

Abstract
The neuropathological hallmarks of Alzheimer's disease (AD) include senile plaques made of Aβ peptide, neurofibrillary tangles containing hyperphosphorylated tau protein and neuronal loss. The pro‐apoptotic kinase PKR can be activated by Aβ and can phosphorylate tau protein via GSK3β kinase activation. The activated form of PKR (pPKR) accumulates in affected neurons and could participate in neuronal degeneration in AD. The mechanism of abnormal PKR activation in AD is not elucidated but could be linked to the PKR activator PACT. PACT stainings, and levels were assessed in the brains of AD patients and in APP/PS1 knock‐in transgenic mice and in cell cultures exposed to stresses. We showed that PACT and pPKR colocalizations are enhanced in AD brains. Their levels are increased and correlated in AD and APP/PS1 knock‐in mice brains. In human neuroblastoma cells exposed to Aβ, tunicamycin or H2O2, PACT and pPKR concentrations are increased. PACT then PKR inhibitions indicate that PACT is upstream of PKR activation. Our findings demonstrate that PACT levels are enhanced in AD brains and could partly be caused by the action of Aβ. In addition, PACT participates in PKR activation. The PACT–PKR pathway represents a potential link between Aβ accumulation, PKR activation and tau phosphorylation.
Keywords: Alzheimer's disease, beta amyloid, neuronal death, PACT, PKR
INTRODUCTION
Senile plaques, neurofibrillary tangles, synaptic and neuronal loss are salient neuropathological features of Alzheimer's disease (AD) (11). The cause of the accumulation of Aβ is not completely elucidated, although a possible increase of Aβ peptide production has been proposed (28). According to the amyloid cascade hypothesis, the accumulation of Aβ peptide or its oligomers could be the initial event responsible for the neurotoxic consequences leading to neurodegeneration (14). However, the mechanisms of this neurotoxicity are unclear and are probably multiple. One of the abnormal pathways detected in AD brains is associated with increased activation of the double‐stranded RNA‐dependant protein kinase (PKR). PKR is an ubiquitous serine‐threonine kinase present at low constitutive levels in cells. It can be activated by dsRNA (virus), IFNγ, TNFα, heparin, PDGF and IL1 13, 27, 33, leading to the activation of different signaling pathways controlling several cellular functions, such as growth regulation, apoptosis and differentiation. The functional kinase domain of PKR contains two important phosphorylation sites, on threonine 446 (pPKRthr446) and threonine 451 (pPKRthr451). Autophosphorylation of PKR on these sites after homodimerization is essential for its activation. Once activated, PKR causes phosphorylation of eukaryotic initiation factor 2 alpha (eIF2α), leading to global inhibition of protein synthesis 13, 25.
In the absence of virus, PKR can be activated by several stresses such as oxidative stress, intracellular calcium or endoplasmic reticulum (ER) stress (10) via a direct protein activator called the PKR associated protein activator (PACT) in humans 23, 25 or via its murine homologue RAX (16). It has been shown that the activation of PKR by PACT under cell stress conditions, can be associated with the phosphorylation of PACT on serine 18 (2), or serine 246 and 287 (24), while Daher et al showed a protein‐protein interaction between PACT and TAR RNA‐binding protein (TRBP), a potent PKR modulator (9). Nevertheless, all studies showed that PACT needs to bind strongly to PKR in order to mediate its activation in vivo. The overexpression of PACT in HeLa cells induces PKR‐mediated apoptosis (22), and the ER‐stress inducer thapsigargin enhances PACT expression and PKR activation (16), suggesting that high cellular levels of PACT can mediate cell degeneration through PKR activation. A recent study showed that tunicamycin exposure can also produce cell apoptosis through the activation of the PACT/PKR pathway (30). At the opposite, PACT can possess a different effect facilitating cell proliferation 18, 26.
In AD brains, activated PKR has been identified in neuronal cytoplasm and in granulo‐vacuolar degeneration, as well as in neuronal nuclei and around senile plaques (5). In cell cultures, PKR can be activated by the Aβ peptide and pharmacological or genetic inhibitions of PKR attenuate Aβ neurotoxicity (21). PKR is also involved in the mechanism of tau phosphorylation and could represent a link between Aβ and tau (3). So far, little is known about mechanisms leading to PKR activation in AD brains. The aim of the present study was to explore the role of PACT in AD using human AD and APP/PS1Ki transgenic mice brains and and SH‐SY5Y neuroblastoma cell line. Our results revealed that PACT could play a major role in PKR activation related to AD.
MATERIALS AND METHODS
Patients and materials
Human brains
We obtained post‐mortem brains for neuropathological analysis from 14 patients (9 paraffin sections, 5 frozen samples) and 15 controls (8 paraffin sections, 7 frozen samples). For all human brains, consents were obtained for using post‐mortem tissues. The post‐mortem intervals never exceeded 24 h. Assessment of pathology associated with dementia was performed by examining histological sections of frontal, temporal, parietal and occipital lobes, corpus striatum, thalamus, midbrain, pons, medulla and cerebellum. Selected sections were immunostained for Aβ, tau and α‐synuclein. All these patients had an history of progressive dementia and satisfied National Institute of Neurological and Communicative Disorders and Stroke–Alzheimer's Disease and Related Disorders (NINCDS‐ADRDA) criteria for probable AD (19) and satisfied neuropathologic criteria for AD according the consortium to establish a registry for Alzheimer's disease protocol (1, 2).
Table 1.
Table shows epidemiological and neuropathological characteristics of Alzheimer's disease (AD) patients and controls brains for immunohistochemistry. The number of the patients, sex, age, post‐mortem interval (PMI) and Braak stages are mentioned.
| Patients | Age | Sex | PMI | Braak |
|---|---|---|---|---|
| AD1 | 79 | F | 3 | 6 |
| AD2 | 85 | F | 5 | 5 |
| AD3 | 95 | M | 8 | 5 |
| AD4 | 92 | F | 17 | 5 |
| AD5 | 87 | M | 7 | 5 |
| Control 1 | 80 | F | 10 | 1 |
| Control 2 | 84 | F | 7 | 2 |
| Control 3 | 97 | F | 8 | 2 |
| Control 4 | 93 | F | 6 | 2 |
| Control 5 | 82 | M | 5 | 2 |
| Control 6 | 78 | M | 12 | 1 |
| Control 7 | 69 | M | 11 | 1 |
Table 2.
