

Abstract
Neurological dysfunction and motor neuron degeneration in amyotrophic lateral sclerosis (ALS) is strongly associated with neuroinflammation reflected by activated microglia and astrocytes in the CNS. In ALS endogenous triggers in the CNS such as aggregated protein and misfolded proteins activate a pathogenic response by innate immune cells. However, there is also strong evidence for a neuroprotective immune response in ALS. Emerging evidence also reveals changes in the peripheral adaptive immune responses as well as alterations in the blood brain barrier that may aid traffic of lymphocytes and antibodies into the CNS. Understanding the triggers of neuroinflammation is key to controlling neuronal loss. Here, we review the current knowledge regarding the roles of non‐neuronal cells as well as the innate and adaptive immune responses in ALS. Existing ALS animal models, in particular genetic rodent models, are very useful to study the underlying pathogenic mechanisms of motor neuron degeneration. We also discuss the approaches used to target the pathogenic immune responses and boost the neuroprotective immune pathways as novel immunotherapies for ALS.
Keywords: ALS, glia, immune regulation, neurodegeneration, protection, therapy
Introduction
Amyotrophic lateral sclerosis (ALS) belongs to the clinical and pathological spectrum of motor neuron disorders and manifests itself with an invariable progression to death from respiratory failure in an average of 3–5 years from onset of symptoms 92. ALS pathology distinctively encompasses distant biological systems including muscle/neuromuscular junction, as well as the spinal cord and brain 72. The origin and propagation of this disorder are still at the centre of extensive investigations and the involvement of different districts may occur either simultaneously or sequentially in the disease process. The main obstacle to gain full insight of the pathogenesis of ALS is the remarkable clinical heterogeneity of the condition and the lack of access to the pre‐symptomatic stage of the disease. Whilst the clinical presentation and the speed of progression have a clear impact in the diagnostic delay, most ALS individuals would come to medical attention when the disease is already advanced with regard to the involvement of motor cell population. It is now acknowledged that a shorter diagnostic latency is a robust risk factor for faster disease progression in ALS 9, 93. Clinical heterogeneity in ALS refers not only to manifestations like motor and bulbar impairment with speech and respiratory involvement either at the outset or later on in the disease progression, but also to the appearance of features of cognitive impairment, with behavioural and semantic changes typical of fronto‐temporal dementia 9. Whilst in rare cases bulbar impairment remains the only clinical feature of the disease from the onset of symptoms (progressive bulbar palsy), the pathological spread of the disease conditions the frequent involvement of other muscle districts with a more obvious clinical and pathological expression of upper and lower motor neuron involvement. Another relevant aspect of the clinical heterogeneity of ALS which is likely to affect the outcome of clinical trials in the absence of biological tools for the clinical stratification of the disease is the difference in rate of clinical progression observed among ALS individuals. Within the same phenotypic appearance, some patients may rapidly progress to end stage in a matter of months, whilst the disease course in others is more prolonged. The ability to define prognostically the disease trajectory as a function of speed of progression in ALS would significantly improve the design of more cost‐effective clinical trials. Recently, the development of assays to monitor the release into biological fluids of neurofilaments, important structural components of neurons and axons, has been an important milestone in the clinical stratification of the disease and provides added value to any new study into disease‐modifying treatments 55.
Pathogenic and Protective Role of Glia
Emerging evidence suggests that while disease progression in ALS is a result of a slow and progressive dysfunction and loss of motor neurons, other non‐neuronal cells in the CNS also play a key role in the disease (Table 1). This is supported by studies in mouse models in which selective deficiency of mutant SOD1 from microglia or astrocytes slows experimental disease progression. Microglia have dual roles in the CNS and specifically in CNS disorders where they not only support neurons but also have an immunological role as innate resident macrophages in the CNS. In ALS the onset of disease in SOD1 mice is associated with microglia activation and production of TNF‐α, IL‐6 and IL‐1β, suggesting that mutant protein triggers a pathogenic response in microglia 28. Yet, microglia also play a protective role by secreting the anti‐inflammatory factors IL‐4 and IL‐10, as well as growth factors, indicating a balance in pathogenic and protective roles 2, 51. Likewise, astrocytes play protective role by secreting growth factors and upregulating GLT1 thereby reducing glutamate. While mutations in TDP‐43, SOD1 and FUS are characteristically described in motor neurons, pathological aggregates of these proteins are also observed in oligodendrocytes that are critical for maintaining axonal and neuronal integrity. The abundance of FUS protein is linked with age of onset of ALS in humans. Such FUS‐containing oligodendrocytes also display myelin abnormalities and myelin degradation 56. Indeed, changes associated with oligodendrocytes are observed prior to the onset of disease in SOD1 mice. In addition, selective deficiency of mutant SOD1 in NG2+ cells also delayed the onset of disease 43, suggesting an additional role for oligodendrocytes in the pathogenesis of ALS.
Table 1.
Innate and adaptive immune systems in ALS. CNS = central nervous system; CSF = cerebrospinal fluid; NMJ = neuromuscular junction; CD = cluster of differentiation; ROS = reactive oxygen species; Cox‐2 = cyclooxygenase‐2; SR‐A = scavenger receptor‐A; TLR = toll‐like receptor; NOD = nucleotide‐binding oligomerization domain; RIG = retinoic acid‐inducible gene; AIM‐2 = absent in melanoma‐2; RAGE = receptor for advanced glycation end‐products; P2X7 = purinergic receptor; NLRP = (NOD)‐like receptor protein; HMGB1 = high‐motility group box 1; LRP4 = lipoprotein receptor‐related protein 4; NF = neurofilament proteins; MCP‐1 = monocyte chemotactic protein‐1; M‐CSF = macrophage‐colony stimulating factor.
| Tissue | Innate immune system | Adaptive immune system | References |
|---|---|---|---|
| CNS | Increase in ROS, Cox‐2, HMGB1, S100β, | Autoantibodies to HMGB1 | 35, 39 |
| Receptors: Increase in TLR2,4, NOD, RIG, AIM‐2, RAGE, P2X7, FasL/FasR, NLR inflammasome: NLRP3 | Cellular: Increase in CD4, CD8 T cells | ||
| Chemokines/Cytokines: Increase in IL‐1β, IL‐18, IL‐6, TNF‐α, IFN‐γ, MCP‐1, M‐CSF, CCL2, | CD markers: CD1a, CD83 | 6, 26, 30 | |
| CD markers: CD11b, CD14, CD18, CD68, SR‐A | |||
| CSF | CNS antigens: NF‐L, S100β | Autoantibodies to LRP4, GM1 ganglioside, NF | 18, 30, 65, 71, 94 |
| Chemokines/Cytokines: MCP‐1, IL‐1β, IL‐6, TNF‐α, | Cellular: CD4 | 89 | |
| CD markers: CD14 |
Cytokines/chemokines: Increase in l17, IL‐23, IFN‐γ |
62, 78 | |
|
Periphery Muscle and NMJ |
Cellular: Microglia, Macrophages | Autoantibodies to LRP4, P/Q‐type calcium channels. | 23, 94 |
|
CD markers: CD11b, CD68. CD169/CD68/Iba1+ |
Cellular: Infiltration of activated macrophages. | 10, 24 | |
| Blood | M1 macrophage: CCR2, CD14+CD16− | Autoantibodies to: LRP4, Voltage‐gated calcium channels: P/Q‐type, N type, L‐type, CD95/Fas‐receptor, GM1 ganglioside, HMGB1, AChR, NF, foetal muscular proteins. | |
| Cellular: NK, CD8, CD4, IL‐13 TNF‐α, IFN‐γ. Decrease in Th2, Treg during disease | 2, 77, 84, 108 |
Contribution of Innate Immunity in ALS
Neuroinflammation that is, innate and adaptive immune responses govern the balance between neuronal repair and neuronal damage in ALS. The innate immune system in ALS can be triggered by aggregated proteins or danger signals and pattern‐recognition signals generated during infection as a result of tissue injury. In broad terms, these are pathogen‐associated molecular patterns (PAMPs) and damage‐associated molecular patterns (DAMPs), respectively. Recent studies have revealed the importance of the transient receptor potential (TRP) channels which recognize DAMPs from the environment 11, 83. These channel ion receptors are activated by high oxidative stress as observed in ALS 31, suggesting the involvement of TRPs in the aetiology of the disease 70.
