

Abstract
Calsenilin is a calcium sensor protein that interacts with presenilin and increases calcium‐triggered neuronal apoptosis, and γ‐secretase activity. Notch is a cell surface receptor that regulates cell‐fate decisions and synaptic plasticity in brain. The aim of the present study was to characterize the role of calsenilin as a regulator of the γ‐secretase cleavage of Notch in ischemic stroke. Here, we determined the modulation of expression level and cellular distribution of calsenilin in neurons subjected to ischemic‐like conditions. The levels of calsenilin and presenilin were increased in primary neurons after oxygen and glucose deprivation. Furthermore, calsenilin was found to enhance the γ‐secretase cleavage of Notch and to contribute to cell death under ischemia‐like conditions. The inhibition of γ‐secretase activity and a presenilin deficiency were both found to protect against calsenilin‐mediated ischemic neuronal death. The expression of calsenilin was found to be increased in brain following experimental ischemic stroke. These findings establish a specific molecular mechanism by which the induction of calsenilin enhances Notch activation in ischemic stroke, and identify calsenilin as an upstream of the γ‐secretase cleavage of Notch.
Keywords: calsenilin, ischemic stroke, neuronal cell death, Notch, γ‐secretase
Introduction
γ‐Secretase is an intramembrane multi‐protein complex that cleaves many type‐I proteins. Some of these proteins are relevant in the brain, such as the N‐ and E‐cadherins, that participate in neuronal development and synapse formation 21. Furthermore, the cleavage of amyloid precursor protein (APP) by γ‐secretase contributes to the generation of amyloid‐β peptide in Alzheimer's disease 10, 12, 15. Moreover, Notch‐1 is cleaved by γ‐secretase to form Notch intracellular domain (NICD), a transcription factor responsible for cell‐fate determination in the developing nervous system 23. γ‐Secretase is composed of four integral membrane proteins, including presenilin (PS) as a catalytic subunit. PS has two isoforms, PS‐1 and PS‐2, that have equivalent catalytic activities within γ‐secretase complex 27, and both isoforms are located in the plasma membrane and in the membranes of the endoplasmic reticulum (ER) and Golgi 4, 13.
Calsenilin is a neuronal Ca2+‐binding protein and a member of the recoverin family, and is distinguished from other family members by the presence of a long N‐terminal domain 29. Calsenilin has been independently described as a binding partner for PS 5, as a gene transcription repressor termed “downstream regulatory element antagonist modulator” 6, and as a potassium channel modulator 1. Although not necessary for γ‐secretase activity, calsenilin binds PS‐C‐terminal fragments (CTF), but its modulation of PS activity seems to be substrate‐dependent. Calsenilin was shown to increase γ‐secretase cleavage of APP in the presence of Ca2+ 16, and this effect of calsenilin was completely abrogated by a γ‐secretase inhibitor. We previously reported that aberrant Notch signaling induced by cerebral ischemia worsens brain damage and functional outcome. In these previous studies, we found that brain damage and post‐ischemic inflammation were significantly attenuated in Notch antisense mice and in normal mice treated with γ‐secretase 2, 26. However, the role of calsenilin as a regulator of γ‐secretase cleavage of Notch in ischemic stroke has not been determined.
Here we show that calsenilin contributes to γ‐secretase‐mediated Notch signaling and to the activations of downstream apoptotic proteins under ischemic stroke conditions. In addition, we found that focal ischemia increases calsenilin levels in the brain, and that these increases are correlated with cell death. Finally, we examined the effect of γ‐secretase inhibition on the activation of apoptotic proteins and on the over‐expression of calsenilin.
Materials and Methods
Middle cerebral artery occlusion (MCAO)
Three‐month‐old C57BL/6 male mice were used for in vivo experiments. The focal cerebral ischemia/reperfusion was induced by MCAO as described previously 2. MCAO was induced by the intraluminal filament technique. In brief, a 6‐0 nylon filament was inserted through the external carotid artery stump and advanced into the right internal carotid artery until it blocked the origin of the middle cerebral artery. After 60 min of MCAO, the filament was withdrawn to restore blood flow. Sham‐operated mice underwent the same surgical procedure without filament insertion. Rectal temperature was monitored throughout the entire duration of the surgical procedure and maintained at 37.5°C with a feedback‐controlled heating pad. These procedures were approved by the University of Queensland Animal Care and Use Committee.
Transient global cerebral ischemia in rats
Animal experiments were performed in accordance with the Guide of Ulsan University College of Medicine for Care and Use of Laboratory Animals. Under halothane anaesthesia, common carotid arteries of male Sprague–Dawley rats weighing 300–350 g were bilaterally occluded with vascular clamps and mean arterial blood pressure was lowered to 50 ± 5 mmHg by withdrawing blood from the femoral artery. After 12 min, the blood perfusion was recovered by removing clips and reinfusion of blood. During and for 2 h after the ischemic surgery, body and brain temperatures were maintained at 37.5°C using a heating blanket and lamp.
Sample processing for brain sections
Brains were fixed by transcardial perfusion with saline followed by perfusion and immersion in 4% paraformaldehyde (PFA) in phosphate‐buffered saline (PBS) and immersed into a sucrose gradient from 10 to 30% for cryopreservation. Serial coronal sections (10‐μm thickness) were cut with a freezing microtome (Leica 1900, Wetzlar, Germany) and collected on slides.
Tissue preparation and cresyl violet stain
At 24, 48 and 72 h after ischemia, brains were removed and quickly frozen in liquid nitrogen. Coronal brain sections containing hippocampus were prepared with cryostat (10‐μm thickness) and put onto the slide glass pre‐coated with poly‐L‐lysine. To identify neuronal death, the brain sections were stained with 0.5% cresyl violet.
Immunohistochemistry
To verify immunocytochemically the expression of calsenilin in brain, sections were exposed to an anti‐calsenilin (Upstate, Lake Placid, NY, USA) antibody. Briefly, after incubating in 0.2% H2O2 for 10 min, brain sections were blocked with 3% goat serum containing 0.3% Triton X‐100 and incubated overnight with anti‐rabbit calsenilin polyclonal antibody diluted at 1:100. After reaction with biotinylated secondary antibody (goat anti‐rabbit immunoglobulin G; Vector Labs, Burlingame, CA, USA), sections were treated with avidin‐horseradish peroxide solution and developed by a nickel‐enriched reaction with diaminobenzidine and hydrogen peroxide. Anti‐calsenilin immunopositive cells in the hippocampus were examined under a light microscope (Olympus, Tokyo, Japan) and photographed with a digital camera (Camedia C2000; Olympus). Calsenilin co‐localization with astrocytes, microglia and endothelium in the mouse stroke brain was performed in 10‐μm cryosections using the following antibodies: rabbit anti‐calsenilin (Abcam), mouse anti‐glial fibrillary acidic protein (GFAP) (Sigma, St. Louis, MO, USA), goat anti‐Iba1 (Abcam, Cambridge, UK) and sheep anti‐Von Willebrand Factor (Abcam). Images were examined under a confocal BX‐61 Olympus microscope.