Table shows epidemiological and neuropathological characteristics of Alzheimer's disease (AD) patients and controls brains used for immunoblotting. The number of the patients, sex, age, post‐mortem interval (PMI) and Braak stages are mentioned.
| Patients | Age | Sex | PMI | Braak |
|---|---|---|---|---|
| AD1 | 87 | M | 15 | 6 |
| AD2 | 58 | M | 8 | 6 |
| AD3 | 82 | M | 6 | 5 |
| AD4 | 96 | F | 10 | 5 |
| AD5 | 72 | M | 9 | 6 |
| AD6 | 81 | F | 11 | 5 |
| AD7 | 73 | F | 5 | 6 |
| AD8 | 83 | F | 15 | 5 |
| AD9 | 88 | F | 6 | 5 |
| Control 1 | 55 | M | 10 | 1 |
| Control 2 | 75 | M | 8 | 1 |
| Control 3 | 87 | F | 5 | 1 |
| Control 4 | 80 | F | 9 | 2 |
| Control 5 | 79 | F | 6 | 2 |
| Control 6 | 80 | F | 9 | 1 |
| Control 7 | 63 | M | 5 | 2 |
For immunohistochemistry, 4 µm sections of hippocampus and temporal cortex from paraffined human brains were provided by the Department of Pathology of Lariboisière Hospital. We examined the brains from 9 AD patients and 8 age‐matched controls (Table 2).
For immunoblotting, frozen brain samples (temporal) from five AD patients and seven age‐matched controls were provided by the Neuropsychiatry Department, University of Geneva, Geneva, Switzerland (Table 1).
Mice brains
All animal experiments were performed in accordance to the guidelines of the French Agriculture and Forestry Ministry for handling animals (decree 87 849, license A75‐05‐22). All transgenic mice and wild‐type were provided by Sanofi‐Aventis. This APP/PS1 knock‐in (APP/PS1 KI) transgenic mouse model carries homozygote M233T/L235P knocked‐in mutations in mouse presenilin‐1 and overexpresses mutated human Aβ amyloid precursor protein (APPSL). Aβx‐42 is the major form of Aβ species present in this model with progressive development of a complex pattern of N‐truncated variants and dimers, similar to those observed in AD brains. Furthermore, at 10 months of age, they display an extensive neuronal loss (>50%), which is already detectable at 6 months of age (4).
For mice study, we used PS1KI (ho)xAPPsl (he) and wild‐type mice. Brains homogenates of 9‐month‐old transgenic mice APP/PS1 KI (n = 6) and 4 age‐matched wild‐type were provided by Sanofi‐Aventis and were established as described in a previous report (4).
Cell cultures
SH‐SY5Y human neuroblastoma cell lines were grown at 37°C and 5% CO2 in complete medium composed of 45% MEM (Eagle's minimum essential medium) (Invitrogen, Carlsbad, CA, USA) containing 2 mM L‐glutamine and 45% F12 (Hams F12 medium, Invitrogen) enriched with 0.1 mM nonessential amino acid (Invitrogen), 1 mM sodium pyruvate (Invitrogen) and 10% fetal bovine serum (FBS) (Invitrogen) and 5% penicillin streptomycin.
Antibodies and reagents
Primary antibodies
PKR: Rabbit anti‐pPKRthr446 (Santa Cruz, Danvers, MA, USA), rabbit anti‐pPKRthr451 (Cell Signaling, Beverly, MA, USA), rabbit anti‐PKR (Cell Signaling), PACT: goat anti‐PACT (N20) (Santa Cruz), Rabbit anti‐Actin (Sigma, St. Louis, MO, USA), mouse anti‐αtubulin (Cell Signaling).
Secondary antibody
For immunohistochemistry, biotinylated antigoat (Abcam, Cambridge, MA, USA) and antirabbit (Vectastain®, Vector Laboratories, Burlingame, CA,USA).
For immunofluorescence, donkey antigoat Alexa Fluor® 488 (Invitrogen Laboratory) or donkey antirabbit Cy3 (Jackson Laboratory, Bar Harbor, Maine, USA) secondary antibodies.
For Western blot, IR Dye® 700DX conjugated antimouse IgG, IR Dye 800CW conjugated antigoat IgG and IR Dye 800CW conjugated antirabbit IgG (Rockland Immunochemical Inc., Gilbertsville, PA, USA).
Reagents
Inhibitor of PKR: The PRI (NeoMPS Polypeptide Laboratories, Strasbourg, France) can bind to the double‐stranded RNA binding site of PKR and prevent its activation (20). PRI peptide was dissolved in DMSO (dimethylsulfoxide).
Stress inducer: Tunicamycin (Tm) (Sigma) is an endoplasmic reticulum stress (ER stress) inducer that inhibits N‐linked glycosylation of lipids and tyrosine incorporation, which ultimately inhibit protein synthesis. Tunicamycin was dissolved in DMSO. We also used Aβ (1‐42) peptide, which is a well‐known inducer of endoplasmic reticulum stress and oxidative stress. We also used Aβ (42‐1) (Tocris, St. Louis, MO, USA) as control peptide. Aβ peptide was dissolved in water and incubated at 37°C for 24 to 48 h to form aggregates prior to use. Finally, to induce oxidative stress, cells were treated with hydrogen peroxide (H2O2) (Calbiochem, Darmstadt, Germany).
