected out with adhering dorsal roots and dorsal root ganglia. Nerves were dissected out, post‐fixed overnight while being stretched, and washed in PBS. Nerves were then incubated in Vectashield for up to four weeks and mounted under glass coverslips. Overview images were obtained with a Zeiss AxioImager L1 Apotome microscope (20× objective) and assembled with the mosaix function of Zeiss AxioVision software. Z‐stacks of 1 μm interval were obtained with a Zeiss LSM510 confocal microscope (20× or 63× objective) and analyzed with Zeiss Zen software. Axonal spheroids were defined as focal axonal caliber protrusions exceeding by at least twofold axonal diameter and quantified by an observer blinded to the genotype.
For immunohistochemistry, deeply anaesthetized mice were perfused with 4% PFA, spinal cords were dissected, post‐fixed overnight and cryoprotected in 30% sucrose for 48 hours. After tissue embedding, 20 µm transverse sections of the spinal cord and adhering DRGs were cut on a Leica‐Jung cryostat and collected on glass slides. Sections were blocked, incubated overnight with primary antibodies, washed, incubated with fluorochrome‐conjugated secondary antibodies and mounted in Vectashield/DAPI.
Light and electron microscopy
Deeply anesthetized mice were transcardially perfused with Sorensen's phosphate buffer pH 7.4 containing 3000 I.U. Heparin per liter, followed by glutaraldehyde 2% (w/v) in cacodylate buffer pH 7.4. Suralis and phrenic nerves as well as lumbar spinal cords with adhering DRGs were dissected, postfixed in glutaraldehyde for 24 hours and washed in cacodylate buffer and then osmificated for 2 hours in 2% OsO4. Nerves were then dehydrated in graded acetone and embedded in Epon 812‐Araldite. For light microscopy analysis, sections were stained with toluidine blue. Ultra‐thin sections were stained with uranyl acetate and lead citrate and observed with a JEOL 1011 electron microscope.
The number of myelinated fibers was determined manually on semi‐thin sections of a whole nerve (magnification ×60) observed under a DXM1200 microscope (Nikon, Tokyo, Japan): after having determined the area of the whole section of the nerve using the software NIS of NIKON DXM1200, we obtained the density of myelinated fibers (fibers/mm2). The density of unmyelinated fibers was determined in three distinct fields of 391 μm2 per nerve.
Biochemical techniques
Brain regions, spinal cords and DRGs were dissected, snap frozen in liquid nitrogen and homogenized in lysis buffer (150 mM NaCl, 50 mM Tris‐HCl, 2 mM EDTA, 1% Triton X‐100, 0.1% SDS, pH 7.4, protease inhibitors). Protein concentration was determined by BCA Kit (Thermo Fisher) and equal amounts or protein (60 µg/mL) were resolved by 10% SDS‐PAGE and transferred to nitrocellulose membranes (Millipore). After blocking with 5% skim milk, membranes were incubated with antibodies specific to TBCE or GAPDH followed by IR‐Dye‐conjugated secondary antibodies (LI‐COR) and protein bands detected using a near‐infrared laser scanning device (Odyssey, LI‐COR).
Recombinant GFP‐TTC GST fusion proteins 7 were purified from Escherichia coli transformed with pGEX plasmids. Briefly, bacterial lysates [1% Triton X‐100, 1% NP‐40, 1 mg/mL lysozyme, complete protease inhibitors (Boehringer)] were ultracentrifuged at 30 000 × g for 10 minutes and the fusion proteins affinity purified using glutathione‐Sepharose B beads. Proteins were eluted by 5 mM reduced glutathione in 50 mM NaCl, dialyzed against PBS and 5% to 10% glycerol. All steps were performed at 4°C. Protein concentrations were determined by spectroscopic Coomassie assay (Pierce) and purity confirmed by SDS‐PAGE.
Cell culture techniques
DRG neurons were isolated from embryonic E15 mice essentially as described by 77. DRGs were dissected from individual embryos, cleaned of their fibers, trypsinized (0.25% in HBSS for 15 minutes), triturated, and filtered through a 100 micron nylon sieve (Becton Dickinson). The cell suspension was incubated for 3 hours on tissue culture dishes containing DMEM plus 15% fetal calf serum, leading to attachment of non‐neuronal cells. Neuronal cells in suspension were pelleted and resuspended in chemically defined medium containing 0.5 mg/mL bovine serum albumin, 5 μg/mL insulin, 50 μg/mL transferrin, 6 ng/mL progesterone, 15 μg/mL putrescine, 5 ng/mL sodium selenite in DMEM. Cells were seeded on 8‐well LabTek plates that had been pretreated with polyornithin (0.1 mg/mL in 0.1 M borate buffer pH 8.4) for 2 hours at room temperature, washed and treated with laminin (2 μg/mL) overnight at 37°C.
Motor neurons were isolated from embryonic E12 mice and cultured as described 52. Genotyping of embryos was performed in parallel to cell isolation using PCR 59.
Microtubule assays
Microtubule densities in suralis nerve cross sections were determined by electron microscopy. Ultrathin sections of the distal suralis nerves were tilted in the electron microscope (Philips CM100, Eindhoven, the Netherlands) by means of a goniometer to obtain exact cross sections. The magnification of the microscope was calibrated against a replica of a diffraction grating (2.160 lines/mm). With the aid of a digitizer tablet, axonal areas and microtubules were counted on electronmicrographs printed to about 60 000 times.
Microtubule growth in cultured DRG neurons was assayed as previously described for motor neurons (Schaefer et al 2007). DRG neurons were allowed to attach for 45 minutes. Microtubules were depolymerized by addition of 10 µM nocodazole for 6 hours at 37°C. Nocodazole was washed out with warm culture medium, and neurons further incubated for 0, 1 or 30 minutes. Cultures were rinsed in PHEM (60 mM PIPES, 25 mM Hepes, 10 mM EGTA, 2 mM MgCl2, 1% formaldehyde, pH 6.9) and extracted for 3 minutes in PHEM containing 0.2% Triton X‐100, and 20 µM Taxol, fixed, blocked and immunostained for βIII‐tubulin and γ‐tubulin to visualize microtubules and centrosomes, respectively. Images were obtained by confocal microscopy in sections of 2.5 µm optical thickness covering the centrosome using identical acquisition parameters. Mean βIII‐tubulin fluorescence and the length of microtubules emanating from the centrosome were measured using Metamorph and NIH ImageJ software, respectively.
Axonal transport assay
GFP‐TTC was cloned and produced as fusion protein in E. coli was done as described 7. Axonal transport was studied in neurons cultured for 48 hours in LabTek chambers. Neurons were incubated with 5 μg/mL GFP‐TTC in 20 mM Tris HCl pH 8, 1 mM CaCl2, 1 mM MgCl2, 0.25% (w/v) BSA in PBS for 15 minutes at 37°C in the CO2 incubator. After several washes, neurons were incubated for 2 hours in culture medium without riboflavin before acquisition. Axonal GFP‐TTC transport was measured with an LSM510 confocal microscope Zeiss equipped with a thermostated and CO2 equilibrated chamber. Time lapse images were acquired over 5 minutes at a rate of one scan per 0.98 second using a Plan‐Apochromat 63× f/1.4 oil objective (Zeiss). The speed of moving GFP‐TTC particles was analyzed with LSM software. The observer was blinded with respect to the genotype of the cultures.
Statistical analyses
Each experiment was repeated at least twice. Data were analyzed with Excel (Microsoft) or GraphPad Prism (GraphPad). Data from two groups each showing Gaussian distribution were analyzed with Student's t‐test; otherwise the Mann–Whitney test was used. Data from more than two groups were analyzed with Kruskal–Willis test and Dunn post hoc test.