Hippocampal and cortical neuron primary cultures
Dissociated cell cultures of neocortical fragments and hippocampi were prepared from 16‐day C57BL/6 mouse embryos as described previously 22. Cells were plated in 35‐ and 60‐mm diameter plastic dishes or 96‐well plates and maintained at 37°C in neurobasal medium containing 25 mM glucose and B‐27 supplements (Invitrogen, Carlsbad, CA, USA), 2 mM L‐glutamine, 0.001% gentamicin sulfate, and 1 mM 4‐(2‐hydroxyethyl)piperazine‐1‐ethanesulfonic acid (HEPES) at pH 7.2. Approximately, 95% of the cells in such cultures were neurons and the remaining cells were astrocytes. Caspase inhibitors, 20 μM Z‐Val‐Ala‐Asp (VAD)‐FMK (carbobenzoxy‐valyl‐alanyl‐aspartyl‐[O‐methyl]‐fluoromethylketone; Enzyme System Products, Livermore, CA, USA) and 10 μM Asp‐Glu‐Val‐Asp (DEVD)‐Asp‐Glu‐ Val‐Asp (CHO) (Calbiochem, Billerica, MA, USA) was added into culture medium 2 h before hypoxia.
Plasmid DNA and siRNA
To construct plasmid expressing calsenilin (calsenilin‐pcDNA3.1) or calsenilin‐enhanced green fluorescence protein fusion construct (calsenilin‐pEGFP), the entire coding region of calsenilin was sub‐cloned into pcDNA3.1 (Invitrogen) or pEGFP‐N1 (Clontech Mountain View, CA, USA) 18. Plasmid DNA was extracted using the Hybrid‐Q plasmid (GeneAll, Seoul, KOREA). Calsenilin siRNA was purchased from Santa Cruz (sc‐42399; Santa Cruz, CA, USA).
Cell culture and transfection
HT‐22 (mouse hippocampal) and mouse embryonic fibroblast (MEF) 15 cells were cultured in Dulbecco's modified Eagle's medium containing 10% fetal bovine serum supplemented with 1% penicillin and streptomycin. Cells were maintained at 37°C in a 5% CO2 atmosphere. For transfections into HT‐22 and MEF cells, cells were plated in 35‐mm dish or 96‐well plate and plasmid DNA and siRNA were transfected using Lipofectamine 2000 (Invitrogen) according to the manufacturer's protocol and then incubated with complete medium for 24 h.
For transfection into primary cortical neurons, cells were plated in 96‐well pates and plasmid DNA was transfected using NeuroFECT (Genlantis, San Diego, CA, USA) according to the manufacturer's protocol and then incubated with neurobasal medium for 24 h.
Oxygen glucose deprivation (OGD)
OGD was performed using glucose‐free Locke's medium consisting of (in mM): 154 NaCl, 5.6 KCl, 2.3 CaCl2, 1 MgCl2, 3.6 NaHCO3 and5 HEPES at pH 7.2 and culture plates were placed in a hypoxic chamber (Stemcell, San Diego, CA, USA). The chamber was flushed with 95% N2 and 5% CO2 for 10 min, sealed and placed in a 37°C incubator for the appropriate duration.
Cytoplasmic and nuclear extraction
Cytoplasmic and nuclear extracts were obtained according to NE‐PER nuclear and cytoplasmic extraction reagents kit (Thermo, Waltham, MA, USA). Briefly, the isolation of nuclear and cytoplasmic fractions using the NE‐PER kit maintains the integrity of the two cellular compartments before extraction. The nuclear extracts were separated from the cytoplasmic extracts by centrifugation at 4°C at 12 000 rpm for 10 min. Supernatants were collected as cytoplasmic extracts. The protein was calculated according to the bicinchoninic acid (BCA) protein assay kit (Thermo).
Immunoblotting
Protein concentrations were determined using the BCA protein assay (Thermo); 20 μg of total protein was separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS/PAGE) and transferred onto polyvinylidene difluoride membranes (PVD) (Millipore, Billerica, MA, USA). Blots were incubated in blocking buffer (5% non‐fat milk) for 1 h at a room temperature. Then the blots were incubated with primary antibodies to NICD (Abcam), calsenilin (Upstate), PS 1 (Chemicon, Temecule, CA, USA), p65 (Cell Signaling, Danvers, MA, USA), p‐p65 (Cell Signaling), Bax (Cell Signaling), Bim (Santa Cruz), cleaved caspase (Cell Signaling), procaspase‐3 (Cell Signaling), Poly [ADP‐ribose] polymerase‐1 (PARP‐1, cell Signaling) α‐tubulin (Epitomics, Burlingame, CA, USA) and β‐actin (Sigma) overnight at 4°C. Protein bands were detected using a secondary antibody conjugated with horseradish peroxidase (Jackson Immunoresearch, Baltimore, PA, USA) and a chemiluminescence detection system (Pierce, Pockford, IL, USA).
Immunofluorescence
Neuronal cells seeded on cover slips were fixed in 4% PFA for 10 min, permeabilized with 0.1% Triton X‐100 in PBS for 5 min. Blocking was performed by incubation in 5% normal serum for 1 h, which the cells were incubated with primary antibodies to NICD (Abcam), mouse calsenilin (Upstate) and microtubule associated protein 2 (MAP‐2; Abcam) overnight at 4°C and then with Alexa 488‐conjugated or Alexa 568‐conjugated secondary antibodies (Molecular Probes, Karlsruhe, Germany) for 1 h at room temperature. Nuclei of immunolabeled specimens were visualized with 4',6‐diamidino‐2‐phenylindol (DAPI) (Molecular Probes). Images were examined under conforcal microscope (LSM510, Carl Zeiss, Göttingen, Germany).