Others reagents: To block protein transcription, we used actinomycin D (1 µg/mL) (Calbiochem) that was dissolved in water. We blocked the proteasome using lactacystin (10 µM) (Sigma) that was dissolved in water
METHODS
Immunohistochemistry for humans brains
All human tissue sections (4 µm) were deparaffinized by incubating the sections in xylene for 10 minutes twice, followed by rehydration with descending concentrations of ethanol (100%, 95%, and 70% for 3 minutes each). Subsequently, the sections were heated in EDTA buffer at 96°C for 40 minutes to retrieve the epitopes, then treated with 3% hydrogen peroxide in distilled water. The brain sections were washed with Ventana reaction buffer then incubated with either pPKRthr451 antibody (dilution 1:200 in DAKO real agent, Dako France, Trappes, France) or PACT N20 (dilution 1:200 in DAKO real agent) overnight at 4°C. Sections were then incubated with either the antirabbit secondary antibody (dilution 1:100 in DAKO real agent) or the antigoat secondary antibody (dilution 1:100 in DAKO real agent) for 30 minutes at room temperature. Slides were then washed twice with Ventana reaction buffer (Ventana, Tucson, AZ, USA) and further incubated with streptavidin peroxidase. The stain was visualized using diaminobenzidine for 5 minutes. Sections were counterstained with hematoxylin for 1 minute mounted with Mowiol® (Invitrogen).
For double confocal immunohistochemistry, sections were incubated with secondary antibodies mentioned above, washed in PBS, mounted with Vectashield mounting medium with DAPI (Vector Laboratories, Burlingame, CA, USA).
Confocal imaging of humans brains sections
Confocal microscopy was performed using an Olympus Fluoview® FV10i confocal microscope. The MetaMorph® software (Roper Scientific, Ottobrun, Germany) was used for image acquisition.
Cells treatment
To induce stress, the cells were treated with tunicamycin 5 µg/mL from 1 h to 8 h or Aβ1‐42 peptide (20 µM) from 4 to 8 h or with 0.5 mM of hydrogen peroxide (H2O2) for 2 h or Aβ42‐1 for 4 h. When necessary, to inhibit the activation of PKR, cells were pretreated one hour (before tunicamycin or Aβ exposure) with the PKR inhibitor PRI peptide (50 µM). Treatments with this inhibitor were repeated every hour. When necessary, control cells were treated with DMSO.
To block transcription, cells were pretreated with actinomycin D for two hours before tunicamycin.
To selectively inhibit the proteasome, cells were pretreated with lactacystin (10 µM). When necessary, control cells were treated with DMSO.
Transfection: cell shRNA PACT
SH‐SY5Y were transfected either with shRNA PACT (5′‐GAACCAGCUUAAUCCUAUU‐3′) or with scramble PACT (5′‐CUACACGCCAGUUUAUUAA‐3) for 48 h using lipofectamine before the treatment of cells with tunicamycin.
Immunoblotting for human and mice brains and cell cultures
Mice brains were homogenized as previously described (4). Temporal cortices of human brains were homogenized with a Potter in Laemmli sample buffer 1:10 (wt/vol) containing 5% sodium dodecyl sulfate (SDS), protease and phosphatase inhibitors cocktail. The lysate was sonicated and then centrifuged at 15 000 g for 15 minutes at 4°C.
To prepare cell lysates for immunoblotting, cells were washed in PBS and then lysed in RIPA buffer containing 25 mM β‐glycerophosphate, 50 mM sodium fluoride, 2 mM sodium pyrophosphate, 1 mM sodium orthovanadate, protease inhibitors (Roche, Penzberg, Germany) and 0.1 mM calyculin (Sigma) as phosphatase inhibitors. They were sonicated and centrifuged at 20 000 G for 10 minutes.
The protein concentration in the supernatant was determined with Micro BCA™ Protein Assay kit (Thermo Scientific, Cergy‐Pontoise, France) using the manufacturer's protocol. The protein samples (20–30 µg for brains, 40–50 µg for cell samples) were used for Western blot analysis. The membranes were blocked in 5% milk in PBS then incubated with primary antibody (anti‐PKR 1/500 or anti‐PACT 1/200), then secondary antibody antirabbit or antigoat (1/5000). Bound proteins were visualized with the Odyssey® Imaging System (LI‐COR Biosciences, Lincoln, NE, USA), and quantified with MultiGauge software (Fuji France, Bois d'Arcy, France). Statistical analysis was performed using Graph Pad Prism version 5 (Graphpad Software Inc. La Jolla, CA, USA). Results were considered significant when P < 0.05 using a nonparametric Mann–Whitney test.
RESULTS
PACT protein expression is increased and colocalizes with activated PKR in AD brains
Using immunohistochemical methods, we showed that in the hippocampus of an AD patient, the neuronal immunostaining of PACT and pPKRthr451 were comparable and consisted in vesicular labelings scattered in the cytoplasm (Figure 1A). Confocal imaging analysis revealed a colocalization of PACT and pPKRthr451 in the cytoplasm of hippocampal neurons (Figure 1B). Merged analysis of dual immunoreactions confirmed diffuse vesicular stainings of the two proteins in neurons of AD, but far less in control brains (Figure 1B). Furthermore, in AD and control brains, we quantified and compared the percentage of neurons with pPKRthr451 and PACT colocalization, and showed a significant difference between both results (Figure 1C).
Figure 1.
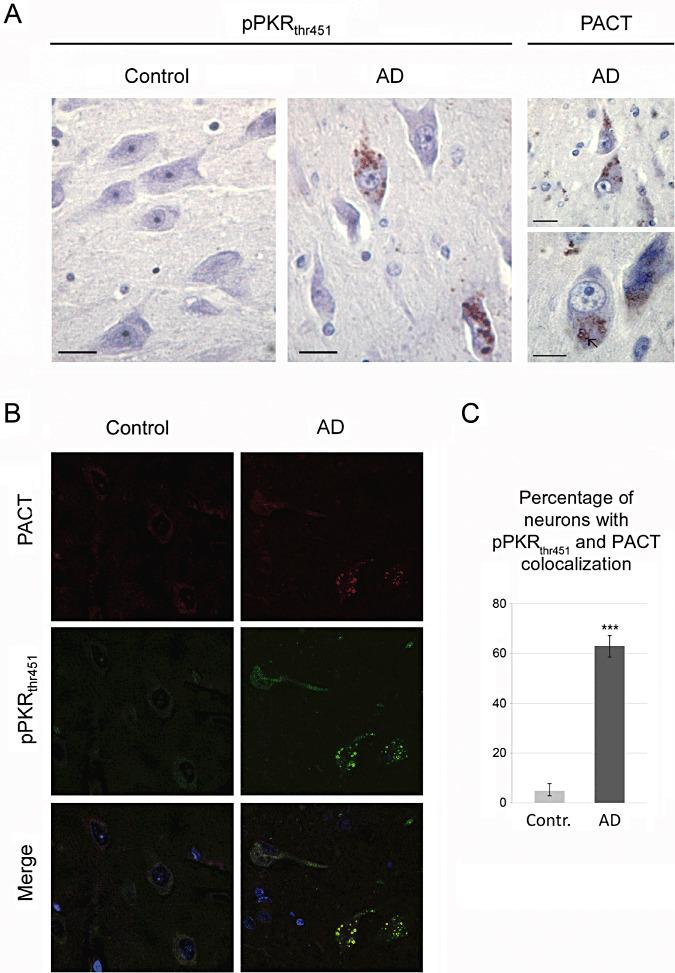
Panels show representative sections of pPKRthr451, PACT‐stained post‐mortem brains of patient with Alzheimer's disease (AD) and control brain (control). Immunohistochemical studies in the hippocampus of AD brains showed that pPKRthr451 (on the left) and PACT (on the right) expression were localized in the cytoplasm of neurons. Horizontal bars: A. 10 µM—(B,C) Confocal analysis and quantification showed a colocalization of pPKRthr451 and PACT in granulovacuolar degeneration in the neurons of entorhinal cortex. Horizontal bars: B. 40 µm. There is a significant difference between the mean numbers of neurons with colocalization in AD compare to control (C).