Endogenous danger signals derived from molecules released by damaged cells, stress‐induced proteins and abnormal protein accumulation, and pathogen‐derived molecules like toxins, complement factors and allergens are well known to activate innate immunity mechanisms. Microglia in the CNS express receptors that sense PAMPs and DAMPs such as Toll‐like receptors (TLRs), NOD, RIG, AIM2‐like receptors, and the receptor for advanced glycation end products (RAGE) to promote autophagy 90 (Table 1).
Importantly, evidence for reactive microglia/macrophages and astrocytes, the main components of the innate immune system in the CNS, is observed in motor regions of the CNS in sporadic and familial ALS 45. One of the important molecules that signal cell damage is the high‐mobility group box 1 (HMGB1) which is activated in response to injury, and triggers a series of events that lead to inflammation. This molecule is upregulated in spinal cords of ALS patients and its binding and signalling through TLR4, expressed by activated microglia and astrocytes, contribute to end‐stage ALS pathology 8, 49. Notably, activated glia in ALS patients, are generally in close interaction to motor neurons.
On the other hand, intracellular inflammasome complexes, mainly expressed in spinal cord astrocytes, mediate inflammatory responses. Increased activation of the nucleotide‐binding domain and leucine‐rich repeat protein‐3 (NLRP3) inflammasome is involved in neuroinflammation in ALS. The NLRP3 inflammasome is key for the activation of caspase‐1 and secretion of IL‐1β and IL‐18 in ALS 39.
As shown in Figure 1, reactive microglia and astrocytes express inflammatory markers including iNOS, ROS and Cox‐2 and produce pro‐inflammatory molecules like prostaglandins, IL‐1β, IL‐6 and TNF‐α. Fibroblast growth factor‐1 (FGF‐1) released by motor neurons during oxidative stress induces activation of astrocytes, which in turn initiate motor neuron apoptosis. Factors that mediate microglia/motor neuron interactions include neurotrophic factors (NT) (e.g., plasminogen, TGF‐β, bFGF, brain‐derived growth factor (BDNF), vascular endothelial growth factor (VEGF), nerve growth factor (NGF), NT‐3 and NT‐4) 61. On the other hand, high levels of neurotrophins, such as insulin‐like growth factor I (IGF‐1), progranulin, and other neurotrophic factors (NTFs) that are released from astrocytes control inflammation. Excitatory aminoacid transporter in astroglial cells (EAAT2) plays a major role in keeping extracellular glutamate concentration below neurotoxic levels. Neuronal excitotoxicity in ALS has been associated with loss of EAAT2 protein and function 82.
Figure 1.
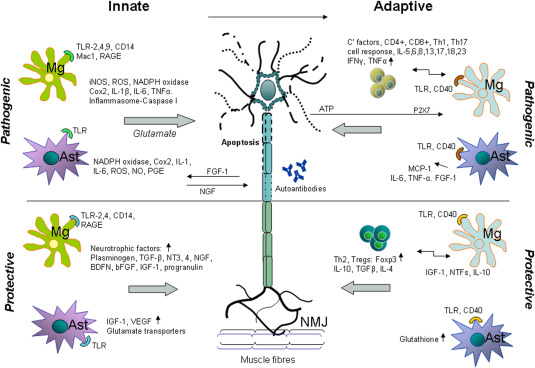
Innate and Adaptive Immunity in ALS. Glia cells are key components modulating the neuroinflammatory response in ALS. During innate activation microglia have a phagocytic role and become antigen‐presenting cells able to actively drive an adaptive immune response. Mg, microglia; Ast, astrocytes; NMJ, neuromuscular junction.
A number of studies indicate that microglial activation occurs previously or concomitantly with the onset of clinical disease, and increases during the disease course 61. Therefore, as the disease progresses, microglia and astrocytes acquire a cytotoxic phenotype. For example, the change to a neurotoxic phenotype is associated with an impairment of the astroglial glutamate‐transporter in ALS that probably contributes to persistent glial activation 19. Continuous inflammation leads to the establishment of adaptive immune responses (Figure 1).
Adaptive Immunity in ALS
While the triggers of adaptive immunity in ALS are unclear, it is well known that PAMPs and DAMPs modulate adaptive immune responses. Apart from glial cell activation, also the infiltration of the CNS by immune cells is a hallmark of ALS and an important role of T cells in the disease process in ALS has been firmly established. T cells are associated with motor neuron death and numbers of cytotoxic CD8+ T cells and natural killer cells are increased in patients with fast progressing disease 29, 77. A first indication that T cells may also play a protective role, however, was reported in 2008 when it was found that CD4+ T cells support glia‐mediated neuroprotection and slow disease progression in experimental ALS 1. Data subsequently collected in ALS patients strengthen the notion that especially CD4+ regulatory T cells (Tregs) are likely to play a crucial protective role in disease, since the numbers of peripheral T cells bearing the typical Treg signature markers CD4, CD25, Gata3 and FoxP3 are reduced in fast progressing of ALS patients.
As illustrated in Figure 1, increased levels of classical complement (C′) C1q, C3, C4 and C5b‐9, and higher numbers of activated microglia, astrocytes, dendritic cells and T cells may all play a role in initiating an adaptive response in ALS 86. CD40 expression on glia cells seems to be key for antigen presentation to T cells. Activated M1 microglia can further enhance pro‐inflammatory responses, including the release of TNF‐α, IL‐6 and IL‐1β, and down‐regulate Treg suppressive functions. Monocyte chemotactic protein‐1 (MCP‐1) released by astrocytes promotes infiltration of macrophages/microglia into affected tissues. A protective role of astrocytes on the other hand, can be mediated by increased glutathione secretion that protects motor neurons against oxidative stress 98, and enhanced expression of glumatate transporters that counteract glutamate excitotoxicity. Tregs and protective M2 microglia also suppress proliferation and cytotoxic functions of Th1 cells 105. Increased levels of FoxP3+ regulatory T cells and production of TGF‐β and IL‐4 are associated with neuroprotection and involved in slowing disease progression of ALS 29.