Cell viability assay
To perform quantification of cell viability in neuronal cells, the CellTiter‐Glo luminescent cell viability assay (Promega, Madison, WI, USA) was used following the manufacturer's instructions. Neuronal cells were seeded in 96‐well plates for luminescence measurements (Berthold Detection Systems, Huntsvile, AL, USA).
pEGFP or Calsenilin‐pEGFP transfected HT‐22 cells were stained with propidium iodide (PI) to differentiate apoptotic and normal cells. The transfected HT‐22 cells were rinsed with PBS and fixed with 4% PFA. The fixed cells were stained for 5 min in 500 nM of PI (Sigma) at room temperature.
Statistical analysis
Statistical analysis was performed using one‐way analysis of variance followed by Newman–Keuls post hoc test using GraphPad Prism, version 5.0 (San Diego, CA, USA). Statistical significance was accepted at the 95% confidence level (P < 0.05).
Results
Calsenilin is expressed predominantly in the adult, but not embryonic, mouse brain
To verify results reported by Zaidi et al 29, expression levels of calsenilin were compared in mouse tissues at different stages of development using an antibody that recognizes the non‐phosphorylated form of this protein around the phosphorylation site at serine 63 (Figure 1A). Absence of calsenilin in the embryonic brain and increased expressions of multiple isoforms at postnatal day 3 confirmed previous results 29. We observed calsenilin expression in embryonic and postnatal hearts and lungs, but in adults, although calsenilin remained strongly expressed in brain, no traces were found in any other tissues studied. Furthermore, when regional localization within the brain was assessed, calsenilin was found to be most highly expressed in the cortex, hippocampus and cerebellum, which is compatible with its functional relevance in these tissues.
Figure 1.
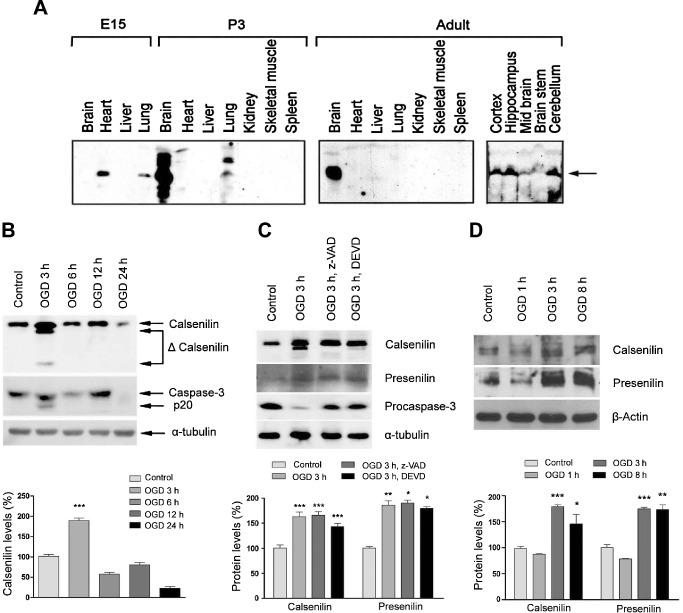
Calsenilin expression was increased by a combination of hypoxia and oxygen glucose deprivation (OGD). A. At embryonic day 15 (E15) calsenilin was present in heart and lung, but not in brain, but at only 3 days after birth (P3) calsenilin was expressed predominantly in the mouse brain (left panel). Only the mouse adult brain expressed calsenilin (middle panel), which was present in all brain regions, but particularly in cortex, hippocampus and cerebellum (right panel). B. Immunoblot of primary hippocampal neuron lysates subjected to OGD in vitro. A strong increase in total and cleaved calsenilin levels was seen 3‐h post‐OGD and at the same time, cleaved caspase‐3 was observed. Quantification of calsenilin levels normalized to α‐tubulin levels as a loading control. Values are the mean with standard error of measurement (SEM). ***P < 0.001 vs. control. C. OGD‐induced cleavage of calsenilin was prevented in the presence of the caspase inhibitors z‐VAD (Val‐Ala‐Asp) or DEVD (Asp‐Glu‐Val‐Asp). Whereas levels of calsenilin increased under OGD conditions, the cleaved form of calsenilin was absent when caspase inhibitors were present. In addition, levels of procaspase‐3, which reduce due to cleavage after OGD, were maintained in the presence of inhibitors. Quantification of casenilin and presenilin levels normalized to α‐tubulin levels as a loading control. Values are the mean with SEM. *P < 0.05, **P < 0.01, ***P < 0.001 vs. control. D. Immunoblot of primary cortical neuron lysates subjected to OGD. Quantification of casenilin and presenilin levels normalized to β‐action levels as a loading control. Values are the mean with SEM. *P < 0.05, **P < 0.01, ***P < 0.001 vs. control.
Neuronal levels of calsenilin were up‐regulated by OGD, and its cleavage during OGD was mediated by caspase‐3
Calsenilin is a pro‐apoptotic protein expressed in neurons, and has been reported to exert its effects under conditions of high Ca2+ concentrations 7 or serum deprivation 20. In the present study, calsenilin levels were up‐regulated in primary hippocampal neurons after only 3 h of OGD, and its cleaved form was apparent (Figure 1B). The active (p20) form of caspase‐3, which is able to cleave calsenilin 8, was also up‐regulated after 3 h of OGD. To verify that calsenilin was cleaved by caspase‐3, cells were subjected to OGD and treated with the caspase inhibitors, z‐VAD or DEVD (Figure 1C). The inhibition of caspase‐3 cleavage, as evidenced by stable levels of procaspase‐3 in treated cells, did not affect levels of full‐length calsenilin protein, but it prevented the appearance of the cleaved, smaller protein product. The up‐regulation of calsenilin in cultured primary cortical neurons during OGD conditions, another in vitro model of ischemic stroke, was confirmed by immunoblotting (Figure 1D). Furthermore, level of PS induced by OGD conditions in cultured primary hippocampal and cortical neurons (Figure 1C,D).
To study the distribution of calsenilin in primary cortical neurons subjected to ischemia‐like conditions, cultures were subjected to GD for up to 24 h and neurons were stained with MAP‐2 (neuronal marker, red) and calsenilin (green) antibodies (Figure 2A). Calsenilin signals in control cells and in the early stages of GD appeared dispersed throughout the cell. However, after 24 h of GD, when most cells are undergoing apoptosis, signals accumulated around the nuclear region of the cells.
Figure 2.