To support and quantify these findings, we analyzed the expression levels of these proteins in the temporal cortex of AD and control brains using Western blot procedures. PACT expression was enhanced in AD brains compared with control brains with nearly no overlapping results (Figure 2A,C). The increase of the PACT mean levels in AD brains was +42.5%, and was statistically significant. Comparable results were found for pPKRthr446 levels (Figure 2A,B) as for pPKRthr451 (data not shown). The statistically significant augmentation reached +28.3% with no overlapping results. A linear regression analysis showed a significant correlation between PACT and pPKRthr446 levels (Figure 2D).
Figure 2.
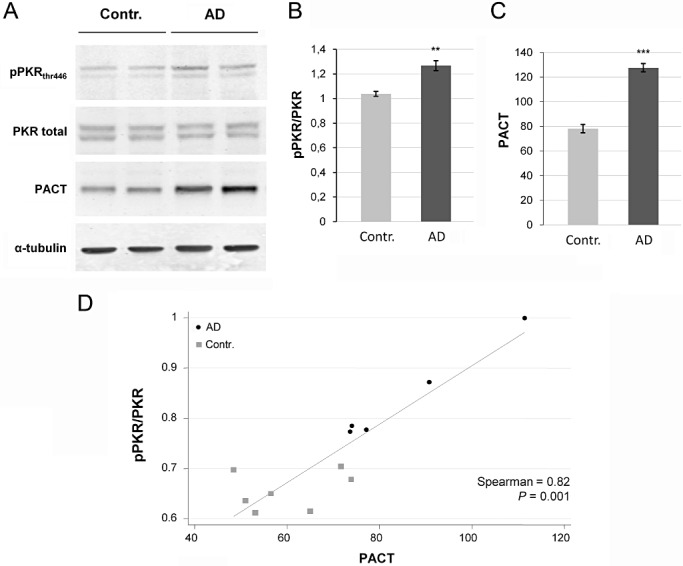
Expression of pPKRthr446, full PKR and PACT in post‐mortem brains of patients with Alzheimer's disease (AD) and control brains (contr). A. Western blotting studies in temporal cortex of AD brains showed that pPKRthr446 and PACT expression were increased compare with expression in the same regions of control brains. B. and C. Diagrams show that pPKRthr446/PKR ratio is increased about 28.3% and 42.5% for PACT. D. pPKRthr446/PKR ratio is significantly correlated to PACT (Spearman P = 0.001).
In summary, we found the same results with two different techniques, with brain samples provided by two different brain banks.
PACT expression is increased in transgenic mice brains
Comparable findings were obtained in the APP/PS1 KI mice previously characterized by widespread neuronal degeneration and pPKR accumulation (21). Molecular evaluation of the brains in these transgenic mice and control littermates revealed increased pPKRthr446 levels (+42.7%) (Figure 3A,B) and PACT levels (+31.2%) in APP/PS1 KI mice compared with control mice (Figure 3A,C). In addition, the levels of PACT and pPKRthr446 in mice brains were correlated (Figure 3D).
Figure 3.
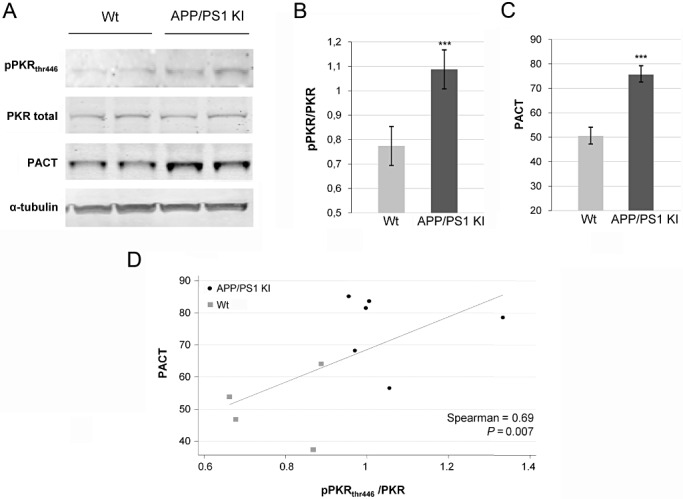
Expression of pPKRthr446, full PKR and PACT in post‐mortem APP/PS1 KI mice and littermates (Wt). A. Western blotting studies of mice brains showed that pPKRthr446 and PACT expression were increased compare to expression in the wild type brains. B. and C. Diagrams show that pPKRthr446/PKR ratio is increased 41.7% and 31.2% for PACT. D. pPKRthr446/PKR ratio is significantly correlated to PACT (Spearman, P = 0.007).
