

Abstract
There is a broad consensus that multiple sclerosis (MS) represents more than an inflammatory disease: it harbors several characteristic aspects of a classical neurodegenerative disorder, that is, damage to axons, synapses and nerve cell bodies. While we are equipped with appropriate therapeutic options to prevent immune‐cell driven relapses, effective therapeutic options to prevent the progressing neurodegeneration are still missing. In this review article, we will discuss to what extent pathology of the progressive disease stage can be modeled in MS animal models. While acute and relapsing–remitting forms of experimental autoimmune encephalomyelitis (EAE), which are T cell dependent, are aptly suited to model relapsing‐remitting phases of MS, other EAE models, especially the secondary progressive EAE stage in Biozzi ABH mice is better representing the secondary progressive phase of MS, which is refractory to many immune therapies. Besides EAE, the cuprizone model is rapidly gaining popularity to study the formation and progression of demyelinating CNS lesions without T cell involvement. Here, we discuss these two non‐popular MS models. It is our aim to point out the pathological hallmarks of MS, and discuss which pathological aspects of the disease can be best studied in the various animal models available.
Keywords: cuprizone, disability, disease progression, EAE, multiple sclerosis, neurodegeneration, PPMS, SPMS, treatment
GENERAL INTRODUCTION
Multiple sclerosis (MS) is an inflammatory disease of the central nervous system (CNS) characterized by large inflammatory plaques of white matter demyelination. Such focal inflammatory lesions are associated with oligodendrocyte destruction, reactive gliosis and axonal degeneration. The composition of established inflammatory infiltrates varies between patients and/or lesion stages but commonly includes T‐lymphocytes and macrophages 78. Furthermore, activated astrocytes and microglia cells participate in lesion development, progression and resolution by the secretion of cytokines, other inflammatory mediators as well as neural growth factors. Inflammatory white matter lesions can arise virtually anywhere in the brain parenchyma although certain predilection sites, such as the periventricular white matter, exist. Besides focal white matter lesions, two additional histopathological features exist which were during the last decades in the focus of clinical and histopathological studies: (i) gray matter demyelination and/or atrophy and (ii) diffuse white matter injury 31, 135. These changes within the white and gray matter are thought to contribute to the clinical picture of MS and, hence, deserve to be studied.
A primary aim of this brief review is to provide an overview of the importance of MS animal models for neurologists, MS specialists and scientists studying MS and related disorders. We specifically focus on progressive experimental autoimmune encephalomyelitis (EAE) models and the cuprizone model. Furthermore, we will summarize important recent advances in our understanding of the underlying pathology of the cuprizone model. It is our aim to point out the pathological hallmarks of MS, and discuss which pathological aspects of the disease can best be studied in the various animal models available (also see Table 1). We consider it important to stress from the very beginning that the perfect animal model for MS does not exist since MS is a purely human disorder. Pathological processes in MS are manifold and heterogeneous, and therefore applied animal models allow us to study different and very distinct aspects of the disease rather than its entire complexity. Thus, the histopathological characteristics of MS are best studied using human tissue samples. Animal models, however, are required to address for example potential pathomechanisms or the efficiency of new compounds.
Table 1.
Different histopathological and clinical characteristics of MS, and whether or not such characteristics can be studies (green) or cannot be studied (red) in different MS animal models. The term Cup/EAE model refers to a recently developed novel MS animal model in our group 115.
| Scientific question | Cuprizone | Cup/EAE | MOG‐EAE | PLP‐EAE | ABH‐EAE |
|---|---|---|---|---|---|
| Acute clinical attack | |||||
| Relapse and remission | |||||
| Focal inflammatory demyelinating lesions | |||||
| Gray matter demyelination | |||||
| Diffuse white matter damage | Moderate | Moderate | |||
| Remyelination | |||||
| Axonal damage | |||||
| Progressive worsening of function (ie, progression) | Not yet known | ||||
| Inside‐out mechanisms | |||||
| Outside‐in mechanisms |
Here, we first highlight the distinct aspects of MS pathology and discuss how these changes relate to clinical symptoms. We then discuss how the clinical and pathology features can best be modeled in experimental animals. Finally, we speculate how the models can be improved to reflect emerging knowledge of MS pathology.
RELAPSE: DEFINITION AND PATHOLOGICAL CORRELATE
Two distinct clinical entities can be defined in patients with MS: First the relapse or the acute clinical attack, second continuous progression of clinical disability (see Figure 1C, lower part). During a relapse, patients experience the rapid occurrence of new or increasing neurologic symptoms. These attacks—also called exacerbations—are followed by periods of partial or even complete recovery (remissions). By definition, there is no apparent progression of the disease between two relapses implying that the level of disability between two attacks remains stable. This phase of the disease course is called Relapsing Remitting MS (RRMS) and initially affects ∼85 percent of patients diagnosed with MS.
Figure 1.
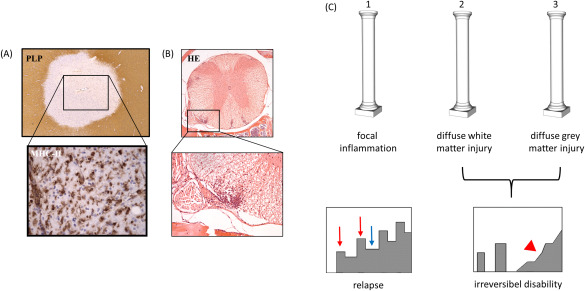
(A) Histopathologic characteristics of an acute, inflammatory MS lesion within the white matter. Demyelination is indicated by a focal, well demarcated loss of anti‐PLP (proteolipid protein) immunoreactivity. The entire lesion is interspersed with activated monocytes and microglia, expressing MHC‐II protein. In (B) the correlate in EAE is shown. Even in HE‐stained sections, inflammatory infiltrates are clearly visible. In EAE, such lesions are mainly found within the spinal cord and cerebellum. (C) The three pathological hallmarks of MS pathology, namely focal inflammation, diffuse white matter injury and gray matter injury are illustrated. While focal inflammation causes acute relapses (see red arrows), diffuse white and gray matter pathology induce neurodegeneration and in consequence accumulation of irreversible clinical disability (red arrowhead). Blue arrow indicates clinical remission.
What happens during a relapse?
It is broadly accepted that the histopathological correlate of acute clinical symptoms is a focal inflammatory demyelinating white matter lesion (see Figure 1A). These focal inflammatory lesions impact on neuronal integrity eventually leading to axonal dysfunction or complete axonal destruction 40, 69. Although the accumulation of peripheral immune cells is one characteristic of such lesions, it is not clear what triggers the influx of immune cells into the CNS, nor is it known what the primary “target” of the (auto)‐inflammatory attack is, that is, is it the axon, neuron, myelin sheath or even glia cells. While it has been suggested that oligodendrocyte stress is an initial trigger of MS lesion development 7, 57, 132, 133, and neuronal damage is the pathological substrate of progressive MS, both hypotheses require further verification. Whatever the trigger, white matter demyelination, axonal damage and peripheral immune cell recruitment are histopathologic hallmarks of the acute relapse. During an acute relapse demyelination, edema formation (mainly in the spinal cord) and, although probably to a lesser extent, acute axonal damage all might contribute to the observed functional deficits. When a demyelinated axon undergoes remyelination and the extent of axonal damage is compensated by, for example, neuronal plasticity complete clinical recovery might occur during the remission phase 62, 89. Notably, most of our knowledge what leads to functional deficits during a relapse, and what is the morphological correlate of recovery comes from experimental animal studies 20. To what extent these findings can be translated to the human situation needs further investigation. We now have a closer look on the pathophysiology of acute exacerbations.
The effects of demyelination have been extensively studied from the cellular level to the whole animal and may be summarized as follows: demyelination leads to attenuated conduction velocity, or in the worst case scenario to a complete conduction block. Why so? During an action potential there is a change in polarity across the cell membrane of the axon and the inside of the axon becomes more positive compared to the outer membrane. The propagation of an action potential generates local currents that depolarize the axonal membrane adjacent to the action potential. When the depolarization caused by the local currents reaches a threshold, a new action potential is produced adjacent to the original one. In unmyelinated or demyelinated axons, the propagation of an action potential occurs in a continuous, sequential fashion. This is obviously a relatively slow way of signal conduction. In myelinated axons however, this action potential propagation jumps from one node to the next node, a phenomenon called saltatory conduction (latin saltare, “to jump”). This form of signal propagation is much faster compared to the continuous one in unmyelinated or demyelinated fibers. Since the function of the myelin sheath is to facilitate the conduction of electrical impulses through the axons, demyelination clearly slows the process of impulse conduction which finally results in functio laesa. Furthermore, demyelination leads to a functional reorganization of ion channels 53, which might as well contribute to relapse‐related functional deficits.
A second pivotal element of impaired neuronal functioning during a relapse is edema formation. It is well known from studies on the peripheral nervous system that the compression of nerve fibers results in a slowing of conduction velocity, or even a complete conduction block 41, 55. Compression of nerve fibers is also operant during tissue edema as observed during inflammatory demyelination, especially in CNS regions with low swelling capacity, such as the spinal cord. A fast recovery of a relapse is, therefore, not only due to remyelination but at the same time due to resolution of tissue edema. To what extent axonal damage contributes to neurological symptoms during a relapse is not clear. Since completely transected axons cannot be repaired, this type of axonal damage results in irreversible clinical disability. However, it has been shown that, in contrast to complete axonal transection, subtle axonal injury is not an irreversible process, and eventually injured axons can recover 97. Therefore, the acute symptoms during inflammatory demyelination might well be due to axonal pathology, and thus recovery after a relapse might be partly mediated by direct axonal recovery.
TREATMENT MODALITIES IN MULTIPLE SCLEROSIS
Treatment of MS has two modalities: therapies to relieve or modify symptoms during an acute relapse, and immunomodulatory therapy to decrease relapse frequency. The adequate management of MS relapses is important as it may help lessen the disability associated with the acute attack yet also reduce the burden associated with recurrent attacks. Historically, treatment of MS relapses was the first approach (and for many years the only approach) to MS treatment. Systemic corticosteroids remain the most established and validated treatment options for a MS relapse 12. The exact mode of action of systemic corticosteroid treatment during an acute attack is incompletely understood but includes stabilization of the blood–brain barrier, activation of anti‐inflammatory pathways, facilitating the apoptosis of activated immune cells and, maybe most importantly, relief of tissue edema and thereby reduction of axonal compression. To what extent remyelination contributes to function recovery during corticosteroid treatment is controversially discussed. Two previous studies examining the effects of corticosteroid treatment on the extent of repair of experimental demyelinating lesions have yielded conflicting results. Methylprednisolone treatment accelerated spontaneous remyelination in the spinal cord after lysolecithin‐induced focal demyelination, which was paralleled by reduced macrophage invasion 105. By contrast, an earlier study using the same lesion model demonstrated a delay in remyelination in rats treated with dexamethasone 127, findings which are in line with other studies 22, 30. To conclude, the positive effect of corticosteroids during an acute relapse is probably due to its anti‐inflammatory rather than regenerative (ie, remyelination) character.