The role of B cells in ALS is likely limited 63 although immunoglobulin and complement deposition is observed in the CNS of ALS patients 15, 16. In addition auto‐antibodies against proteins of the CNS cells are frequently found in ALS patients and correlate with severity of disease course. Autoantibodies recognise actin, desmin and neurofilament light 64, 74. Likewise, autoantibodies to P/Q‐type voltage‐gated calcium channel and AChR in presynaptic motor nerve terminals and at NMJs have been found to participate in muscle denervation 23, 60, 67. Also, autoantibodies against HMGB1 are elevated in serum of patients with ALS, suggesting they may serve as biomarkers for the diagnosis of the disease and to monitor disease progression 35 (Table 1).
Blood CNS Barriers
The potential contribution of an altered blood‐CNS and cerebrospinal fluid (CSF) barrier (BCBs) to ALS initiation and propagation (Figure 2) has emerged from human pathology and a variety of experimental observations in animal models of the disease 58, 100. This body of work has revealed changes in post‐mortem tissues and systemic effects that a malfunctioning biological interface between nervous tissue and biological fluids is likely to create 20. Structural and functional impairment in post‐mortem gray and white matter microvessels of medulla and spinal cord from sALS patients and from SOD1 animal models of the disease are strongly suggestive of pervasive barrier damage. Yet, these findings pertain mostly to commonly recognised alterations in barrier damage including endothelial cell degeneration, capillary leakage, perivascular oedema, down regulation of tight junction proteins and microhemorrhages, while ALS‐specific changes have yet to emerge 17, 100. Among the systemic features of an altered BCB in ALS are reduced numbers of circulating endothelial cells in peripheral blood of ALS patients. This indicates the occurrence of endothelial damage and/or impaired endothelium repair in ALS, leading to BCB dysfunction 21.
Figure 2.
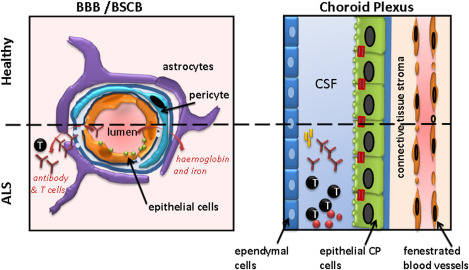
The Blood‐CNS barriers in ALS. The blood‐brain barrier (BBB), blood‐spinal cord barrier (BSCB) limits potential pathogenic antibodies and T cells entering the CNS in health. Evidence suggests that also pathogenic antibodies directed to neuronal structures and pathogenic T cells also enter the brain via the choroid plexus. However, as well as pathogenic immunity, regulatory T cells entering the brain may also play a protective role in ALS (see text for details).
Prospective lines of investigations on BCB dysfunction in ALS would require detailed study of the immunopathological signature of this condition, including the widespread activation of dendritic cells and microglia/macrophages in affected tissues and the up‐regulation of a number of inflammatory phase reactants including circulating cytokines, MCP‐1 and ferritine 58, 87. For example, perivascular iron deposition has long been recognised in neurological disorders including multiple sclerosis and may play a part also in ALS, where ferritine regulation in blood could result from altered storage or from an anti‐oxidant protective effect 87.
Recent studies indicate that apart from the BBB, the choroid plexus, i.e. the CSF‐blood barrier plays a protective role in experimental models of ALS. In the SOD1 mouse model, the influx of anti‐inflammatory macrophages and Tregs into the CNS was markedly enhanced following immunization with a myelin peptide. Thus, the choroid plexus may actively recruit Tregs to attenuate disease progression and improved survival during experimental ALS 47.
Therapies Targeting Glia
As key effector cells of innate immune responses in the CNS and critical players in ALS 3, 4, 75, 79, microglia and astrocytes are attractive therapeutic targets. Rather than only mounting a secondary response to degeneration of motor neurons, glial cells play an active role in controlling disease progression, and may even play a primary driving role in ALS 85. Such a primary pathogenic role for glia cells would be well in line with the observation that transgenic expression of mutant SOD1 only in motor neurons does not trigger experimental ALS in mice 53, 73. Several mechanisms have been identified that drive important, and potentially even primary pathogenic functions of microglia and astrocytes in ALS 79, 105. Interference especially with the soluble factors that control innate glial responses during ALS offers several therapeutic possibilities. Apart from a number of obvious but more general immune‐modulatory approaches that are based on interfering with cellular migration or cytokine‐mediated signalling of glial cells that are also relevant to other neurodegenerative conditions, more selective approaches based on soluble factors that are ALS specific may be envisaged as well, as explained below.
While the appearance of intracellular protein inclusions of TDP‐43, SOD1 or FUS in motor neurons and astrocytes are the well‐known pathological hallmarks of ALS, these proteins are also secreted into the extracellular space 33, 44, 95, 104. As soluble extracellular proteins in the CNS, they may be equally relevant to the disease process in ALS as the extracellular aggregates of tau protein that drive proteopathic spreading of neurodegeneration during disorders such as Alzheimer's disease. Evidence has recently started to accumulate that like extracellular tau, extracellular forms of abnormal TDP‐43 and SOD1 can trigger prion‐like spreading of pathogenic protein aggregation during ALS 25, 50, 88. Such spreading is likely aggravated by the fact that extracellular SOD1 or TDP‐43 additionally activates microglia. They do so through a CD14‐restricted receptor‐mediated pathway, involving one or more family members of the Toll‐like receptor family and/or scavenger receptors 8, 49, 54. Microglia activation by altered forms of SOD1 or TDP‐43 triggers the release of neurotoxic pro‐inflammatory factors including IL‐1β, TNF‐α, and reactive nitrogen and oxygen species 80, 106, 107. These, in turn promote aberrant localization and aggregation of proteins in other glial cells as well as in motor neurons 12, thus aggravating spreading of protein abnormalities within the CNS. When astrocytes are targeted by the above pro‐inflammatory microglial factors, accumulation of abnormal forms of SOD1 or TDP‐43 changes them to become pro‐inflammatory themselves as well 39. They lose their normal neurotrophic capacity 34, and release factors that are directly toxic to motor neurons 27, 76, 81, 91. At multiple levels, therefore, the presence of extracellular ALS‐associated abnormal forms of SOD1 or TDP‐43 can play a pivotal role in the rapid progression of ALS.