PACT expression is increased and upstream of PKR in stressed human neuroblastoma
To determine whether ER stress, Aβ1‐42 or oxidative stress could induce PACT protein expression and PKR activation, we exposed human neuroblastoma cells SH‐SY5Y to tunicamycin, H2O2 or to Aβ1‐42. These exposures triggered increased PACT and pPKRthr446 protein expression in human neuroblastoma cells (4, 5). To determine if PACT is upstream of downstream of PKR, SH‐SY5Y cells were exposed to 20 µmol of Aβ1‐42 for 4 and 8 h with or without PRI pretreatment (Figure 5A–C). Results revealed that Aβ1‐42 treatment triggered both increase of PACT expression and PKR phosphorylation. Importantly, PRI reduced significantly PKR phosphorylation, but not PACT expression, suggesting that Aβ1‐42 triggers a PACT/PKR pathway, in which PACT acts upstream of PKR. To support a specific role for Aβ1‐42 in driving enhanced PACT/PKR interaction, we performed a control with Aβ42‐1 that did not modify PACT and pPKR/PKR levels (Figure 5D–F).
Figure 4.
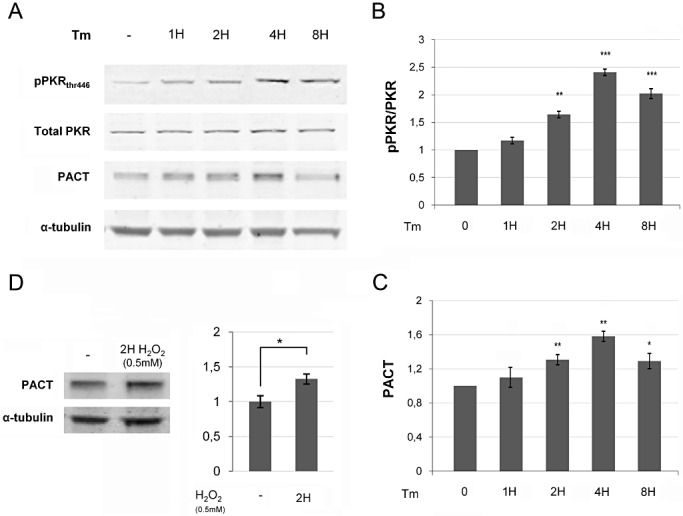
(A) Immunoblot analysis of tunicamycin (tm) treatment in SH‐SY5Y cells showed that, after Tm exposure, PACT and pPKRthr446 begin to increase at 1 h, and this augmentation becomes significant at 2 h, then progressively increased over time with peaks after 4 h (B,C), while full PKR and tubulin are unchanged. Results were obtained from at least three independent experiments. (*P < 0.05; **P < 0.001; ***P < 0.0001). (D) Immunoblot analysis of H2O2 treatment in SH‐SY5Y cells showed that PACT and pPKR are increased after H2O2 exposure.
Figure 5.
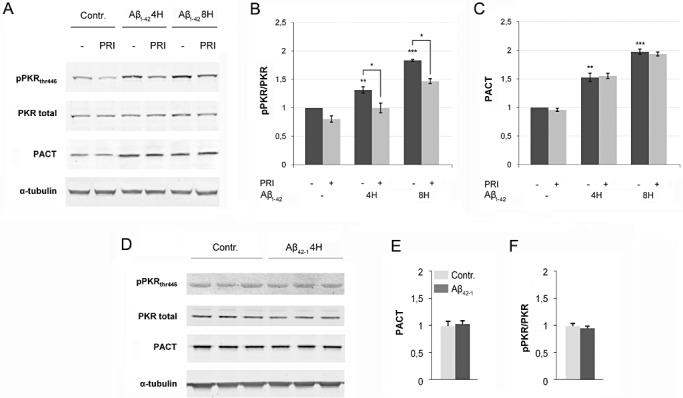
A, B and C immunoblot analysis of Aβ exposure and PRI peptide in SH‐SY5Y cells. PACT and pPKRthr446 progressively increased over time after Aβ1‐42 treatment with peaks after 8 h. PKR activation is decreased with PRI treatment, but not PACT expression, while full PKR and tubulin are stable. Results were obtained from five independent experiments (*P < 0.05; **P < 0.001; ***P < 0.0001). D, E and F PACT and pPKRthr446 levels are stable after nontoxic Aβ42‐1 treatment that supports specific role of Aβ1‐42 in enhanced PACT/PKR interaction.
To confirm that PACT is upstream of PKR, we silenced PACT expression with PACT short hairpin (sh) RNA or used the PKR inhibitor PRI to block PKR activation. Transfection of SH‐SY5Y cells with the PACT shRNA significantly attenuated PACT protein expression, as expected, and resulted in a dramatic inhibition of PKR phosphorylation. This inhibition was already present in the control cells and was more pronounced in cells treated with tunicamycin (Figure 6A–C), confirming that PACT controls PKR phosphorylation in cells exposed to ER stress.
Figure 6.
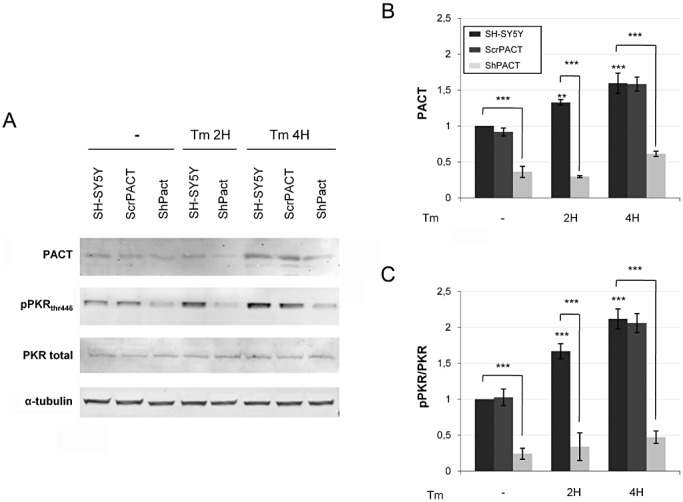
Immunoblot analysis of tunicamycin exposure in SH‐SY5Y cells controls, or with scramble or with shRNA PACT. PACT and pPKRthr446 levels dramatically decreased when cells were transfected with shRNA PACT. Results were obtained from three independent experiments (*P < 0.05; **P < 0.001; ***P < 0.0001).
