

Abstract
Huntington's disease (HD) is a polyglutamine disease and characterized neuropathologically by degeneration of the striatum and select layers of the neo‐ and allocortex. In the present study, we performed a systematic investigation of the cerebellum in eight clinically diagnosed and genetically confirmed HD patients. The cerebellum of all HD patients showed a considerable atrophy, as well as a consistent loss of Purkinje cells and nerve cells of the fastigial, globose, emboliform and dentate nuclei. This pathology was obvious already in HD brains assigned Vonsattel grade 2 striatal atrophy and did not correlate with the extent and distribution of striatal atrophy. Therefore, our findings suggest (i) that the cerebellum degenerates early during HD and independently from the striatal atrophy and (ii) that the onset of the pathological process of HD is multifocal. Degeneration of the cerebellum might contribute significantly to poorly understood symptoms occurring in HD such as impaired rapid alternating movements and fine motor skills, dysarthria, ataxia and postural instability, gait and stance imbalance, broad‐based gait and stance, while the morphological alterations (ie ballooned neurons, torpedo‐like axonal inclusions) observed in the majority of surviving nerve cells may represent a gateway to the unknown mechanisms of the pathological process of HD.
Keywords: cerebellum, Huntington's disease, neurodegeneration, pathoanatomy, polyglutamine diseases
Introduction
Huntington's disease (HD) is an autosomal dominantly inherited, progressive polyglutamine or CAG‐repeat disease 15, 20, 48, 49, 56, 67, 68. Identified in 1993, the HD gene on chromosome 4p16.3 harbors an elongated and meiotically unstable CAG‐repeat 15, 49, 56, which encodes the disease protein huntingtin 34, 48, 60, 61, 67, 68, whose physiological role is only poorly understood 15, 61. The normal HD gene comprises 6–35 CAG‐repeats and is prone to meiotic instability when containing sequences of 28 or more CAG‐repeats. HD occurs when the critical threshold of about 35 CAG‐repeats is exceeded 15, 34, 56, 60, 61, 67, 68.
Onset of HD commonly is around the age of 40 years 12, 15, 34, 56, 60, 61, 62, 67, 68 with motor symptoms (“clumsiness,” “tremor,” “balance trouble,” “jerkiness”) 12, 15, 34, 49 or progressive cognitive impairments 12, 15, 22, 29, 34, 49, 67, 68. Choreatic movements may also be early and progressive HD symptoms, but may plateau and disappear in the advanced clinical stages of HD 15, 22, 34, 49, 67, 68. Additional somatomotor (ie, bradykinesia, akinesia, dystonia, hypotonia, rigidity, dysarthria, dysphagia) 12, 15, 29, 34, 49, 67, 68 and oculomotor symptoms 32, executive and sensory dysfunctions, personality changes, psychiatric disturbances (ie, depression, schizophrenia‐like symptoms) 12, 15, 22, 29, 34, 49, 67, 68 and severe weight loss 6, 15, 29, 43, 47 may develop during the further course of HD. Along with dysarthria, additional symptoms possibly pointing to cerebellar damage may occur in HD patients: impaired rapid alternating movements and fine motor skills, postural and gait instability, as well as ataxia 27, 29, 34, 67, 68.
Gradual worsening degeneration of the striatum is the pathological hallmark of HD 6, 21, 22, 30, 31, 38, 47, 48, 57, 60, 61, 62; it represents the basis of a neuropathological grading system for HD 15, 60, 61, 62, which in the past was widely used to explanation the large spectrum of HD disease symptoms, including oculomotor and choreatic symptoms 3, 22, 29, 57. As most of the current ideas about the function of the human striatum, however, are derived from clinical observations in HD, these one‐sided explanations may at least in part represent circular statements and have therefore been gradually qualified 36, 68. Along with degeneration of the striatum, in a further step, laminar degeneration of select regions of the cerebral neo‐ and allocortex has been accepted as an additional neuropathological hallmark of HD 3, 21, 22, 30, 31, 47, 60, 61 and is now considered as a plausible and specific pathoanatomical explanation for the occurrence of cognitive and psychiatric symptoms 3, 21, 22, as well as visual 21 and electrophysiological dysfunctions 22 in HD.
Detailed knowledge of additional and consistent brain targets of the pathological process of HD is mandatory to establish pending exact clinico‐pathological HD correlations and the exact role of extrastriatal brain sites in the pathogenesis of clinical HD symptoms. It will help to (i) detect the initial events of the pathological process of HD and to trace its spread through the brain; (ii) understand the shared characteristics and intrinsic connections of neuronal dropout at vulnerable brain sites; and (iii) evaluate the validity of engineered cellular or animal models of HD. The cerebellum is among the extrastriatal brain sites whose involvement in HD and relevance for the clinical picture of HD is still controversial 6, 11, 25, 46, 47, 48, 61, 62. Therefore, we performed the first systematic study of the cerebellum of clinically well‐characterized and genetically confirmed HD patients and correlated the symptoms with pathological changes involving the cerebellum.
Patients and Methods
Patients
We studied the cerebellum of eight patients clinically diagnosed and genetically confirmed with HD (age at death, 67.3 ± 11.3 years; Table 1) and, for comparison, the cerebellum of 11 individuals without any neurological or neuropsychiatric diseases (four females, seven males; age at death, 60.8 ± 15.0 years). The causes of death of the HD patients included starvation (patients 1, 2, 4, 8; Table 1), subdural hematoma (patient 3; Table 1), aspiration pneumonia (patients 5, 7; Table 1), ruptured aortic aneurysm (patient 6; Table 1) and pneumonia (patient 7; Table 1). The causes of death in the control cases included pancreatitis, myocardial infarction, sarcoidosis, lung cancer, pericarditis, stomach carcinoma, arrhythmia and lymphoma. Upon routine, neuropathological investigation atrophic changes, cerebral tumors, vascular and inflammatory diseases, as well as white matter damage were excluded in the brains of the control individuals. The Alzheimer's disease (AD)‐related cortical tau (τ) cytoskeletal pathology of the control individuals ranged from Braak and Braak stages 0–III 2, 5. The examination of the HD and control brains was approved by the ethical board of the Faculty of Medicine at the Goethe University of Frankfurt/Main.
Table 1.
Age at death (years), time interval (TI) between the last neurological investigation and death (months), gender (F, female; M, male), number of expanded CAG‐repeats (CAG) in the mutated Huntington allele, Vonsattel grade (grade of striatal atrophy according to Vonsattel et al) 60, 61, 62, age at onset of initial symptoms (years), initial disease symptom and duration of Huntington's disease (years)
| Patient | Age | TI | Gender | CAG | Vonsattel grade | Onset of initial symptoms | Initial disease symptoms | Duration of disease |
|---|---|---|---|---|---|---|---|---|
| 1 | 51 | 60 | M | 49 | 4 | 33 | Personality changes | 18 |
| 2 | 61 | 6 | F | 45 | 3 | 35 | Chorea | 26 |
| 3 | 64 | 3 | M | 41 | 2 | 55 | Chorea | 9 |
| 4 | 65 | 36 | M | 45 | 4 | 53 | Ataxia | 12 |
| 5 | 68 | 18 | F | 46 | 3 | 40 | Cognitive decline | 28 |
| 6 | 68 | 29 | M | 40 | 3 | 60 | Cognitive decline | 8 |
| 7 | 70 | 4 | F | 44 | 2 | 50 | Chorea | 20 |
| 8 | 91 | 72 | M | 42 | 2 | 55 | Chorea | 36 |
In all HD patients investigated here, the clinical diagnosis was confirmed by molecular genetical investigation disclosing the presence of expanded CAG‐repeat sequences in the mutated HD allele of all HD patients (Table 1) 56.
The predominant initial disease symptoms of the HD patients studied or listed in Table 1 and their symptoms during the further disease course related to extracerebellar damage, as well as those possibly related to cerebellar damage are provided in Table 2.
Table 2.