A possible therapeutic strategy to counteract this role of extracellular SOD1 or TDP‐43 aggregates in ALS is to sequester them by molecular chaperones. Potentially therapeutic chaperones targeting misfolded SOD1 and/or TDP‐43 may include macrophage migration inhibitory factor 37, or chemically synthesized compounds designed to selectively bind to misfolded versions of certain proteins 22. Some therapeutic compounds such as arimoclomol may help augment the natural induction of chaperones 40. Yet, the stress‐inducible natural chaperones of the human CNS may also be used as therapeutic agents themselves. As ATP‐independent molecular chaperones, members of the family of small heat shock proteins such as HSP27 (HspB1) or alpha B‐crystallin (HspB5) appear to be best suited to counteract the detrimental effects of extracellular proteins during ALS. HspB1 and HspB5 are able bind to a wide range of abnormally folded proteins and protein aggregates in vivo as well as in vitro, including mutant or chemically altered SOD1 and TDP‐43 52, 99, 101. Indeed, endogenous HspB5 acts as a protective factor during experimental ALS in mice 59, and recombinant human HspB5 has recently been successfully tested as a therapeutic protein in multiple sclerosis 97. When supplied as extracellular proteins, HspB5 and HspB1 will bind to abnormal extracellular proteins, eliminate their neurotoxicity, prevent continued protein aggregation and proteopathic spreading, and promote their phagocytic clearance by microglia 32, 57, 101. Furthermore, HspB5 acts as a ligand for CD14 and TLR2 expressed by microglia and macrophages. While being taken up via these receptors, HspB5 activates a broad neuroprotective and anti‐inflammatory TLR2‐mediated response in microglia as well as in any infiltrated macrophages, and promotes their reversal from a pro‐inflammatory M1 to an anti‐inflammatory and protective M2 state of activation 5, 96. Extracellular HspB5 also activates astrocytes, and promote increased production of neuroprotective factors as well as strongly enhanced expression of the glutamate transporter EAAT2 that can help normalize levels of extracellular glutamate (van Noort et al, unpublished data). Such comprehensive actions by small heat‐shock proteins like HspB5 may well help counteract the detrimental effects of extracellular SOD1 and TDP‐43 on glial cells at various levels, and selectively target what is likely a critical pathogenic pathway during ALS.
Therapies Targeting the Immune System
Largely based on a significant body of animal model data on the role of Tregs in experimental ALS, it now seems likely that Tregs infiltrating the CNS engage in intimate cross‐talk with local glial cells. By releasing regulatory cytokines such as IL‐4, Tregs help microglia to maintain their protective M2 phenotype. In turn, M2 microglia help Tregs to maintain their suppressive functions. At the early stages of disease, their mutually supported functions counteract pathogenic mechanisms (reviewed by Zhao et al [105]). As disease progresses, however, the protective balance between microglia and Tregs appears to become increasingly difficult to maintain. As local glial cells gradually acquire a more proinflammatory state of activation due to chronic activation, also Tregs gradually lose their capacity to locally control damage. When supplied at the rapidly progressive stage of experimental ALS in mice, Tregs no longer suppress disease 2. This erosion of Treg functions during ALS suggests that therapeutically augmenting the activity or numbers of Tregs may well produce benefit at the earlier stages of ALS, but may fail to do so in advanced disease. After all, being apparently dependent on productive cross‐talk with early‐stage M2‐like microglia, Tregs might well fail to protect when confronted with primarily M1‐like microglia at later stages of disease.
A large body of encouraging data suggest that cell‐based therapies may be exploited to replace not only corrupted glia cells or dying motor neurons during ALS, but also to augment Treg functions at early stages of disease 46, 79. Via soluble mediators including for example prostaglandin E2 and transforming growth factor β, mesenchymal stem cells for example promote the development and function of Tregs. As a result, intrathecal mesenchymal stem cells stimulate CNS entry by CD4+CD25+Foxp3+ Tregs and their local production of anti‐inflammatory cytokines such as IL‐4, IL‐10 and TGF‐β 48. A fundamentally different route to stimulate the entry of beneficial Tregs into the CNS during experimental ALS was recently illustrated by Michal Schwartz and colleagues. By immunizing mice with a myelin peptide, the influx of IL‐10‐producing anti‐inflammatory macrophages and Tregs into the CNS was markedly enhanced. By their anti‐inflammatory actions, and while promoting the local production of neuroprotective factors, Tregs recruited into the CNS by myelin peptide immunization led to attenuation of disease progression and improved survival during experimental ALS 47. These findings illustrate that therapeutic approaches aimed at strengthening Treg functions during ALS hold promise for the future.
Therapies Targeting the BBB
The blood‐brain and ‐spinal cord barriers are critical factors for any effort to develop effective treatments for neurological diseases like ALS, due to their ability to protect the CNS from potentially harmful substances. It is speculated that when penetration through these barriers increases as a result of neurological as well as systemic conditions, drugs may be in a better position to reach those areas involved in the pathological process 14. However, studies have shown that during ALS levels of ABC transporters P‐glycoprotein (P‐gp) and breast cancer resistance protein (BCRP) increase in the CNS. Their inability to penetrate in the CNS may well be one reason why many drugs have failed in ALS. Selective increase of two ABC drug efflux transporters at the blood‐spinal cord barrier suggests induced pharmacoresistance in ALS 38. Nevertheless, compounds with a potential immunomodulatory effect have been considered for disease modification. The rationale for testing nonsteroidal anti‐inflammatory drugs like cyclooxygenase‐2 inhibitors and prostaglandins for example, draws on their CNS penetrability, on their anti‐inflammatory effect but also on the observation that these molecules are already endogenously expressed in neurons and glia cells under normal conditions 102. Nevertheless, treating ALS with the wide range of immune‐regulating drugs so far contemplated in the field of neurodegeneration may not be that simple.
ALS lags behind disorders like multiple sclerosis in the understanding of the role played by BBB integrity in disease progression, of the cross‐talk between a systemic inflammatory state and brain homeostasis, and in the role of immune responses in disease initiation and progression. In MS, ways to restore BBB function and promote its immune quiescence have been tested as novel therapeutic regimes that specifically reduce leukocyte entry into the central nervous system but also restore brain homeostasis with regard to CNS penetration of T cells, B cells and macrophages 42. However, whilst this strategy remains pivotal in those conditions where exchange of immunological factors between the CNS and systemic biological systems may be the main driving force in the establishment and development of the disease, it is not clear whether the same therapeutic approach may be effective in ALS. Novel druggable targets which partake in the complex network of molecular players that leads to BBB dysfunction are being investigated, including the role of microRNAs (miRNAs) in controlling the function of the barrier endothelium 41. Whilst this biological framework is being investigated in MS, any relevant finding may be translated to other neurodegenerative conditions like ALS, where the pathobiology of miRNAs is progressively uncovered 7. The recent report of a significantly lower level of circulating Tregs patients with ALS displaying a faster disease progression rate has opened new treatment avenues but also raised questions on the biological significance of this observation 2. Reduced Tregs levels may effectively imply altered immune‐tolerance and an overall change of the way the brain deals with autoimmunity. It is also not clear how and whether the systemic change in this subset of T cells is mirrored by a similar development in the brain. It is not clear how the decline of Tregs takes place in ALS and whether this biological alteration occurs simultaneously or in sequence in the CNS and in the peripheral circulation. Any potential repurposing or use of new drugs for the correction of this imbalance in ALS will have to take into account the role of the BBB in allowing or preventing the flow of T cells across compartments.
Conclusions
Increasing evidence supports the role of pathogenic immune responses in ALS pathology. Here, we highlight the key roles of immune responses and the role of non‐neuronal cells in the development of ALS. Microglia and astrocytes as the major glial cell types represent the first line of defence that maintains homeostasis in the healthy CNS. Glia‐neuron and glia‐T lymphocytes interactions control signalling pathways and the differentiation of microglia and astrocytes and hence, the balance between neuroprotection and neuronal death. During ALS emerging data suggests the switch from neuroprotective Tregs/M2 to neurotoxic Th1/M1 cells defines progression and the final outcome of the disease. Also, compelling data underscores the importance of impairment of the blood‐CNS barrier as a key factor in promoting motor neuron damage. Restoring the balance between pathogenic responses and protective immune responses, as well as the understanding the role of non‐neuronal cells i.e. oligodendrocytes and blood‐CNS barriers offers novel targets for therapeutic approaches in ALS.