Immunomodulatory therapies are beneficial during the relapsing–remitting phase of MS, where such treatment reduces relapse frequency and severity of clinical attacks. As of August 2016, 13 disease modifying therapies (DMTs) have been approved by the Food and Drug Administration (FDA). Of these, seven are delivered by injection, three are oral and three are delivered by infusion. Currently approved drugs include Interferon beta‐1a (Avonex, Rebif), Interferon beta‐1b (Betaseron, Extavia), Peginterferon beta‐1a (Plegridy), Glatiramer acetate (Copaxone, Glatopa), Natalizumab (Tysabri), Mitoxantrone, Fingolimod (Gilenya), Teriflunomide (Aubagio), Dimethyl fumarate (Tecfidera), Alemtuzumab (Lemtrada) and Daclizumab (Zinbryta). These DMTs have quite different mechanisms of action, efficacy, safety and tolerability profiles. It is out of the scope of this review article to elaborate on the mode of action of these drugs, and several excellent review articles are recommended 77, 101.
PROGRESSION: DEFINITION AND PATHOLOGICAL CORRELATE
As we have now discussed what happens during a relapse, and the options that are available to treat relapses and to reduce their frequency, we now discuss disease progression, what are the underlying mechanisms and why it is so important to understand these mechanisms to efficiently develop new therapeutic modalities. As pointed out above, during the initial RR stage of MS the symptoms appear (relapse) and then resolve either partially or completely (remission). Histopathologically, white matter inflammatory demyelination blocks axonal conduction, and relieve of inflammation results in recovery. If remyelination is incomplete and/or axons are destroyed, functional recovery might be incomplete as well. Most patients (85%) diagnosed with RRMS will develop a secondary progressive course, called SPMS. In SPMS, there is a progressive worsening of neurologic function (accumulation of disability) over time, while acute relapses become less frequent. Thus, SPMS occurs as a second phase of the disease for many individuals, and may be seen as a long‐term outcome of RRMS. It is the therapy against continuous progression and accumulation of clinical disability that is an urgent unmet clinical need. Sufficient to say, to date no treatment option has proven to be effective during the SPMS disease stage, nor for primary progressive MS patients that develop accumulating disability from disease onset.
Why is disease progression in SPMS (probably) not due to focal inflammation?
Results of several clinical studies strongly suggest that focal, autoimmune‐driven inflammation is not the underlying mechanism driving disease progression in SPMS patients. Also, while available DMTs reduce relapse frequency 15, 128, these approaches do not prevent long‐term neurologic disability 65, 103. Of note the two most widely prescribed therapies for MS (β‐interferon and glatiramer acetate) have no effect on the progressive forms of the disease (primary or secondary MS), although relapse rates may be reduced by about one‐third in some patients 103. In line with these findings, a recent study showed that MS subjects with no evidence of disease activity during the first two years of the study had long‐term outcomes that were no different from the cohort as a whole 33. Furthermore, in women, the effect of pregnancy on relapse remission is impressive, however, disability progression is not influenced by the reduced relapse rate during pregnancy 32. Finally, treatments such as natalizumab, which are effective in reducing immune cell invasion into the CNS, may not reduce the disease progression 92, 140. An important, yet unresolved question is: Why are anti‐inflammatory therapies not effective during the progressive disease stage? One simple explanation would be that inflammation is not the main mediator of disease progression in SPMS and PPMS. Against this superficial assumption are results from a recent histopathological study 44. The authors performed an extensive and detailed quantification of different inflammatory cells in relation to axonal injury, one of the best predictors of Expanded Disability Status Scale (EDSS)‐rated clinical disability in MS 124. Those lesions with pronounced axonal injury, irrespective of being present in RRMS, SPMS or PPMS, clearly showed inflammation, suggesting a close association between inflammation and neurodegeneration also during the progressive disease stage. One possible explanation for therapeutic failure in the progressive disease stage is that this inflammatory process becomes trapped within the CNS compartment behind a closed blood–brain barrier and blood–liquor barrier. Furthermore, it has been suggested that a chronic inflammatory process, as present in the CNS of progressive MS patients, creates a microenvironment supporting persistence of inflammatory cells 90. The presence of B‐cell follicle‐like structures within the meninges, especially during progressive disease stages, should be noted in this context.
In summary, the clinical course of the progressive stage of the disease remains largely unpredictable, underlying mechanisms of tissue destruction are poorly understood, but autoimmune‐mediated inflammatory demyelination is presumably not the major force driving disease progression in SPMS patients.
Disease progression in SPMS is due to neurodegeneration
The pathological substrate of disease progression is neurodegeneration. It is widely accepted that axonal loss occurs in MS, and is at least in part responsible for the permanent disability characterizing the later chronic progressive stage of the disease. While most studies focused on axonal pathology, all aspects of neurons undergo degenerative changes including cell somata, dendrites, spines and neurotransmitter metabolism 6, 104, 134, 137. Our knowledge regarding the mechanisms leading to progressive neurodegeneration is scant at best, but a number of potential contributors are discussed including microglia activation, reactive oxygen species and mitochondrial dysfunction 83, 102, 117. For example, it has been shown that neuroinflammation enhances glutamate transmission and promotes synaptopathy, which occurs in the early phase of EAE and is associated with the release of inflammatory cytokines, such as TNFα and IL‐1β from activated microglia 19, 84. In addition, mitochondrial structural changes, altered mitochondrial gene expression and enzyme activities, increased free radical production and oxidative damage have all been reported in patients with MS 86. As demonstrated by Nave's lab, myelinating oligodendrocytes release lactate through the monocarboxylate transporter MCT1. Lactate is then utilized by axons for mitochondrial ATP generation 46. Thus, demyelination and subtle oligodendrocyte loss are believed to contribute to neurodegeneration by a lack of trophic axonal support usually provided by oligodendrocytes. To summarize, although MS is considered to be a classical autoimmune inflammatory disorder, it harbors at the same time important elements of a classical neurodegenerative disease. It is the extent of neurodegeneration which is responsible for the accumulation of irreversible clinical disability in MS patients. Of note, to what extent inflammatory processes interfere with this ongoing neurodegenerative process remains to be clarified.
WHAT TRIGGERS NEURODEGENERATION IN MS: PATHOLOGY OF THE NAWM AND GRAY MATTER
As discussed above focal inflammatory demyelination accounts for the relapse‐related clinical disability whereas diffuse neurodegeneration accounts for the irreversible clinical progression, most evident during the progressive disease stage (ie, SPMS). Since focal inflammatory lesions are a less prominent feature of SPMS compared to RRMS, the question arises what triggers the neurodegeneration in progressive MS.
In MS, brain white matter is not only damaged within the typical and apparent focal demyelinated lesions but also within the so‐called normal appearing white matter (NAWM), so called since the white matter appears normal in routine myelin stains, such as anti‐proteolipid protein (PLP) immunohistochemistry or LFB‐PAS histochemistry. Detailed histopathological studies, however, have demonstrated that this white matter is not normal at all. Histopathologic findings include diffuse gliosis, microglial activation, vascular fibrosis, perivascular cuffing by inflammatory cells, perivascular lipofuscin deposition, abnormal endothelial tight junctions, blood–brain barrier breakdown or vessels containing proliferating endothelial cells. Electron microscopy revealed increased numbers of lysosomes, particularly, in astrocytes. Notably, axonal loss is observed as well in the NAWM 39, 71. In addition, the term “NAWM” is used in the context of white matter parts appearing normal in imaging techniques such as T1 and T2‐weighted MRI. There is now substantial evidence from advanced MRI techniques (including measures of magnetization transfer, diffusion, relaxation times and spectroscopic metabolite concentrations) showing that significant in vivo abnormalities exist in MS WM that appears normal by conventional MRI 43. In contrast to inflammatory white matter lesions, NAWM pathology is not a focal but a diffuse, widespread process.
Several mechanisms leading to NAWM pathology have been proposed although it is still debatable as to which are involved in MS. These include oligodendrocyte stress that may lead to dysfunctional myelination and impaired neuronal support 46, 106, Wallerian degeneration of axons transected by focal demyelination within near or distant focal white matter lesions 38, tissue hypoperfusion 42 or decrease in iron concentration 54 among other factors. Because there appears to be, at best, a modest relationship between these widespread white matter abnormalities and the focal inflammatory lesion load as defined by MRI 125 it is widely considered that NAWM abnormalities are (at least partly) independent of focal inflammatory lesions, thus being a distinct or separate disease process. Another contributor to NAWM abnormalities is cortical pathology 95. Cortical lesion load correlates with diffuse white matter injury in MS patients. Furthermore, there is a significant correlation between the histopathological extent of cortical demyelination and diffuse changes in the NAWM 71. Underlying mechanisms of this interplay are not completely understood. Most of the proteins, vesicles and organelles required for axon survival have to be continuously synthesized in the cell body, and are then transported along the microtubule network along the axon to their final destination. It appears feasible that even a modest impairment of this neuronal synthesis and transport machinery, due to cortical demyelination, might finally result in axonal pathology. Of note, NAWM findings are clearly clinical relevant because, in contrast to focal WM lesions, they correlate better with disability and cognitive impairment 93, and less NAWM pathology is detected in patients presenting with a benign disease course 35.
Similar to the diffuse white matter damage, tissue damage in the gray matter is also a key component of the disease process, especially during SPMS, and the number of studies investigating gray matter damage in MS has increased exponentially during the past few years. Like NAWM, gray matter involvement can also be extensive involving both the cortex and subcortical gray matter such as the hippocampus or thalamus 47, 49, 79. Results from clinical studies suggest that neuronal damage or neuronal loss critically contributes to functional deficits in MS patients. It has been shown that disability progression is associated with whole brain atrophy 48, 89, and reduced levels of brain N‐acetyl‐aspartate (NAA) levels, a marker believed to reflect neuronal health, correlate with cognitive impairment in MS 87. Virtually every element of the neuron can be damaged or even destroyed during gray matter demyelination including cell soma 82, 108, dendritic spines 64, synapses 37, 91, 137 and neurites 108. Of note, there is evidence that neuronal structures are not “simply” destroyed but in parallel neuronal plasticity is impaired in the CNS of MS patients 98.