Increased PACT protein expression is due to increased synthesis of PACT
We determined whether the increased PACT level was the result of increased PACT protein synthesis or decreased PACT protein degradation. Cell cultures were exposed to actinomycin D to inhibit PACT synthesis or lactacystin to inhibit proteasome. We showed that without tunicamycin, pretreatment with actinomycin D or lactacystin, at the dose used, did not modify PACT levels. When cells were treated with tunycamicin, actinomycin D, but not lactacystin, reduced PACT protein levels. These results suggest that the augmentation of PACT level is due to an induction of PACT synthesis. Tunicamycin and ER stress increased the production of PACT, but did not alter the proteasome activity (Figure 7A–C).
Figure 7.
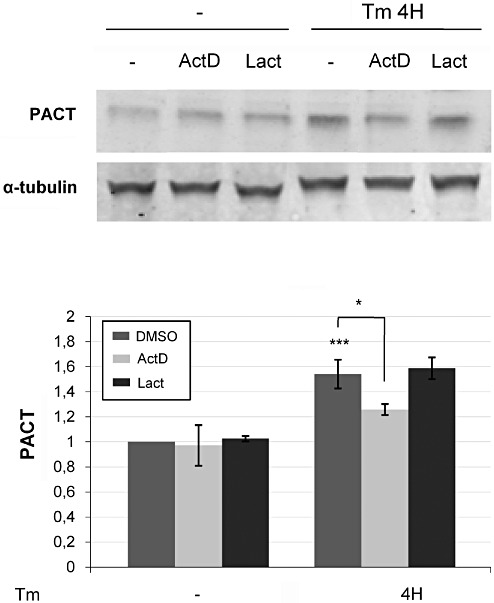
Effect of actinomycin D (ActD) and lactacystin (Lact) on PACT protein expression after tunicamycin exposure. (A,B) After tunicamycin treatment, cctinomycin D decreases the PACT protein levels, while there is no modiciation observed with lactacystin treatment or in absence of ER stress. Results were obtained from three independent experiments (*P < 0.05; **P < 0.00; P < 0.0001).
DISCUSSION
Our study showed that the PKR activator PACT was overexpressed in the brains of AD patients and colocalizes with phosphorylated PKR in hippocampal neurons. PACT and phosphorylated PKR levels were statistically correlated in AD brains. Comparable results were found in the brains of APP/PS1 KI transgenic mice characterized by a widespread neuronal degeneration. In neuroblastoma cell cultures, Aβ1‐42 or tunicamycin or H2O2 induced an increased PACT expression that was associated with PKR activation. PACT protein depletion (using PACT shRNA) dramatically decreased PKR activation. At the opposite, PKR inhibition did not show any effect on PACT expression. These effects allow the conclusion that PACT acts upstream of PKR. Thus, PACT expression levels represent a critical point in AD, since its enhanced expression could lead to PKR activation and neuronal degeneration.
In AD brains, the colocalization PACT‐pPKR suggests that PACT may be part of the PKR activation process detected in these neurons. These results are consistent with several previous studies showing that PACT domain 3 interacts with the kinase domain of PKR to lead to PKR homodimerization and autophosphorylation, resulting in translation blockade and apoptosis 9, 13, 25. Although the exact mechanism of this interaction is not yet completely known (phosphorylation, protein‐protein interaction or both), it is now well admitted that PACT and PKR are strongly linked. Our findings highlight the colocalization of PACT and PKR in AD. Previous studies on the mechanism of PKR activation by PACT have suggested a role in the cellular stress that could induce an increase in the PACT protein levels (17) or lead to PACT phosphorylation on serine 246 and 287 (24). Both mechanisms are not mutually exclusive. Further studies using specific antibodies toward the phosphorylation motifs of PACT will illuminate the mode of activation of PKR by PACT.
Previous reports have shown that PACT expression can be enhanced in vitro by ER stress induced by tunicamycin 16, 22, 24, and that PKR/RAX interaction is enhanced by oxidative stress (32). In addition, it is well admitted that Aβ neurotoxicity may be associated with ER and oxidative stresses (31). Our biochemical results in SH‐SY5Y showed that Aβ1‐42, tunicamycin and H2O2 treatment lead to an increased PACT and pPKR protein levels, and that PACT is upstream of PKR. Furthermore, we confirmed the increase of PACT/pPKR augmentations in animal models and in human AD brains. All these results are consistent with previous in vitro studies 16, 22, 24, 32 and suggest that in AD, cellular stresses lead to increased PACT expression level and to PKR phosphorylation.
To determine whether increased PACT levels take place at the transcriptional or posttranslational level, we used pharmacological inhibitors to block these different steps. We demonstrated that the increase in PACT expression is not a consequence of proteasome impairment but might be due the activation of PACT gene expression in our cell lines.
A previous report has studied thaspigargin‐induced ER stress in HEK293A and HeLa cells, and has revealed that PACT mRNA expression was enhanced by cell stress (17). We have explored this possibility in our cell culture system. Induction of PACT expression upon Tunicamycin treatment was inhibited by actinomycin D, thus confirming that ER stress increases PACT expression at the mRNA level. The exact mechanisms triggering PACT gene expression remains to be determined, but it is already known that the expression of PACT is regulated by the SP1 transcription factor (12), and that ER stress pathways induced by tunicamycin or thaspigargin can activate SP transcription factors, including SP1 protein (1). It is not yet clear whether SP1 mRNA and protein levels are increased or not in AD brains 7, 29. If this was the case, sp1 may participate in the PACT/PKR triggering pathway. Regarding these results, we can hypothesize that in AD, Aβ neurotoxiticity and others stresses lead to the augmentation of PACT expression, which in turn activates PKR leading to neuronal death.
The PACT/PKR interaction has been shown to be controlled by another cellular protein, TRBP (TAR RNA‐binding protein). TRBP inhibits the activity of PKR and binds to PACT through their respective double‐stranded RNA binding domains, which may influence their activity on PKR. Stresses dissociated the TRBP‐PACT interaction and increased PACT‐induced PKR activation, demonstrating the relevance of this control in a physiological context. Daher et al showed that in cells, TRBP controls PACT activation of PKR, an activity that is reversed by stress. These results suggest that an increase in the expression levels of PACT in response to stress may not be sufficient to trigger cell death because TRBP can modulate PACT/PKR interaction. Such a mechanism could occur in our models and AD brains. Further studies are needed to assess the fate of TRBP metabolism in Aβ‐exposed cell cultures, as well as in AD brains or in the brains of transgenic mice.