Results
YFP imaging reveals sensory axon degeneration in suralis nerves of pmn mice
Axon degeneration is characterized by breakdown of the cytoskeleton leading to swollen axon segments (spheroids), axonal discontinuities and granular degeneration, and by axonal demyelination resulting in the formation of myelin remnants (ovoids) 27, 74.
To detect signs of sensory axon degeneration in pmn mice, we crossed these mice with thy1.2‐YFP16 mice (termed in the following YFP) which express high non‐toxic levels of fluorescent YFP in the axoplasm of > 80% of all DRG neurons including all major DRG subsets 16. We choose to focus our analyses on the suralis nerve for several reasons: this sensory nerve innervates the hindlimb territory which is affected early in pmn mice 60; it represents a long distal nerve likely to be affected by axonal dying back in pmn 60 and nerve biopsies of the sural nerve are widely used in the differential diagnosis of human neuropathies 43.
We demonstrate that isolated suralis nerves of 25‐day old wildtype (wt) YFP mice contain a large number of YFP‐positive axons of different calibers which show uniform labeling in internodes (Figure 1A1). By contrast, we find that suralis nerves of pmn YFP mice display prominent axonal caliber irregularities and bead‐like spheroids (Figure 1A2). These latter qualitatively resemble those in the pmn phrenic nerve (Figure 1A3), which contains a vast majority (69%) of motor axons 37. The axonal caliber irregularities and spheroids occur across the whole pmn × YFP suralis nerve and are not restricted to specific nerve fascicles (Figure 1A2). This rules out that they are specific to the few motor axons present in the suralis nerves in rodents 53, 70 which are known to innervate the deep lumbrical and interosseus muscles of the hind foot 46.
Figure 1.
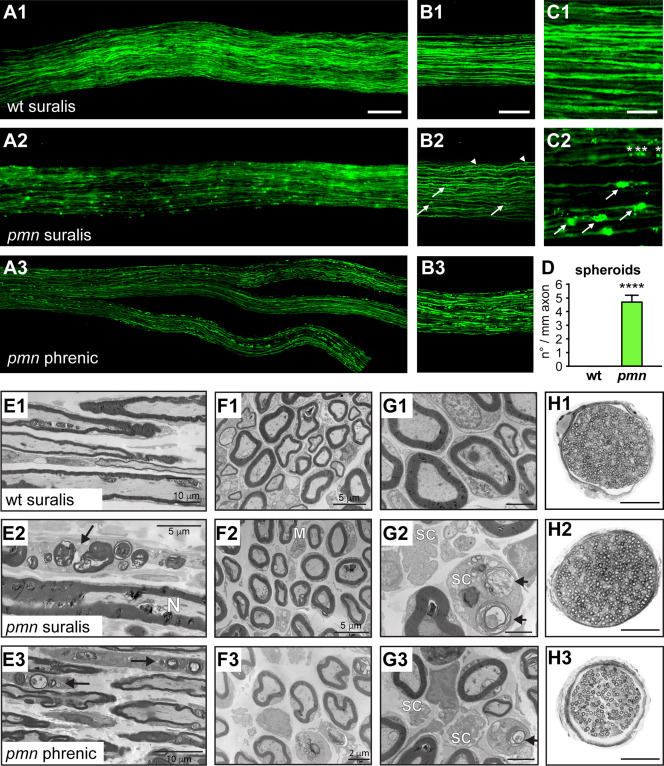
Sensory axon degeneration in suralis nerves of pmn mice revealed by axonal YFP imaging and electron microscopy. A. Images of suralis nerves from 25‐day old wildtype (wt) thy1‐YFP‐16 mice (A1) and pmn thy1‐YFP‐16 mice (A2) expressing YFP in axons. A pmn phrenic motor nerve with its distal branches (A3) is shown for comparison. Scale: 100 µm. B. Images of confocal z‐stacks showing axon caliber irregularities and axonal spheroids in peripheral nerves of pmn YFP mice (B2) as compared to wt (B1). In suralis nerves of pmn YFP mice, axonal spheroids occur in both large caliber axons (arrows) and small caliber axons (arrowheads). Signs of axon degeneration in suralis sensory axons are qualitatively similar to those in phrenic motor axons (B3) of pmn YFP mice but quantitatively less pronounced. Scale: 100 μm. C. High power microscopic images showing axonal spheroids in axons of pmn YFP suralis nerves (C2, arrows) but not in corresponding wt YFP axons (C1). Also note the presence of multiple small axonal YFP foci (asterisks) in the same axon. Scale: 30 μm. D. Quantitation of YFP spheroids in pmn suralis nerves. The number of spheroids per axon segment is about 100 fold higher in pmn YFP axons (4.69 ± 0.47) than in wt YFP axons (0.047 ± 0.013). Axons were monitored on single confocal sections and traced along > 200 μm length in z‐stacks of 20–30 μm depth. Number of axon blebs per mm axon segment is represented as mean of means per nerve ± sd. A total of 2701 axons (wt YFP: 203 to 427 per nerve, pmn YFP: 112 to 278 per nerve) was analyzed in five nerves per genotype. Statistical significance ****, P < 1.7 × 10−23 by Student's t‐test, unpaired, unequal variance. E. Electron microscopy images of longitudinal nerve sections. In comparison to the wt suralis nerve (E1), the pmn suralis nerve (E2) presents some damaged myelinated axons. These myelinated fibers are now a row of several aligned ovoids, consisting of myelin debris and disrupted axons (arrow). N: normal axon. Similar lesions are seen in the pmn phrenic motor nerve where two Schwann cells contain numerous myelin and axonal debris (ovoids, arrows) (E3). Scale bars see images. F. Electron microscopy of transverse sections. The density of myelinated axons is similar in wt (F1) and pmn (F2) suralis nerves. A macrophage (M) in the pmn suralis nerve is loaded with some myelin debris. Macrophages have only a plasma membrane but no basal membrane and present elongated cytoplasmic expansions. Schwann cells have a plasma and a basal membrane, and no elongated cytoplasmic expansions. Scale bars see images. G. High power electron microscopy images of transverse sections. In the pmn suralis nerve (G2) note myelin and axonal debris inside a Schwann cell (SC) cytoplasm (ovoid, arrow) and disappearance of unmyelinated fibers in the other Schwann cell. Ovoids are also seen in the pmn phrenic nerve (G3, arrow). Scale: 2 μm. H. Entire nerve cross sections (semi‐thin sections) show normal number of myelinated fibers in pmn (H2) as compared to wt suralis nerves (H1). Note severe degeneration of myelinated fibers in the pmn phrenic motor nerve (H3). Scale: 50 μm.
Figure 2.
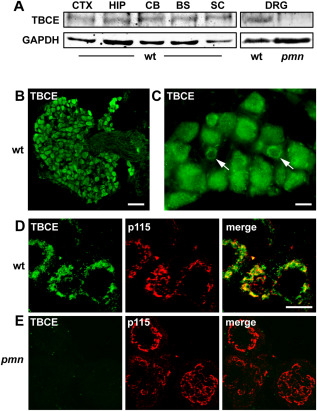
TBCE expression in DRG neurons of wildtype and pmn mice. A. Immunoblot showing expression of TBCE (∼59 kD) at similar level in cortex, hippocampus, cerebellum, brainstem, spinal cord and DRGs of 35‐day‐old wt mice. Note loss of TBCE expression in DRGs of pmn mice as compared to wt. Loading control: GAPDH. B. Immunofluorescence labeling of a lumbar DRG from a wt mouse shows strong TBCE expression in neuronal cell bodies but not in axons. Scale: 50 µm. C. Immunofluorescence labeling shows cytosolic and perinuclear (arrows) distribution of TBCE. Scale: 10 µm. D, E. Confocal imaging showing TBCE protein expression at the p115‐labeled Golgi membrane in wt DRG neurons (D) and the loss of TBCE from the Golgi membrane in pmn DRG neurons (E). Scale: 10 µm.