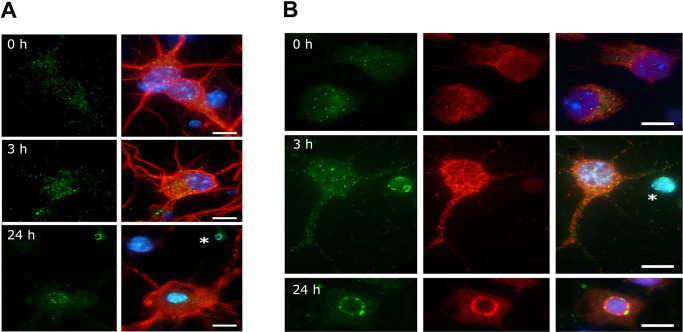
The distribution of calsenilin and presenilin (PS) 1 in primary cortical neurons subjected to ischemia‐like conditions. A. Calsenilin immunofluorescence in mouse primary cortical neurons, showing calsenilin (green), the neuronal marker MAP2 (red) and 4’,6‐diamidino‐2‐phenylindol (DAPI) (blue). Calsenilin staining showed a small, non‐significant increase after 3 h of glucose deprivation (GD). At 24 h, calsenilin congregated at nuclear peripheries. Staining was prominent around apoptotic bodies (asterisk). B. Calsenilin (green) and PS1 (red) immunofluorescence in mouse primary cortical neurons, both showing a small increase with GD. Calsenilin, but not PS1, congregated around apoptotic bodies (GD for 3 h, asterisk). At 24 h, calsenilin and PS1 signals co‐localized around nucleus. Scale bar: 10 μm.
To determine whether calsenilin and PS‐1 co‐localized in situ, primary cortical neurons subjected to GD were labeled with calsenilin (green) and PS‐1 (red) antibodies (Figure 2B). No obvious co‐localization was found during the early stages of GD, but at 24 h strong, the perinuclear calsenilin and PS‐1 staining was observed in many cells. These results suggest the up‐regulated clasenilin under ischemic condition increase γ‐secretase activity through interaction with PS‐1.
Calsenilin overexpression enhanced Notch‐1 cleavage and signals downstream of Notch‐1
We previously reported that γ‐secretase‐mediated Notch signaling induces neuronal cell death in ischemic stroke 2, 3. To test the hypothesis that calsenilin enhances the γ‐secretase cleavage of Notch‐1, HT‐22 cells were transfected with calsenilin and then assessed by immunofluorescence and immunoblotting for NICD levels. Figure 3 shows that overexpression of calsenilin increased the NICD signal (red) after immunostaining with a specific antibody for NICD that does not bind to full length Notch‐1. When these cells were subjected to OGD for 6 h, NICD staining increased even more dramatically, whereas transfection with vector only did not increase the NICD signal in OGD condition (Figure 3A). These results were confirmed by immunoblotting, where overexpression of calsenilin (verified in the top panel) increased the levels of NICD, and this increase was even more dramatic after 6 h of OGD (Figure 3B,C). NICD has been shown to increase levels of the NF‐κB activated subunit, phospho‐p65, in neurons in response to ischemia‐like conditions 26, and consistently, the overexpression of calsenilin contributes to an increase in phospho‐p65 under both normal and OGD conditions (Figure 3B,C).
Figure 3.
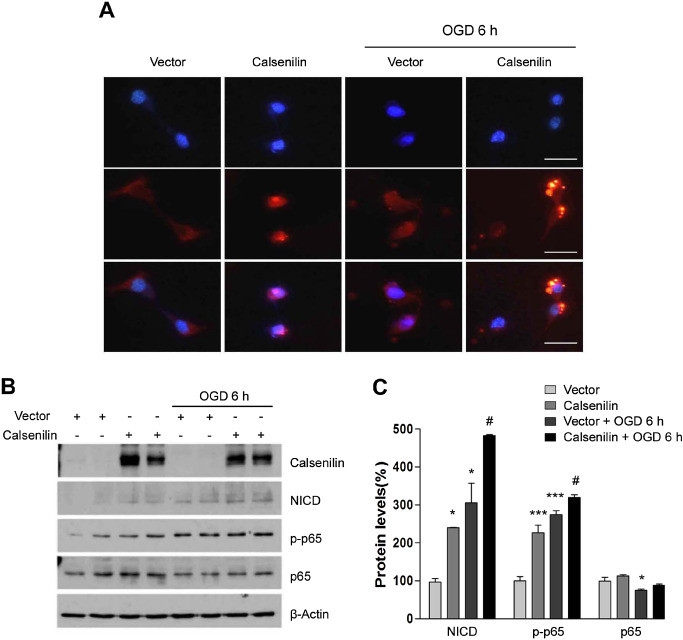
Calsenilin affected Notch cleavage in HT‐22 cells subjected to oxygen glucose deprivation (OGD). A. The overexpression of calsenilin in standard conditions (left panels) increased Notch intracellular domain (NICD) (red) levels. Under OGD (right panels), calsenilin overexpression dramatically increased NICD (red) levels. Original magnification: 1000×. Scale bar: 50 μm. B. The overexpression of calsenilin potentiated OGD‐induced cell death. HT‐22 cells were transfected with either vector or calsenilin constructs. Calsenilin overexpression increased NICD, and the phosphorylation of p65 (p‐p65) in cultured cells. After 6 h of OGD, NICD and phospho‐p65 levels increased further. C. Quantification of NICD, p‐p65 and p65 levels normalized to β‐actin levels as a loading control. Values are the mean with standard error of measurement. *P < 0.05, ***P < 0.001 vs. vector, # P < 0.05 vs. vector + OGD 6 h.
Calsenilin‐regulated apoptotic neuronal death under ischemia‐like conditions
Calsenilin is a pro‐apoptotic protein, and has been shown to increase cell death in response to the toxic Alzheimer's disease‐related by‐product, amyloid‐β 18. We found that calsenilin also increases cell death under ischemia‐like conditions, in that calsenilin‐transfected primary cortical neurons and HT‐22 cells subjected to OGD showed a 40% higher cell death rate than control cells (Figure 4A,B). Furthermore, treatment with DAPT (N‐[N‐(3,5‐Difluorophenacetyl)‐L‐alanyl]‐S‐phenylglycine t‐butyl ester) significantly decreased cell death, indicating that γ‐secretase, at least in part, mediates this effect of calsenilin. These results were also confirmed by PI staining. Cell death was assessed by PI staining in the GFP‐positive cells. Calsenlin‐GFP positive HT‐22 cells showed significantly more PI staining by OGD, whereas treatment with DAPT reduced co‐localization of PI and calsenilin‐GFP (Figure 4C). Immunoblotting with specific antibodies showed that the pathways activated by calsenilin under OGD conditions involve the up‐regulations of the pro‐apoptotic proteins Bim, Bcl‐2–associated X protein (Bax) and the cleavage of caspase‐3 (Figure 4D,E). None of these pathways appeared to be affected by γ‐secretase inhibition by DAPT, indicating the complexity of the interaction between calsenilin, γ‐secretase and cell death pathways (Figure 4D,E).