An earlier report has shown that activated PKR was also increased in the brains of AD transgenic APP/PS1 KI mice (21). Using Western blot, we demonstrated increased levels of PACT and phosphorylated PKR and a statistically significant correlation between the levels of these proteins in these mice brains. These mice are characterized by a widespread neuronal degeneration, eg, located in the hippocampus (4), and enhanced levels of PACT and phosphorylated PKR could participate in the detrimental cellular process leading to neuronal loss.
It is interesting to notice that PKR activation can be associated with tau phosphorylation. An earlier report has linked PKR, GSK3β activation and tau phosphorylation, and PKR inhibition could reduce tau phosphorylation induced by Aβ exposure in cell cultures (3). In this report, showing that Aβ is able to trigger PACT protein expression and PKR activation, we can proposed that the cell stress induced by Aβ could be at the origin of a cascade of signaling events initiating PACT accumulation and leading to tau phosphorylation.
Taken together our results allow drawing several conclusions (Figure 8). 1‐PACT levels are increased in AD brains and APP/PS1 KI mice brains, and PACT colocalize with neuronal activated PKR. This augmented PACT expression could be at the origin of activated PKR pathway detected in AD brains. 2‐Enhanced PACT protein levels can be induced in cells by Aβ exposure, ER stress or oxidative stress, all toxic events present in AD brains. 3‐Disturbed PACT gene expression could be responsible for neuronal PACT protein accumulation and could be a consequence of transcription factor activation, such as SP1, linked to cell stress. Knowing the possible role of PACT, modulation of PACT activity could be a new molecular target to attenuate PKR activation and neurodegeneration in AD.
Figure 8.
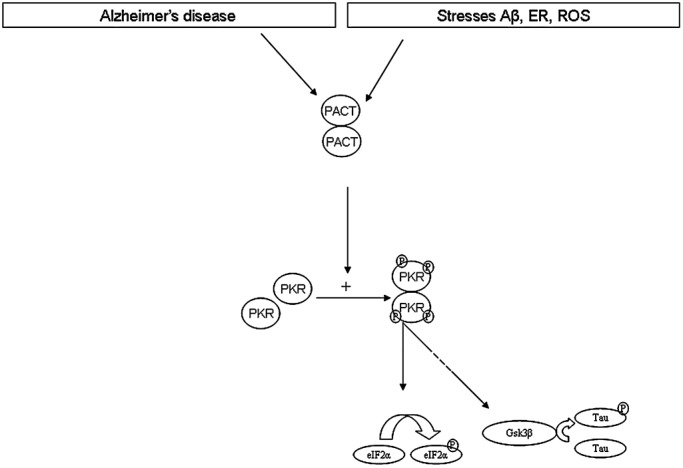
Hypothesis about PACT/PKR involvement in AD brain. Cellular stresses (Stress Aβ, ER stress or oxidative stress) increase PACT expression. According to previous studies, there is a protein–protein interaction with another molecule of PACT (9), leading to phosphorylation of PKR then activation of several pathways as eiF2α or GSK3β then tau.
To our knowledge, no previous study has reported that PACT protein levels were increased in the brain of AD patients compared with controls. The mechanism leading to this increase is not known, but this modified expression could explain the concurrent enhanced PKR phosphorylation. Whether or not this PKR activation could explain neuronal degeneration is difficult to assess in AD patients. We used cell culture models to explore the mechanism that could lead to increased PACT protein levels. Based on cell models, we designed a diagram representing the possible abnormalities of this pathway in AD (Figure 8).
ACKNOWLEDGMENTS
The authors thank Dr Jean‐Philippe Brouland, Patrice Castagnet, Francis Bernard, Agnès Marquet, Claudine Poiron and Katia Dossou for technical help, Hervé Enslen and Eliane Meurs for advice. Marion Yger and Mariko Taga for technical help. Dr Julien Dumurgier for statistical analysis.
Conflict of interest: Transgenic APP/PS1 knock‐in mice were provided by Sanofi‐Aventis. Dr Laurent Pradier is staff of Sanofi‐Aventis. No other conflict of interest
REFERENCES
- 1. Abdelrahim M, Liu S, Safe S (2005) Induction of endoplasmic reticulum‐induced stress genes in Panc‐1 pancreatic cancer cells is dependent on Sp proteins. J Biol Chem 280:16508–16513. [DOI] [PubMed] [Google Scholar]
- 2. Bennett RL, Blalock WL, May WS (2004) Serine 18 phosphorylation of RAX, the PKR activator, is required for PKR activation and consequent translation inhibition. J Biol Chem 279:42687–42693. [DOI] [PubMed] [Google Scholar]
- 3. Bose A, Mouton‐Liger F, Paquet C, Mazot P, Vigny M, Gray F, Hugon J (2011) Modulation of Tau Phosphorylation by the Kinase PKR: implications in Alzheimer's disease. Brain Pathol 21:189–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Casas C, Sergeant N, Itier JM, Blanchard V, Wirths O, van der Kolk N et al (2004) Massive CA1/2 neuronal loss with intraneuronal and N‐terminal truncated Abeta42 accumulation in a novel Alzheimer transgenic model. Am J Pathol 165:1289–1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chang RC, Suen KC, Ma CH, Elyaman W, Ng HK, Hugon J (2002) Involvement of double‐stranded RNA‐dependent protein kinase and phosphorylation of eukaryotic initiation factor‐2alpha in neuronal degeneration. J Neurochem 83:1215–1225. [DOI] [PubMed] [Google Scholar]
- 6. Chang RC, Wong AK, Ng HK, Hugon J (2002) Phosphorylation of eukaryotic initiation factor‐2alpha (eIF2alpha) is associated with neuronal degeneration in Alzheimer's disease. Neuroreport 13:2429–2432. [DOI] [PubMed] [Google Scholar]
- 7. Citron BA, Dennis JS, Zeitlin RS, Echeverria V (2008) Transcription factor Sp1 dysregulation in Alzheimer's disease. J Neurosci Res 86:2499–2504. [DOI] [PubMed] [Google Scholar]