Remarkably, post‐mortem analyses revealed widespread loss of dendritic spines in the cortex of patients with MS that similarly affected demyelinated and non‐demyelinated cortex regions, and preceded loss of cortical axons 64. Another important element of cortical gray matter injury in progressive MS patients is meningeal inflammation. The formation of lymphoid structures resembling B‐cell follicles in the inflamed CNS meninges in a subset of patients with progressive MS and in relapsing–remitting EAE has been shown, and this form of meningeal inflammation might contribute to cortical demyelination 68, 81, 82. Importantly, cases with primary progressive MS with extensive meningeal immune cell infiltration exhibited a more severe clinical course, including a shorter disease duration and younger age at death 26.
To sum up, focal inflammation leads to a relapse with focal neurodegeneration whereas diffuse white matter and gray matter pathology results in diffuse neurodegeneration and in consequence accumulation of irreversible clinical disability (see Figure 1C) 45, 74. Since NAWM and gray matter pathology is a diffuse process, a significant number of neurons can be damaged leading to global neurodegeneration and finally irreversible clinical disability. Although this view might well be an oversimplification (pathological studies have clearly shown a close association between inflammation and neurodegeneration 45), and inflammation might well play an important role during the progressive disease stage, focal inflammation and diffuse gray/white matter pathology should be considered as separate disease elements and, in consequence, separately addressed in pre‐clinical studies.
ANIMAL MODELS TO STUDY DISTINCT DISEASE PROCESSES IN MULTIPLE SCLEROSIS
As pointed out above, the three distinct pathogenetic processes in MS are focal inflammatory lesions, diffuse white matter injury in the NAWM and diffuse gray matter pathology. All three processes are associated with neurodegeneration to a certain extent, and might finally lead to permanent clinical disability. To investigate these pathogenetic entities distinct animal models can be used. We focus here on EAE models in which the role of recurrent episodes of neuroinflammation as well as ageing promote progressive neurological disease, and the cuprizone model in which primary oligodendrocyte damage leads to innate immune activation within the CNS, prior to demyelination.
Experimental autoimmune encephalomyelitis
EAE is the most commonly used animal model to study inflammation and autoimmune‐mediated diseases in the CNS. In this model, experimental animals (commonly rodents) are immunized with a CNS‐related antigen administered in a strong adjuvant, usually complete Freud's adjuvant (CFA). Although it is out of the scope of this review article to describe the various existing protocols to reliably induce EAE in rodents, the combination of the peptide used and the mouse strain determine the disease course, that is, relapsing or chronic clinical disease 4, 131. For example, immunization of C57BL6 mice with myelin oligodendrocyte glycoprotein (MOG)35–55 peptide results in a monophasic chronic disease (Figure 2), whereas immunization of SJL mice with PLP139–151 peptide results in a relapsing–remitting disease course. Especially the MOG35–55 peptide model is frequently used since many transgenic and mutant mice are bred on the C57BL6 background. Following immunization, the CNS antigens are phagocytized by local professional antigen‐presenting cells (ie, Langerhans dendritic cells of the skin). These antigen‐presenting cells travel via the lymphatic route to local lymph nodes or the spleen, and interact there with lymphocytes which trigger the formation of encephalitogenic Th1‐ and Th17‐cells. These encephalitogenic T‐cells leave the peripheral lymphoid organs, invade the brain via the blood–brain barrier or blood–CSF barrier, and induce inflammation in the spinal cord (see Figure 1B) and to a certain extent in the cerebellum and optic nerve 100. Generally, the forebrain including the cortex, corpus callosum and subcortical structures are largely spared in EAE 115. Of note, mouse EAE lesions are dominated by extensive axonal degeneration and very little primary demyelination, which is fundamentally different from what is seen in MS. Indeed, development of marked demyelination requires autoantibodies to myelin components specifically those directed to MOG 96. A closer look at these inflammatory lesions reveals besides demyelination acute axonal damage, which can be reversible or permanent 97. Like augmentation of myelin damage, autoantibodies to neuronal components augment axonal damage in acute EAE 60, 76. It is important to notice that these models are not progressive models. For example in MOG35–55‐induced EAE, animals develop after 10–14 days a rapid paralysis of the tail and hindlimb, however after a brief plateau‐phase and slight recovery there is no clinical progression (see Figure 2).
Figure 2.
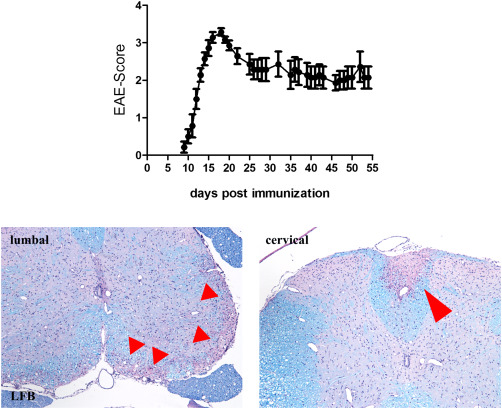
EAE clinical disease score in C57BL/6 mice immunized with MOG35–55 peptide is shown. After 9–10 days post‐immunization, inflammatory demyelination of the spinal cord results in overt clinical disability of experimental animals. After the peak of the disease (around day 15 post‐immunization) animals slightly recover, but from then on do not progress any more. Figure adapted from J Mol Neurosci. 2016 Sep;60(1):102‐14. Arrows highlight inflammatory lesions within the spinal cord.
The EAE model is an excellent tool to study mechanism associated with T cell infiltration which is central to development of acute monophasic EAE and relapsing–remitting disease. These models have proven useful in developing and understanding currently approved DMTs including natalizumab 10, fingolimod 25, dimethylfumarate 24 or alemtuzumab 129. Notably, the two approved medications, glatiramer acetate and natalizumab, were developed directly from studies in the EAE model. Furthermore, anti‐B cell therapies, which are currently tested for their effectiveness in MS in several clinical trials, have shown beneficial effects in classical EAE models 113. However, while these current therapies for MS reduce the frequency of relapses by modulating adaptive immune responses they fail to limit the irreversible neurodegeneration driving progressive disability.
Progressive EAE models
To address the contribution of adaptive immune responses in neurodegeneration, different EAE models working with Biozzi ABH mice have been developed. EAE in Biozzi ABH mice immunized with spinal cord homogenate recapitulates clinical features of MS including relapsing–remitting episodes and secondary‐progressive disability 3, 107 (see Figure 3 for histopathology). In addition, following immunization with the neuronal antigen NF‐L protein, Biozzi ABH mice develop spastic paresis and paralysis concomitant with axonal degeneration and inflammation primarily in the dorsal column of the spinal cord 59. Notably, age of experimental animals at the time of disease induction appears to be critical. While in young animals one can observe a relapsing–remitting (RR‐EAE) followed by a secondary‐progressive (SP‐EAE) phenotype, we have recently reported that EAE induction in old ABH mice is characterized by progressive disease from onset associated with pronounced axonal damage 107. Thus, not only does the extent of neurodegeneration depend on the immunizing antigen, but as well on the age during disease induction. This result is especially noteworthy because it has been shown that age is a strong predictor for RRMS conversion to SPMS 130. While the NF‐L and aged‐mouse EAE models required further validation to examine therapies applicable to SPMS, SP‐EAE has already been used to investigate the pathogenic mechanisms associated with progressive disease as well as test therapies 2, 112. Of note is that these studies clearly reveal that SP‐EAE continues despite T‐cells ablation indicating that progression is a T‐cell independent process in this model 112.
Figure 3.
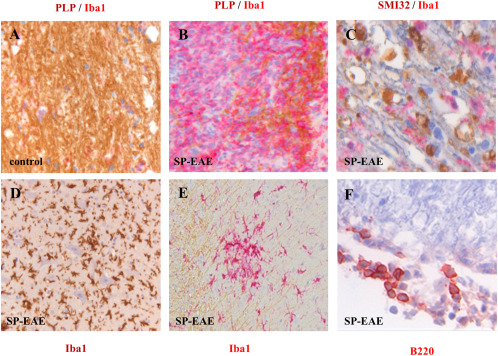
Histopathology of secondary progressive EAE in Biozzi ABH mice. Compared to aged‐matched controls in which myelin integrity is intact and in which few if any Iba‐1+ cells are present (A), in SP‐EAE demyelination is demarcated by the loss of PLP associated with highly immunereactive Iba‐1+ microglia/macrophages (B). In white matter, SMI32 depicting damaged axons is prominent in regions of highly reactive Iba‐1+ cells (C). In addition to Iba‐1 positive cells in the white matter, large infiltrates are also observed in gray matter in close association with motor neurons (D). Like MS, clusters of activated microglia are observed in normal appearing white matter in mice with SP‐EAE (E). In comparison to acute EAE in Biozzi ABH mice, a sparse number of adaptive immune cells are observed during SP‐EAE, and when observed are generally restricted to the leptomeninges (F).
Neurodegeneration and diffuse pathology in EAE
Using EAE one can investigate the mechanisms operant during T‐cell priming, how encephalitogenic T‐cells and other peripheral immune cells cross the blood–brain barrier, the cell types involved in orchestrating local T‐cell reactivation within the brain parenchyma and how inflammation, demyelination and axonal pathology are related to each other. As an example, the Kerschensteiner lab demonstrated that focal axonal degeneration is a reversible event, and that demyelination—a hallmark of MS—is not a prerequisite for this form of axon damage 97. As another example, it has recently been demonstrated in the EAE model how T‐cell trafficking between the leptomeninges and the cerebrospinal fluid is regulated 116. Besides focal pathology, certain EAE models allow us to investigate underlying mechanisms of diffuse pathology. For example, by means of neurophysiological recordings from single neurons, it was found in mice with EAE that central neurons undergo dramatic changes in glutamate‐mediated transmission, starting in the presymptomatic phase of the disease and evolving independently of demyelination or axonal injury 19. From a morphological point of view, electrophysiological abnormalities were found to be associated with marked dendritic and spine pathology. Additional important histopathological characteristics of MS that are not adequately replicated in EAE include the inflammatory white matter demyelination of the forebrain, diffuse NAWM pathology and gray matter demyelination 21, 115. In summary, although MS is a uniquely human disease, many pathological features can be induced in EAE models and thus be studied. Of note, EAE is a heterogeneous group of models and each of the various subtypes might be well suited or not for a particular scientific question.