Acknowledgments
All individuals listed as authors contributed substantially to the design and writing of the paper and agreed to the final version submitted. Several of our studies on the role of glia cells in neurodegenerative disorders have been financially supported by the Netherlands Foundation for MS Research and the Multiple Sclerosis Society of Great Britain and Northern Ireland.
J.M. van Noort holds equity in Delta Crystallon BV. The other authors declare no conflicts of interests.
References
- 1. Beers DR, Henkel JS, Zhao W, Wang J, Appel SH (2008) CD4+ T cells support glial neuroprotection, slow disease progression, and modify glial morphology in an animal model of inherited ALS. Proc Natl Acad Sci U S A 105:15558–15563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Beers DR, Henkel JS, Zhao W, Wang J, Huang A, Wen S, Liao B, Appel SH (2011) Endogenous regulatory T lymphocytes ameliorate amyotrophic lateral sclerosis in mice and correlate with disease progression in patients with amyotrophic lateral sclerosis. Brain 134:1293–1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Brettschneider J, Libon DJ, Toledo JB, Xie SX, McCluskey L, Elman L et al (2012) Microglial activation and TDP‐43 pathology correlate with executive dysfunction in amyotrophic lateral sclerosis. Acta Neuropathol 123:395–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Brettschneider J, Toledo JB, Van Deerlin VM, Elman L, McCluskey L, Lee VM, Trojanowski JQ (2011) Microglial activation correlates with disease progression and upper motor neuron clinical symptoms in amyotrophic lateral sclerosis. PLoS One 7:e39216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bsibsi M, Holtman IR, Gerritsen WH, Eggen BJ, Boddeke E, van der Valk P et al (2013) Alpha‐B‐crystallin induces an immune‐regulatory and antiviral microglial response in preactive multiple sclerosis lesions. J Neuropathol Exp Neurol 72:970–979. [DOI] [PubMed] [Google Scholar]
- 6. Butovsky O, Siddiqui S, Gabriely G, Lanser AJ, Dake B, Murugaiyan G et al (2012) Modulating inflammatory monocytes with a unique microRNA gene signature ameliorates murine ALS. J Clin Invest 122:3063–3087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Campos‐Melo D, Droppelmann CA, He Z, Volkening K, Strong MJ (2013) Altered microRNA expression profile in Amyotrophic Lateral Sclerosis: a role in the regulation of NFL mRNA levels. Mol Brain 6:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Casula M, Iyer AM, Spliet WG, Anink JJ, Steentjes K, Sta M, Troost D, Aronica E (2011) Toll‐like receptor signaling in amyotrophic lateral sclerosis spinal cord tissue. Neuroscience. 179:233–243. [DOI] [PubMed] [Google Scholar]
- 9. Cellura E, Spataro R, Taiello AC, La Bella V (2012) Factors affecting the diagnostic delay in amyotrophic lateral sclerosis. Clin Neurol Neurosurg 114:550–554. [DOI] [PubMed] [Google Scholar]
- 10. Chiu IM, Phatnani H, Kuligowski M, Tapia JC, Carrasco MA, Zhang M, Maniatis T, Carroll MC (2009) Activation of innate and humoral immunity in the peripheral nervous system of ALS transgenic mice. Proc Natl Acad Sci U S A 106:20960–20965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Clapham DE (2003) TRP channels as cellular sensors. Nature 426:517–524. [DOI] [PubMed] [Google Scholar]
- 12. Correia AS, Patel P, Dutta K, Julien JP (2015) Inflammation Induces TDP‐43 Mislocalization and Aggregation. PLoS One 10:e0140248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Couratier P, Yi FH, Preud'homme JL, Clavelou P, White A, Sindou P et al (1998) Serum autoantibodies to neurofilament proteins in sporadic amyotrophic lateral sclerosis. J Neurol Sci 154:137–145. [DOI] [PubMed] [Google Scholar]
- 14. Dominguez A, Alvarez A, Suarez‐Merino B, Goni‐de‐Cerio F (2014) Neurological disorders and the blood‐brain barrier. Strategies and limitations for drug delivery to the brain. Rev Neurol 58:213–224. [PubMed] [Google Scholar]
- 15. Donnenfeld H, Kascsak RJ, Bartfeld H (1984) Deposits of IgG and C3 in the spinal cord and motor cortex of ALS patients. J Neuroimmunol 6:51–57. [DOI] [PubMed] [Google Scholar]
- 16. Engelhardt JI, Appel SH (1990) IgG reactivity in the spinal cord and motor cortex in amyotrophic lateral sclerosis. Arch Neurol 47:1210–1216. [DOI] [PubMed] [Google Scholar]
- 17. Evans MC, Couch Y, Sibson N, Turner MR (2013) Inflammation and neurovascular changes in amyotrophic lateral sclerosis. Mol Cell Neurosci 53:34–41. [DOI] [PubMed] [Google Scholar]
- 18. Fialova L, Svarcova J, Bartos A, Ridzon P, Malbohan I, Keller O, Rusina R (2010) Cerebrospinal fluid and serum antibodies against neurofilaments in patients with amyotrophic lateral sclerosis. Eur J Neurol 17:562–566. [DOI] [PubMed] [Google Scholar]
- 19. Foran E, Trotti D (2009) Glutamate transporters and the excitotoxic path to motor neuron degeneration in amyotrophic lateral sclerosis. Antioxid Redox Signal 11:1587–1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Garbuzova‐Davis S, Sanberg PR (2014) Blood‐CNS Barrier Impairment in ALS patients versus an animal model. Front Cell Neurosci 8:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Garbuzova‐Davis S, Woods RL, 3rd , Louis MK, Zesiewicz TA, Kuzmin‐Nichols N, Sullivan KL et al (2010) Reduction of circulating endothelial cells in peripheral blood of ALS patients. PLoS One 5:e10614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Getter T, Zaks I, Barhum Y, Ben‐Zur T, Boselt S, Gregoire S et al (2015) A chemical chaperone‐based drug candidate is effective in a mouse model of amyotrophic lateral sclerosis (ALS). ChemMedChem 10:850–861. [DOI] [PubMed] [Google Scholar]
- 23. Gonzalez LE, Kotler ML, Vattino LG, Conti E, Reisin RC, Mulatz KJ et al (2011) Amyotrophic lateral sclerosis‐immunoglobulins selectively interact with neuromuscular junctions expressing P/Q‐type calcium channels. J Neurochem 119:826–838. [DOI] [PubMed] [Google Scholar]
- 24. Graber DJ, Hickey WF, Harris BT (2010) Progressive changes in microglia and macrophages in spinal cord and peripheral nerve in the transgenic rat model of amyotrophic lateral sclerosis. J Neuroinflammation 7:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Grad LI, Cashman NR (2014) Prion‐like activity of Cu/Zn superoxide dismutase: implications for amyotrophic lateral sclerosis. Prion 8:33–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Graves MC, Fiala M, Dinglasan LA, Liu NQ, Sayre J, Chiappelli F et al (2004) Inflammation in amyotrophic lateral sclerosis spinal cord and brain is mediated by activated macrophages, mast cells and T cells. Amyotroph Lateral Scler Other Motor Neuron Disord 5:213–219. [DOI] [PubMed] [Google Scholar]