Figure 4.
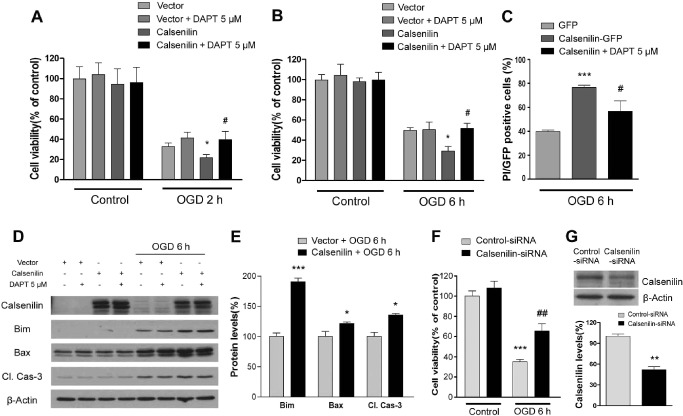
Calsenilin regulated oxygen glucose deprivation (OGD)‐induced neuronal cell death and the levels of associated signaling proteins. A. Primary cortical neurons were transfected with either vector or calsenilin constructs. Transfected primary cortical neurons pretreated with 5 μM DAPT for 12 h. Primary cortical neurons were exposed to 2 h of OGD. The overexpression of calsenilin increased OGD‐induced cell death as determined by luminescent cell viability assay. Values are the mean with standard error of measurement (SEM). (n = 3). *P < 0.05 vs. vector + OGD 2 h, # P < 0.05 vs. calsenilin + OGD 2 h. B. Overexpressed HT‐22 cells were exposed to 6 h of OGD. The overexpression of calsenilin significantly increased cell death caused by OGD, but the γ‐secretase inhibitor DAPT reduced this cell death. Values are the mean with SEM. (n = 3). *P < 0.05 vs. vector + OGD 6 h, # P < 0.05 vs. calsenilin + OGD 6 h. C. Only green fluorescence protein (GFP)‐positive dead cells were counted and cell death measured by propidium iodide (PI) staining. Values are the mean with SEM. (n = 4). ***P < 0.001 vs. GFP, # P < 0.05 calsenilin‐GFP. D. Immunoblot showing the levels of proteins involved in OGD‐induced neuronal death. The top panel confirms calsenilin overexpression. The pro‐apoptotic proteins Bim, Bax, and cleaved caspase‐3, whose levels were increased by OGD, were further increased by calsenilin overexpression. Whereas, these changes were independent of γ‐secretase inhibition by DAPT. E. Quantification of D normalized to β‐actin levels as a loading control. Values are the mean with SEM. *P < 0.05, ***P < 0.001 vs. calsenilin + OGD6 h. F. Silencing of calsenilin enhanced cell viability in OGD‐induced HT‐22 cells as detected by luminescent cell viability assay. Values the mean with SEM. (n = 3). ***P < 0.001 vs. control‐siRNA + control. ## P < 0.01 vs. control‐siRNA + OGD 6 h. G. Down‐regulation of calsenilin protein was confirmed by performing ummunoblot analyses. For quantification calsenilin levels were normalized to β‐actin levels as a loading control. Values are the mean with SEM. **P < 0.01 vs. control‐siRNA.
Next, we evaluated the neuroprotective effect of calsenilin knockdown in neuronal cell death following OGD. Calsenilin siRNA, but not control scrambled siRNA, attenuated cell death in OGD‐induced HT‐22 cells (Figure 4F). We also performed Western blotting with antibody against calsenilin to establish the down‐regulation of calsenilin expression. Treatment of calsenilin‐siRNA reduced the protein level to less than 50% of control (Figure 4G).
PS mediated the apoptotic effects of calsenilin
To further understand the interplay between calsenilin and PS in cells subjected to ischemia‐like conditions, PS‐1, 2 double knockout (PSKO) MEFs were subjected to OGD for 6 h after transfection with calsenilin or vector only. The results (Figure 5A) obtained showed that absence of PS significantly decreased the rate of cell death caused by the overexpression of calsenilin during OGD. The localization of calsenilin to different cellular organelles has been shown to affect its activity as a partner of PS and as a transcriptional regulator 28. To determine whether the absence of PS affects the cellular distribution of calsenilin, PSKO MEFs subjected to 6 h of OGD were immunostained for calsenilin (Figure 5B). A dramatic increase in non‐nuclear calsenilin staining was seen in cells in response to OGD in wild‐type MEFs. In addition, the levels of calsenilin in the cytoplasmic fraction increased compared with control in OGD‐induced WT MEF (Figure 5C,D). Calsenilin in the nucleus also increased compared with WT MEFs in PSKO MEFs (Figure 5B–D). These results indicate that calsenilin accumulates in the nucleus of PSKO MEFs, which would thereby prevent it from reaching the ER and Golgi where it exerts its pro‐apoptotic function.
Figure 5.
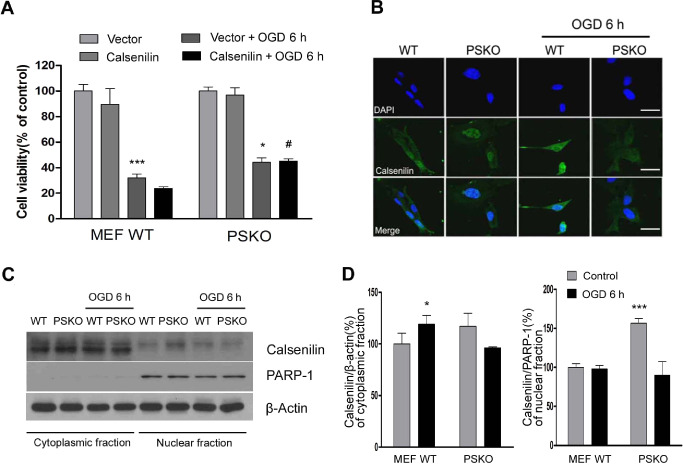
Calsenilin over‐expression in PS‐1, 2 double knockout (PSKO) cells reduces OGD‐induced cell death. A. No significant cell death rate difference was observed between WT and PSKO mouse embryonic fibroblast (MEFs) subjected to OGD. However, after cells had been transfected with calsenilin, PSKO MEFs showed significantly lower death rates than WT MEFs. Cell viability determined by luminescent cell viability assay. Values are the mean with standard error of measurement (SEM). (n = 4). *P < 0.05, ***P < 0.001 vs. vector + control, # P < 0.05 MEF WT calsenilin + OGD6h. B. PSKO MEFs, exposed to OGD for 6 h and then immunostained with anti‐calsenilin, showed a cell‐wide calsenilin distribution whereas in WT MEFs, calsenilin was restricted to the nucleus. Original magnification, 1000×. Scale bar: 50 μm. C. Representative immunoblot of cytoplasmic and nuclear fractions of WT and PSKO MEF cells showed that nuclear localization of calsenilin was significantly increased in PSKO MEF cells compared with WT MEF cells, however nuclear calsenilin of PSKO MEF cells decreased significantly after OGD. D. Quantification of calsenilin levels normalized to a loading control (β‐actin for cytoplasmic fraction; Poly [ADP‐ribose] polymerase‐1 (PARP‐1) for nuclear fraction). Values are the mean with SEM. (n = 3) *P < 0.01, ***P < 0.001 vs. MEF WT + control.