The cuprizone model
Another model to study demyelination and related pathological changes in MS is the cuprizone model 52, 66, 110. Several important differences exist between the EAE and the cuprizone model. Maybe most importantly, in contrast to EAE T‐cells are not relevant for cuprizone‐induced demyelination. This is best documented by the finding that RAG‐deficient mice are fully susceptible to cuprizone‐induced demyelination and oligodendrocyte pathology 58. Furthermore, breakdown of the blood–brain barrier, a hallmark of MS and EAE, is not a characteristic feature of the cuprizone model 5. Although the exact mode of action of cuprizone‐intoxication is not well understood, it is believed that the copper‐chelation action of cuprizone inhibits mitochondrial enzymes of the respiratory chain which require copper as co‐factor. This leads to oxidative stress 36 and in consequence to primary oligodendrocyte apoptosis which is closely followed by microglia and astrocyte activation (see Figure 4A). The first signs of oligodendrocyte apoptosis can be seen as early as two days after initiation of the cuprizone‐diet, paralleled by a massive reduction of oligodendrocyte‐specific mRNA species 18. Demyelination is complete after 4–5 weeks cuprizone intoxication, paralleled by massive microgliosis, astrocytosis and axonal damage (see Figure 4B). Of note, robust endogenous remyelination occurs if the cuprizone‐intoxication is ceased, and the animals are provided normal chow. Due to this endogenous repair process, the cuprizone model is frequently used to study mechanisms operant during remyelination 72, 119, 121. There are several similarities between cuprizone‐induced pathology and histopathological alterations as described in post‐mortem MS material. For example, the density of mitochondria within demyelinated axons is increased in both, active MS lesions 138, and cuprizone‐induced white matter lesions 102. Besides, dying‐back oligodendrogliopathy, which manifests primarily at the most distal processes of the oligodendrocyte, has been described in MS 114 and the cuprizone model 80. Finally, the presence of active caspase‐3 expressing, pre‐apoptotic oligodendrocytes during lesions formation has been reported in MS 111 and cuprizone‐induced demyelination 18. Thus, understanding mechanisms operant during cuprizone‐induced oligodendrocyte loss and subsequent demyelination might help to understand what happens in the CNS of MS patients.
Figure 4.
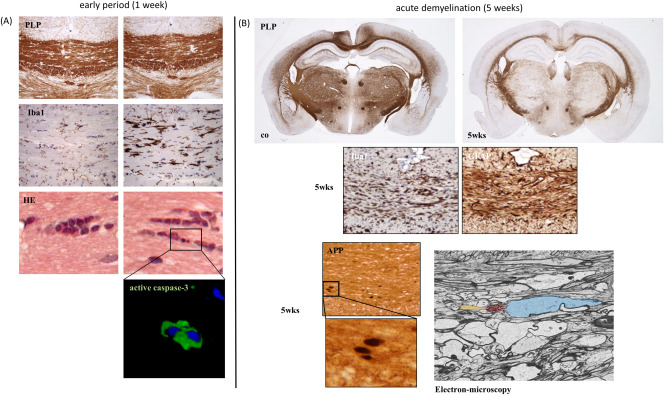
(A) Histopathological characteristics of early (1 week treatment) cuprizone‐induced lesions. While the myelination appears still normal in an anti‐PLP stain, microglia activation and oligodendrocyte apoptosis is clearly evident. Left column control, right column 1 week cuprizone‐intoxication. (B) The diffuse pathology after prolonged (5 weeks treatment) cuprizone intoxication is illustrated. Besides the midline of the corpus callosum, the cortex, hippocampus and diverse subcortical areas are affected. Demyelination is paralleled by intense microglia (Iba1) and astrocyte (GFAP) activation. Furthermore, acute axonal damage can be seen in brain sections processed for amyloid precursor protein (APP)‐immunohistochemistry. APP is a membrane‐spanning glycoprotein which is transported from neuron cell bodies to axon terminals by fast anterograde axonal transport. In healthy axons, APP is below the immunohistochemical detection limit. Under pathological conditions, for example cytoskeletal disruption, anterograde axonal transport is disturbed and APP accumulates as ovoid spheroids 70. Accumulation of such axonal spheroids can be directly demonstrated by electron microscopy. In yellow, the intact part of the axon is highlighted, red shows the axonal spheroid. Blue shows severe axonal enlargement.
Brain intrinsic degeneration as trigger of inflammatory lesion development
In recent years, the MS community gained interest in the underlying mechanisms of cuprizone‐induced pathology. Presumably, this is due to major clinical and histopathological advances of the last decade, which shaped our understanding of MS pathology. While in the year 2000 there were 116 reports on cuprizone listed in pubmed, ten years later, this number has increased to 269. Today (August 2016) 543 publications are listed in the pubmed database which either used the cuprizone model as an experimental tool (mainly as remyelination model), or investigated the mode of action underlying cuprizone‐induced pathology. As already stated above, histopathological hallmarks after short‐term cuprizone intoxication are oligodendrocyte apoptosis, normal appearing myelin on the histological level and focal microglia activation without T‐cell infiltration. Such histopathological characteristics have been described during early MS lesion formation by the Prineas lab 57. In their study, twenty‐six active lesions from 11 patients with early MS were histopathologically characterized. They observed extensive oligodendrocyte apoptosis and microglial activation associated with few lymphocytes and phagocytes in regions of myelin preservation. This was followed by the disappearance of oligodendrocytes and the presence of intramyelinic edema with tissue vacuolation. Finally, the myelin sheaths were fragmented and phagocytosed by macrophages in the presence of infiltrating T‐cells. Although it was speculated that the case(s) described by Barnett and Prineas may have been neuromyelitis optica rather than MS 17 , there is clear evidence from other studies, conducted in other laboratories, showing that demyelination (either as new lesion of at the rim of established lesions) in MS can start with initial oligodendrocyte degeneration with concomitant microglia activation. The nomenclature of this phenomenon is unfortunately rather heterogeneous including the terms “preactive” lesions 132, slowly expanding lesions 14, smoldering lesions 45 or prephagocytic areas 73. Although these lesions have different pathological characteristics, all these terms describe white matter prone to demyelination eventually with signs of oligodendrocyte degeneration and microglia activation. The predominance of T‐cells, in contrast, is not a characteristic feature of such areas. By some authors it was even speculated that MS relapses are primarily caused by metabolic changes influencing glial and/or neuronal function that secondary lead to a breakdown in the blood–brain barrier integrity and consequently peripheral immune cell recruitment 23, 122.
An important question thus is: Can brain intrinsic, degenerative events in the CNS trigger peripheral immune cell recruitment 122. To answer this question, the cuprizone model has proven to be extremely helpful. In a recently published work, our group demonstrated that primary oligodendrocyte stress can act as a potent trigger for peripheral immune cell recruitment into the brain 115. In this study, mice were fed cuprizone for 3 weeks, followed by a period of 2 weeks on normal chow to induce primary oligodendrocyte apoptosis, followed by the formation of degenerative forebrain lesions. Subsequent immunization with MOG35–55, which induces myelin autoreactive T‐cells in peripheral lymphoid organs, resulted in massive immune cell recruitment into the mouse cerebrum. On the histopathological level, such infiltrates were characterized by destruction of the perivascular glia limitans, monocyte and lymphocyte extravasation as well as demyelination (see Figure 5). In line with reports from other groups 11, 13, these results clearly illustrate the significance of brain‐intrinsic degenerative cascades for immune cell recruitment and MS lesion formation. Since activated immune cells show enhanced migration into the brain parenchyma, one might speculate that two triggers have to occur simultaneously during the formation of new inflammatory lesions: intracerebral oligodendrocyte/myelin damage and peripheral immune activation. Additional studies have now to address the signaling cascades and mechanistic processes that form the top‐down communication between the affected brain area, neurovascular unit and peripheral immune cells. Furthermore, additional studies have to investigate whether, and if so, under which conditions oligodendrocyte degeneration is sufficient to trigger an adaptive autoimmune response against myelin 126.
Figure 5.
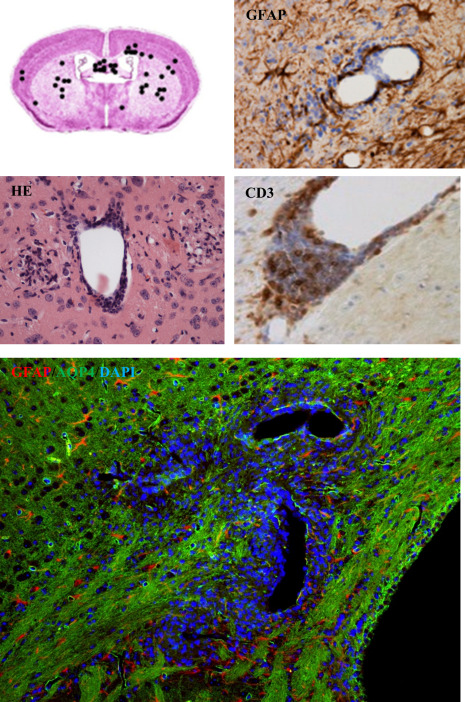
Distribution and characteristics of inflammatory forebrain lesions in the Cup/EAE model is shown 115. Infiltrates were found widespread within the forebrain, including the cortex, corpus callosum and subcortical regions (upper‐left part). Anti‐GFAP stains show the breakdown of the glia limitans perivascularis. Anti‐CD3 stains demonstrate that a significant number of perivascular cells are lymphocytes. Double staining for GFAP and the water channel protein AQP4 (aquaporin‐4) clearly demonstrate functional impairment of astrocytes around such inflammatory lesions.
Diffuse white matter injury in the cuprizone model
To what extent brain intrinsic degenerative changes are at the root of MS lesion formation has to be investigated more in detail in the future. However, without a doubt diffuse white and gray matter pathology is an important pathological event in MS. Since the presence of T‐cells is less prominent in these diffusely damaged areas, but rather oligodendrocyte pathology and microglia activation can be found, the cuprizone model appears to be a valuable tool to study such processes. First, there is diffuse demyelination in the cuprizone model. We and others have shown that cuprizone provokes demyelination in several brain regions among the hippocampus 99, cerebellum 51, caudate putamen, corpus callosum 1 and ventral part of the caudate nucleus 109. Interestingly, distinct gray matter sub‐regions in the cortex 120, within the hippocampus, and the striatal complex are also affected. In a recent study, our group was able to show that microglia activation is less intense in demyelinated gray compared to white matter areas. Myelin debris might play a critical role in this context, not just in the cuprizone model but as well in human gray matter lesions 28. A second reason why the cuprizone model is a valuable tool to study diffuse tissue damage in MS is the fact that even after prolonged cuprizone exposure there are white matter areas, such as the hippocampal fimbria region, parts of the corpus callosum or the anterior commissure, that are not demyelinated but rather present classical abnormalities of the NAWM (ie, minor myelin deficits, microglia activation and oligodendrocyte stress) 50, 67. Why some regions display overt demyelination in the cuprizone model, whereas others do not is currently not known, however it allows us to study NAWM pathology.