- 27. Haidet‐Phillips AM, Hester ME, Miranda CJ, Meyer K, Braun L, Frakes A et al (2011) Astrocytes from familial and sporadic ALS patients are toxic to motor neurons. Nat Biotechnol 29:824–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Heneka MT, Kummer MP, Latz E (2014) Innate immune activation in neurodegenerative disease. Nat Rev Immunol. 14:463–477. [DOI] [PubMed] [Google Scholar]
- 29. Henkel JS, Beers DR, Wen S, Rivera AL, Toennis KM, Appel JE et al (2013) Regulatory T‐lymphocytes mediate amyotrophic lateral sclerosis progression and survival. EMBO Mol Med 5:64–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Henkel JS, Engelhardt JI, Siklos L, Simpson EP, Kim SH, Pan T et al (2004) Presence of dendritic cells, MCP‐1, and activated microglia/macrophages in amyotrophic lateral sclerosis spinal cord tissue. Ann Neurol 55:221–235. [DOI] [PubMed] [Google Scholar]
- 31. Hermosura MC, Garruto RM (2007) TRPM7 and TRPM2‐Candidate susceptibility genes for Western Pacific ALS and PD? Biochim Biophys Acta 1772:822–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hochberg GK, Ecroyd H, Liu C, Cox D, Cascio D, Sawaya MR et al (2014) The structured core domain of alphaB‐crystallin can prevent amyloid fibrillation and associated toxicity. Proc Natl Acad Sci U S A 111:E1562–E1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hosokawa M, Arai T, Yamashita M, Tsuji H, Nonaka T, Masuda‐Suzukake M et al (2014) Differential diagnosis of amyotrophic lateral sclerosis from Guillain‐Barre syndrome by quantitative determination of TDP‐43 in cerebrospinal fluid. Int J Neurosci 124:344–349. [DOI] [PubMed] [Google Scholar]
- 34. Huang C, Huang B, Bi F, Yan LH, Tong J, Huang J et al (2014) Profiling the genes affected by pathogenic TDP‐43 in astrocytes. J Neurochem 129:932–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hwang CS, Liu GT, Chang MD, Liao IL, Chang HT (2013) Elevated serum autoantibody against high mobility group box 1 as a potent surrogate biomarker for amyotrophic lateral sclerosis. Neurobiol Dis 58:13–18. [DOI] [PubMed] [Google Scholar]
- 36. Ilzecka J (2009) Serum‐soluble receptor for advanced glycation end product levels in patients with amyotrophic lateral sclerosis. Acta Neurol Scand 120:119–122. [DOI] [PubMed] [Google Scholar]
- 37. Israelson A, Ditsworth D, Sun S, Song S, Liang J, Hruska‐Plochan M et al (2015) Macrophage migration inhibitory factor as a chaperone inhibiting accumulation of misfolded SOD1. Neuron 86:218–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Jablonski MR, Jacob DA, Campos C, Miller DS, Maragakis NJ, Pasinelli P et al (2012) Selective increase of two ABC drug efflux transporters at the blood‐spinal cord barrier suggests induced pharmacoresistance in ALS. Neurobiol Dis 47:194–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Johann S, Heitzer M, Kanagaratnam M, Goswami A, Rizo T, Weis J, Troost D, Beyer C (2015) NLRP3 inflammasome is expressed by astrocytes in the SOD1 mouse model of ALS and in human sporadic ALS patients. Glia 63:2260–2273. [DOI] [PubMed] [Google Scholar]
- 40. Kalmar B, Lu CH, Greensmith L (2014) The role of heat shock proteins in Amyotrophic Lateral Sclerosis: The therapeutic potential of Arimoclomol. Pharmacol Ther 141:40–54. [DOI] [PubMed] [Google Scholar]
- 41. Kamphuis WW, Derada Troletti C, Reijerkerk A, Romero IA, de Vries HE (2014) The blood‐brain barrier in multiple sclerosis: microRNAs as key regulators. CNS Neurol Disord Drug Targets 14:157–167. [DOI] [PubMed] [Google Scholar]
- 42. Kanda T (2014) Molecular targeted therapy against the blood brain barrier in multiple sclerosis. Clin Exp Neuroimmunol 5:28–34. [Google Scholar]
- 43. Kang SH, Li Y, Fukaya M, Lorenzini I, Cleveland DW, Ostrow LW et al (2013) Degeneration and impaired regeneration of gray matter oligodendrocytes in amyotrophic lateral sclerosis. Nat Neurosci 16:571–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kasai T, Tokuda T, Ishigami N, Sasayama H, Foulds P, Mitchell DJ et al (2009) Increased TDP‐43 protein in cerebrospinal fluid of patients with amyotrophic lateral sclerosis. Acta Neuropathol 117:55–62. [DOI] [PubMed] [Google Scholar]
- 45. Kawamata T, Akiyama H, Yamada T, McGeer PL (1992) Immunologic reactions in amyotrophic lateral sclerosis brain and spinal cord tissue. Am J Pathol 140:691–707. [PMC free article] [PubMed] [Google Scholar]
- 46. Kim C, Lee HC, Sung JJ (2014) Amyotrophic lateral sclerosis ‐ cell based therapy and novel therapeutic development. Exp Neurobiol 23:207–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kunis G, Baruch K, Miller O, Schwartz M (2015) Immunization with a myelin‐derived antigen activates the brain's choroid plexus for recruitment of immunoregulatory cells to the CNS and attenuates disease progression in a mouse model of ALS. J Neurosci 35:6381–6393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kwon MS, Noh MY, Oh KW, Cho KA, Kang BY, Kim KS et al (2014) The immunomodulatory effects of human mesenchymal stem cells on peripheral blood mononuclear cells in ALS patients. J Neurochem 131:206–218. [DOI] [PubMed] [Google Scholar]
- 49. Lee JY, Lee JD, Phipps S, Noakes PG, Woodruff TM (2015) Absence of toll‐like receptor 4 (TLR4) extends survival in the hSOD1 G93A mouse model of amyotrophic lateral sclerosis. J Neuroinflammation 12:90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lee S, Kim HJ (2015) Prion‐like Mechanism in Amyotrophic Lateral Sclerosis: are Protein Aggregates the Key? Exp Neurobiol 24:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Lewis CA, Manning J, Rossi F, Krieger C (2012) The neuroinflammatory response in ALS: The roles of microglia and T cells. Neurol Res Int 2012:803701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Lin WL, Castanedes‐Casey M, Dickson DW (2009) Transactivation response DNA‐binding protein 43 microvasculopathy in frontotemporal degeneration and familial Lewy body disease. J Neuropathol Exp Neurol 68:1167–1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Lino MM, Schneider C, Caroni P (2002) Accumulation of SOD1 mutants in postnatal motoneurons does not cause motoneuron pathology or motoneuron disease. J Neurosci 22:4825–4832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Liu Y, Hao W, Dawson A, Liu S, Fassbender K (2009) Expression of amyotrophic lateral sclerosis‐linked SOD1 mutant increases the neurotoxic potential of microglia via TLR2. J Biol Chem 284:3691–3699. [DOI] [PubMed] [Google Scholar]