Disease symptoms of the Huntington's disease patients
| Patient | Disease symptoms related to extracerebellar damage | Disease symptoms possibly related to cerebellar damage |
|---|---|---|
| 1 | Choreatic movements, dysphagia, weight loss, executive dysfunctions, personality changes, cognitive decline | Unsteady and broad‐based gait, dysarthria, slowed smooth pursuit eye movements |
| 2 | Choreatic movements, dysphagia, weight loss, cognitive decline | — |
| 3 | Choreatic movements, executive dysfunctions, cognitive decline | Broad‐based gait, dysarthria |
| 4 | Choreatic movements, bradykinesia, dysphagia, weight loss, cognitive decline | Ataxia, dysarthria |
| 5 | Choreatic movements, dysphagia, weight loss, cognitive decline | — |
| 6 | Choreatic movements, weight loss, cognitive decline | Gait imbalance, impaired gait coordination |
| 7 | Choreatic movements, dysphagia, weight loss, executive dysfunctions, cognitive decline | Unsteady and broad‐based gait, gait imbalance, dysarthria, slowed smooth pursuit eye movements |
| 8 | Choreatic movements, weight loss, executive dysfunctions, cognitive decline | — |
Tissue treatment
Autopsy was performed in the HD patients within 10.9 ± 5.2 h and in the control individuals within 14.2 ± 12.94 h post‐mortem. The time interval between the last neurological examination and death was 28.5 ± 26.2 months in the HD patients (Table 1).
The brains of the HD patients and the control individuals were fixed in a 4% aqueous formaldehyde solution. Atrophic changes of the cerebral hemisphere, cerebellum and brainstem of the HD patients were assessed macroscopically, and degeneration of their striatum was graded according to Vonsattel et al 60, 61, 62. Subsequent to this assessment, the right cerebellum was embedded in polyethylene glycol [polyethylene glycol (PEG) 1000, Merck, Darmstadt, Germany] 54 and completely cut into seven sets of equidistant serial 100 μm thick sagittal sections (distance between these sets, 600 μm).
The first set of serial sections from the HD patients and control individuals was stained for lipofuscin pigment (aldehyde‐fuchsin)/Nissl material (Darrow red) 4, 50, 52 and used for the identification of the three layers of the cerebellar cortex (ie, molecular, Purkinje and granular cell layers) and the four deep nuclei of the cerebellum (ie, fastigial, globose, emboliform and dentate nuclei) 14, 16, 53, 63, 64, 65, as well as for the examination of their neuronal loss in the HD patients. In each instance, the second set of cerebellar tissue sections was processed according to a modified Heidenhain procedure to visualize the cerebellar white matter 24, while the third set was immunostained with a rabbit polyclonal antibody against glial fibrillary acidic protein (GFAP) (1:500; Dako, Hamburg, Germany) to visualize reactive astrogliosis. The fourth set of cerebellar sections from the HD patients and from eight control individuals was immunolabeled with calbindin (1:1000; Chemicon International, Temecula, CA, USA), a selective and reliable marker for cerebellar Purkinje cells 69. These immunolabeled tissue sections were used for confirmation of Purkinje cell loss in the vermal portion (at the level of the fastigial nucleus) and cerebellar hemisphere (at the level of the dentate nucleus), which was already evident in the pigment‐Nissl stained cerebellar sections from the HD patients. The specificity of the immunolabelings was assessed by omission of the primary antibodies. Incubation with the primary antibodies was performed for 12 h at room temperature followed by incubation with biotinylated anti‐rabbit immunoglobulins for 1.5 h at room temperature. Bound antigens were visualized with the AB‐complex (Vectastain, Vector Laboratories, Burlingame, CA, USA) and 3,3‐diaminobenzidine‐tetra‐HCl/H2O2 [diaminobenzidine (DAB) D5637 Sigma, Taufkirchen, Germany]. In each instance, approximately eight pigment‐Nissl stained, GFAP and calbindin immunostained tissue sections were studied.
The pathological changes in the deep cerebellar nuclei and cerebellar cortex, and reactive astrogliosis were semi‐quantitatively assessed. Neuronal loss in the deep cerebellar nuclei: none discernible (–), obvious as compared with control cases and presence of extraneuronal lipofuscin granules (+), nucleus devoid or nearly devoid of nerve cells and presence extraneuronal lipofuscin granules (++); loss of cerebellar Purkinje cells: none discernible (–), obvious loss of Purkinje cells in a given cerebellar lobule or lobular area as compared with control cases (+) cerebellar lobule or lobular area devoid or nearly devoid of Purkinje cells (++); reactive astrogliosis: none discernible (–), a few reactive astrocytes present (+), an abundance of reactive astrocytes present (++) Tables 3, 4, 5, 6 6). Kendall's rank correlation coefficient τ was used to analyze the correlation between the loss of Purkinje cells and the loss of neurons of the deep nuclei on the one hand, and the length of the CAG‐repeats, age at onset of symptoms, duration of the disease, age at death and Vonsattel grade of striatal atrophy on the other hand 1.
Table 3.
Severity of loss of Purkinje cells in lobules III–X of the vermal portion of the cerebellum of the Huntington's disease patients
| Patient | I | II | III | IV | V | VI | VII | VIII | IX | X |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | + | + | ++ | ++ | ++ | + | + | + | + | + |
| 2 | + | + | + | + | + | + | + | + | + | + |
| 3 | + | + | + | + | + | + | + | + | + | + |
| 4 | + | + | + | + | + | + | + | + | + | + |
| 5 | + | + | + | + | + | + | + | + | + | + |
| 6 | + | + | + | + | + | + | + | + | + | + |
| 7 | + | + | + | + | + | + | ++ | + | + | + |
| 8 | + | + | + | + | + | + | ++ | ++ | + | + |
− = none discernible; + = obvious loss of Purkinje cells in a given cerebellar lobule or lobular area as compared with control cases; ++ = cerebellar lobule or lobular area devoid or nearly devoid of Purkinje cells.
Table 4.
Severity of of Purkinje cells in lobules III–X of the cerebellar hemisphere of the Huntington's disease patients
| Patient | III | IV | V | VI | Cr1 | Cr2 | VIIB | VIIIAA | VIIIB | IX | X |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | + | + | + | + | + | ++ | ++ | ++ | ++ | + | + |
| 2 | + | + | + | + | + | + | + | + | + | + | + |
| 3 | ++ | + | + | + | + | + | + | + | + | − | − |
| 4 | − | − | − | + | + | + | + | + | + | − | − |
| 5 | + | + | + | + | + | + | ++ | ++ | ++ | ++ | ++ |
| 6 | + | + | + | + | + | + | ++ | ++ | ++ | ++ | ++ |
| 7 | + | + | + | + | + | + | + | + | + | + | + |
| 8 | − | + | + | + | + | + | ++ | + | + | + | + |
− = none discernible; + = obvious loss of Purkinje cells in a given cerebellar lobule or lobular area as compared to control cases. ++ = cerebellar lobule or lobular area devoid or nearly devoid of Purkinje cells.
Table 5.
Severity of neuronal loss in the deep cerebellar nuclei of the Huntington's disease patients
| Patient | Fastigial nucleus | Globose nucleus | Emboliform nucleus | Dentate nucleus |
|---|---|---|---|---|
| 1 | ++ | ++ | ++ | + |
| 2 | ++ | + | ++ | + |
| 3 | ++ | + | ++ | + |
| 4 | ++ | + | ++ | + |
| 5 | ++ | ++ | ++ | + |
| 6 | + | + | + | + |
| 7 | ++ | ++ | ++ | ++ |
| 8 | ++ | + | + | + |
− = none discernible; + = obvious as compared to control cases and presence of extraneuronal lipofuscin granules; ++ = nucleus devoid or nearly devoid of nerve cells and/or extraneuronal lipofuscin granules.
Table 6.
Severity of GFAP‐immunoreactive astrogliosis in the cerebellar cortex, white matter and deep nuclei of the Huntington's disease patients.
| Patient | Molecular cell layer | Purkinje cell layer | Granular cell layer | Cerebellar white matter | Fastigial nucleus | Globose nucleus | Emboliform nucleus | Dentate nucleus |
|---|---|---|---|---|---|---|---|---|
| 1 | + | + | + | ++ | + | + | + | + |
| 2 | + | + | + | ++ | + | ++ | + | + |
| 3 | + | + | + | ++ | + | + | + | + |
| 4 | + | + | + | ++ | + | − | − | − |
| 5 | + | + | + | ++ | + | ++ | ++ | + |
| 6 | + | + | + | ++ | + | + | + | + |
| 7 | + | + | + | ++ | ++ | ++ | + | + |
| 8 | + | + | + | ++ | + | + | + | + |
− = none discernible; + = a few reactive astrocytes present; ++ = an abundance of reactive astrocytes present.