The signature and magnitude of diffuse neurodegeneration in the cuprizone model needs further studies
Another critical question is whether diffuse neurodegeneration can be observed in the cuprizone model. The answer is yes, although systematic studies having addressed this important aspect are rare. First, it has been shown by many groups that axonal damage can be observed in demyelinated white matter areas such as the corpus callosum 61, 121. In an elegant study Bruce Trapp's lab was able to show that disturbed mitochondrial mobility within demyelinated axons is involved in axonal degeneration in this model 102. Interestingly, results of a recent study clearly show that even after completed remyelination, axonal degeneration continues to progress at a low level leading to substantial clinical deficits 85. Second, Hamada and Kole were able to show that apical dendrites of cortical pyramidal cells are shorter after cuprizone‐intoxication 53. Besides, when comparing cuprizone‐treated groups to control, it was found that chronic demyelination induced an increase in number of dendritic branches. In the same study, the authors also showed axon initial segment pathology, the axonal domain responsible for action potential initiation. Of note, axon initial segment pathology was as well demonstrated in the non‐inflammed cortex of EAE‐induced mice 27, strongly suggesting that such changes are not directly related to autoimmune‐driven, focal inflammation. Third, it has been shown that glutamate‐signaling is altered in the brains of cuprizone‐treated animals 123. To sum up, although there is good evidence that neurodegeneration occurs in the cuprizone model, gold standard methods such as design‐based stereology, dendritic spine imaging or serial block‐face scanning electron microscopy should be applied in the future to give a precise picture about the magnitude and kinetic of cuprizone‐induced neuronal pathology. Some of these studies are currently performed in our laboratory.
SUMMARY
It was our aim to present a brief overview about preclinical models available to study MS pathology, and to develop new therapeutic options. One of the most frequently applied MS animal models is EAE, and from the various EAE protocols, MOG35–55‐induced disease in C57BL6 mice is in many laboratories the first method of choice. As of this day (August 2016), 1015 publications are listed in the pubmed database using MOG35–55‐induced EAE whereas just 54 reports are listed when entering the search key “Biozzi AND EAE.” Despite its heavy use it is however questionable to what extent the C57BL6 model will prove helpful for the development of novel therapeutic strategies for progressive MS. In our opinion, other animal models should be taken into account, and some of them have been listed herein. Disease progression is not a feature of MOG35–55 136 nor is it characteristic of PLP139–151‐induced EAE 139 and as such might, thus, be best studied in the Biozzi ABH EAE model. Another option is to use MOG‐induced EAE in the non‐obese diabetic (NOD) mouse strain 8, 34, 88, where continuous progression has been reported after the initial inflammatory attack. Disease progression can as well be studied in the cuprizone model, although this is less well appreciated by the community. As pointed out elsewhere one hallmark of the cuprizone model is spontaneous remyelination after experimentally induced acute demyelination. This is paralleled by an impaired motor performance and, during remyelination, functional recovery 75, 85. However, long after remyelination is completed (approximately 6 months after the last demyelinating episode), locomotor performance again declines in remyelinated animals as compared to age‐matched controls. This functional decline is accompanied by brain atrophy and callosal axonal loss. Furthermore, the number of acutely damaged axons is still significantly elevated in long‐term remyelinated animals as compared to age‐matched controls 66, 85. To conclude, diffuse pathology is as well evident after demyelination episodes in the cuprizone model, and neuroprotective compounds might be tested using this experimental paradigm.
Not in extenso discussed in this review article are EAE models in which myelin damage is augmented with antibodies directed to CNS components, nor was the relevance of B‐cells discussed 9. Although MS is still dominated by concepts of myelin reactive autoaggressive T‐cells, analogy to murine EAE models, and a belief that CNS immunoglobulins, including oligoclonal bands, represent meaningless, nonsense antibodies, the effectiveness of B‐cell depletion therapy (such as rituximab) clearly points toward an important function of antibodies and/or B‐cells in MS pathogenesis 56. Of note, B‐cells are extremely diverse members of the universe of adaptive immunity, and have numerous effector functions independent of their differentiation from antibody‐secreting plasma cells. In this context it is important to mention that peptide immunization models are B‐cell independent because the B‐cell receptor that binds mostly conformational rather than short linear epitopes is not involved in antigen capture.
Another aim of this review article was to highlight the significance of the cuprizone model to understand pathological processes operant in MS. If we understand which factors link oligodendrocyte degeneration, microglia activation and neurodegeneration, we may well get an insight into cellular and molecular processes of NAWM and gray matter pathology in MS. For example, it has been shown that the chemokine CXCL10, but not CCL2 or CCL3, actively participates in the initiation of early microglial activation in the cuprizone model 29. Just as EAE, the cuprizone model can be used to identify pathogenic mechanisms and therapeutics. For example, it has recently been shown that Laquinimod prevents cuprizone‐induced demyelination 16. Recent findings from the Phase III study “ALLEGRO” indicate that Laquinimod has even more pronounced effects on sustained disability progression as well as on brain atrophy compared to its effect on inflammatory relapses. The finding that cuprizone‐induced oligodendrocyte damage is ameliorated by Laquinimod, and that brain‐intrinsic degenerative cascades can trigger the formation of new inflammatory brain lesions raises the question whether the observed anti‐inflammatory effects of Laquinimod are secondary due to amelioration of diffuse white matter pathology.
As announced at the very beginning of this review article, we briefly would like to discuss how these models can be improved to reflect emerging knowledge of MS pathology. There is clinical evidence that the clinical course of MS is age related, and age is a good predictor for entering the secondary progressive disease stage. Moreover, while in patients younger than 30 years of age, the disease typically begins as RRMS, in patients greater than 40 years of age, the disease often begins as PPMS. In contrast, most experimental studies use young animals to reveal underlying mechanisms of neurodegeneration in MS. There is clear evidence that regenerative processes decrease with age 94, 118 strongly indicating that experiments performed in aged animals will reveal new findings which will help to understand underlying mechanisms of neurodegeneration is SPMS and, eventually, PPMS. Furthermore, parameters used in clinical studies to measure neurodegeneration (eg, brain or cortical atrophy) should be implemented in preclinical studies to allow a better understanding what the radiological parameters mean. The first studies covering this area have already been published 63, 118 and certainly more of them will follow in the future.
CONFLICT OF INTEREST
The authors report no conflict of interest.
REFERENCES
- 1. Acs P, Kipp M, Norkute A, Johann S, Clarner T, Braun A et al (2009) 17Beta‐estradiol and progesterone prevent cuprizone provoked demyelination of corpus callosum in male mice. Glia 57:807–814. [DOI] [PubMed] [Google Scholar]
- 2. Al‐Izki S, Pryce G, Hankey DJ, Lidster K, von Kutzleben SM, Browne L et al (2014) Lesional‐targeting of neuroprotection to the inflammatory penumbra in experimental multiple sclerosis. Brain 137:92–108. [DOI] [PubMed] [Google Scholar]
- 3. Al‐Izki S, Pryce G, O'Neill JK, Butter C, Giovannoni G, Amor S, Baker D (2012) Practical guide to the induction of relapsing progressive experimental autoimmune encephalomyelitis in the Biozzi ABH mouse. Mult Scler Relat Disord 1:29–38. [DOI] [PubMed] [Google Scholar]
- 4. Baker D, Amor S (2014) Experimental autoimmune encephalomyelitis is a good model of multiple sclerosis if used wisely. Mult Scler Relat Disord 3:555–564. [DOI] [PubMed] [Google Scholar]
- 5. Bakker DA, Ludwin SK (1987) Blood–brain barrier permeability during Cuprizone‐induced demyelination. Implications for the pathogenesis of immune‐mediated demyelinating diseases . J Neurol Sci 78:125–137. [DOI] [PubMed] [Google Scholar]
- 6. Baranzini SE, Srinivasan R, Khankhanian P, Okuda DT, Nelson SJ, Matthews PM et al (2010) Genetic variation influences glutamate concentrations in brains of patients with multiple sclerosis. Brain 133:2603–2611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Barnett MH, Prineas JW (2004) Relapsing and remitting multiple sclerosis: pathology of the newly forming lesion. Ann Neurol 55:458–468. [DOI] [PubMed] [Google Scholar]
- 8. Basso AS, Frenkel D, Quintana FJ, Costa‐Pinto FA, Petrovic‐Stojkovic S, Puckett L et al (2008) Reversal of axonal loss and disability in a mouse model of progressive multiple sclerosis. J Clin Invest 118:1532–1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Batoulis H, Wunsch M, Birkenheier J, Rottlaender A, Gorboulev V, Kuerten S (2015) Central nervous system infiltrates are characterized by features of ongoing B cell‐related immune activity in MP4‐induced experimental autoimmune encephalomyelitis. Clin Immunol 158:47–58. [DOI] [PubMed] [Google Scholar]