Calsenilin levels increased in vivo in response to focal ischemia and correlated with increased cell death and with the up‐regulations of apoptosis‐related proteins
Given that calsenilin levels increase in ischemia‐like conditions in vitro in parallel with the levels of the apoptosis‐related protein, Bim and the phosphorylated form of NF‐κB p65, we investigated calsenilin and NICD levels in the brains of animals following ischemic stroke. In mouse brain, an increase in calsenilin levels was observed across all time points post‐focal ischemia, but peaking at 24 h. Furthermore, this correlated with the sustained activation of p65 and increased levels of NICD, particularly at 12 h post‐reperfusion, indicating that calsenilin protein expression is similarly enhanced following cerebral ischemia in vivo (Figure 6A,B). This was further confirmed by immunohistochemistry of calsenilin in a rat global ischemic model. Figure 6C (top row of panels) shows a small increase in calsenilin, particularly in the dentate gyrus, 24 h post‐reperfusion and a dramatic increase throughout the hippocampus 72‐h post‐reperfusion. The expression pattern of calsenilin in the dentate gyrus was similar to that in CA1‐3 regions. We also investigated also calsenilin levels in non‐neuronal cells (astrocytes, microglia, endothelia) of ischemic stroke tissues. Astrocytes marginally showed expression of calsenilin, but calsenilin were not found in microglial and endothelial cells (Figure 6D).
Figure 6.
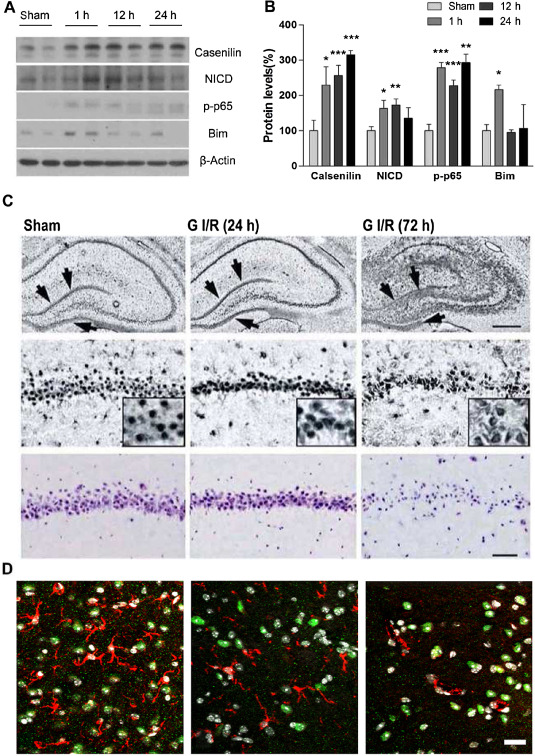
Role of calsenilin in vivo. A and B. Immunoblots (A) and quantitative densitometric analysis (B) showing significant increases in calsenilin, Notch intracellular domain, p‐p65 and Bim levels of whole brain after cerebral ischemia/reperfusion: from sham (no ischemia) to after 1 h ischemia and 1, 12 and 24 h of reperfusion. Values are the mean with standard error of measurement (n = 5 mice/group). *P < 0.05, **P < 0.01, ***P < 0.001 vs. sham. C. Calsenilin immunohistochemistry in the hippocampus of rats subjected to a sham‐operation or transient global ischemia after 24 or 72 h of reperfusion. Top panels: magnification 100×, hippocampus, middle panels: magnification 200×, close‐up of CA1 pyramidal neurons, and inset: magnification, 200×. Scale bar, 200 μm; arrows show calsenilin. Bottom: cresyl violet staining of hippocampal CA1 neurons from sham‐operated and ischemic rats after 24 or 72 h of reperfusion. Scale bar, 100 μm. D. Confocal images of ipsilateral peri‐infarct tissue sections showing green fluorescent signals for calsenilin and red fluorescent signals for astrocyte (left), microglia (middle) and endothelial cells (right). Only astrocytes showed very weak calsenilin signals. Original magnification: 200×. Scale bar: 20 μm.
Discussion
The results of this study provide evidence that calsenilin is an important protein constitutively present primarily in the adult brain, and that its expression is up‐regulated under ischemic conditions in mouse and rat models. Calsenilin is known to be a pro‐apoptotic factor, and the present study demonstrates this action in several in vitro models of stroke, including a glucose deprivation model and a model of combined hypoxia and glucose deprivation (i.e. OGD). Furthermore, calsenilin is known to interact with PS, the catalytic subunit of the multi‐protein enzyme γ‐secretase, and in the present study, it was found that it increases cell death by increasing the activity of γ‐secretase, and that this is primarily driven by other PS‐dependent mechanisms.
Calsenilin was previously shown to be pro‐apoptotic in an in vitro model of Alzheimer's Disease 18, in which it not only increased the rate of apoptotic cell death, but also up‐regulated the generation of Aβ42, the form of Aβ responsible for neuronal damage in Alzheimer's disease. In addition, both of these effects of calsenilin were found to be mediated by γ‐secretase. Furthermore, the overexpression of calsenilin was found to enhance γ‐secretase activity, including the cleavage that leads to Aβ42, and this effect was blocked by γ‐secretase inhibitors 17. The importance of γ‐secretase and its substrate Notch‐1, which is cleaved to form the transcription factor NICD 19, has been established in the contexts of neuronal damage and cell death in stroke 2. The present study also shows that calsenilin overexpression increases the production of NICD, and of downstream proteins related to cell damage and neuronal death. However, to our surprise, only a fraction of these effects can be accounted for by the actions of γ‐secretase, because in the presence of γ‐secretase inhibitors, calsenilin‐mediated cell death was only partially diminished. These results suggest that γ‐secretase‐independent pathways mediate much of the calsenilin‐dependent effect on OGD‐induced neuronal death.