Additional quantitative analysis was performed to confirm loss of Purkinje cells in the HD cases. Therefore, in all control individuals and HD patients, two calbindin immunostained tissue sections through identical levels of the vermal cerebellar portion and two calbindin immunostained tissue sections through identical levels of the hemispheral portion of the cerebellum were investigated using a wide‐field stereomicroscope at 40× magnification and blinded to the clinical diagnosis. A 3 × 3 mm transparent counting grid was superimposed onto the cerebellar tissue sections and starting at a random crossing point, all Purkinje cells with a clearly immunostained cytoplasm and proximal processes hitting the crossing points of the grid were counted. Differences in the Purkinje cells counted in four representative tissue sections were analyzed by means of the Mann–Whitney U‐test (BiAS for Windows, version 9.14, Epsilon, Darmstadt, Germany).
For the identification and subcellular localization of HD‐associated neuronal protein aggregates in the cerebellar cortex, deep nuclei and white matter, we applied the following antibodies to vermal and hemispheral cerebellar tissue sections from three control individuals and the following HD patients: the monoclonal anti‐polyglutamine antibody 1C2 (1:1000; Chemicon, Billerica, MA, USA) (patients 1–8; Table 1), which selectively detects polyglutamine sequences of 38 and more glutamines 59 and a monoclonal anti‐huntingtin antibody (1:1000; Chemicon, Billerica, MA, USA) (patients 1, 2 and 4; Table 1) 58. Primary incubation was performed for 20 h at room temperature and a secondary, biotin‐conjugated antibody was applied for 1.5 h at room temperature. The AB‐complex and 3,3‐diaminobenzidine‐tetra‐HCl/H2O2 were used to visualize positive immunoreactions. In each instance, 1C2 and huntingtin immunoreactions were examined in approximately six cerebellar tissue sections.
Recommended cerebral tissue sections from all HD patients were immunostained with the anti‐τ antibody AT8 (1:2000; Innogenetics, Ghent, Belgium) to highlight AD‐related τ cortical cytoskeletal burden 2, 5 and were used for the neuropathological classification of the AD‐related cortical cytoskeletal pathology according to the Braak and Braak AD staging procedure 5.
Results
Macroscopic aspects of the cerebellum in HD
Upon macroscopical inspection, the following abnormalities were observed in the HD brains: atrophy of the frontal and temporal lobes (patients 1–8; Table 1), pericentral cortical atrophy (patients 1–8; Table 1), atrophy of the occipital lobe (patients 1, 2, 4–8; Table 1), atrophy of the cerebellum (patients 1–8; Table 1; Figure 1) and atrophy of the striatum (patients 1–8; Table 1). The extent of striatal atrophy of the HD patients ranged from Vonsattel grades 2–4 60, 61, 62 (Table 1).
Figure 1.
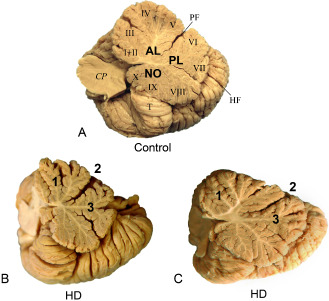
Macroscopic aspects of the cerebellum in Huntington's disease (HD). A. Medial view of the right cerebellar hemisphere of a 68‐year‐old male control individual. B. Atrophic right cerebellar hemisphere of a female HD patient (age at death, 68 years; onset of HD, 40 years; number of CAG‐repeats in the mutated HD allele, 46) (HD patient 5; Table 1): reduced arbor vitae, atrophic lobules of the anterior (1) and posterior lobes (3), and widened primary fissure (2). C. Atrophic right cerebellar hemisphere of a male HD patient (age at death, 68 years; onset of HD, 60 years; number of CAG‐repeats in the mutated HD allele, 40) (HD patient 6; Table 1): reduced arbor vitae, atrophic lobules of the anterior (1) and posterior lobes (3), and widened primary fissure (2). AL = anterior lobe; lobules I, II and III = lingula and central lobule; lobules IV and V = culmen; lobule VI = declive; lobule VII = tuber and folium; lobule VIII = pyramis; lobule IX = uvula; lobule X ‐= nodule; CP = cerebellar peduncles; NO = nodule; PL = posterior lobe; HF = horizontal fissure; PF = primary fissure; T = cerebellar tonsil.
Microscopic cerebellar findings in HD
Light microscopic examination disclosed a widespread neuron loss in the cerebellar cortex and deep nuclei of all eight HD patients studied (Tables 1, 2, 3, 4, 5, 6; Figures 2, 3, 4, 5, 6).
Figure 2.

The cerebellar Purkinje cell layer in Huntington's disease (HD). A. Sagittal section through the vermal portion of lobule X of a 91‐year‐old male HD patient (CAG‐repeats in the mutated HD allele, 42; age at HD onset, 55 years; duration of HD, 36 years; Vonsattel grade of striatal atrophy, 2; patient 8; Tables 1 and 3): intact Purkinje cell layer without loss of these cerebellar output neurons. Framed area is depicted at higher magnification at top left and shows axonal torpedo‐like inclusion. B. Sagittal section through the vermal portion of lobule V of a 70‐year‐old female HD patient (CAG‐repeats in the mutated HD allele, 44; age at HD onset, 50 years; duration of HD, 20 years; Vonsattel grade of striatal atrophy, 2; patient 7; Tables 1 and 3). The Purkinje cell layer is well preserved. These cerebellar nerve cells, however, show torpedo‐like inclusions in their axons (framed area and inset at top left). C. Sagittal section through the hemispheral portion of lobule VI of a 65‐year‐old female HD patient (CAG‐repeats in the mutated HD allele, 45; age at HD onset, 53 years; duration of HD, 12 years; Vonsattel grade of striatal atrophy, 4; patient 4; Tables 1 and 4). Intact segments of the Purkinje cell layer (asterisk) are intermingled with sections showing severe Purkinje cell loss (arrow). D. Sagittal section through the hemispheral portion of lobule VI of a 91‐year‐old male HD patient (CAG‐repeats in the mutated HD allele, 42; age at HD onset, 55 years; duration of HD, 36 years; Vonsattel grade of striatal atrophy, 2; patient 8; Tables 1 and 4): intact segments of the Purkinje cell layer (asterisk) and degenerated segments with severe loss Purkinje cells (arrow). E. Sagittal section through the hemispheral portion of lobule VI of a 91‐year‐old male HD patient (CAG‐repeats in the mutated HD allele, 42; age at HD onset, 55 years; duration of HD, 36 years; Vonsattel grade of striatal atrophy, 2; patient 8; Tables 1 and 4): along with intact Purkinje cell layer segments (asterisk) portions with severe loss of Purkinje cells (arrow) prevail. F. Sagittal section through the hemispheral portion of lobule IX of a 91‐year‐old male HD patient (CAG‐repeats in the mutated HD allele, 42; age at HD onset, 55 years; duration of HD, 36 years; Vonsattel grade of striatal atrophy, 2; patient 8; Tables 1 and 4): portions with no Purkinje cell loss, but dendritic alterations (asterisk) alternate with regions affected by severe loss of Purkinje nerve cells (arrow). Note the remaining and pathologically altered dendritic trees (arrowheads). G,H. Remaining Purkinje cells in the vermal portion of the cerebellum of a 91‐year‐old male HD patient (CAG‐repeats in the mutated HD allele, 42; age at HD onset, 55 years; duration of HD, 36 years; Vonsattel grade of striatal atrophy, 2; patient 8; Table 1) with fine and granular dispersed cytoplasmic protein aggregates (asterisks), as well as dot‐like neuronal intranuclear inclusions (arrows). (A–F) Anti‐calbindin immunostaining; (G,H) 1C2 immunostaining; 100 μm polyethylene glycol sections. GL = granular layer of the cerebellum; ML = molecular layer of the cerebellum.
Figure 3.
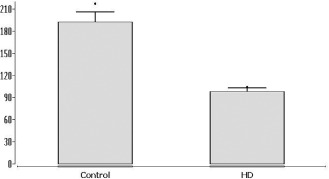
Number of cerebellar Purkinje cells in Huntington's disease (HD). Number of calbindin immunostained cerebellar Purkinje cells (bar, mean; whisker, standard deviation) counted at identical levels of the vermal and hemispheral portions of the cerebellum in the control individuals (control) and HD patients. Controls, 192 ± 44 immunolabeled Purkinje cells (range, 173–217); HD, 100 ± 16 immunolabeled Purkinje cells (range, 88–104) (P < 0.0002; Mann–Whitney U‐test).