- 10. Bauer M, Brakebusch C, Coisne C, Sixt M, Wekerle H, Engelhardt B, Fassler R (2009) Beta1 integrins differentially control extravasation of inflammatory cell subsets into the CNS during autoimmunity. Proc Natl Acad Sci USA 106:1920–1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Baxi EG, DeBruin J, Tosi DM, Grishkan IV, Smith MD, Kirby LA et al (2015) Transfer of myelin‐reactive th17 cells impairs endogenous remyelination in the central nervous system of cuprizone‐fed mice. J Neurosci 35:8626–8639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Berkovich R (2013) Treatment of acute relapses in multiple sclerosis. Neurotherapeutics 10:97–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Boretius S, Escher A, Dallenga T, Wrzos C, Tammer R, Bruck W et al (2012) Assessment of lesion pathology in a new animal model of MS by multiparametric MRI and DTI. Neuroimage 59:2678–2688. [DOI] [PubMed] [Google Scholar]
- 14. Bramow S, Frischer JM, Lassmann H, Koch‐Henriksen N, Lucchinetti CF, Sorensen PS, Laursen H (2010) Demyelination versus remyelination in progressive multiple sclerosis. Brain 133:2983–2998. [DOI] [PubMed] [Google Scholar]
- 15. Brown MG, Kirby S, Skedgel C, Fisk JD, Murray TJ, Bhan V, Sketris IS (2007) How effective are disease‐modifying drugs in delaying progression in relapsing‐onset MS? Neurology 69:1498–1507. [DOI] [PubMed] [Google Scholar]
- 16. Bruck W, Pfortner R, Pham T, Zhang J, Hayardeny L, Piryatinsky V et al (2012) Reduced astrocytic NF‐kappaB activation by laquinimod protects from cuprizone‐induced demyelination. Acta Neuropathol 124:411–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bruck W, Popescu B, Lucchinetti CF, Markovic‐Plese S, Gold R, Thal DR, Metz I (2012) Neuromyelitis optica lesions may inform multiple sclerosis heterogeneity debate. Ann Neurol 72:385–394. [DOI] [PubMed] [Google Scholar]
- 18. Buschmann JP, Berger K, Awad H, Clarner T, Beyer C, Kipp M (2012) Inflammatory response and chemokine expression in the white matter corpus callosum and gray matter cortex region during cuprizone‐induced demyelination. J Mol Neurosci 48:66–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Centonze D, Muzio L, Rossi S, Cavasinni F, De Chiara V, Bergami A et al (2009) Inflammation triggers synaptic alteration and degeneration in experimental autoimmune encephalomyelitis. J Neurosci 29:3442–3452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Cerina M, Narayanan V, Gobel K, Bittner S, Ruck T, Meuth P, Herrmann AM, Stangel M, Gudi V, Skripuletz T, Daldrup T, Wiendl H, Seidenbecher T, Ehling P, Kleinschnitz C, Pape HC, Budde T, Meuth SG (2017) The quality of cortical network function recovery depends on localization and degree of axonal demyelination. Brain Behav Immun 59:103–117. [DOI] [PubMed] [Google Scholar]
- 21. Chanaday NL, Roth GA (2016) Microglia and astrocyte activation in the frontal cortex of rats with experimental autoimmune encephalomyelitis. Neuroscience 314:160–169. [DOI] [PubMed] [Google Scholar]
- 22. Chari DM, Zhao C, Kotter MR, Blakemore WF, Franklin RJ (2006) Corticosteroids delay remyelination of experimental demyelination in the rodent central nervous system. J Neurosci Res 83:594–605. [DOI] [PubMed] [Google Scholar]
- 23. Chaudhuri A, Behan PO (2004) Multiple sclerosis is not an autoimmune disease. Arch Neurol 61:1610–1612. [DOI] [PubMed] [Google Scholar]
- 24. Chen H, Assmann JC, Krenz A, Rahman M, Grimm M, Karsten CM et al (2014) Hydroxycarboxylic acid receptor 2 mediates dimethyl fumarate's protective effect in EAE. J Clin Invest 124:2188–2192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Choi JW, Gardell SE, Herr DR, Rivera R, Lee CW, Noguchi K et al (2011) FTY720 (fingolimod) efficacy in an animal model of multiple sclerosis requires astrocyte sphingosine 1‐phosphate receptor 1 (S1P1) modulation. Proc Natl Acad Sci USA 108:751–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Choi SR, Howell OW, Carassiti D, Magliozzi R, Gveric D, Muraro PA et al (2012) Meningeal inflammation plays a role in the pathology of primary progressive multiple sclerosis. Brain 135:2925–2937. [DOI] [PubMed] [Google Scholar]
- 27. Clark KC, Josephson A, Benusa SD, Hartley RK, Baer M, Thummala S et al (2016) Compromised axon initial segment integrity in EAE is preceded by microglial reactivity and contact. Glia 64:1190–1209. [DOI] [PubMed] [Google Scholar]
- 28. Clarner T, Diederichs F, Berger K, Denecke B, Gan L, van der Valk P et al (2012) Myelin debris regulates inflammatory responses in an experimental demyelination animal model and multiple sclerosis lesions. Glia 60:1468–1480. [DOI] [PubMed] [Google Scholar]
- 29. Clarner T, Janssen K, Nellessen L, Stangel M, Skripuletz T, Krauspe B et al (2015) CXCL10 triggers early microglial activation in the cuprizone model. J Immunol 194:3400–3413. [DOI] [PubMed] [Google Scholar]
- 30. Clarner T, Parabucki A, Beyer C, Kipp M (2011) Corticosteroids impair remyelination in the corpus callosum of cuprizone‐treated mice. J Neuroendocrinol 23:601–611. [DOI] [PubMed] [Google Scholar]
- 31. Comi G (2008) Is it clinically relevant to repair focal multiple sclerosis lesions? J Neurol Sci 265:17–20. [DOI] [PubMed] [Google Scholar]
- 32. Confavreux C, Hutchinson M, Hours MM, Cortinovis‐Tourniaire P, Moreau T (1998) Rate of pregnancy‐related relapse in multiple sclerosis. Pregnancy in Multiple Sclerosis Group. N Engl J Med 339:285–291. [DOI] [PubMed] [Google Scholar]
- 33. Cree BA, Gourraud PA, Oksenberg JR, Bevan C, Crabtree‐Hartman E, Gelfand JM, Goodin DS, Graves J, Green AJ, Mowry E, Okuda DT, Pelletier D, von Budingen HC, Zamvil SS, Agrawal A, Caillier S, Ciocca C, Gomez R, Kanner R, Lincoln R, Lizee A, Qualley P, Santaniello A, Suleiman L, Bucci M, Panara V, Papinutto N, Stern WA, Zhu AH, Cutter GR, Baranzini S, Henry RG, Hauser SL (2016) Long‐term evolution of multiple sclerosis disability in the treatment era. Ann Neurol 80(4):499–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Dang PT, Bui Q, D'Souza CS, Orian JM (2015) Modelling MS: chronic‐relapsing EAE in the NOD/Lt mouse strain. Curr Top Behav Neurosci 26:143–177. [DOI] [PubMed] [Google Scholar]
- 35. De Stefano N, Battaglini M, Stromillo ML, Zipoli V, Bartolozzi ML, Guidi L et al (2006) Brain damage as detected by magnetization transfer imaging is less pronounced in benign than in early relapsing multiple sclerosis. Brain 129:2008–2016. [DOI] [PubMed] [Google Scholar]
- 36. Draheim T, Liessem A, Scheld M, Wilms F, Weissflog M, Denecke B, Kensler TW, Zendedel A, Beyer C, Kipp M, Wruck CJ, Fragoulis A, Clarner T (2016) Activation of the astrocytic Nrf2/ARE system ameliorates the formation of demyelinating lesions in a multiple sclerosis animal model. Glia 64(12):2219–2230. [DOI] [PubMed] [Google Scholar]
- 37. Dutta R, Chang A, Doud MK, Kidd GJ, Ribaudo MV, Young EA et al, (2011) Demyelination causes synaptic alterations in hippocampi from multiple sclerosis patients. Ann Neurol 69:445–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Dziedzic T, Metz I, Dallenga T, Konig FB, Muller S, Stadelmann C, Bruck W (2010) Wallerian degeneration: a major component of early axonal pathology in multiple sclerosis. Brain Pathol 20:976–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Evangelou N, Esiri MM, Smith S, Palace J, Matthews PM (2000) Quantitative pathological evidence for axonal loss in normal appearing white matter in multiple sclerosis. Ann Neurol 47:391–395. [PubMed] [Google Scholar]
- 40. Ferguson B, Matyszak MK, Esiri MM, Perry VH (1997) Axonal damage in acute multiple sclerosis lesions. Brain 120:393–399. [DOI] [PubMed] [Google Scholar]
- 41. Fern R, Harrison PJ (1991) The effects of compression upon conduction in myelinated axons of the isolated frog sciatic nerve. J Physiol 432:111–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Fernando KT, McLean MA, Chard DT, MacManus DG, Dalton CM, Miszkiel KA et al (2004) Elevated white matter myo‐inositol in clinically isolated syndromes suggestive of multiple sclerosis. Brain 127:1361–1369. [DOI] [PubMed] [Google Scholar]
- 43. Filippi M, Campi A, Dousset V, Baratti C, Martinelli V, Canal N et al (1995) A magnetization transfer imaging study of normal‐appearing white matter in multiple sclerosis. Neurology 45:478–482. [DOI] [PubMed] [Google Scholar]
- 44. Frischer JM, Bramow S, Dal‐Bianco A, Lucchinetti CF, Rauschka H, Schmidbauer M et al (2009) The relation between inflammation and neurodegeneration in multiple sclerosis brains. Brain 132:1175–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Frischer JM, Weigand SD, Guo Y, Kale N, Parisi JE, Pirko I et al (2015) Clinical and pathological insights into the dynamic nature of the white matter multiple sclerosis plaque. Ann Neurol 78:710–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Funfschilling U, Supplie LM, Mahad D, Boretius S, Saab AS, Edgar J et al (2012) Glycolytic oligodendrocytes maintain myelin and long‐term axonal integrity. Nature 485:517–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Geurts JJ, Bo L, Roosendaal SD, Hazes T, Daniels R, Barkhof F et al (2007) Extensive hippocampal demyelination in multiple sclerosis. J Neuropathol Exp Neurol 66:819–827. [DOI] [PubMed] [Google Scholar]
- 48. Geurts JJ, Calabrese M, Fisher E, Rudick RA (2012) Measurement and clinical effect of grey matter pathology in multiple sclerosis. Lancet Neurol 11:1082–1092. [DOI] [PubMed] [Google Scholar]
- 49. Gilbert JJ, Sadler M (1983) Unsuspected multiple sclerosis. Arch Neurol 40:533–536. [DOI] [PubMed] [Google Scholar]
- 50. Goldberg J, Clarner T, Beyer C, Kipp M (2015) Anatomical distribution of cuprizone‐induced lesions in C57BL6 mice. J Mol Neurosci 57:166–175. [DOI] [PubMed] [Google Scholar]
- 51. Groebe A, Clarner T, Baumgartner W, Dang J, Beyer C, Kipp M (2009) Cuprizone treatment induces distinct demyelination, astrocytosis, and microglia cell invasion or proliferation in the mouse cerebellum. Cerebellum 8:163–174. [DOI] [PubMed] [Google Scholar]