We also observed that calsenilin and PS co‐localize at the perinuclear region of primary cortical neurons subjected to 24 h of glucose deprivation in vitro. PS is not only localized on the plasma membrane, where it acts as part of γ‐secretase, but it also resides to a great extent on the ER membrane, where it regulates intracellular Ca2+ signaling 24, and calsenilin has been reported to participate in both of these processes 11, 16. Furthermore, these two roles of PS are not independent, as γ‐secretase activity was found to be up‐regulated in response to increases in cytosolic Ca2+ 9. In addition, it has been demonstrated that modulation by PS of Ca2+ release from the ER is independent of γ‐secretase 25. It is therefore plausible that the increased cell death caused by the overexpression of calsenilin observed in this study occurred via the activation of PS by calsenilin to modulate Ca2+ release from the ER. To examine this mechanism further, calsenilin‐overexpressing PSKO MEFs were subjected to OGD, and it was found that the pro‐apoptotic effects of calsenilin were mediated by PS more so than by γ‐secretase. This finding can be explained by the effects of calsenilin on γ‐secretase–independent, but PS‐dependent actions, such as Ca2+ release from the ER 25. In this regard, it is known that calsenilin increases the ability of PS‐2 to reduce ER Ca2+ concentrations 11, but on the other hand, it has been observed that PSs affect calsenilin trafficking because in cells overexpressing PS‐2, calsenilin accumulates in the perinuclear regions of the ER and Golgi 28. Consistent with this, we found that calsenilin accumulates in the nucleus of PSKO MEFs, which would thereby prevent it from reaching the ER and Golgi where it exerts its pro‐apoptotic function. This study also shows that in primary cortical neurons subjected to ischemia‐like conditions for 24 h, calsenilin and PS‐1 co‐localize in perinuclear regions, possibly in the ER and Golgi. Interestingly, apoptotic bodies in this region display calsenilin, but not PS. It is evident that the role of calsenilin in neuronal injury is complex, for example calsenilin has also been reported to exert PS‐independent functions such as transcriptional repression in the nucleus 6.
Our finding that calsenilin is cleaved by caspase‐3 concurs with previous reports, and it has been proposed that this process may have a regulatory function 8, 14. Full‐length calsenilin localizes to membrane fractions where it can interact with PS and exert its Ca2+‐modulatory and γ‐secretase activating roles, but cleaved calsenilin remains in the cytosol where it loses these activities. Therefore, the activity of γ‐secretase appears to be regulated by calsenilin's subcellular localization.
Considering its roles in apoptosis and Ca2+ modulation in response to stress stimuli, such as OGD, we propose that calsenilin participates in stroke‐induced neuronal damage. Such a role is supported by our finding that calsenilin is up‐regulated in vivo in focal ischemia and that this up‐regulation is correlated with the activation of other ischemia‐induced factors, such as phosphorylated p65 and Bim.
The role played by calsenilin in ischemia‐induced neuronal injury is complex, as are its mechanisms of action, which include transcriptional repression, PS regulation, and the modulation of potassium channels. Calsenilin also modulates the catalytic subunit of γ‐secretase and intracellular Ca2+ signaling, which are both involved in stroke‐induced injury. Here, we provide novel evidence regarding the relevance of calsenilin in ischemic injury, its up‐regulation in animal models of focal ischemia, and its activation of cell death mediated by apoptotic factors. The pro‐apoptotic effects of calsenilin seem to be partly mediated by the γ‐secretase activity of PS and by other roles of PS, such as its modulation of Ca2+ ER levels. Furthermore, the mediation of calsenilin‐activated cell death by PS seems to involve changes in the intracellular distribution of calsenilin. We hope that these findings open new avenues to the effective clinical treatment of stroke.
Acknowledgments
This research was supported by the National Research Foundation of Korea (NRF) funded by the Ministry of Education, Science and Technology (2009–0069827) (2012R1A1A2009093), the Korea Healthcare technology R&D Project, Ministry for Health, Welfare & Family Affairs (A092042), the Korea Polar Research Institute, KOPRI, under a project PE12040, the Australian National Health and Medical Research Council grant (NHMRC APP1008048) and an Australian Research Council Future Fellowship to TVA (ARC FT100100427).