To further characterize and quantify the axonal spheroids, we performed confocal microscopy. Analysis of confocal z‐stacks revealed that the spheroids are present in both large and small caliber pmn sensory axons (Figure 1B2, arrows and arrowheads). Longitudinal tracing of axons on consecutive single confocal sections was used to quantify the degenerative alterations (Figure 1C1,C2). Analysis of a total of 2701 axons in 5 suralis nerves per genotype revealed that axonal spheroids occur at a mean frequency of 4.69 ± 0.47 per mm in pmn YFP suralis axons, which is about 100‐fold higher than in wt YFP suralis axons (0.047 ± 0.013 per mm, means of means ± sd, student's t‐test, 1.7 × 10E23, Figure 1D). Longitudinal tracing of pmn YFP suralis nerves further demonstrated that these spheroids often manifest repeatedly on the same axon, like “beads on a string,” suggesting a multifocal degenerative process (Figure 1C2, asterisks). These data unravel the degeneration of long sensory axons in pmn mice.
Ultrastructural axonal lesions in pmn suralis nerves
To further characterize the sensory nerve degeneration in pmn mice, we studied ultrathin longitudinal (Figure 1E) and transverse sections (Figure 1F–G) from day 25 wt and pmn suralis nerves by electron microscopy. These ultrastructural studies demonstrated signs of axonal destruction in myelinated fibers of pmn suralis nerves (Figur 1E2) as compared to wildtype (Figure 1E1). Signs of axonal destruction include collapsed and destroyed myelin sheaths, and disappearance of axons in these myelin debris (Figure 1E2, arrows). These lesions, called ovoids, are constituted of various types of lipid structures; such as myelin and axon remnants are often located in Schwann cell cytoplasms or inside macrophages in the endoneurium (Figure 1F2,G2). A few unmyelinated axons also have disappeared and some non‐myelinating Schwann cells do not contain any axon (Figure 1G2). Such lesions are absent in pmn lumbar dorsal roots (not shown) but are present in the distal phrenic motor nerve (Figure 1E3, F3, G3). These data suggest that the sensory neuropathy starts distally, similarly to the motor neuronopathy of pmn mice.
Figure 3.
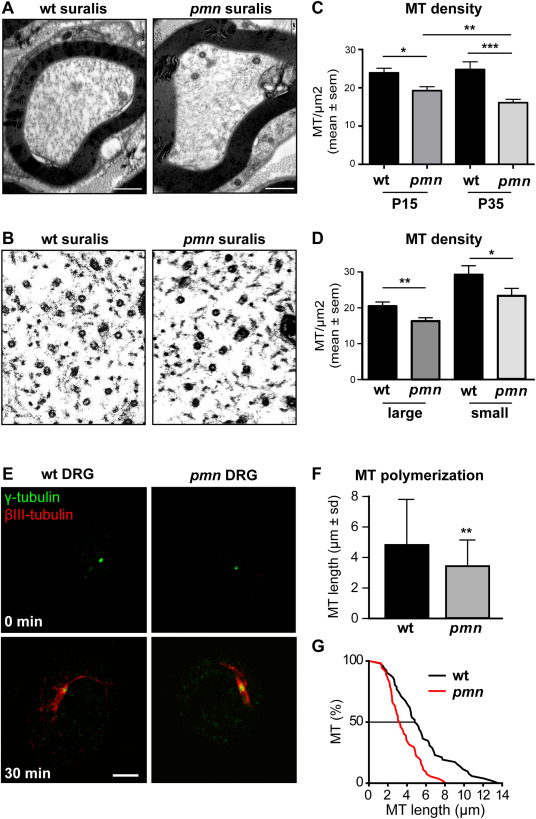
Progressive microtubule loss and defective microtubule polymerization in pmn sensory neurons. A. Electron micrographs showing microtubules in suralis nerve axons from wildtype and pmn mice aged 35 days. B. High power magnifications showing individual microtubules in suralis nerve axons. C. Kinetics of microtubule loss. Microtubule densities (mean ± sem) in myelinated pmn suralis axons are significantly reduced at day 15 (19.3 ± 1.2 vs. 23.9 ± 1.2 MT/μm2, *P = 0.0056) and at day 35 (16.1 ± 0.9 vs. 24.8 ± 2 MT/μm2, ***P = 0.0001) with a significant difference between both time points (**P = 0.0072). Cross‐sectional area of axons does not significantly differ between wt and pmn (day 15: wt 3.59 ± 1.1 μm2, pmn: 3.22 ± 1.2 μm2). Analyses at day 15 were done on a total of 50 axons (wt) and 51 axons (pmn) from n = 4 nerves per genotype. Analyses at day 35 were done on a total of 50 axons (wt) and 64 axons (pmn) from n = 4 wt and n = 5 pmn nerves. Statistical significance was determined by Mann–Whitney test. D. Axonal microtubule loss. In comparison to wt suralis nerves at day 15, microtubule densities (mean ± sem) in pmn suralis nerves are reduced by 20% in both large caliber axons (> 2 μm diameter) and small caliber axons (≤ 2 μm diameter). Note the higher density of microtubules in small caliber axons. **P < 0.001 by Student's t‐test, *P < 0.05 by Student's t‐test. Microtubule densities were calculated from total numbers of microtubules and cross sectional areas of axons. A total of n = 30 large diameter axons and n = 20 small diameter axons from 5 suralis nerves were analyzed per genotype. E. Images showing microtubule regrowth in DRG neurons purified from wt or pmn E15 embryos. Neurons were treated with nocodazole to depolymerize microtubules (0 minutes) and allowed to re‐grow microtubules for 30 minutes after nocodazole washout. Microtubules and centrosomes were labeled with βIII‐tubulin‐ and γ‐tubulin‐specific antibodies, respectively. Scale: 5 µm. F. Histogram showing reduced microtubule length in pmn DRG neurons 30 minutes after nocodazole washout (mean ± sd, n > 70 microtubules per genotype pooled from two independent experiments, P = 0.007 by Mann–Whitney test). G. Cumulative plot of microtubule length 30 minutes after nocodazole washout.
Despite these lesions, the number of large myelinated sensory fibers in pmn suralis nerves does not significantly differ between wt and pmn at day 35 corresponding to disease endstage (Figure 1H1,H2, wt: 303 ± 41; pmn: 349 ± 56, n = 6 nerves per genotype, mean ± sd). This confirms earlier data 1. The mean density of unmyelinated fibers in pmn suralis nerves (0.17 fibers per μm2, 25/75 quartiles: 0.16/0.18) is also close to that in wt (0.19 fibers per μm2, 25/75 quartiles: 0.11/0.27, difference nonsignificant by Mann–Whitney test, n = 6 analyses from n = 2 mice per genotype). This contrasts with the massive loss of motor fibers in the pmn phrenic nerve [Figure 1H3 lower panel, Supporting Information Fig. 1, and 60].
Loss of TBCE expression in DRG neurons of pmn mice
We had previously shown that TBCE mRNA is ubiquitously expressed in neuronal and non‐neuronal tissues and organs 40, reflecting the essential role of TBCE in tubulin folding, microtubule polymerization and scavenging of soluble tubulins 48, 71.
To begin to understand the origin of sensory axon degeneration in pmn mice, we compared TBCE protein expression levels in DRGs and various brain regions by immunoblot (Figure 2A). We demonstrate that TBCE protein is expressed at a similar level in DRG, cortex, hippocampus and brainstem. By contrast, we find that TBCE expression is strongly reduced in DRG neurons of pmn mice (Figure 2A).