- 52. Gudi V, Gingele S, Skripuletz T, Stangel M (2014) Glial response during cuprizone‐induced de‐ and remyelination in the CNS: lessons learned. Front Cell Neurosci 8:73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Hamada MS, Kole MH (2015) Myelin loss and axonal ion channel adaptations associated with gray matter neuronal hyperexcitability. J Neurosci 35:7272–7286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Hametner S, Wimmer I, Haider L, Pfeifenbring S, Bruck W, Lassmann H (2013) Iron and neurodegeneration in the multiple sclerosis brain. Ann Neurol 74:848–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Hargens AR, Botte MJ, Swenson MR, Gelberman RH, Rhoades CE, Akeson WH (1993) Effects of local compression on peroneal nerve function in humans. J Orthop Res 11:818–827. [DOI] [PubMed] [Google Scholar]
- 56. Hauser SL, Waubant E, Arnold DL, Vollmer T, Antel J, Fox RJ et al (2008) B‐cell depletion with rituximab in relapsing‐remitting multiple sclerosis. N Engl J Med 358:676–688. [DOI] [PubMed] [Google Scholar]
- 57. Henderson AP, Barnett MH, Parratt JD, Prineas JW (2009) Multiple sclerosis: distribution of inflammatory cells in newly forming lesions. Ann Neurol 66:739–753. [DOI] [PubMed] [Google Scholar]
- 58. Hiremath MM, Chen VS, Suzuki K, Ting JP, Matsushima GK (2008) MHC class II exacerbates demyelination in vivo independently of T cells. J Neuroimmunol 203:23–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Huizinga R, Heijmans N, Schubert P, Gschmeissner S, t Hart BA, Herrmann H, Amor S (2007) Immunization with neurofilament light protein induces spastic paresis and axonal degeneration in Biozzi ABH mice. J Neuropathol Exp Neurol 66:295–304. [DOI] [PubMed] [Google Scholar]
- 60. Huizinga R, Linington C, Amor S (2008) Resistance is futile: antineuronal autoimmunity in multiple sclerosis. Trends Immunol 29:54–60. [DOI] [PubMed] [Google Scholar]
- 61. Irvine KA, Blakemore WF (2006) Age increases axon loss associated with primary demyelination in cuprizone‐induced demyelination in C57BL/6 mice. J Neuroimmunol 175:69–76. [DOI] [PubMed] [Google Scholar]
- 62. Irvine KA, Blakemore WF (2008) Remyelination protects axons from demyelination‐associated axon degeneration. Brain 131:1464–1477. [DOI] [PubMed] [Google Scholar]
- 63. Jelescu IO, Zurek M, Winters KV, Veraart J, Rajaratnam A, Kim NS et al (2016) In vivo quantification of demyelination and recovery using compartment‐specific diffusion MRI metrics validated by electron microscopy. Neuroimage 132:104–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Jurgens T, Jafari M, Kreutzfeldt M, Bahn E, Bruck W, Kerschensteiner M, Merkler D (2016) Reconstruction of single cortical projection neurons reveals primary spine loss in multiple sclerosis. Brain 139:39–46. [DOI] [PubMed] [Google Scholar]
- 65. Kappos L, Polman C, Pozzilli C, Thompson A, Beckmann K, Dahlke F (2001) Final analysis of the European multicenter trial on IFNbeta‐1b in secondary‐progressive MS. Neurology 57:1969–1975. [DOI] [PubMed] [Google Scholar]
- 66. Kipp M, Clarner T, Dang J, Copray S, Beyer C (2009) The cuprizone animal model: new insights into an old story. Acta Neuropathol 118:723–736. [DOI] [PubMed] [Google Scholar]
- 67. Kipp M, Norkus A, Krauspe B, Clarner T, Berger K, van der Valk P et al (2011) The hippocampal fimbria of cuprizone‐treated animals as a structure for studying neuroprotection in multiple sclerosis. Inflamm Res 60:723–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Kooi EJ, Geurts JJ, van Horssen J, Bo L, van der Valk P (2009) Meningeal inflammation is not associated with cortical demyelination in chronic multiple sclerosis. J Neuropathol Exp Neurol 68:1021–1028. [DOI] [PubMed] [Google Scholar]
- 69. Kornek B, Storch MK, Weissert R, Wallstroem E, Stefferl A, Olsson T et al (2000) Multiple sclerosis and chronic autoimmune encephalomyelitis: a comparative quantitative study of axonal injury in active, inactive, and remyelinated lesions. Am J Pathol 157:267–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Kuhlmann T, Lingfeld G, Bitsch A, Schuchardt J, Bruck W (2002) Acute axonal damage in multiple sclerosis is most extensive in early disease stages and decreases over time. Brain 125:2202–2212. [DOI] [PubMed] [Google Scholar]
- 71. Kutzelnigg A, Lucchinetti CF, Stadelmann C, Bruck W, Rauschka H, Bergmann M et al (2005) Cortical demyelination and diffuse white matter injury in multiple sclerosis. Brain 128:2705–2712. [DOI] [PubMed] [Google Scholar]
- 72. Kuypers NJ, Bankston AN, Howard RM, Beare JE, Whittemore SR (2016) Remyelinating oligodendrocyte precursor cell miRNAs from the sfmbt2 cluster promote cell cycle arrest and differentiation. J Neurosci 36:1698–1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Lassmann H (2011) Review: the architecture of inflammatory demyelinating lesions: implications for studies on pathogenesis. Neuropathol Appl Neurobiol 37:698–710. [DOI] [PubMed] [Google Scholar]
- 74. Lassmann H, van Horssen J, Mahad D (2012) Progressive multiple sclerosis: pathology and pathogenesis. Nat Rev Neurol 8:647–656. [DOI] [PubMed] [Google Scholar]
- 75. Liebetanz D, Merkler D (2006) Effects of commissural de‐ and remyelination on motor skill behaviour in the cuprizone mouse model of multiple sclerosis. Exp Neurol 202:217–224. [DOI] [PubMed] [Google Scholar]
- 76. Lindner M, Ng JK, Hochmeister S, Meinl E, Linington C (2013) Neurofascin 186 specific autoantibodies induce axonal injury and exacerbate disease severity in experimental autoimmune encephalomyelitis. Exp Neurol 247:259–266. [DOI] [PubMed] [Google Scholar]
- 77. Longbrake EE, Cross AH (2016) Effect of multiple sclerosis disease‐modifying therapies on B cells and humoral immunity. JAMA Neurol 73:219–225. [DOI] [PubMed] [Google Scholar]
- 78. Lucchinetti C, Bruck W, Parisi J, Scheithauer B, Rodriguez M, Lassmann H (2000) Heterogeneity of multiple sclerosis lesions: implications for the pathogenesis of demyelination. Ann Neurol 47:707–717. [DOI] [PubMed] [Google Scholar]
- 79. Lucchinetti CF, Popescu BF, Bunyan RF, Moll NM, Roemer SF, Lassmann H et al (2011) Inflammatory cortical demyelination in early multiple sclerosis. N Engl J Med 365:2188–2197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Ludwin SK, Johnson ES (1981) Evidence for a “dying‐back” gliopathy in demyelinating disease. Ann Neurol 9:301–305. [DOI] [PubMed] [Google Scholar]
- 81. Magliozzi R, Howell O, Vora A, Serafini B, Nicholas R, Puopolo M et al (2007) Meningeal B‐cell follicles in secondary progressive multiple sclerosis associate with early onset of disease and severe cortical pathology. Brain 130:1089–1104. [DOI] [PubMed] [Google Scholar]
- 82. Magliozzi R, Howell OW, Reeves C, Roncaroli F, Nicholas R, Serafini B et al (2010) A Gradient of neuronal loss and meningeal inflammation in multiple sclerosis. Ann Neurol 68:477–493. [DOI] [PubMed] [Google Scholar]
- 83. Mahad DH, Trapp BD, Lassmann H (2015) Pathological mechanisms in progressive multiple sclerosis. Lancet Neurol 14:183–193. [DOI] [PubMed] [Google Scholar]
- 84. Mandolesi G, Musella A, Gentile A, Grasselli G, Haji N, Sepman H et al (2013) Interleukin‐1beta alters glutamate transmission at purkinje cell synapses in a mouse model of multiple sclerosis. J Neurosci 33:12105–12121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Manrique‐Hoyos N, Jurgens T, Gronborg M, Kreutzfeldt M, Schedensack M, Kuhlmann T et al (2012) Late motor decline after accomplished remyelination: impact for progressive multiple sclerosis. Ann Neurol 71:227–244. [DOI] [PubMed] [Google Scholar]
- 86. Mao P, Reddy PH (2010) Is multiple sclerosis a mitochondrial disease? Biochim Biophys Acta 1802:66–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Mathiesen HK, Jonsson A, Tscherning T, Hanson LG, Andresen J, Blinkenberg M et al (2006) Correlation of global N‐acetyl aspartate with cognitive impairment in multiple sclerosis. Arch Neurol 63:533–536. [DOI] [PubMed] [Google Scholar]
- 88. Mayo L, Trauger SA, Blain M, Nadeau M, Patel B, Alvarez JI et al (2014) Regulation of astrocyte activation by glycolipids drives chronic CNS inflammation. Nat Med 20:1147–1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Mei F, Lehmann‐Horn K, Shen YA, Rankin KA, Stebbins KJ, Lorrain DS et al (2016) Accelerated remyelination during inflammatory demyelination prevents axonal loss and improves functional recovery. Elife 5:e18246. doi: 10.7554/eLife.18246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Meinl E, Krumbholz M, Derfuss T, Junker A, Hohlfeld R (2008) Compartmentalization of inflammation in the CNS: a major mechanism driving progressive multiple sclerosis. J Neurol Sci 274:42–44. [DOI] [PubMed] [Google Scholar]
- 91. Michailidou I, Willems JG, Kooi EJ, van Eden C, Gold SM, Geurts JJ et al (2015) Complement C1q‐C3‐associated synaptic changes in multiple sclerosis hippocampus. Ann Neurol 77:1007–1026. [DOI] [PubMed] [Google Scholar]
- 92. Miller DH, Khan OA, Sheremata WA, Blumhardt LD, Rice GP, Libonati MA et al (2003) A controlled trial of natalizumab for relapsing multiple sclerosis. N Engl J Med 348:15–23. [DOI] [PubMed] [Google Scholar]
- 93. Miller DH, Thompson AJ, Filippi M (2003) Magnetic resonance studies of abnormalities in the normal appearing white matter and grey matter in multiple sclerosis. J Neurol 250:1407–1419. [DOI] [PubMed] [Google Scholar]
- 94. Miron VE, Boyd A, Zhao JW, Yuen TJ, Ruckh JM, Shadrach JL et al (2013) M2 microglia and macrophages drive oligodendrocyte differentiation during CNS remyelination. Nat Neurosci 16:1211–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Mistry N, Abdel‐Fahim R, Mougin O, Tench C, Gowland P, Evangelou N (2014) Cortical lesion load correlates with diffuse injury of multiple sclerosis normal appearing white matter. Mult Scler (Houndmills, Basingstoke, England) 20:227–233. [DOI] [PubMed] [Google Scholar]
- 96. Morris‐Downes MM, Smith PA, Rundle JL, Piddlesden SJ, Baker D, Pham‐Dinh D et al (2002) Pathological and regulatory effects of anti‐myelin antibodies in experimental allergic encephalomyelitis in mice. J Neuroimmunol 125:114–124. [DOI] [PubMed] [Google Scholar]
- 97. Nikic I, Merkler D, Sorbara C, Brinkoetter M, Kreutzfeldt M, Bareyre FM et al (2011) A reversible form of axon damage in experimental autoimmune encephalomyelitis and multiple sclerosis. Nat Med 17:495–499. [DOI] [PubMed] [Google Scholar]