Figure 4.
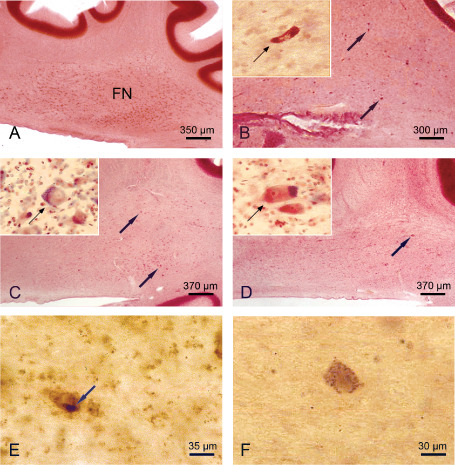
The cerebellar fastigial nucleus in Huntington's disease (HD). A. Sagittal section through the right cerebellum of a representative 82‐year‐old male control individual depicting the roof ridge‐like fastigial nucleus (FN). B. Complete neuronal loss in the FN of a 91‐year‐old male HD patient (CAG‐repeats in the mutated HD allele, 42; age at HD onset, 55 years; duration of HD, 36 years; Vonsattel grade of striatal atrophy, 2; patient 8; Tables 1 and 5). Arrows point to remaining FN nerve cells and inset shows severely shrunken FN nerve cell (arrow). C. Severe neuronal loss in the FN of a 70‐year‐old female HD patient (CAG‐repeats in the mutated HD allele, 44; age at HD onset, 50 years; duration of HD, 20 years; Vonsattel grade of striatal atrophy, 2; patient 7; Tables 1 and 5). Arrows mark remaining FN nerve cells and inset shows ballooned FN nerve cell (arrow): enlarged cytoplasm with basophilic center, peripheral displaced nucleus, Nissl substance and lipofuscin granules. D. The FN of a 65‐year‐old male HD patient with a severe loss of nerve cells (CAG‐repeats in the mutated HD allele, 45; age at HD onset, 53 years; duration of HD, 12 years; Vonsattel grade of striatal atrophy, 4; patient 4; Tables 1 and 5). Arrows indicate surviving FN nerve cells and inset shows ballooned surviving FN nerve cells (arrow). E. Surviving FN nerve cell of a 64‐year‐old male HD patient (CAG‐repeats in the mutated HD allele, 41; age at HD onset, 55 years; duration of HD, 9 years; Vonsattel grade of striatal atrophy, 2; patient 3; Table 1) showing a large compact cytoplasmic protein aggregate (arrow). F. Remaining FN nerve cell of a 68‐year‐old female HD patient (CAG‐repeats in the mutated HD allele, 46; age at HD onset, 40 years; duration of HD, 28 years; Vonsattel grade of striatal atrophy, 3; patient 5; Table 1) depicting granular cytoplasmic protein aggregates. (A–D) Aldehyde‐fuchsin Darrow red staining; (E,F) 1C2 immunostaining; 100 μm polyethylene glycol sections.
Figure 5.
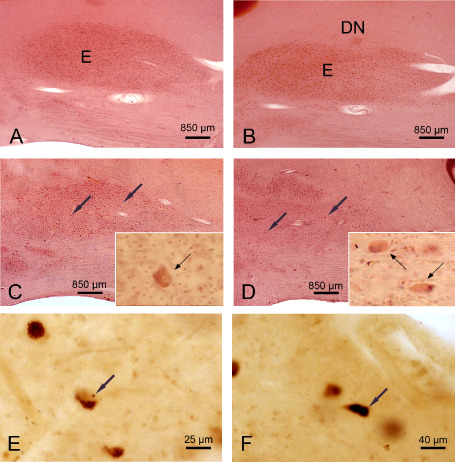
The cerebellar emboliform nucleus in Huntington's disease (HD). A. Sagittal section through the right cerebellum of a representative 72‐year‐old male control individual depicting the medial portion of the cigar‐shaped emboliform nucleus (E). B. Sagittal section through the right cerebellum of a typical 82‐year‐old male control individual depicting the lateral portion of the cigar‐shaped E and the most medial portion of the dentate nucleus (DN). C. Marked neuronal loss in the medial portion of the E of a 61‐year‐old male HD patient (CAG‐repeats in the mutated HD allele, 45; age at HD onset, 35 years; duration of HD, 26 years; Vonsattel grade of striatal atrophy, 3; patient 3; Tables 1 and 5). Arrows point to remaining E nerve and inset shows remaining ballooned E nerve cell (arrow): bloated cytoplasm with space‐consuming basophilic centre, peripheral displaced nucleus, Nissl material, and lipofuscin granules. D. Severe neuronal dropout in the lateral portion of the E of a 64‐year‐old male HD patient, as well as marked neuronal dropout in the most medial portion of its dentate nucleus (DN) (CAG‐repeats in the mutated HD allele, 41; age at HD onset, 55 years; duration of HD, 9 years; Vonsattel grade of striatal atrophy, 2; patient 3; Tables 1 and 5). Arrows points to remaining E nerve cells of this HD patient and inset shows ballooned surviving E nerve cells (arrows). E. Remaining E nerve cell of a 68‐year‐old female HD patient (CAG‐repeats in the mutated HD allele, 46; age at HD onset, 40 years; duration of HD, 28 years; Vonsattel grade of striatal atrophy, 3; patient 5; Table 1) with an intranuclear inclusion (arrow) and granular cytoplasmic aggregates. F. Remaining E nerve cell of the same HD patient with a compact cytoplasmic inclusion (arrow). (A–D) Aldehyde‐fuchsin Darrow red staining; (E,F) 1C2 immunostaining; 100 μm polyethylene glycol sections.
Figure 6.

The cerebellar dentate nucleus in Huntington's disease (HD). A. Sagittal section through the right cerebellum of 82‐year‐old male control individual with no history of neuropsychiatric diseases depicting the folded dentate nucleus (DN). B. Marked neuronal loss of the DN of a 64‐year‐old male HD patient CAG‐repeats in the mutated HD allele, 41; age at HD onset, 55 years; duration of HD, 9 years; Vonsattel grade of striatal atrophy, 2; patient 3; Tables 1 and 5). Inset at top left points to surviving DN nerve cells: ballooned nerve cells with bloated cytoplasm and space‐consuming basophilic centre, peripheral displaced nucleus, Nissl material, and lipofuscin granules (arrows), extraneuronal lipofuscin granules (arrowheads). C. Considerable neuronal loss in the DN of a 61‐year‐old female HD patient (CAG‐repeats in the mutated HD allele, 45; age at HD onset, 35 years; duration of HD, 26 years; Vonsattel grade of striatal atrophy, 3; patient 2; Tables 1 and 5). Inset at top left shows remaining nerve cells: ballooned nerve cells (arrows), extraneuronal lipofuscin granules (arrowheads). D. Remaining DN nerve cell of a 70‐year‐old female HD patient (CAG‐repeats in the mutated HD allele, 44; age at HD onset, 50 years; duration of HD, 20 years; Vonsattel grade of striatal atrophy, 2; patient 4; Table 1) with a compact cytoplasmic inclusion (arrow). E. Surviving DN nerve cell of the same HD patient with granular cytoplasmic inclusions. (A–C) Aldehyde‐fuchsin Darrow red staining; (D,E) 1C2 immunostaining; 100 μm polyethylene glycol sections.