We previously showed that TBCE localizes to cis‐Golgi membranes and that its Golgi association is critical for microtubule polymerization and axonal routing 40. We therefore studied TBCE protein expression in lumbar DRG neurons which project to the suralis nerve. Immunostaining demonstrated that TBCE is strongly expressed in the cell bodies of DRG neurons (including large, intermediate and small size neurons) but not in their axons (Figure 2B,C). Confocal Imaging demonstrates that TBCE localizes predominantly to the p115‐labeled membrane of the Golgi apparatus (Figure 2D), which is a major site of microtubule polymerization in mature neurons 4, 57. In DRG neurons from pmn mice however, TBCE expression is undetectable at the Golgi apparatus (Figure 2E). These results show that TBCE protein is strongly expressed at the Golgi apparatus of wt DRG neurons and reduced in its levels in pmn DRG neurons.
Progressive microtubule loss in sensory axons of pmn mice
Given the critical role of TBCE in axonal tubulin routing and microtubule maintenance 59 and its loss in DRG neurons of pmn mice (Figure 2A,E), we then analyzed axonal microtubules densities in suralis nerves. Using electron microscopy, microtubules were counted in whole axon cross sections of distal suralis nerves from wt and pmn mice at several disease stages (Figure 3A–C).
At day 15, when pmn mice manifest first signs of motor neuron disease 60, mean microtubule densities in pmn suralis axons were already significantly reduced by 19% (P = 0.0056 by Mann–Whitney test) (Figure 3C). At day 35 corresponding to pmn endstage, axonal microtubule densities were further reduced to 35% of wt values (P < 0.0001 by Mann–Whitney test) (Figure 3C). A similar reduction was seen at both stages for the total number of cross‐sectioned microtubules in pmn suralis axons, as compared to wt (not shown). These data indicate a progressive loss of microtubules in sensory axons of pmn mice.
Our observation of degenerative signs in both large and small fibers of pmn suralis nerves by YFP imaging (Figure 1B2) and electron microscopy (Figure 1E2,F2,G2) then prompted us to compare the microtubule densities in small axons (≤ 2‐µm diameter) and large axons (> 2‐µm diameter). In wt nerves, small axons displayed higher mean microtubule densities than large axons, in agreement with previous reports 14. In suralis nerves of pmn mice, microtubule densities were significantly reduced in both small and large axons (Figure 3D). In sum, these findings indicate that loss of TBCE in pmn sensory neurons causes progressive microtubule loss and pathology in large and small sensory axons.
Reduced microtubule polymerization in DRG neurons of pmn mice
We then investigated microtubule polymerization in cultured DRG neurons under defined in vitro conditions. DRG neurons were isolated from E15 pmn and wt embryos, seeded, allowed to attach for 45 minutes and incubated with the microtubule‐depolymerizing drug nocodazole (10 µM, 6 hours, 37°C). This treatment resulted in complete microtubule depolymerization (Figure 3E, upper panels). Thirty minutes following nocodazole washout, we monitored microtubule polymerization from the centrosome, the major site of microtubule nucleation in immature neurons 66 (Figure 3E, lower panels). Immunolabeling of centrosomal microtubules by βIII‐tubulin/γ‐tubulin revealed significantly reduced microtubule length in pmn as compared to wt DRG neurons (Figure 3F,G). These data show that the pmn mutation compromises microtubule polymerization in DRG neurons.
The pmn mutation impedes microtubule‐dependent axonal transport in DRG neurons
The occurrence of axonal spheroids in several neurodegenerative diseases has been attributed to focal defects in axonal transport of proteins, vesicles and organelles 17, 67. To search for defects in axonal microtubule‐based transport in pmn sensory neurons, we used live imaging of GFP‐tagged tetanus toxin C‐fragment (GFP‐TTC). We and others had previously shown that GFP‐TTC, a genetic fusion protein between GFP and the C‐terminal fragment of the tetanus toxin heavy chain (TTC), is taken up by cultured neurons 7, internalized into vesicles and retrogradely transported along microtubules 19, 54.
We cultured wt and pmn DRG neurons (Figure 4A) and verified that internalization of GFP‐TTC is not affected by the pmn mutation (Figure 4B). Confocal imaging and particle tracking revealed that the mean axonal velocity of GFP‐TTC particles is significantly reduced by 19% in pmn DRG neurons in comparison to wt DRG neurons (0.79 vs. 0.95 μm/sec, Figure 4C). Histogram analysis of axonal velocities further demonstrated that the maximum velocity of GFP‐TTC particles is lower in pmn (1.2 μm/sec) than in wt (2.0 μm/sec) sensory axons (Figure 4D).
Figure 4.
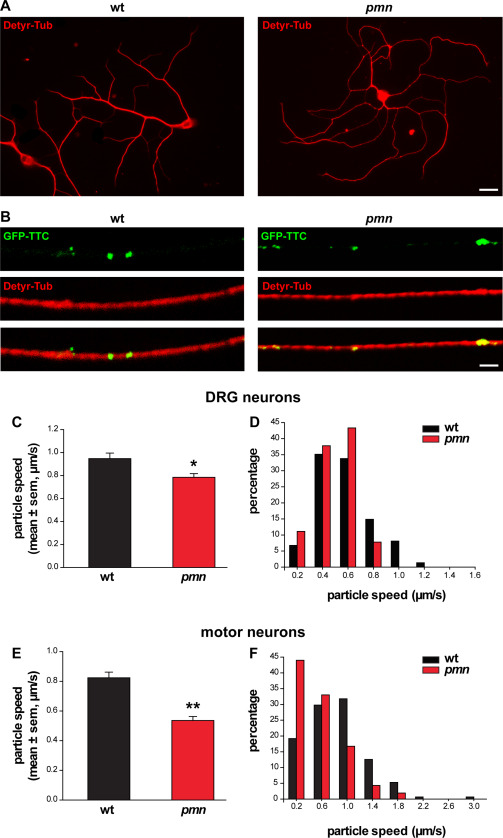
Reduced axonal transport in DRG neurons of pmn mice. A. Images of DRG neurons showing embryonic wt and pmn mice in culture after microtubule labeling with antibodies against detyrosinated tubulin (Detyr‐Tub): Scale: 50 µm. B. Confocal images show neuronal axons after internalization of GFP‐TTC into particles and labeling for detyrosinated tubulin (Detyr‐Tub): Scale: 1 µm. C. Mean velocity of GFP‐TTC axonal transport in DRG neurons [n = 74 (wt), n = 90 (pmn), *P = 0.033 by Mann–Whitney test]. D. Histogram analysis of GFP‐TTC axonal transport velocities reveals a global shift and a reduction of maximal particle velocities in pmn as compared to wt DRG neurons. E. Mean velocity of GFP‐TTC axonal transport in motor neurons [n = 151 (wt), n = 209 (pmn), **P < 0.001 by Mann–Whitney test]. The mean velocity is significantly lower in pmn motor neurons as compared to pmn DRG neurons (C), *P = 0.033 by Mann–Whitney test. F. Histogram analysis of GFP‐TTC axonal transport velocities in wt and pmn motor neurons.
Finally, we analyzed axonal transport in primary motor neurons from pmn and wt mice using GFP‐TTC. We found that the mean particle velocity in pmn motor neurons is reduced by 37% (Figure 4E), that is, to a significantly greater extent than in pmn DRG neurons (Figure 4C). Together, these results show that reduced TBCE function impairs microtubule‐based axonal transport in pmn DRG neurons albeit to a lower extent than in pmn motor neurons.
Discussion
We here report that reduced expression of the tubulin‐specific chaperone TBCE leads to defective microtubule polymerization and impaired microtubule‐dependent axonal transport in sensory DRG neurons of pmn mice. In pmn suralis nerves, the microtubule loss is progressive, affects both large and small caliber sensory axons, and is associated with the occurrence of axonal spheroids, caliber irregularities and myelin ovoids. The prominent sensory neuropathy of pmn mice indicates that the degenerative process is not restricted to motor neurons, similar to the situation in human ALS which is now increasingly recognized as a multisystem disease 68, 75.