- 55. Lu CH, Petzold A, Topping J, Allen K, Macdonald‐Wallis C, Clarke J et al (2015) Plasma neurofilament heavy chain levels and disease progression in amyotrophic lateral sclerosis: insights from a longitudinal study. J Neurol Neurosurg Psychiatry 86:565–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Mackenzie IR, Ansorge O, Strong M, Bilbao J, Zinman L, Ang LC et al (2011) Pathological heterogeneity in amyotrophic lateral sclerosis with FUS mutations: two distinct patterns correlating with disease severity and mutation. Acta Neuropathol 122:87–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Mainz A, Peschek J, Stavropoulou M, Back KC, Bardiaux B, Asami S et al (2015) The chaperone alphaB‐crystallin uses different interfaces to capture an amorphous and an amyloid client. Nat Struct Mol Biol. 22:898–905. [DOI] [PubMed] [Google Scholar]
- 58. Malaspina A, Puentes F, Amor S (2015) Disease origin and progression in amyotrophic lateral sclerosis: an immunology perspective. Int Immunol. 27:117–129. [DOI] [PubMed] [Google Scholar]
- 59. Marino M, Papa S, Crippa V, Nardo G, Peviani M, Cheroni C et al (2015) Differences in protein quality control correlate with phenotype variability in 2 mouse models of familial amyotrophic lateral sclerosis. Neurobiol Aging 36:492–504. [DOI] [PubMed] [Google Scholar]
- 60. Mehanna R, Patton EL, Jr. , Phan CL, Harati Y (2012) Amyotrophic lateral sclerosis with positive anti‐acetylcholine receptor antibodies. Case report and review of the literature. J Clin Neuromuscul Dis. 14:82–85. [DOI] [PubMed] [Google Scholar]
- 61. Moisse K, Strong MJ (2006) Innate immunity in amyotrophic lateral sclerosis. Biochim Biophys Acta 1762:1083–1093. [DOI] [PubMed] [Google Scholar]
- 62. Moreau C, Devos D, Brunaud‐Danel V, Defebvre L, Perez T, Destee A, Tonnel AB, Lassalle P, Just N (2005) Elevated IL‐6 and TNF‐alpha levels in patients with ALS: inflammation or hypoxia? Neurology 65:1958–1960. [DOI] [PubMed] [Google Scholar]
- 63. Naor S, Keren Z, Bronshtein T, Goren E, Machluf M, Melamed D (2009) Development of ALS‐like disease in SOD‐1 mice deficient of B lymphocytes. J Neurol 256:1228–1235. [DOI] [PubMed] [Google Scholar]
- 64. Niebroj‐Dobosz I, Dziewulska D, Janik P (2006) Auto‐antibodies against proteins of spinal cord cells in cerebrospinal fluid of patients with amyotrophic lateral sclerosis (ALS). Folia Neuropathol 44:191–196. [PubMed] [Google Scholar]
- 65. Niebroj‐Dobosz I, Janik P, Kwiecinski H (2004) Serum IgM anti‐GM1 ganglioside antibodies in lower motor neuron syndromes. Eur J Neurol 11:13–16. [DOI] [PubMed] [Google Scholar]
- 66. Offen D, Halevi S, Orion D, Mosberg R, Stern‐Goldberg H, Melamed E et al (1998) Antibodies from ALS patients inhibit dopamine release mediated by L‐type calcium channels. Neurology 51:1100–1103. [DOI] [PubMed] [Google Scholar]
- 67. Okuyama Y, Mizuno T, Inoue H, Kimoto K (1997) Amyotrophic lateral sclerosis with anti‐acetylcholine receptor antibody. Intern Med 36:312–315. [DOI] [PubMed] [Google Scholar]
- 68. Ordonez G, Sotelo J (1989) Antibodies against fetal muscle proteins in serum from patients with amyotrophic lateral sclerosis. Neurology 39:683–686. [DOI] [PubMed] [Google Scholar]
- 69. Pagani MR, Gonzalez LE, Uchitel OD (2011) Autoimmunity in amyotrophic lateral sclerosis: past and present. Neurol Res Int 2011:497080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Park HS, Hong C, Kim BJ, So I (2014) The Pathophysiologic Roles of TRPM7 Channel. Korean J Physiol Pharmacol 18:15–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Pestronk A, Adams RN, Clawson L, Cornblath D, Kuncl RW, Griffin D, Drachman DB (1988) Serum antibodies to GM1 ganglioside in amyotrophic lateral sclerosis. Neurology. 38:1457–1461. [DOI] [PubMed] [Google Scholar]
- 72. Peters OM, Ghasemi M, Brown RH, Jr. (2015) Emerging mechanisms of molecular pathology in ALS. J Clin Invest. 125:1767–1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Pramatarova A, Laganiere J, Roussel J, Brisebois K, Rouleau GA (2001) Neuron‐specific expression of mutant superoxide dismutase 1 in transgenic mice does not lead to motor impairment. J Neurosci 21:3369–3374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Puentes F, Topping J, Kuhle J, van der Star BJ, Douiri A, Giovannoni G, Baker D, Amor S, Malaspina A (2013) Immune reactivity to neurofilament proteins in the clinical staging of amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry 85:274–278. [DOI] [PubMed] [Google Scholar]
- 75. Radford RA, Morsch M, Rayner SL, Cole NJ, Pountney DL, Chung RS (2015) The established and emerging roles of astrocytes and microglia in amyotrophic lateral sclerosis and frontotemporal dementia. Front Cell Neurosci 9:414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Re DB, Le Verche V, Yu C, Amoroso MW, Politi KA, Phani S et al (2014) Necroptosis drives motor neuron death in models of both sporadic and familial ALS. Neuron 81:1001–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Rentzos M, Evangelopoulos E, Sereti E, Zouvelou V, Marmara S, Alexakis T, Evdokimidis I (2011) Alterations of T cell subsets in ALS: a systemic immune activation? Acta Neurol Scand 125:260–264. [DOI] [PubMed] [Google Scholar]
- 78. Rentzos M, Rombos A, Nikolaou C, Zoga M, Zouvelou V, Dimitrakopoulos A et al (2010) Interleukin‐17 and interleukin‐23 are elevated in serum and cerebrospinal fluid of patients with ALS: a reflection of Th17 cells activation? Acta Neurol Scand 122:425–429. [DOI] [PubMed] [Google Scholar]
- 79. Rizzo F, Riboldi G, Salani S, Nizzardo M, Simone C, Corti S, Hedlund E (2014) Cellular therapy to target neuroinflammation in amyotrophic lateral sclerosis. Cell Mol Life Sci 71:999–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Roberts K, Zeineddine R, Corcoran L, Li W, Campbell IL, Yerbury JJ (2013) Extracellular aggregated Cu/Zn superoxide dismutase activates microglia to give a cytotoxic phenotype. Glia 61:409–419. [DOI] [PubMed] [Google Scholar]
- 81. Rojas F, Cortes N, Abarzua S, Dyrda A, van Zundert B (2014) Astrocytes expressing mutant SOD1 and TDP43 trigger motoneuron death that is mediated via sodium channels and nitroxidative stress. Front Cell Neurosci 8:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Rothstein JD, Van Kammen M, Levey AI, Martin LJ, Kuncl RW (1995) Selective loss of glial glutamate transporter GLT‐1 in amyotrophic lateral sclerosis. Ann Neurol 38:73–84. [DOI] [PubMed] [Google Scholar]