The eight cerebella showed widespread cortical and deep nuclei neuronal loss. The Purkinje cell layer was selectively and diffusely affected throughout the lobules. In contrast, the molecular and granular cell layers were spared (Tables 1, 3, 4; Figure 2A–F). In all HD patients, loss of cerebellar Purkinje cells was present in all lobules of the vermis and cerebellar hemispheres and was rated as marked or severe (Tables 1, 3, 4; Figure 2A–F). Typically, however, loss of Purkinje cells was not evenly distributed over the affected cerebellar lobules. Instead, it was rather present at circumscribed predilections sites, which were intermingled between well‐preserved and unremarkable segments of cerebellar lobules (Figure 2C–F). Surviving Purkinje cells often appeared ballooned or shrunken. In all HD patients, although torpedo‐like inclusions were observable in the axons of surviving Purkinje cells of the vermal as well as hemispheral cerebellar portions (Figure 2A,B), the overall frequency of these cerebellar axonal inclusions was low. Although rare, torpedoes were found in all HD cerebella. Quantitative analysis clearly confirmed loss of Purkinje cells in our HD patients. While 192 ± 44 (range, 173–217) calbindin immunostained Purkinje cells were counted in the control individuals only 100 ± 16 (range, 88–104) immunolabeled Purkinje cells were present in the HD patients (P < 0.0002; Mann–Whitney U‐test; Figure 3).
The four deep cerebellar (ie, fastigial, globose, emboliform and dentate nuclei) of all HD patients underwent a marked to serious neuronal loss (patients 1–8; Tables 1 and 5; Figures 4A–D, 5A–F and 6A–C). In all patients, the fastigial nucleus was most severely affected and nearly devoid of nerve cells (Tables 1 and 5; Figure 4A–D). Most neurons left were either swollen or shrunken (Figure 4B–D). These ballooned fastigial nerve cells had a rounded and massively enlarged cytoplasm with a homogenous central basophilic substance. Their Nissl substance, lipofuscin granules and flattened nucleus were concentrated along the cytoplasmic membrane mimicking central chromatolysis (Figure 4C,D). The shrunken fastigial neurons displayed a pale, slender and arrowhead‐like cytoplasm. Their central and pale nucleus was broader than and stretched the abnormal cytoplasm, which was nearly devoid of Nissl substance (Figure 4B). Also the emboliform nucleus was consistently affected and underwent a severe neuronal dropout in the majority of the HD patients (Table 5; Figure 5A–D). As with the affected fastigial nucleus, the majority of emboliform neurons were ballooned or shrunken (Figure 5C,D). Although neuronal loss in the globose and dentate nuclei of most of the HD patients was less severe than in their fastigial or emboliform nuclei, both nuclei showed a clearly reduced nerve cell density and numerous surrounding lipofuscin granules in the neuropil (Table 5; Figure 6B,C). Lipofuscin granules are normal cytoplasmic constituents of nerve and glial cells and are structurally stable post‐mortem even in delayed or suboptimal fixation. Owing to the low affinity of glial and the high affinity of neuronal lipofuscin granules to the aldehyde‐fuchsin dye, lipofuscin granules in the neuropil can serve as reliable markers of the former position of lipofuscin‐laden neuronal perikarya that have disappeared from tissue 4. Along with a reduced nerve cell density and the presence of lipofuscin granules in the neuropil, the globose and dentate nuclei of all HD patients exhibited additional signs of neurodegeneration: a high percentage of their remaining nerve cells were ballooned or shrunken (Figure 6A–C).
Calculation of Kendalls's rank correlation coefficient τ revealed no significant nonparametric correlations between the extent of Purkinje cell loss and neuronal loss in the deep cerebellar nuclei of the HD patients on the one hand and the length of the CAG‐repeats in the mutated HD alleles, age at disease onset, age at death, duration of HD and the Vonsattel grade of striatal atrophy on the other (all P‐values > 0.10).
Despite careful and systematical investigation of serial cerebellar tissue sections, the remaining cerebellar nerve cells did not display morphological features (ie, chromatin condensation, nuclear fragmentation, apoptotic bodies) associated with the occurrence of apoptosis 17 in all HD patients studied here.
Abnormalities of the cerebellar white matter were observed only in two of our HD patients (patients 5, 7; Table 1). They included myelin pallor in the deep cerebellar white matter around the dentate nucleus (patients 5 and 7; Table 1) and in the peripheral portions of the cerebellar folia (patients 5; Table 1).
Associated tissue change
In contrast to the control cases, GFAP‐immunopositive astrocytes were present in all cerebellar components of all eight HD patients (Table 6). They were most frequent in the cerebellar white matter, but were also consistently present in the three layers of the cerebellar cortex, as well as in all four deep cerebellar nuclei (Table 6).
The cerebella of all control individuals studied were free of 1C2 and/or anti‐huntingtin immunopositive neuronal aggregates. In the cerebella of all HD patients, however, a wealth of 1C2 immunopositive neuronal aggregation was present. Nearly all of the remaining Purkinje cells showed fine and granular dispersed cytoplasmic protein aggregates, while dot‐like neuronal intranuclear inclusions (NI) occurred only occasionally (Figure 2G,H). In the granular cell layer and underlying white matter, numerous compact aggregates with presumed axonal localization were observed. In the deep cerebellar nuclei, the overwhelming majority of surviving nerve cells exhibited one of three types of aggregates. In the remaining nerve cells of the fastigial and globose nuclei compact cytoplasmic, granular cytoplasmic aggregates and NI were equally frequent (Figure 4E,F). In the nerve cells of the embolifom nucleus, compact cytoplasmic aggregates clearly dominated, while granular cytoplasmic aggregates and NI were only occasionally observed (Figure 5E,F). In the dentate nucleus, granular cytoplasmic aggregates or compact cytoplasmic aggregates were most prevalent, whereas NI were only rarely encountered (Figure 6D,E). The anti‐huntingtin immunostainings revealed a few compact neuropil aggregates in the molecular cell layer, numerous compact neuropil aggregates in the granule cell layer and deep white matter, as well as few NI in remaining nerve cells of the Purkinje cell layer, globose and dentate nuclei.
The distribution and severity of the accompanying AD‐related cortical τ cytoskeletal pathology in the HD patients corresponded to Braak and Braak stages 0 (patients 5, 7; Table 1), I (patient 3, Table 1), II (patients 1, 2; Table 1) or III (patients 4, 6, 8; Table 1).
Discussion
Degeneration of the cerebellum in and pathoanatomy of HD
The affection of the cerebellum during HD has been controversially discussed for several decades of neuropathological HD research 6, 11, 25, 46, 47, 48, 61, 62. In the present HD study with thick serial tissue sections, we could for the first time unequivocally show that the cerebellum, an important somatomotor and oculomotor brain component of the posterior cranial fossa, undergoes multiple neurodegenerative features in clinically diagnosed and genetically confirmed HD patients. According to our findings, cerebellar degeneration in HD is not confined to the Purkinje cell layer and/or the dentate nucleus as previously assumed 6, 25, 46, 47, 48, 49, 61, 62. Instead, our study has revealed that the features of cerebellar neurodegeneration in our HD patients comprise not only macroscopically visible atrophy of the cerebellum and widespread loss of Purkinje cells, but also considerable and consistent demise of nerve cells in all deep cerebellar nuclei (ie, fastigial, globose, emboliform and dentate nucleus). Associated with, in part, other genetical, molecular biological and clinical features, the polyglutamine disease HD according to our findings shows cerebellar degeneration that is highly reminiscent to cerebellar damage occurring in autosomal dominantly inherited polyglutamine ataxias (eg, spinocerebellar ataxias types 1, 2 and 3). Accordingly, HD represents a multisystem degenerative disease affecting the striatum, cerebral neo‐and allocortex, thalamus and cerebellum 3, 21, 22, 25, 30, 31, 38, 60, 61, 62 and thus appears to have more neuropathological similarities with these polyglutamine ataxias than previously thought 33, 44, 50, 51. The atrophy of the cerebellum and neuronal loss in the cerebellar Purkinje cell layer and deep cerebellar nuclei was even present in our HD patients who showed initial and still mild Vonsattel grade 2 striatal atrophy (patients 3, 7, 8; Tables 1, 3, 4, 5, 6) that did not correlate with the Vonsattel grade of striatal atrophy. These findings suggest that damage to the cerebellum is an early event during the pathological process of HD and does not develop simultaneously with, but independently, from the well‐known pathognomonic lesions in the striatum. Our neuropathological findings conform to results from recent in vivo magnetic resonance imaging (MRI) studies 11, 48, but are at variance with the current opinion that the cerebellum becomes affected only during the terminal phases of striatal degeneration (ie, Vonsattel grades 3 and 4) 60, 61, 62. Moreover, in view of the independent development of cerebellar damage, it is conceivable that the pathological process of HD does not primarily and selectively affect the striatum and subsequently other brain grays in a second step, but is characterized by a polytopic or multifocal onset 60. This means that the striatum is not the sole induction site of the underlying pathological process.