These findings are particularly noteworthy as pmn has been initially described as a pure motor neuron disease, characterized by degeneration of motor endplates, axonal dying back of spinal and rubrospinal motor neurons 60 and loss of facial motor neurons 61. Our findings fit with recent studies showing that pmn mice also manifest a progressive hearing loss 79 caused by degeneration of cochlear outer hair cells 51, indicating central sensory symptoms.
How does loss of axonal microtubules cause sensory axon degeneration? We hypothesize that axonal transport defects along microtubules may be involved (for review, 44). It is indeed well known that inhibiting axonal microtubule‐dependent transport, for instance by genetic knockout of the anterograde motor Kinesin Light Chain 1 15 or transgenic disruption of the retrograde motor complex Dynein/Dynactin 36, leads to axonal spheroids similar to those observed in pmn sensory axons. Similarly, Vincristine, a cytostatic drug that causes severe sensory neuropathy in humans 9, leads to disorganization of axonal microtubules, strongly reduces fast axonal transport 38 and impedes axonal growth 64. Vincristine‐treated rats display reduced microtubule densities in myelinated 73 and unmyelinated axons 69 in association with axonal caliber swellings and accumulation of vesicles 56. These defects are however not associated with significant loss of axons 69, 73, similar to the pmn sensory neuropathy. Interestingly however, in pmn suralis axons neither swollen axons nor accumulation of organelles, which might represent a functional pre‐degenerative change of altered axonal transport, were prominent by EM (not shown).
Why are sensory neurons in pmn mice less severely affected than spinal motor neurons? We propose several potential explanations which are not mutually exclusive.
First, the microtubule defects seem to be milder in sensory than in motor axons. We indeed demonstrate that the microtubule densities are reduced by 19% in pmn suralis axons at day 15 which is significantly less than the 60% reduction in distal phrenic motor axons at this age (wt 15.2 ± 2.3 vs. pmn 5.97 ± 1.6 microtubules/μm2 [mean ± sd), see also Schaefer et al 59]. Similarly, the axonal transport defects are significantly less pronounced in cultured DRG neurons, where mean velocities are reduced by 17% (Figure 4C), than in motor neurons, where mean velocities are reduced by 37% (Figure 4E).
Second, microtubule loss may have different consequences depending on the neuronal cell types. Interestingly, the pronounced loss of microtubules in pmn auditory nerves does not give rise to fiber loss or reported signs of degeneration 51. Similarly, a mutation in the Caenorhabditis elegans homologue of the TBCE‐like gene (COEL) which is required for tubulin stability 3 does not cause axonal degeneration 47.
Third, neuronal microtubules are functionally heterogenous. Despite their uniform structure and assembly from α/β‐tubulin dimers, microtubules differ in their tubulin code, that is, their composition of tubulin isoforms expressed by multiple α‐tubulin and β‐tubulin genes and their post‐translational tubulin modifications 33. Interestingly, the lower vulnerability of pmn sensory as compared to pmn motor neurons (this study) is associated with differential expression of several α‐tubulin genes, as shown in a comparison of publicly available gene expression data of DRG neurons and spinal motor neurons (Supporting Information Figure S2). Similarly, defects in post‐translational tubulin modifications that affect microtubule stability have distinct outcomes in different cell types. For example, cortico‐thalamic neurons are preferentially affected in knockout mice for TTL (tubulin tyrosine ligase) where detyrosinated (dynamic) microtubules accumulate 13. Mechanoreceptor neurons are affected in C. elegans mutated in α‐tubulin acetyltransferase (mec‐17), a modification associated with stable microtubules 47.
Fourth, the long peripheral axons of DRG neurons functionally correspond to dendrites and their rate of slow microtubule‐depedent axonal transport differs from the corresponding rate in the central axons of DRG neurons.
Are cytoskeletal defects also involved in sensory neuropathies linked to further motor neuron diseases?
A recent study in mutant SOD1 G93A mice, the most widely studied ALS mouse model, reported increased expression of the Peripherin isoform p56 in small DRG neurons, leading to an accumulation of non‐assembled neurofilaments and a decrease in assembled neurofilaments 58. This may contribute to the degeneration of small DRG neurons and the loss of sensory axon endings in skin 58. Whether cytoskeletal defects also underlie the degeneration of large sensory axons 24, 78 or dorsal root axons 18 remains to be determined.
Defects in the actin cytoskeleton may be involved in the sensory neuropathy of spinal muscular atrophy (SMA). Patients with severe SMA type I indeed display electrophysiological signs of sensory axonopathy 49 and axonal degeneration in suralis nerves including empty myelin sheaths and atrophic axons 55. In mice with severe SMA (Smn‐/‐; Tg SMN2 mice), sensory nerve terminals in the skin were smaller and the reduced growth of sensory axons was associated with a reduction of β‐actin mRNA and protein 32.
Finally, cytoskeletal alterations may also underlie sensory neuropathies in sporadic forms of ALS. Some sporadic forms of ALS have been linked to mutations in the PFN1 gene encoding Profilin‐1, a protein that regulates filamentous F‐actin growth through its binding to monomeric G‐actin 29, 72. Indeed, Profilin‐1 catalyzes the exchange of ADP for ATP on G‐actin which replenishes the pool of polymerization‐competent ATP‐actin, and provides profilin‐actin complexes that join the barbed end of the growing actin filament 12. As Profilin‐1 is ubiquitously expressed 82 including in sensory neurons 2, PFN1 mutations may affect the actin cytoskeleton in sensory axons thereby causing sensory neuropathy. Furthermore, it is conceivable that some of the epigenetic risk factors suspected in sporadic ALS such as exposure to lead, pesticides and agricultural chemicals 34, cigarette smoking 20, 80 or strong electromagnetic fields 30 exert damage to both sensory and motor axons.
Taken together, data from SOD1‐linked and sporadic ALS, SMA and pmn thus suggest that sensory neuropathy involving alterations in cytoskeletal elements such as microtubules, neurofilaments and actin filaments is more common than usually suspected in motor neuron disease.
Supporting information
Additional Supporting Information may be found in the online version of this article at the publisher's web‐site:
Supplemental Figure S1. Motor axon degeneration in pmn mice. Cross sections of distal distal phrenic motor nerves show severe loss of myelinated fibers in 35‐day old pmn mice as compared to wildtype litter mice. Scale: 50 μm.
Supplemental Figure S2. Isoforms of α‐tubulins are differentially expressed between motor neurons and DRG neurons. Histograms showing expression data for α‐tubulin gene isoforms retrieved from the Gene Expression Omnibus (GEO) database of high‐throughput data. Shown are α‐tubulin gene expression values relative to the housekeeping gene RPL13. Similar results were obtained with the housekeeping gene PPIA (cyclophilin A, data not shown). Data for motor neuron subpopulations (LMM: lateral motor neurons, MMM: medial motor neurons, IMN: intermediolateral motor neurons) and DRG neurons were retrieved from the data set record GDS1455 (1) and GDS1634 (2), respectively. Motor neurons (n = 5 animals/subpopulation) and DRG neurons (n = 5 animals) were sampled by laser capture microdissection. Statistical differences between samples were calculated by Mann Whitney test (*P < 0.05; **P < 0.001; ***P < 0.0001).