- 83. Santoni G, Cardinali C, Morelli MB, Santoni M, Nabissi M, Amantini C (2015) Danger‐ and pathogen‐associated molecular patterns recognition by pattern‐recognition receptors and ion channels of the transient receptor potential family triggers the inflammasome activation in immune cells and sensory neurons. J Neuroinflammation 12:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Shi N, Kawano Y, Tateishi T, Kikuchi H, Osoegawa M, Ohyagi Y, Kira J (2007) Increased IL‐13‐producing T cells in ALS: positive correlations with disease severity and progression rate. J Neuroimmunol. 182:232–235. [DOI] [PubMed] [Google Scholar]
- 85. Sloan SA, Barres BA (2013) Glia as primary drivers of neuropathology in TDP‐43 proteinopathies. Proc Natl Acad Sci U S A 110:4439–4440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Sta M, Sylva‐Steenland RM, Casula M, de Jong JM, Troost D, Aronica E, Baas F (2011) Innate and adaptive immunity in amyotrophic lateral sclerosis: evidence of complement activation. Neurobiol Dis 42:211–220. [DOI] [PubMed] [Google Scholar]
- 87. Su XW, Clardy SL, Stephens HE, Simmons Z, Connor JR (2015) Serum ferritin is elevated in amyotrophic lateral sclerosis patients. Amyotroph Lateral Scler Frontotemporal Degener 16:102–107. [DOI] [PubMed] [Google Scholar]
- 88. Sundaramoorthy V, Walker AK, Yerbury J, Soo KY, Farg MA, Hoang V, Zeineddine R, Spencer D, Atkin JD (2013) Extracellular wildtype and mutant SOD1 induces ER‐Golgi pathology characteristic of amyotrophic lateral sclerosis in neuronal cells. Cell Mol Life Sci 70:4181–4195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Sussmuth SD, Sperfeld AD, Hinz A, Brettschneider J, Endruhn S, Ludolph AC, Tumani H (2010) CSF glial markers correlate with survival in amyotrophic lateral sclerosis. Neurology 74:982–987. [DOI] [PubMed] [Google Scholar]
- 90. Tang D, Kang R, Coyne CB, Zeh HJ, Lotze MT (2012) PAMPs and DAMPs: signal 0s that spur autophagy and immunity. Immunol Rev 249:158–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Tong J, Huang C, Bi F, Wu Q, Huang B, Liu X, Li F, Zhou H, Xia XG (2013) Expression of ALS‐linked TDP‐43 mutant in astrocytes causes non‐cell‐autonomous motor neuron death in rats. EMBO J 32:1917–1926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Traxinger K, Kelly C, Johnson BA, Lyles RH, Glass JD (2013) Prognosis and epidemiology of amyotrophic lateral sclerosis: Analysis of a clinic population, 1997‐2011. Neurol Clin Pract 3:313–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Turner MR, Benatar M (2015) Ensuring continued progress in biomarkers for amyotrophic lateral sclerosis. Muscle Nerve 51:14–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Tzartos JS, Zisimopoulou P, Rentzos M, Karandreas N, Zouvelou V, Evangelakou P et al (2014) LRP4 antibodies in serum and CSF from amyotrophic lateral sclerosis patients. Ann Clin Transl Neurol 1:80–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Urushitani M, Sik A, Sakurai T, Nukina N, Takahashi R, Julien JP (2006) Chromogranin‐mediated secretion of mutant superoxide dismutase proteins linked to amyotrophic lateral sclerosis. Nat Neurosci 9:108–118. [DOI] [PubMed] [Google Scholar]
- 96. van Noort JM, Bsibsi M, Nacken PJ, Gerritsen WH, Amor S, Holtman IR et al (2013) Activation of an immune‐regulatory macrophage response and inhibition of lung inflammation in a mouse model of COPD using heat‐shock protein alpha B‐crystallin‐loaded PLGA microparticles. Biomaterials 34:831–840. [DOI] [PubMed] [Google Scholar]
- 97. van Noort JM, Bsibsi M, Nacken PJ, Verbeek R, Venneker EH (2015) Therapeutic Intervention in Multiple Sclerosis with Alpha B‐Crystallin: A Randomized Controlled Phase IIa Trial. PLoS One 10:e0143366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Vargas MR, Johnson JA (2010) Astrogliosis in amyotrophic lateral sclerosis: role and therapeutic potential of astrocytes. Neurotherapeutics 7:471–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Wang J, Xu G, Li H, Gonzales V, Fromholt D, Karch C et al (2005) Somatodendritic accumulation of misfolded SOD1‐L126Z in motor neurons mediates degeneration: alphaB‐crystallin modulates aggregation. Hum Mol Genet 14:2335–2347. [DOI] [PubMed] [Google Scholar]
- 100. Winkler EA, Sengillo JD, Sullivan JS, Henkel JS, Appel SH, Zlokovic BV (2013) Blood‐spinal cord barrier breakdown and pericyte reductions in amyotrophic lateral sclerosis. Acta Neuropathol 125:111–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Yerbury JJ, Gower D, Vanags L, Roberts K, Lee JA, Ecroyd H (2013) The small heat shock proteins alphaB‐crystallin and Hsp27 suppress SOD1 aggregation in vitro. Cell Stress Chaperones. 18:251–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Yermakova A, O'Banion MK (2000) Cyclooxygenases in the central nervous system: implications for treatment of neurological disorders. Curr Pharm Des. 6:1755–1776. [DOI] [PubMed] [Google Scholar]
- 103. Yi FH, Lautrette C, Vermot‐Desroches C, Bordessoule D, Couratier P, Wijdenes J et al (2000) In vitro induction of neuronal apoptosis by anti‐Fas antibody‐containing sera from amyotrophic lateral sclerosis patients. J Neuroimmunol 109:211–220. [DOI] [PubMed] [Google Scholar]
- 104. Zetterstrom P, Andersen PM, Brannstrom T, Marklund SL (2011) Misfolded superoxide dismutase‐1 in CSF from amyotrophic lateral sclerosis patients. J Neurochem 117:91–99. [DOI] [PubMed] [Google Scholar]
- 105. Zhao W, Beers DR, Appel SH (2013) Immune‐mediated mechanisms in the pathoprogression of amyotrophic lateral sclerosis. J Neuroimmune Pharmacol 8:888–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Zhao W, Beers DR, Bell S, Wang J, Wen S, Baloh RH, Appel SH (2015) TDP‐43 activates microglia through NF‐kappaB and NLRP3 inflammasome. Exp Neurol 273:24–35. [DOI] [PubMed] [Google Scholar]
- 107. Zhao W, Beers DR, Henkel JS, Zhang W, Urushitani M, Julien JP, Appel SH (2010) Extracellular mutant SOD1 induces microglial‐mediated motoneuron injury. Glia 58:231–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Zhao W, Xie W, Xiao Q, Beers DR, Appel SH (2006) Protective effects of an anti‐inflammatory cytokine, interleukin‐4, on motoneuron toxicity induced by activated microglia. J Neurochem. 99:1176–1187. [DOI] [PubMed] [Google Scholar]