Degeneration of the cerebellum and its possible relevance for the clinical symptoms of HD
The human cerebellum is crucial for the initiation and termination of movements, for the smoothly integrated coordination and fine adjustment of limb movements, trunk and eye movements, as well as for the regulation of posture and balance, by adjusting the programs of the major descending cortical and subcortical motor brain systems (ie, pyramidal system, corticopontine tract) 14, 16, 65. Its flocculonodular lobe (ie, vestibulocerebellum) governs body equilibrium by controlling axial muscles via its afferent and efferent connections, its vermal and paravermal region (ie, spinocerebellum) play a major role in controlling the ongoing execution of limb movements and its hemispheral portion (ie, pontocerebellum) is essentially involved in initiating, planning and timing of movements 14, 16, 63, 65. Damage to these three phylogenetically and functionally defined longitudinal cerebellar zones has been linked to the occurrence of distinct characteristic clinical syndromes: (i) damage to the flocculonodular lobe is correlated with disturbances of equilibrium, gait ataxia, wide‐based standing and gait position; (ii) damage to the vermal and paravermal portion of the cerebellum and the fastigial nucleus is correlated with dysarthria, impairment of axial and truncal control, gait and stance ataxia, hypotonia, imbalance as well as reduced activity of gamma motor neurons of the spinal cord; and (iii) damage to the hemispheral portion of the cerebellum and globose, emboliform and/or dentate nucleus is correlated with ataxia, delay in initiating and terminating movements, hypotonia, dysmetria, dysdiadochokinesia, dysarthria, disorders of movement coordination and impaired fine movements of the hands and fingers 14, 16, 45, 63.
The pathological affection of the cerebellum has been discussed for several decades 6, 11, 25, 46, 47, 48, 61, 62, but only a few clinical reports of cerebellar dysfunctions are currently available in HD patients. Although rare, the clinical reports in existence, provide suitable arguments for the validity of our concept about cerebellar affection in HD. Thus, clinical HD studies have demonstrated that minor motor abnormalities including impairments of rapid alternating movements and fine motor skills of the upper limbs may precede the clinical manifestation of the well‐known extrapyramidal symptoms by some years and may already be observed in patients in the very early clinical phases of HD, as well as in at‐risk individuals 29, 68. Furthermore, a distinctive pattern denominated as ‘Huntington's ataxia’ that includes dysarthria, postural instability and ataxia, as well as gait abnormalities (ie, impaired gait balance, lateral swaying, broad‐based gait and stance) have also been identified upon neurological examination in patients suffering from HD 27, 34. In these patients, gait disturbances partially emerged independently from choreatic movements and ultimately resulted in severe disability and frequent falls 29. The medical reports of our HD patients are in agreement with these clinical studies and mentioned dysarthria, gait imbalance, broad‐based gait, imbalance, as well as impaired fine motor skills of the upper limbs as early HD symptoms. In view of the current knowledge about the neurobiology and pathophysiology of the cerebellum, the aforementioned disease symptoms indicate that the cerebellum is indeed affected in HD and support our view that cerebellar damage occurs early during HD and independently from striatal degeneration. Along with the neuropathological findings, the described disease symptoms provide additional clinical evidence that the polyglutamine disease HD is more closely related to the polyglutamine ataxias than believed to date.
Impairments of smooth pursuit eye movements are well‐known oculomotor dysfunctions of HD patients 32. However, the morphological correlate of these dysfunctions still remain unclear 52. The lobules VI and VII of the vermal portion of the cerebellum and the fastigial nuclei form the so‐called “oculomotor vermis” and are concerned with the control and adaption of smooth pursuit eye movements 23, 45, 64, 66. The consistent loss of Purkinje cells in these cerebellar lobules and the severe neuronal loss in the fastigial nucleus detected in our HD patients for the first time provide a plausible explanation for impaired smooth pursuits in HD patients. The fastigial nucleus also controls the neural activity of the gamma motor neurons in the spinal cord and drives the fusimotor and afferent activity of the muscle spindles via its descending projections in the reticulospinal tract. Loss of these descending projections can explain the hypotonia of HD patients as well as the impairments of their Hoffmann reflex 8, 28, 35, 37, 39, 70. The observation that the impairment of this reflex occurs early during the course of HD again supports our concept of an early affection of the cerebellum in HD.
Degeneration of the cerebellum and its potential gateway to the pathological mechanisms underlying HD
Despite systematical investigation of serial tissue sections from our HD patients, we found no signs of apoptotic cell death in cerebellar nerve cells. These findings conform to recent HD studies suggesting that apoptosis does not play a significant role for neurodegeneration occurring in HD patients or in transgenic HD models 15, 17, 42. They rather point to other mechanisms of nerve cell death in HD. The vast majority of remaining cerebellar nerve cells of our HD patients was morphologically abnormal and appeared either as ballooned or shrunken nerve cells. They were surrounded by lipofuscin granules in the neuropil marking the former position of declined lipofuscin‐laden nerve cells 4. In view of these consistent observations in both the cerebellar cortex and the deep cerebellar nuclei of our HD patients, we suggest the following sequence or phases of morphological changes associated with the demise of cerebellar nerve cells in HD. Subsequent to initial and unknown pathological events that cannot yet be detected light microscopically (phase 1), cerebellar nerve cells ballooned (phase 2), lost the balance between pathological and protective mechanisms competing in these vulnerable nerve cells, underwent—after exhaustion of their protective mechanisms—loss of cellular organelles (ie, mitochondria, Nissl substance) (phase 3) and severe shrinkage of their cytoplasm (phase 4), died ultimately (phase 5) and left structurally stable lipofuscin granules in the neuropil, which represent reliable markers of the former position of demised nerve cells (phase 6).
Although, we described the widespread occurrence of ballooned nerve cells for the first time in HD brains, these morphologically abnormal nerve cells cannot be regarded as pathognomonic for HD. Occurring in a large quantity at multiple brain sites ballooned nerve cells are established, but not disease‐specific neuropathological, features of (i) AD; (ii) argyrophilic grains disease (AGD); (iii) corticobasal degeneration (CBD); and (iv) progressive supranuclear palsy (PSP) 10, 13, 18, 26, 55. The massive enlargement of the perikarya of the neurons is linked to structural damage to their axons and/or functional impairment of intraaxonal mechanisms 13, 26. Axonal pathologies observed in HD brains along with the occurrence of torpedo‐like inclusions described in the present study also include the presence of axonal aggregates of the disease protein huntingtin in nerve cells of the cerebral cortex and striatum 9, 15, 19, 49. Concentration of mutant huntingtin within axonal aggregates may point to a strain of the neuronal protein quality control mechanisms in HD leading to an ineffective proteasomal elimination of this abnormal protein 7, 15, 19, 49. In view of the axonal pathologies occurring in the brain of HD patients and the known association between axonal damage and the appearance of ballooned nerve cells, it appears promising to study the possible consequences of ineffective proteasomal elimination of huntingtin for the occurrence of axonal pathologies, as well as the role of the HD‐associated axonal pathology for the pathogenesis of ballooned nerve cells in more detail. Clarification of the significance and role of this axonal pathology may lead to the identification of early and crucial steps in the cascades of pathological events that lead to destabilization, enlargement and ultimately death of susceptible nerve cells in HD. These attempts may help to develop concrete and empirically based ideas about the pathological mechanisms resulting in the destruction of nerve cells in HD. In search for such mechanisms, it should be remembered that ballooned neurons occurring in other neurodegenerative diseases show an upregulation and an increased amount of cytoplasmic heat shock proteins including alpha beta crystallin 13. Heat shock proteins are important components of the neuronal protein quality control machinery, facilitate refolding of abnormal proteins and prevent their neuronal aggregation 13, 42, 49. Therefore, further biochemical investigations should also consider the presence of cytoplasmic heat shock proteins in ballooned neurons of HD tissue. Identification of upregulated heat shock proteins as a protective mechanism in HD may be an initial step on the way to influence their cytoplasmic level by means drugs and may pave the way for novel therapeutic strategies providing external intervention into the pathological cascades leading to nerve cell death in HD.