- 8. Costa RO, Ferreiro E, Cardoso SM, Oliveira CR, Pereira CM (2010) ER stress‐mediated apoptotic pathway induced by Abeta peptide requires the presence of functional mitochondria. J Alzheimers Dis 20:625–636. [DOI] [PubMed] [Google Scholar]
- 9. Daher A, Laraki G, Singh M, Melendez‐Pena CE, Bannwarth S, Peters AH et al (2009) TRBP control of PACT‐induced phosphorylation of protein kinase R is reversed by stress. Mol Cell Biol 29:254–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Der SD, Yang YL, Weissmann C, Williams BR (1997) A double‐stranded RNA‐activated protein kinase‐dependent pathway mediating stress‐induced apoptosis. Proc Natl Acad Sci U S A 94:3279–3283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Duyckaerts C, Delatour B, Potier MC (2009) Classification and basic pathology of Alzheimer disease. Acta Neuropathol 118:5–36. [DOI] [PubMed] [Google Scholar]
- 12. Fasciano S, Kaufman A, Patel RC (2007) Expression of PACT is regulated by Sp1 transcription factor. Gene 388:74–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Garcia MA, Meurs EF, Esteban M (2007) The dsRNA protein kinase PKR: virus and cell control. Biochimie 89:799–811. [DOI] [PubMed] [Google Scholar]
- 14. Hardy JA, Higgins GA (1992) Alzheimer's disease: the amyloid cascade hypothesis. Science 256:184–185. [DOI] [PubMed] [Google Scholar]
- 15. Hoozemans JJ, Veerhuis R, Van Haastert ES, Rozemuller JM, Baas F, Eikelenboom P, Scheper W (2005) The unfolded protein response is activated in Alzheimer's disease. Acta Neuropathol 110:165–172. [DOI] [PubMed] [Google Scholar]
- 16. Ito T, Yang M, May WS (1999) RAX, a cellular activator for double‐stranded RNA‐dependent protein kinase during stress signaling. J Biol Chem 274:15427–15432. [DOI] [PubMed] [Google Scholar]
- 17. Lee ES, Yoon CH, Kim YS, Bae YS (2007) The double‐strand RNA‐dependent protein kinase PKR plays a significant role in a sustained ER stress‐induced apoptosis. FEBS Lett 581:4325–4332. [DOI] [PubMed] [Google Scholar]
- 18. Lee Y, Hur I, Park SY, Kim YK, Suh MR, Kim VN (2006) The role of PACT in the RNA silencing pathway. EMBO J 25:522–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM (1984) Clinical diagnosis of Alzheimer's disease: report of the NINCDS‐ADRDA work group under the auspices of the Department of Health and Human Services Task Force on Alzheimer's disease. Neurology 34:939–944. [DOI] [PubMed] [Google Scholar]
- 20. Nekhai S, Bottaro DP, Woldehawariat G, Spellerberg A, Petryshyn R (2000) A cell‐permeable peptide inhibits activation of PKR and enhances cell proliferation. Peptides 21:1449–1456. [DOI] [PubMed] [Google Scholar]
- 21. Page G, Rioux Bilan A, Ingrand S, Lafay‐Chebassier C, Pain S, Perault Pochat MC et al (2006) Activated double‐stranded RNA‐dependent protein kinase and neuronal death in models of Alzheimer's disease. Neuroscience 139:1343–1354. [DOI] [PubMed] [Google Scholar]
- 22. Patel CV, Handy I, Goldsmith T, Patel RC (2000) PACT, a stress‐modulated cellular activator of interferon‐induced double‐stranded RNA‐activated protein kinase, PKR. J Biol Chem 275:37993–37998. [DOI] [PubMed] [Google Scholar]
- 23. Patel RC, Sen GC (1998) PACT, a protein activator of the interferon‐induced protein kinase, PKR. EMBO J 17:4379–4390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Peters GA, Li S, Sen GC (2006) Phosphorylation of specific serine residues in the PKR activation domain of PACT is essential for its ability to mediate apoptosis. J Biol Chem 281:35129–35136. [DOI] [PubMed] [Google Scholar]
- 25. Peters GA, Dickerman B, Sen GC (2009) Biochemical analysis of PKR activation by PACT. Biochemistry 48:7441–7447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Peters GA, Seachrist DD, Keri RA, Sen GC (2009) The double‐stranded RNA‐binding protein, PACT, is required for postnatal anterior pituitary proliferation. Proc Natl Acad Sci U S A 106:10696–10701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Proud CG (1995) PKR: a new name and new roles. Trends Biochem Sci 20:241–246. [DOI] [PubMed] [Google Scholar]
- 28. Querfurth HW, LaFerla FM (2010) Alzheimer's disease. N Engl J Med 362:329–344. [DOI] [PubMed] [Google Scholar]
- 29. Santpere G, Nieto M, Puig B, Ferrer I (2006) Abnormal Sp1 transcription factor expression in Alzheimer disease and tauopathies. Neurosci Lett 397:30–34. [DOI] [PubMed] [Google Scholar]
- 30. Singh M, Fowlkes V, Handy I, Patel CV, Patel RC (2009) Essential role of PACT‐mediated PKR activation in tunicamycin‐induced apoptosis. J Mol Biol 385:457–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sultana R, Perluigi M, Butterfield DA (2009) Oxidatively modified proteins in Alzheimer's disease (AD), mild cognitive impairment and animal models of AD: role of Abeta in pathogenesis. Acta Neuropathol 118:131–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wang X, Fan Z, Wang B, Luo J, Ke ZJ (2007) Activation of double‐stranded RNA‐activated protein kinase by mild impairment of oxidative metabolism in neurons. J Neurochem 103:2380–2390. [DOI] [PubMed] [Google Scholar]
- 33. Williams BR (1997) Role of the double‐stranded RNA‐activated protein kinase (PKR) in cell regulation. Biochem Soc Trans 25:509–513. [DOI] [PubMed] [Google Scholar]