Acknowledgments
We gratefully acknowledge the expert help of Drs. Alain Bernadac and Pascal Weber (CNRS, Marseille, France) in confocal microscopy and cellular imaging, the help of Drs. Emmanuelle Buhler and Gilbert Baillat in genotyping and the contribution of Prof. Dr. Henning Schmalbruch (University of Copenhagen, Denmark) to an early stage of this work. We are grateful to Dr. Annie Andrieux (INSERM Grenoble, France) for providing essential reagents, Prof. Dr. Benoît Funalot (CHU Henri‐Mondor, Créteil, France) for helpful discussions, and Prof. Dr. Richard Ribchester (University of Edinburgh, UK) for expert advice on the presence of motor fibers in mouse suralis nerves. Work in G. Haase's laboratory was supported by grants from Association Française contre les Myopathies (AFM), Agence Nationale pour la Recherche (ANR), ERANET Neuron, INSERM and CNRS. Work in M. Schaefer's laboratory was supported by Deutsche Forschungsgemeinschaft. Work in J.M. Vallat's laboratory was supported by a grant from the Limoges University hospital and the French Ministry of Health (Department of rare diseases). S. Bellouze was supported by PhD fellowships from Fondation pour la Recherche Médicale (FRM) and AFM. A. Jacquier was supported by PhD fellowships from INSERM. S. Schaller received salary support from ANR.
[Correction added on 18 January 2017, after first online publication: the name of Prof. Dr. Benoît Funalot was accidentally omitted from the Acknowledgments, and has now been added.]
Conflict of interest
The authors declare no conflict of interest
References
- 1. Amin MR, Harris D, Cassel SG, Grimes E, Heiman‐Patterson T (2006) Sensory testing in the assessment of laryngeal sensation in patients with amyotrophic lateral sclerosis. Ann Otol Rhinol Laryngol 115:528–534. [DOI] [PubMed] [Google Scholar]
- 2. Bali KK, Venkataramani V, Satagopam VP, Gupta P, Schneider R, Kuner R (2013) Transcriptional mechanisms underlying sensitization of peripheral sensory neurons by granulocyte‐/granulocyte‐macrophage colony stimulating factors. Mol Pain 9:1744–8069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bartolini F, Tian G, Piehl M, Cassimeris L, Lewis SA, Cowan NJ (2005) Identification of a novel tubulin‐destabilizing protein related to the chaperone cofactor E. J Cell Sci 118:1197–1207. [DOI] [PubMed] [Google Scholar]
- 4. Bellouze S, Schafer MK, Buttigieg D, Baillat G, Rabouille C, Haase G (2014) Golgi fragmentation in pmn mice is due to a defective ARF1/TBCE cross‐talk that coordinates COPI vesicle formation and tubulin polymerization. Hum Mol Genet 23:5961–5975. [DOI] [PubMed] [Google Scholar]
- 5. Bento‐Abreu A, Van Damme P, Van Den Bosch L, Robberecht W (2010) The neurobiology of amyotrophic lateral sclerosis. Eur J Neurosci 31:2247–2265. [DOI] [PubMed] [Google Scholar]
- 6. Bommel H, Xie G, Rossoll W, Wiese S, Jablonka S, Boehm T et al (2002) Missense mutation in the tubulin‐specific chaperone E (Tbce) gene in the mouse mutant progressive motor neuronopathy, a model of human motoneuron disease. J Cell Biol 159:563–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bordet T, Castelnau‐Ptakhine L, Fauchereau F, Friocourt G, Kahn A, Haase G (2001) Neuronal targeting of cardiotrophin‐1 by coupling with tetanus toxin C fragment. Mol Cell Neurosci 17:842–854. [DOI] [PubMed] [Google Scholar]
- 8. Braak H, Brettschneider J, Ludolph AC, Lee VM, Trojanowski JQ, Del Tredici K (2013) Amyotrophic lateral sclerosis–a model of corticofugal axonal spread. Nat Rev Neurol 9:708–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bradley WG, Lassman LP, Pearce GW, Walton JN (1970) The neuromyopathy of vincristine in man. Clinical, electrophysiological and pathological studies. J Neurol Sci 10:107–131. [DOI] [PubMed] [Google Scholar]
- 10. Chio A, Canosa A, Gallo S, Moglia C, Ilardi A, Cammarosano S et al (2012) Pain in amyotrophic lateral sclerosis: a population‐based controlled study. Eur J Neurol 19:551–555. [DOI] [PubMed] [Google Scholar]
- 11. Dalla Bella E, Lombardi R, Porretta‐Serapiglia C, Ciano C, Gellera C, Pensato V et al (2016) Amyotrophic lateral sclerosis causes small fiber pathology. Eur J Neurol 23:416–420. [DOI] [PubMed] [Google Scholar]
- 12. Dominguez R (2009) Actin filament nucleation and elongation factors–structure‐function relationships. Crit Rev Biochem Mol Biol 44:351–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Erck C, Peris L, Andrieux A, Meissirel C, Gruber AD, Vernet M et al (2005) A vital role of tubulin‐tyrosine‐ligase for neuronal organization. Proc Natl Acad Sci USA 102:7853–7858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Fadic R, Vergara J, Alvarez J (1985) Microtubules and caliber of central and peripheral processes of sensory axons. J Comp Neurol 236:258–264. [DOI] [PubMed] [Google Scholar]
- 15. Falzone TL, Stokin GB, Lillo C, Rodrigues EM, Westerman EL, Williams DS et al (2009) Axonal stress kinase activation and tau misbehavior induced by kinesin‐1 transport defects. J Neurosci 29:5758–5767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Feng G, Mellor RH, Bernstein M, Keller‐Peck C, Nguyen QT, Wallace M et al (2000) Imaging neuronal subsets in transgenic mice expressing multiple spectral variants of GFP. Neuron 28:41–51. [DOI] [PubMed] [Google Scholar]
- 17. Ferreirinha F, Quattrini A, Pirozzi M, Valsecchi V, Dina G, Broccoli V et al (2004) Axonal degeneration in paraplegin‐deficient mice is associated with abnormal mitochondria and impairment of axonal transport. J Clin Invest 113:231–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Fischer LR, Culver DG, Davis AA, Tennant P, Wang M, Coleman M et al (2005) The WldS gene modestly prolongs survival in the SOD1G93A fALS mouse. Neurobiol Dis 19:293–300. [DOI] [PubMed] [Google Scholar]
- 19. Gallistel CR, Tucci V, Nolan PM, Schachner M, Jakovcevski I, Kheifets A et al (2014) Cognitive assessment of mice strains heterozygous for cell‐adhesion genes reveals strain‐specific alterations in timing. Philos Trans R Soc Lond B Biol Sci 369:20120464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gallo V, Bueno‐De‐Mesquita HB, Vermeulen R, Andersen PM, Kyrozis A, Linseisen J et al (2009) Smoking and risk for amyotrophic lateral sclerosis: analysis of the EPIC cohort. Ann Neurol 65:378–385. [DOI] [PubMed] [Google Scholar]
- 21. Georgesco M, Salerno A, Camu W (1997) Somatosensory evoked potentials elicited by stimulation of lower‐limb nerves in amyotrophic lateral sclerosis. Electroencephalogr Clin Neurophysiol 104:333–342. [DOI] [PubMed] [Google Scholar]
- 22. Geser F, Martinez‐Lage M, Kwong LK, Lee VM, Trojanowski JQ (2009) Amyotrophic lateral sclerosis, frontotemporal dementia and beyond: the TDP‐43 diseases. J Neurol 256:1205–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gregory R, Mills K, Donaghy M (1993) Progressive sensory nerve dysfunction in amyotrophic lateral sclerosis: a prospective clinical and neurophysiological study. J Neurol 240:309–314. [DOI] [PubMed] [Google Scholar]