Mitochondria are further structural components that may accumulate in ballooned nerve cells and contribute to the massive enlargement and change in shape of the affected neuronal perikarya. The accumulation of mitochondria, which most probably results from an impaired anterograde axonal transport, by axonal protein aggregates providing a suitable explanation for the disturbed energy situation described in HD patients 15, 19, 40, 41, 49. Therefore, improvement of the protein quality control machinery may also facilitate the anterograde transport, improve the energy situation in HD and eventually elicit positive effects on the progression of HD.
In conclusion, our neuropathological study clearly demonstrates neuronal degeneration in the cerebellum as a consistent feature of HD. In initial stages, the cerebellar pathology may be mild and thus cause only discrete disease symptoms that may be masked by the more prominent disease symptoms (eg, chorea, dystonia, rigidity, dysarthria, weight loss). These findings should be taken into consideration by clinicians as well as by researchers investigating experimental animal models for HD.
Disclosure Statement
All authors have no actual or potential conflicts of interest to disclose, including financial, personal, or other relationships with other people or organizations, within three years of beginning the work submitted.
Acknowledgments
This study was supported by grants from the Dr. Senckenbergische Stiftung (Frankfurt/Main, Germany) and the Deutsche Huntington‐Hilfe e.V.. The skillful assistance of M. Babl, M. Bouzrou (tissue processing and immunohistochemistry) and D. von Meltzer (secretary) is thankfully acknowledged. This work is dedicated to Christiane Lohkamp and her family.
Udo Rüb and Franziska Hoche are joint first authors.
References
- 1. Bortz J, Lienert GA, Boehnke K (1990) Verteilungsfreie Methoden in der Biostatistik. Springer: Berlin. [Google Scholar]
- 2. Braak E, Braak H, Mandelkow EM (1994) A sequence of cytoskeleton changes related to the formation of neurofibrillary tangles and neuropil threads. Acta Neuropathol 87:554–567. [DOI] [PubMed] [Google Scholar]
- 3. Braak H, Braak E (1992) The human entorhinal cortex: normal morphology and lamina‐specific pathology in various diseases. Neurosci Res 15:6–31. [DOI] [PubMed] [Google Scholar]
- 4. Braak H, Rüb U, Del Tredici K (2003) Involvement of precerebellar nuclei in multiple system atrophy. Neuropathol Appl Neurobiol 29:60–76. [DOI] [PubMed] [Google Scholar]
- 5. Braak H, Alafuzoff I, Arzberger T, Kretzschmar H, Del Tredici K (2006) Staging of Alzheimer disease‐associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol 112:389–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bruyn GW, Bots GTAM, Dom R (1979) Huntington's chorea: current neuropathological status. Adv Neurol 23:83–93. [Google Scholar]
- 7. Davies JE, Sarkar S, Rubinsztein DC (2007) The ubiquitin proteasome system in Huntington's disease and the spinocerebellar ataxias. BMC Biochem 8(Suppl.):1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Deuschl G, Lücking CH, Schenck E (1989) Hand muscle reflexes following electrical stimulation in choreatic movement disorders. J Neurol Neurosurg Psychiatry 52:755–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. De Vos KJ, Grierson AJ, Ackerley S, Miller CCJ (2008) Role of axonal transport in neurodegenerative diseases. Annu Rev Neurosci 31:1151–1173. [DOI] [PubMed] [Google Scholar]
- 10. Dickson DW (1999) Neuropathologic differentiation of progressive supranuclear palsy and corticobasal degeneration. J Neurol 246(Suppl. 2):6–15. [DOI] [PubMed] [Google Scholar]
- 11. Fennema‐Notestine C, Archibald SL, Jacobson MW, Corey‐Bloom J, Paulsen JS, Peavy GM et al (2004) In vivo evidence of cerebellar atrophy and cerebral white matter loss in Huntington disease. Neurology 63:989–995. [DOI] [PubMed] [Google Scholar]
- 12. Folstein SE (1989) Huntington's Disease: A Disorder of Families. Johns Hopkins University: Baltimore. [Google Scholar]
- 13. Fujino Y, DeLucia MW, Davies P, Dickson DW (2004) Ballooned neurons in the limbic lobe are associated with Alzheimer type pathology and lack diagnostic specificity. Neuropathol Appl Neurobiol 30:676–682. [DOI] [PubMed] [Google Scholar]
- 14. Ghez C (1991) The cerebellum. In: Principles of Neural Science, 3rd edn. Kandel ER, Schwartz JH, Jessell TM (eds), pp. 596–607. Elsevier: New York. [Google Scholar]
- 15. Gil JM, Rego AC (2008) Mechanisms of neurodegeneration in Huntington's disease. Eur J Neurosci 27:2803–2820. [DOI] [PubMed] [Google Scholar]
- 16. Gilman S (1992) Cerebellum and motor dysfunction. In: Diseases of the Nervous System: Clinical Neurobiology, 2nd edn. Asbury AK, McKhann GM, McDonald WI (eds), pp. 368–389. Saunders: Philadelphia. [Google Scholar]
- 17. Graeber MB, Moran LB (2002) Mechanisms of cell death in neurodegenerative diseases: fashion, fiction, and facts. Brain Pathol 12:385–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Grinberg LT, Heinsen H (2009) Argyrophilic grain disease. An update on a frequent cause of dementia. Dement Neuropsych 3:2–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gunawardena S, Goldstein LSB (2005) Polyglutamine diseases and transport problems. Arch Neurol 62:46–51. [DOI] [PubMed] [Google Scholar]
- 20. Harper PS (1992) The epidemiology of Huntington's disease. Hum Genet 89:365–376. [DOI] [PubMed] [Google Scholar]
- 21. Heinsen H, Rüb U (1997) Quantitative investigations of neural circuits in Huntington's disease. J Neural Transm 104:1139–1140. [Google Scholar]
- 22. Heinsen H, Strik M, Bauer M, Luther K, Ulmar G, Gangnus D et al (1994) Cortical and striatal neurone number in Huntington's disease. Acta Neuropathol 88:320–333. [DOI] [PubMed] [Google Scholar]
- 23. Horn AK, Büttner U, Büttner‐Ennever JA (1999) Brainstem and cerebellar structures for eye movement generation. Adv Otorhinolaryngol 55:1–25. [DOI] [PubMed] [Google Scholar]
- 24. Hutchins B, Weber JT (1983) A rapid myelin stain for frozen sections: modification of the Heidenhain procedure. J Neurosci Methods 7:289–294. [DOI] [PubMed] [Google Scholar]
- 25. Jeste DV, Barban L, Parisi J (1984) Reduced Purkinje cell density in Huntington's disease. Exp Neurol 85:78–86. [DOI] [PubMed] [Google Scholar]
- 26. Kato S, Hirano A, Umahara T, Llena JF, Herz F, Ohama E (1992) Ultrastructural and immunohistochemical studies on ballooned cortical neurons in Creutzfeldt‐Jakob disease: expression of αB‐crystallin, ubiquitin and stress‐response protein 27. Acta Neuropathol 84:443–448. [DOI] [PubMed] [Google Scholar]
- 27. Koller WC, Trimble J (1985) The gait abnormality of Huntington's disease. Neurology 35:1450–1454. [DOI] [PubMed] [Google Scholar]
- 28. Knikou M (2008) The H‐reflex as a probe: pathways and pitfalls. J Neurosci Methods 171:1–12. [DOI] [PubMed] [Google Scholar]
- 29. Kremer B, Weber B, Hayden MR (1992) New insights into the clinical features, pathogenesis and molecular genetics of Huntington disease. Brain Pathol 2:321–335. [DOI] [PubMed] [Google Scholar]
- 30. Lange H, Thorner G, Hopf A, Schroder KF (1976) Morphometric studies of the neuropathological changes in choreatic diseases. J Neurol Sci 28:401–425. [DOI] [PubMed] [Google Scholar]