- 98. Nistico R, Mori F, Feligioni M, Nicoletti F, Centonze D (2014) Synaptic plasticity in multiple sclerosis and in experimental autoimmune encephalomyelitis. Philos Trans R Soc Lond B Biol Sci 369:20130162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Norkute A, Hieble A, Braun A, Johann S, Clarner T, Baumgartner W et al (2009) Cuprizone treatment induces demyelination and astrocytosis in the mouse hippocampus. J Neurosci Res 87:1343–1355. [DOI] [PubMed] [Google Scholar]
- 100. O'Neill JK, Baker D, Morris MM, Gschmeissner SE, Jenkins HG, Butt AM et al (1998) Optic neuritis in chronic relapsing experimental allergic encephalomyelitis in Biozzi ABH mice: demyelination and fast axonal transport changes in disease. J Neuroimmunol 82:210–218. [DOI] [PubMed] [Google Scholar]
- 101. Oh J, O'Connor PW (2015) Established disease‐modifying treatments in relapsing‐remitting multiple sclerosis. Curr Opin Neurol 28:220–229. [DOI] [PubMed] [Google Scholar]
- 102. Ohno N, Chiang H, Mahad DJ, Kidd GJ, Liu L, Ransohoff RM et al (2014) Mitochondrial immobilization mediated by syntaphilin facilitates survival of demyelinated axons. Proc Natl Acad Sci USA 111:9953–9958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Panitch H, Miller A, Paty D, Weinshenker B (2004) Interferon beta‐1b in secondary progressive MS: results from a 3‐year controlled study. Neurology 63:1788–1795. [DOI] [PubMed] [Google Scholar]
- 104. Papadopoulos D, Dukes S, Patel R, Nicholas R, Vora A, Reynolds R (2009) Substantial archaeocortical atrophy and neuronal loss in multiple sclerosis. Brain Pathol 19:238–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Pavelko KD, van Engelen BG, Rodriguez M (1998) Acceleration in the rate of CNS remyelination in lysolecithin‐induced demyelination. J Neurosci 18:2498–2505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Peferoen L, Kipp M, van der Valk P, van Noort JM, Amor S (2014) Oligodendrocyte‐microglia cross‐talk in the central nervous system. Immunology 141:302–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Peferoen LA, Breur M, van de Berg S, Peferoen‐Baert R, Boddeke EH, van der Valk P, Pryce G, van Noort JM, Baker D, Amor S (2016) Ageing and recurrent episodes of neuroinflammation promote progressive experimental autoimmune encephalomyelitis in Biozzi ABH mice. Immunology 149(2):146–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Peterson JW, Bo L, Mork S, Chang A, Trapp BD (2001) Transected neurites, apoptotic neurons, and reduced inflammation in cortical multiple sclerosis lesions. Ann Neurol 50:389–400. [DOI] [PubMed] [Google Scholar]
- 109. Pott F, Gingele S, Clarner T, Dang J, Baumgartner W, Beyer C, Kipp M (2009) Cuprizone effect on myelination, astrogliosis and microglia attraction in the mouse basal ganglia. Brain Res 1305:137–149. [DOI] [PubMed] [Google Scholar]
- 110. Praet J, Guglielmetti C, Berneman Z, Van der Linden A, Ponsaerts P (2014) Cellular and molecular neuropathology of the cuprizone mouse model: clinical relevance for multiple sclerosis. Neurosci Biobehav Rev 47:485–505. [DOI] [PubMed] [Google Scholar]
- 111. Prineas JW, Parratt JD (2012) Oligodendrocytes and the early multiple sclerosis lesion. Ann Neurol 72:18–31. [DOI] [PubMed] [Google Scholar]
- 112. Pryce G, O'Neill JK, Croxford JL, Amor S, Hankey DJ, East E et al (2005) Autoimmune tolerance eliminates relapses but fails to halt progression in a model of multiple sclerosis. J Neuroimmunol 165:41–52. [DOI] [PubMed] [Google Scholar]
- 113. Ray A, Basu S, Williams CB, Salzman NH, Dittel BN (2012) A novel IL‐10‐independent regulatory role for B cells in suppressing autoimmunity by maintenance of regulatory T cells via GITR ligand. J Immunol 188:3188–3198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Rodriguez M, Scheithauer B (1994) Ultrastructure of multiple sclerosis. Ultrastruct Pathol 18:3–13. [DOI] [PubMed] [Google Scholar]
- 115. Scheld M, Ruther BJ, Grosse‐Veldmann R, Ohl K, Tenbrock K, Dreymuller D et al (2016) Neurodegeneration triggers peripheral immune cell recruitment into the forebrain. J Neurosci 36:1410–1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Schlager C, Korner H, Krueger M, Vidoli S, Haberl M, Mielke D et al (2016) Effector T‐cell trafficking between the leptomeninges and the cerebrospinal fluid. Nature 530:349–353. [DOI] [PubMed] [Google Scholar]
- 117. Shemer A, Jung S (2015) Differential roles of resident microglia and infiltrating monocytes in murine CNS autoimmunity. Semin Immunopathol 37:613–623. [DOI] [PubMed] [Google Scholar]
- 118. Sim FJ, Zhao C, Penderis J, Franklin RJ (2002) The age‐related decrease in CNS remyelination efficiency is attributable to an impairment of both oligodendrocyte progenitor recruitment and differentiation. J Neurosci 22:2451–2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Skripuletz T, Hackstette D, Bauer K, Gudi V, Pul R, Voss E et al (2013) Astrocytes regulate myelin clearance through recruitment of microglia during cuprizone‐induced demyelination. Brain 136:147–167. [DOI] [PubMed] [Google Scholar]
- 120. Skripuletz T, Lindner M, Kotsiari A, Garde N, Fokuhl J, Linsmeier F et al (2008) Cortical demyelination is prominent in the murine cuprizone model and is strain‐dependent. Am J Pathol 172:1053–1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Slowik A, Schmidt T, Beyer C, Amor S, Clarner T, Kipp M (2015) The sphingosine 1‐phosphate receptor agonist FTY720 is neuroprotective after cuprizone‐induced CNS demyelination. Br J Pharmacol 172:80–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Stys PK, Zamponi GW, van Minnen J, Geurts JJ (2012) Will the real multiple sclerosis please stand up? Nat Rev Neurosci 13:507–514. [DOI] [PubMed] [Google Scholar]
- 123. Suhs KW, Gudi V, Eckermann N, Fairless R, Pul R, Skripuletz T, Stangel M (2016) Cytokine regulation by modulation of the NMDA receptor on astrocytes. Neurosci Lett 629:227–233. [DOI] [PubMed] [Google Scholar]
- 124. Tallantyre EC, Bo L, Al‐Rawashdeh O, Owens T, Polman CH, Lowe JS, Evangelou N (2010) Clinico‐pathological evidence that axonal loss underlies disability in progressive multiple sclerosis. Mult Scler (Houndmills, Basingstoke, England) 16:406–411. [DOI] [PubMed] [Google Scholar]
- 125. Traboulsee A, Dehmeshki J, Peters KR, Griffin CM, Brex PA, Silver N et al (2003) Disability in multiple sclerosis is related to normal appearing brain tissue MTR histogram abnormalities. Mult Scler 9:566–573. [DOI] [PubMed] [Google Scholar]
- 126. Traka M, Podojil JR, McCarthy DP, Miller SD, Popko B (2016) Oligodendrocyte death results in immune‐mediated CNS demyelination. Nat Neurosci 19:65–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Triarhou LC, Herndon RM (1986) The effect of dexamethasone on l‐alpha‐lysophosphatidyl choline (lysolecithin)‐induced demyelination of the rat spinal cord. Arch Neurol 43:121–125. [DOI] [PubMed] [Google Scholar]
- 128. Tsivgoulis G, Katsanos AH, Grigoriadis N, Hadjigeorgiou GM, Heliopoulos I, Papathanasopoulos P et al (2015) The effect of disease modifying therapies on disease progression in patients with relapsing‐remitting multiple sclerosis: a systematic review and meta‐analysis. PLoS One 10:e0144538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Turner MJ, Pang PT, Chretien N, Havari E, LaMorte MJ, Oliver J et al (2015) Reduction of inflammation and preservation of neurological function by anti‐CD52 therapy in murine experimental autoimmune encephalomyelitis. J Neuroimmunol 285:4–12. [DOI] [PubMed] [Google Scholar]
- 130. Tutuncu M, Tang J, Zeid NA, Kale N, Crusan DJ, Atkinson EJ et al (2013) Onset of progressive phase is an age‐dependent clinical milestone in multiple sclerosis. Mult Scler 19:188–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. van der Star BJ, Vogel DY, Kipp M, Puentes F, Baker D, Amor S (2012) In vitro and in vivo models of multiple sclerosis. CNS Neurol Disord Drug Targets 11:570–588. [DOI] [PubMed] [Google Scholar]
- 132. van der Valk P, Amor S (2009) Preactive lesions in multiple sclerosis. Curr Opin Neurol 22:207–213. [DOI] [PubMed] [Google Scholar]
- 133. van Noort JM, van den Elsen PJ, van Horssen J, Geurts JJ, van der Valk P, Amor S (2011) Preactive multiple sclerosis lesions offer novel clues for neuroprotective therapeutic strategies. CNS Neurol Disord Drug Targets 10:68–81. [DOI] [PubMed] [Google Scholar]
- 134. Vercellino M, Merola A, Piacentino C, Votta B, Capello E, Mancardi GL et al (2007) Altered glutamate reuptake in relapsing‐remitting and secondary progressive multiple sclerosis cortex: correlation with microglia infiltration, demyelination, and neuronal and synaptic damage. J Neuropathol Exp Neurol 66:732–739. [DOI] [PubMed] [Google Scholar]
- 135. Vrenken H, Geurts JJ (2007) Gray and normal‐appearing white matter in multiple sclerosis: an MRI perspective. Expert Rev Neurother 7:271–279. [DOI] [PubMed] [Google Scholar]
- 136. Wagenknecht N, Becker B, Scheld M, Beyer C, Clarner T, Hochstrasser T, Kipp M (2016) Thalamus degeneration and inflammation in two distinct multiple sclerosis animal models. J Mol Neurosci 60:102–114. [DOI] [PubMed] [Google Scholar]
- 137. Wegner C, Esiri MM, Chance SA, Palace J, Matthews PM (2006) Neocortical neuronal, synaptic, and glial loss in multiple sclerosis. Neurology 67:960–967. [DOI] [PubMed] [Google Scholar]
- 138. Witte ME, Bo L, Rodenburg RJ, Belien JA, Musters R, Hazes T et al (2009) Enhanced number and activity of mitochondria in multiple sclerosis lesions. J Pathol 219:193–204. [DOI] [PubMed] [Google Scholar]
- 139. Youssef S, Stuve O, Patarroyo JC, Ruiz PJ, Radosevich JL, Hur EM et al (2002) The HMG‐CoA reductase inhibitor, atorvastatin, promotes a Th2 bias and reverses paralysis in central nervous system autoimmune disease. Nature 420:78–84. [DOI] [PubMed] [Google Scholar]
- 140. Zivadinov R, Hojnacki D, Bergsland N, Kennedy C, Hagemeier J, Melia R et al (2016) Effect of natalizumab on brain atrophy and disability progression in multiple sclerosis patients over 5 years. Eur J Neurol 23:1101–1109. [DOI] [PubMed] [Google Scholar]