- 24. Guo YS, Wu DX, Wu HR, Wu SY, Yang C, Li B et al (2009) Sensory involvement in the SOD1‐G93A mouse model of amyotrophic lateral sclerosis. Exp Mol Med 41:140–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Haase G, Pettmann B, Bordet T, Villa P, Vigne E, Schmalbruch H et al (1999) Therapeutic benefit of ciliary neurotrophic factor in progressive motor neuronopathy depends on the route of delivery. Ann Neurol 45:296–304. [PubMed] [Google Scholar]
- 26. Hammad M, Silva A, Glass J, Sladky JT, Benatar M (2007) Clinical, electrophysiologic, and pathologic evidence for sensory abnormalities in ALS. Neurology 69:2236–2242. [DOI] [PubMed] [Google Scholar]
- 27. Hirano A (1996) Neuropathology of ALS: an overview. Neurology 47:S63–S66. [DOI] [PubMed] [Google Scholar]
- 28. Iglesias C, Sangari S, El Mendili MM, Benali H, Marchand‐Pauvert V, Pradat PF (2015) Electrophysiological and spinal imaging evidences for sensory dysfunction in amyotrophic lateral sclerosis. BMJ Open 5:007659. (2015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ingre C, Landers JE, Rizik N, Volk AE, Akimoto C, Birve A et al (2013) A novel phosphorylation site mutation in profilin 1 revealed in a large screen of US, Nordic, and German amyotrophic lateral sclerosis/frontotemporal dementia cohorts. Neurobiol Aging 34:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ingre C, Roos PM, Piehl F, Kamel F, Fang F (2015) Risk factors for amyotrophic lateral sclerosis. Clin Epidemiol 7:181–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Isaacs JD, Dean AF, Shaw CE, Al‐Chalabi A, Mills KR, Leigh PN (2007) Amyotrophic lateral sclerosis with sensory neuropathy: part of a multisystem disorder? J Neurol Neurosurg Psychiatry 78:750–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Jablonka S, Karle K, Sandner B, Andreassi C, von Au K, Sendtner M (2006) Distinct and overlapping alterations in motor and sensory neurons in a mouse model of spinal muscular atrophy. Hum Mol Genet 15:511–518. [DOI] [PubMed] [Google Scholar]
- 33. Janke C (2014) The tubulin code: molecular components, readout mechanisms, and functions. J Cell Biol 206:461–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kamel F, Umbach DM, Bedlack RS, Richards M, Watson M, Alavanja MC et al (2012) Pesticide exposure and amyotrophic lateral sclerosis. Neurotoxicology 33:457–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kawamura Y, Dyck PJ, Shimono M, Okazaki H, Tateishi J, Doi H (1981) Morphometric comparison of the vulnerability of peripheral motor and sensory neurons in amyotrophic lateral sclerosis. J Neuropathol Exp Neurol 40:667–675. [DOI] [PubMed] [Google Scholar]
- 36. LaMonte BH, Wallace KE, Holloway BA, Shelly SS, Ascano J, Tokito M et al (2002) Disruption of dynein/dynactin inhibits axonal transport in motor neurons causing late‐onset progressive degeneration. Neuron 34:715–727. [DOI] [PubMed] [Google Scholar]
- 37. Langford LA, Schmidt RF (1983) An electron microscopic analysis of the left phrenic nerve in the rat. Anat Rec 205:207–213. [DOI] [PubMed] [Google Scholar]
- 38. LaPointe NE, Morfini G, Brady ST, Feinstein SC, Wilson L, Jordan MA (2013) Effects of eribulin, vincristine, paclitaxel and ixabepilone on fast axonal transport and kinesin‐1 driven microtubule gliding: implications for chemotherapy‐induced peripheral neuropathy. Neurotoxicology 37:231–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Marangi G, Traynor BJ (2015) Genetic causes of amyotrophic lateral sclerosis: new genetic analysis methodologies entailing new opportunities and challenges. Brain Res 14:75–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Martin N, Jaubert J, Gounon P, Salido E, Haase G, Szatanik M et al (2002) A missense mutation in Tbce causes progressive motor neuronopathy in mice. Nat Genet 32:443–447. [DOI] [PubMed] [Google Scholar]
- 41. Matsumoto A, Kawashima A, Doi S, Moriwaka F, Tashiro K (1999) [The spinal somatosensory evoked potentials in amyotrophic lateral sclerosis in relation to the spinal cord conduction velocities]. No to Shinkei 51:41–47. [PubMed] [Google Scholar]
- 42. McCluskey L, Vandriel S, Elman L, Van Deerlin VM, Powers J, Boller A et al (2014) ALS‐Plus syndrome: non‐pyramidal features in a large ALS cohort. J Neurol Sci 345:118–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Mellgren SI, Lindal S (2011) Nerve biopsy–some comments on procedures and indications. Acta Neurol Scand Suppl 191:64–70. [DOI] [PubMed] [Google Scholar]
- 44. Millecamps S, Julien JP (2013) Axonal transport deficits and neurodegenerative diseases. Nat Rev Neurosci 14:161–176. [DOI] [PubMed] [Google Scholar]
- 45. Mulder DW, Bushek W, Spring E, Karnes J, Dyck PJ (1983) Motor neuron disease (ALS): evaluation of detection thresholds of cutaneous sensation. Neurology 33:1625–1627. [DOI] [PubMed] [Google Scholar]
- 46. Nakanishi T, Norris FH (1970) Motor fibers in rat sural nerve. Exp Neurol 26:433–435. [DOI] [PubMed] [Google Scholar]
- 47. Neumann B, Hilliard MA (2014) Loss of MEC‐17 leads to microtubule instability and axonal degeneration. Cell Rep 6:93–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Nithianantham S, Le S, Seto E, Jia W, Leary J, Corbett KD et al (2015) Tubulin cofactors and Arl2 are cage‐like chaperones that regulate the soluble alphabeta‐tubulin pool for microtubule dynamics. Elife 24:e08811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Omran H, Ketelsen UP, Heinen F, Sauer M, Rudnik‐Schoneborn S, Wirth B et al (1998) Axonal neuropathy and predominance of type II myofibers in infantile spinal muscular atrophy. J Child Neurol 13:327–331. [DOI] [PubMed] [Google Scholar]
- 50. Pugdahl K, Fuglsang‐Frederiksen A, de Carvalho M, Johnsen B, Fawcett PR, Labarre‐Vila A et al (2007) Generalised sensory system abnormalities in amyotrophic lateral sclerosis: a European multicentre study. J Neurol Neurosurg Psychiatry 78:746–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Rak K, Frenz S, Radeloff A, Groh J, Jablonka S, Martini R et al (2013) Mutation of the TBCE gene causes disturbance of microtubules in the auditory nerve and cochlear outer hair cell degeneration accompanied by progressive hearing loss in the pmn/pmn mouse. Exp Neurol 250:333–340. [DOI] [PubMed] [Google Scholar]
- 52. Raoul C, Buhler E, Sadeghi C, Jacquier A, Aebischer P, Pettmann B et al (2006) Chronic activation in presymptomatic amyotrophic lateral sclerosis (ALS) mice of a feedback loop involving Fas, Daxx, and FasL. Proc Natl Acad Sci USA 103:6007–6012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ribchester RR, Thomson D, Wood NI, Hinks T, Gillingwater TH, Wishart TM et al (2004) Progressive abnormalities in skeletal muscle and neuromuscular junctions of transgenic mice expressing the Huntington's disease mutation. Eur J Neurosci 20:3092–3114. [DOI] [PubMed] [Google Scholar]
- 54. Roux S, Colasante C, Saint Cloment C, Barbier J, Curie T, Girard E et al (2005) Internalization of a GFP‐tetanus toxin C‐terminal fragment fusion protein at mature mouse neuromuscular junctions. Mol Cell Neurosci 30:572–582. [PubMed] [Google Scholar]
- 55. Rudnik‐Schöneborn S, Goebel HH, Schlote W, Molaian S, Omran H, Ketelsen U et al (2003) Classical infantile spinal muscular atrophy with SMN deficiency causes sensory neuronopathy. Neurology 60:983–987. [DOI] [PubMed] [Google Scholar]