- 31. Lange HW (1981) Quantitative changes of telencephalon, diencephalon, and mesencephalon in Huntington's chorea, postencephalitic, and idiopathic Parkinsonism. Verh Anat Ges 73:923–925. [Google Scholar]
- 32. Lasker AG, Zee DS (1997) Ocular motor abnormalities in Huntington's disease. Vision Res 37:3639–3645. [DOI] [PubMed] [Google Scholar]
- 33. Lastres‐Becker I, Rüb U, Auburger G (2008) Spinocerebellar ataxia 2 (SCA2). Cerebellum 7:115–124. [DOI] [PubMed] [Google Scholar]
- 34. Margolis RL, Ross CA (2003) Diagnosis of Huntington disease. Clin Chem 49:1726–1732. [DOI] [PubMed] [Google Scholar]
- 35. McLeod JG (1969) H‐reflex studies in patients with cerebellar disorders. J Neurol Neurosurg Psychiatry 32:21–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Mink JW (1996) The basal ganglia: focused selection and inhibition of competing motor programs. Prog Neurobiol 50:381–425. [DOI] [PubMed] [Google Scholar]
- 37. Misiaszek JE (2003) The H‐reflex as a tool in neurophysiology: its limitations and uses in understanding nervous system function. Muscle Nerve 28:144–160. [DOI] [PubMed] [Google Scholar]
- 38. Myers RH, Vonsattel JP, Stevens TJ, Cupples LA, Richardson EP, Martin JB, Bird ED (1988) Clinical and neuropathologic assessment of severity in Huntington's disease. Neurology 38:341–347. [DOI] [PubMed] [Google Scholar]
- 39. Oepen G, Hillesheimer W (1981) Qualitative H‐reflex testing in Huntington's disease. Arch Psychiatr Nervenkr 230:221–226. [DOI] [PubMed] [Google Scholar]
- 40. Oliveira JMA (2010) Nature and cause of mitochondrial dysfunction in Huntington's disease: focusing on huntingtin and the striatum. J Neurochem 114:1–12. [DOI] [PubMed] [Google Scholar]
- 41. Pandey M, Mohanakumar KP, Usha R (2010) Mitochondrial functional alterations in relation to pathophysiology of Huntington's disease. J Bioenerg Biomembr 42:217–226. [DOI] [PubMed] [Google Scholar]
- 42. Pattison LR, Kotter MR, Fraga D, Bonelli RM (2006) Apoptotic cascades as possible targets for inhibiting cell death in Huntington's disease. J Neurol 253:1137–1142. [DOI] [PubMed] [Google Scholar]
- 43. Petersen A, Gil J, Maat‐Schieman ML, Bjorkqvist M, Tanila H, Araujo IM et al (2005) Orexin loss in Huntington's disease. Hum Mol Genet 14:39–47. [DOI] [PubMed] [Google Scholar]
- 44. Riess O, Rüb U, Pastore A, Bauer P, Schöls L (2008) SCA3: neurological features, pathogenesis and animal models. Cerebellum 7:125–137. [DOI] [PubMed] [Google Scholar]
- 45. Robinson FR, Fuchs AF (2001) The role of the cerebellum in voluntary eye movements. Annu Rev Neurosci 24:981–1004. [DOI] [PubMed] [Google Scholar]
- 46. Rodda RA (1981) Cerebellar atrophy in Huntington's disease. J Neurol Sci 50:147–157. [DOI] [PubMed] [Google Scholar]
- 47. Roos RAC (1986) Neuropathology of Huntington's chorea. In: Handbook of Clinical Neurology Vol. 5. Extrapyramidal Disorders, Vinken PJ, Bruyn GW, Klawans HL (eds), pp. 315–325. Elsevier Science Publishers: Amsterdam. [Google Scholar]
- 48. Rosas HD, Koroshetz WJ, Chen YI, Skeuse C, Vangel M, Cudkowicz ME, Kaplan K (2003) Evidence for more widespread cerebral pathology in early HD: an MRI‐based morphometric analysis. Neurology 60:1615–1620. [DOI] [PubMed] [Google Scholar]
- 49. Ross CA, Tabrizi SJ (2011) Huntington's disease: from molecular pathogenesis to clinical treatment. Lancet Neurol 10:83–98. [DOI] [PubMed] [Google Scholar]
- 50. Rüb U, Brunt ER, Gierga K, Seidel K, Schultz C, Schöls L et al (2005) Spinocerebellar ataxia type 7 (SCA7): first report of a systematic neuropathological study of the brain of a patient with a very short expanded CAG‐repeat. Brain Pathol 15:287–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Rüb U, Brunt ER, Deller T (2008) New insights into the pathoanatomy of spinocerebellar ataxia type 3 (Machado‐Joseph disease). Curr Opin Neurol 21:111–116. [DOI] [PubMed] [Google Scholar]
- 52. Rüb U, Heinsen H, Brunt ER, den Landwehrmeyer B, Dunnen WF, Gierga K, Deller T (2009) The human premotor oculomotor brainstem system—can it help to understand oculomotor symptoms in Huntington's disease? Neuropathol Appl Neurobiol 35:4–15. [DOI] [PubMed] [Google Scholar]
- 53. Schmahmann JD, Doyon J, McDonald D, Holmes C, Lavoie K, Hurwitz AS et al (1999) Three‐dimensional MRI atlas of the human cerebellum in proportional stereotaxic space. Neuroimage 10:233–260. [DOI] [PubMed] [Google Scholar]
- 54. Smithson KG, MacVicar BA, Hatton GI (1983) Polyethylene glycol embedding: a technique compatible with immunocytochemistry, enzyme histochemistry, histofluorescence and intracellular staining. J Neurosci Methods 7:27–41. [DOI] [PubMed] [Google Scholar]
- 55. Stover NP, Watts RL (2001) Corticobasal degeneration. Semin Neurol 21:49–58. [DOI] [PubMed] [Google Scholar]
- 56. The Huntington's Disease Collaborative Research Group (1993) A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. Cell 72:971–983. [DOI] [PubMed] [Google Scholar]
- 57. Thorner GW, Lange HW, Hopf A (1977) Quantitative studies of human basal ganglia in extrapyramidal motor disorders. Verh Anat Ges 71:99–101. [PubMed] [Google Scholar]
- 58. Trottier Y, Devys D, Imbert G, Saudou F, An I, Lutz Y et al (1995) Cellular localization of the Huntington's disease protein and discrimination of the normal and mutated form. Nat Genet 10:104–110. [DOI] [PubMed] [Google Scholar]
- 59. Trottier Y, Lutz Y, Stevanin G, Imbert G, Devys D, Cancel G et al (1995) Polyglutamine expansion as a pathological epitope in Huntington's disease and four dominant cerebellar ataxias. Nature 378:403–406. [DOI] [PubMed] [Google Scholar]
- 60. Vonsattel JP (2008) Huntington disease models and human neuropathology: similarities and differences. Acta Neuropathol 115:55–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Vonsattel JP, DiFiglia M (1998) Huntington disease. J Neuropathol Exp Neurol 57:369–384. [DOI] [PubMed] [Google Scholar]
- 62. Vonsattel JP, Myers RH, Stevens TJ, Ferrante RJ, Bird ED, Richardson EP Jr (1985) Neuropathological classification of Huntington's disease. J Neuropathol Exp Neurol 44:559–577. [DOI] [PubMed] [Google Scholar]
- 63. Voogd J (2003) The human cerebellum. J Chem Neuroanat 26:243–252. [DOI] [PubMed] [Google Scholar]
- 64. Voogd J, Barmack NH (2006) Oculomotor cerebellum. Prog Brain Res 151:231–268. [DOI] [PubMed] [Google Scholar]
- 65. Voogd J, Feirabend HKP, Schoen JHR (1990) Cerebellum and precerebellar nuclei. In: The Human Central Nervous System, Paxinos G (ed.), pp. 321–386. Academic Press: San Diego. [Google Scholar]
- 66. Voogd J, van der Schraa‐Tam CKL, Geest JN, De Zeeuw CI (2012) Visuomotor cerebellum in human and nonhuman primates. Cerebellum 11:392–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Walker FO (2007) Huntington's disease. Lancet 369:218–228. [DOI] [PubMed] [Google Scholar]
- 68. Walker FO (2007) Huntington's disease. Semin Neurol 27:143–150. [DOI] [PubMed] [Google Scholar]
- 69. Whitney ER, Kemper TL, Rosene DL, Bauman ML, Blatt GJ (2008) Calbindin‐D28k is a more reliable marker of human Purkinje cells than standard Nissl stains: a stereological experiment. J Neurosci Methods 168:42–47. [DOI] [PubMed] [Google Scholar]
- 70. Yanagisawa N (1992) The spectrum of motor disorders in Huntington's disease. Clin Neurol Neurosurg 94(Suppl.):182–184. [DOI] [PubMed] [Google Scholar]