References
- 1. An WF, Bowlby MR, Betty M, Cao J, Ling HP, Mendoza G et al (2000) Modulation of A‐type potassium channels by a family of calcium sensors. Nature 403:553–556. [DOI] [PubMed] [Google Scholar]
- 2. Arumugam TV, Chan SL, Jo DG, Yilmaz G, Tang SC, Cheng A et al (2006) Gamma secretase‐mediated Notch signaling worsens brain damage and functional outcome in ischemic stroke. Nat Med 12:621–623. [DOI] [PubMed] [Google Scholar]
- 3. Arumugam TV, Cheng YL, Choi Y, Choi YH, Yang S, Yun YK et al (2011) Evidence that gamma‐secretase‐mediated Notch signaling induces neuronal cell death via the nuclear factor‐kappaB‐Bcl‐2‐interacting mediator of cell death pathway in ischemic stroke. Mol Pharmacol 80:23–31. [DOI] [PubMed] [Google Scholar]
- 4. Baulac S, LaVoie MJ, Kimberly WT, Strahle J, Wolfe MS, Selkoe DJ, Xia W (2003) Functional gamma‐secretase complex assembly in Golgi/trans‐Golgi network: interactions among presenilin, nicastrin, Aph1, Pen‐2, and gamma‐secretase substrates. Neurobiol Dis 14:194–204. [DOI] [PubMed] [Google Scholar]
- 5. Buxbaum JD, Choi EK, Luo Y, Lilliehook C, Crowley AC, Merriam DE, Wasco W (1998) Calsenilin: a calcium‐binding protein that interacts with the presenilins and regulates the levels of a presenilin fragment. Nat Med 4:1177–1181. [DOI] [PubMed] [Google Scholar]
- 6. Carrion AM, Link WA, Ledo F, Mellstrom B, Naranjo JR (1999) DREAM is a Ca2+‐regulated transcriptional repressor. Nature 398:80–84. [DOI] [PubMed] [Google Scholar]
- 7. Chang JW, Choi H, Kim HJ, Jo DG, Jeon YJ, Noh JY et al (2007) Neuronal vulnerability of CLN3 deletion to calcium‐induced cytotoxicity is mediated by calsenilin. Hum Mol Genet 16:317–326. [DOI] [PubMed] [Google Scholar]
- 8. Choi EK, Zaidi NF, Miller JS, Crowley AC, Merriam DE, Lilliehook C et al (2001) Calsenilin is a substrate for caspase‐3 that preferentially interacts with the familial Alzheimer's disease‐associated C‐terminal fragment of presenilin 2. J Biol Chem 276:19197–19204. [DOI] [PubMed] [Google Scholar]
- 9. Choi YH, Gwon AR, Jeong HY, Park JS, Baik SH, Arumugam TV, Jo DG (2010) Contribution of gamma‐secretase to calcium‐mediated cell death. Neurosci Lett 469:425–428. [DOI] [PubMed] [Google Scholar]
- 10. De Strooper B, Saftig P, Craessaerts K, Vanderstichele H, Guhde G, Annaert W et al (1998) Deficiency of presenilin‐1 inhibits the normal cleavage of amyloid precursor protein. Nature 391:387–390. [DOI] [PubMed] [Google Scholar]
- 11. Fedrizzi L, Lim D, Carafoli E, Brini M (2008) Interplay of the Ca2+‐binding protein DREAM with presenilin in neuronal Ca2+ signaling. J Biol Chem 283:27494–27503. [DOI] [PubMed] [Google Scholar]
- 12. Gwon AR, Park JS, Arumugam TV, Kwon YK, Chan SL, Kim SH et al (2012) Oxidative lipid modification of nicastrin enhances amyloidogenic gamma‐secretase activity in Alzheimer's disease. Aging Cell 11:559–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hendriks L, Thinakaran G, Harris CL, De Jonghe C, Martin JJ, Sisodia SS, Van Broeckhoven C (1997) Processing of presenilin 1 in brains of patients with Alzheimer's disease and controls. Neuroreport 8:1717–1721. [DOI] [PubMed] [Google Scholar]
- 14. Jin JK, Choi JK, Wasco W, Buxbaum JD, Kozlowski PB, Carp RI et al (2005) Expression of calsenilin in neurons and astrocytes in the Alzheimer's disease brain. Neuroreport 16:451–455. [DOI] [PubMed] [Google Scholar]
- 15. Jo DG, Arumugam TV, Woo HN, Park JS, Tang SC, Mughal M et al (2010) Evidence that gamma‐secretase mediates oxidative stress‐induced beta‐secretase expression in Alzheimer's disease. Neurobiol Aging 31:917–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Jo DG, Chang JW, Hong HS, Mook‐Jung I, Jung YK (2003) Contribution of presenilin/gamma‐secretase to calsenilin‐mediated apoptosis. Biochem Biophys Res Commun 305:62–66. [DOI] [PubMed] [Google Scholar]
- 17. Jo DG, Jang J, Kim BJ, Lundkvist J, Jung YK (2005) Overexpression of calsenilin enhances gamma‐secretase activity. Neurosci Lett 378:59–64. [DOI] [PubMed] [Google Scholar]
- 18. Jo DG, Kim MJ, Choi YH, Kim IK, Song YH, Woo HN et al (2001) Pro‐apoptotic function of calsenilin/DREAM/KChIP3. FASEB J 15:589–591. [DOI] [PubMed] [Google Scholar]
- 19. Kopan R (2002) Notch: a membrane‐bound transcription factor. J Cell Sci 115:1095–1097. [DOI] [PubMed] [Google Scholar]
- 20. Lilliehook C, Chan S, Choi EK, Zaidi NF, Wasco W, Mattson MP, Buxbaum JD (2002) Calsenilin enhances apoptosis by altering endoplasmic reticulum calcium signaling. Mol Cell Neurosci 19:552–559. [DOI] [PubMed] [Google Scholar]
- 21. Marambaud P, Shioi J, Serban G, Georgakopoulos A, Sarner S, Nagy V et al (2002) A presenilin‐1/gamma‐secretase cleavage releases the E‐cadherin intracellular domain and regulates disassembly of adherens junctions. EMBO J 21:1948–1956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Okun E, Arumugam TV, Tang SC, Gleichmann M, Albeck M, Sredni B, Mattson MP (2007) The organotellurium compound ammonium trichloro(dioxoethylene‐0,0') tellurate enhances neuronal survival and improves functional outcome in an ischemic stroke model in mice. J Neurochem 102:1232–1241. [DOI] [PubMed] [Google Scholar]
- 23. Song W, Nadeau P, Yuan M, Yang X, Shen J, Yankner BA (1999) Proteolytic release and nuclear translocation of Notch‐1 are induced by presenilin‐1 and impaired by pathogenic presenilin‐1 mutations. Proc Natl Acad Sci U S A 96:6959–6963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Supnet C, Bezprozvanny I (2011) Presenilins function in ER calcium leak and Alzheimer's disease pathogenesis. Cell Calcium 50:303–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tu H, Nelson O, Bezprozvanny A, Wang Z, Lee SF, Hao YH et al (2006) Presenilins form ER Ca2+ leak channels, a function disrupted by familial Alzheimer's disease‐linked mutations. Cell 126:981–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wei Z, Chigurupati S, Arumugam TV, Jo DG, Li H, Chan SL (2011) Notch activation enhances the microglia‐mediated inflammatory response associated with focal cerebral ischemia. Stroke 42:2589–2594. [DOI] [PubMed] [Google Scholar]
- 27. Wolfe MS, Xia W, Ostaszewski BL, Diehl TS, Kimberly WT, Selkoe DJ (1999) Two transmembrane aspartates in presenilin‐1 required for presenilin endoproteolysis and gamma‐secretase activity. Nature 398:513–517. [DOI] [PubMed] [Google Scholar]
- 28. Woo HN, Chang JW, Choi YH, Gwon AR, Jung YK, Jo DG (2008) Characterization of subcellular localization and Ca2+ modulation of calsenilin/DREAM/KChIP3. Neuroreport 19:1193–1197. [DOI] [PubMed] [Google Scholar]
- 29. Zaidi NF, Berezovska O, Choi EK, Miller JS, Chan H, Lilliehook C et al (2002) Biochemical and immunocytochemical characterization of calsenilin in mouse brain. Neuroscience 114:247–263. [DOI] [PubMed] [Google Scholar]