- 56. Sahenk Z, Brady ST, Mendell JR (1987) Studies on the pathogenesis of vincristine‐induced neuropathy. Muscle Nerve 10:80–84. [DOI] [PubMed] [Google Scholar]
- 57. Sanders AA, Kaverina I (2015) Nucleation and Dynamics of Golgi‐derived Microtubules. Front Neurosci 9:431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Sassone J, Taiana M, Lombardi R, Porretta‐Serapiglia C, Freschi M, Bonanno S et al (2016) ALS mouse model SOD1G93A displays early pathology of sensory small fibers associated to accumulation of a neurotoxic splice variant of peripherin. Hum Mol Genet 25:1588–1599. [DOI] [PubMed] [Google Scholar]
- 59. Schäfer MKE, Schmalbruch H, Buhler E, Lopez C, Martin N, Guenet J‐L et al (2007) Progressive Motor Neuronopathy: A Critical Role of the Tubulin Chaperone TBCE in Axonal Tubulin Routing from the Golgi Apparatus. J Neurosci 27:8779–8789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Schmalbruch H, Jensen HJ, Bjaerg M, Kamieniecka Z, Kurland L (1991) A new mouse mutant with progressive motor neuronopathy. J Neuropathol Exp Neurol 50:192–204. [DOI] [PubMed] [Google Scholar]
- 61. Sendtner M, Schmalbruch H, Stockli KA, Carroll P, Kreutzberg GW, Thoenen H (1992) Ciliary neurotrophic factor prevents degeneration of motor neurons in mouse mutant progressive motor neuronopathy. Nature 358:502–504. [DOI] [PubMed] [Google Scholar]
- 62. Shefner JM, Tyler HR, Krarup C (1991) Abnormalities in the sensory action potential in patients with amyotrophic lateral sclerosis. Muscle Nerve 14:1242–1246. [DOI] [PubMed] [Google Scholar]
- 63. Sheng L, Ma H, Zhong J, Shang H, Shi H, Pan P (2015) Motor and extra‐motor gray matter atrophy in amyotrophic lateral sclerosis: quantitative meta‐analyses of voxel‐based morphometry studies. Neurobiol Aging 36:3288–3299. [DOI] [PubMed] [Google Scholar]
- 64. Silva A, Wang Q, Wang M, Ravula SK, Glass JD (2006) Evidence for direct axonal toxicity in vincristine neuropathy. J Peripher Nerv Syst 11:211–216. [DOI] [PubMed] [Google Scholar]
- 65. Smith BN, Ticozzi N, Fallini C, Gkazi AS, Topp S, Kenna KP et al (2014) Exome‐wide rare variant analysis identifies TUBA4A mutations associated with familial ALS. Neuron 84:324–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Stiess M, Maghelli N, Kapitein LC, Gomis‐Ruth S, Wilsch‐Brauninger M, Hoogenraad CC et al (2010) Axon extension occurs independently of centrosomal microtubule nucleation. Science 327:704–707. [DOI] [PubMed] [Google Scholar]
- 67. Stokin GB, Lillo C, Falzone TL, Brusch RG, Rockenstein E, Mount SL et al (2005) Axonopathy and transport deficits early in the pathogenesis of Alzheimer's disease. Science 307:1282–1288. [DOI] [PubMed] [Google Scholar]
- 68. Swinnen B, Robberecht W (2014) The phenotypic variability of amyotrophic lateral sclerosis. Nat Rev Neurol 10:661–670. [DOI] [PubMed] [Google Scholar]
- 69. Tanner KD, Levine JD, Topp KS (1998) Microtubule disorientation and axonal swelling in unmyelinated sensory axons during vincristine‐induced painful neuropathy in rat. J Comp Neurol 395:481–492. [PubMed] [Google Scholar]
- 70. Taxt T (1983) Cross‐innervation of fast and slow‐twitch muscles by motor axons of the sural nerve in the mouse. Acta Physiol Scand 117:331–341. [DOI] [PubMed] [Google Scholar]
- 71. Tian G, Huang MC, Parvari R, Diaz GA, Cowan NJ (2006) Cryptic out‐of‐frame translational initiation of TBCE rescues tubulin formation in compound heterozygous HRD. Proc Natl Acad Sci USA 103:13491–13496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Tiloca C, Ticozzi N, Pensato V, Corrado L, Del Bo R, Bertolin C et al (2013) Screening of the PFN1 gene in sporadic amyotrophic lateral sclerosis and in frontotemporal dementia. Neurobiol Aging 34:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Topp KS, Tanner KD, Levine JD (2000) Damage to the cytoskeleton of large diameter sensory neurons and myelinated axons in vincristine‐induced painful peripheral neuropathy in the rat. J Comp Neurol 424:563–576. [PubMed] [Google Scholar]
- 74. Trojanowski JQ, Lee VM, Schlaepfer WW (1984) Neurofilament breakdown products in degenerating rat and human peripheral nerves. Ann Neurol 16:349–355. [DOI] [PubMed] [Google Scholar]
- 75. Turner MR, Swash M (2015) The expanding syndrome of amyotrophic lateral sclerosis: a clinical and molecular odyssey. J Neurol Neurosurg Psychiatry 86:667–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. van der Graaff MM, de Jong JM, Baas F, de Visser M (2009) Upper motor neuron and extra‐motor neuron involvement in amyotrophic lateral sclerosis: a clinical and brain imaging review. Neuromuscul Disord 19:53–58. [DOI] [PubMed] [Google Scholar]
- 77. Varon S, Skaper SD, Manthorpe M (1981) Trophic activities for dorsal root and sympathetic ganglionic neurons in media conditioned by Schwann and other peripheral cells. Brain Res 227:73–87. [DOI] [PubMed] [Google Scholar]
- 78. Vaughan SK, Kemp Z, Hatzipetros T, Vieira F, Valdez G (2015) Degeneration of proprioceptive sensory nerve endings in mice harboring amyotrophic lateral sclerosis‐causing mutations. J Comp Neurol 523:2477–2494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Volkenstein S, Brors D, Hansen S, Berend A, Mlynski R, Aletsee C et al (2009) Auditory development in progressive motor neuronopathy mouse mutants. Neurosci Lett 465:45–49. [DOI] [PubMed] [Google Scholar]
- 80. Wang H, O'Reilly EJ, Weisskopf MG, Logroscino G, McCullough ML, Thun MJ et al (2011) Smoking and risk of amyotrophic lateral sclerosis: a pooled analysis of 5 prospective cohorts. Arch Neurol 68:207–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Weis J, Katona I, Muller‐Newen G, Sommer C, Necula G, Hendrich C et al (2011) Small‐fiber neuropathy in patients with ALS. Neurology 76:2024–2029. [DOI] [PubMed] [Google Scholar]
- 82. Witke W, Sutherland JD, Sharpe A, Arai M, Kwiatkowski DJ (2001) Profilin I is essential for cell survival and cell division in early mouse development. Proc Natl Acad Sci USA 98:3832–3836. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting Information may be found in the online version of this article at the publisher's web‐site:
Supplemental Figure S1. Motor axon degeneration in pmn mice. Cross sections of distal distal phrenic motor nerves show severe loss of myelinated fibers in 35‐day old pmn mice as compared to wildtype litter mice. Scale: 50 μm.
Supplemental Figure S2. Isoforms of α‐tubulins are differentially expressed between motor neurons and DRG neurons. Histograms showing expression data for α‐tubulin gene isoforms retrieved from the Gene Expression Omnibus (GEO) database of high‐throughput data. Shown are α‐tubulin gene expression values relative to the housekeeping gene RPL13. Similar results were obtained with the housekeeping gene PPIA (cyclophilin A, data not shown). Data for motor neuron subpopulations (LMM: lateral motor neurons, MMM: medial motor neurons, IMN: intermediolateral motor neurons) and DRG neurons were retrieved from the data set record GDS1455 (1) and GDS1634 (2), respectively. Motor neurons (n = 5 animals/subpopulation) and DRG neurons (n = 5 animals) were sampled by laser capture microdissection. Statistical differences between samples were calculated by Mann Whitney test (*P < 0.05; **P < 0.001; ***P < 0.0001).