

Abstract
IgG4‐related disease is an immune‐mediated disease with manifestations in most organ systems among them the pituitary gland. To date, few cases of histologically confirmed cases of IgG‐related hypophysitis have been reported. The aim of this study was to retrospectively determine the prevalence of IgG4‐related hypophysitis among cases previously diagnosed as primary hypophysitis (lymphocytic hypophysitis, granulomatous hypophysitis and hypophysitis not otherwise specified). Histological and immunohistochemical analysis revealed that 12 of 29 cases (41.4%) previously diagnosed as primary hypophysitis fulfilled the criteria for IgG4‐related disease and, thus, IgG4‐related hypophysitis should always be considered in the differential diagnosis of primary hypophysitis. All cases of IgG4‐related hypophysitis showed a dense lymphoplasmacytic infiltrate with more than 10 IgG4‐positive cells per high power field and a ratio of IgG4/IgG‐positive cells of more than 40%, whereas storiform fibrosis was an inconsistent histological feature and was also seen in few cases of non‐IgG‐related hypophysitis, thus lacking sensitivity and specificity. Obliterative phlebitis was not seen in any case. Thus, histological criteria defined for IgG4‐related disease in other organs should be modified for IgG4‐related hypophysitis, accordingly.
Keywords: IgG4, histological criteria, hypophysitis, prevalence
Introduction
Inflammatory processes account for less than 1% of surgical specimen from the sellar region and are most often secondary to intrasellar lesions like Rathke's cleft cyst, histiocytosis and tumors like craniopharyngeoma and germinoma. Primary inflammations comprising mainly lymphocytic and granulomatous hypophysitis are rare and have to be differentiated from the involvement of the pituitary gland in generalized autoimmune diseases 16, 38.
IgG4‐related disease is an immune‐mediated disease first described in 2001 as the underlying cause of a subset of autoimmune pancreatitis 10, 21, 22 and has since been reported in most organ systems including lung, kidney, biliary tree, liver, salivary glands, thyroid gland, prostate orbit, retroperitoneum, skin, orbit, meninges and pituitary gland 23. IgG4‐related disease is characterized by a dense lymphoplasmacytic infiltrate rich in IgG4 plasma cells that may be accompanied by elevated serum IgG4 levels 8 and is treatable by glucocorticoid therapy in its inflammatory stage 23. Histopathological criteria supporting the diagnosis of IgG4‐related disease comprise a dense lymphoplasmacytic infiltrate with a percentage of IgG4‐positive of all IgG‐positive cells exceeding 40% and—depending on the organ involved—absolute numbers of IgG4‐positive plasma cells exceeding at least 10 per high power field (HPF), accompanied by an at least focally storiform fibrosis and obliterative phlebitis 8. The pituitary gland is rarely (0%–8% of cases) involved in IgG4‐related disease 9, 27, 29, 30 and, thus, few cases of histologically confirmed IgG4‐related hypophysitis have been reported. Leporati et al proposed three criteria for the diagnosis of IgG4‐related hypophysitis with the fulfillment of any of these sufficing to diagnose IgG4‐related hypophysitis but only one of these criteria involves histological proof in a biopsy of the pituitary whereas the other criteria rely on histological proof of IgG4‐related disease in other organs, clinical, laboratory and imaging data only and according to these criteria, IgG4‐related hypophysitis can be diagnosed without histological proof in any organ 26. Adhering to the criteria of Leporati et al, 39 cases of IgG4‐related hypophysitis have been described in the English literature to our best knowledge 1, 2, 3, 6, 11, 12, 13, 14, 15, 18, 19, 20, 24, 25, 26, 31, 32, 33, 34, 35, 36, 37, 39, 41, 42, 43, 44, 45, 46, 47, of which only 14 were histologically confirmed 1, 3, 13, 15, 26, 32, 35, 39, 43, 46.
Considering the importance of early diagnosing IgG4‐related disease in a potentially curable stage of the disease, the aim of this study was to determine the prevalence of IgG4‐related hypophysitis among histologically confirmed primary hypophysitis and to verify whether diagnostic criteria established for other organ systems are applicable in the diagnosis of IgG‐related hypophysitis.
Materials and Methods
Selection of cases
The archives of the German pituitary adenoma registry and the Institutes of Neuropathology of the University Medical Center Hamburg‐Eppendorf and the University of Erlangen were screened for diagnosed inflammatory lesions of the pituitary gland. Cases spanning the years 2001–2016 were retrieved from the archives and reviewed by two independent pathologists for confirmation of the previous diagnosis. Cases with inflammation secondary to Rathke's cleft cysts, tumors, previous operations or infectious agents were excluded from further analysis. The remaining 29 cases of primary hypophysitis comprised lymphocytic hypophysitis, granulomatous hypophysitis and hypophysitis, not otherwise specified (NOS) according to standard histomorphological criteria. Available clinical data were restricted to the tentative diagnosis, differential diagnoses and secondary diagnoses submitted by the surgeon together with the specimen. Laboratory data and clinical data on follow‐up including response to immunotherapy were not available.
Histological and immunohistochemical analyses
For histomorphological analysis of lymphoplasmacytic infiltrate, storiform fibrosis and obliterative phlebitis 3‐µm‐thick paraffin sections were cut on a microtome and stained with hematoxylin and eosin according to standard laboratory protocols.
For immunohistochemical analysis with antibodies against total IgG and IgG4, consecutive 3‐µm‐thick sections were immunostained to allow evaluation of corresponding visual fields. Slides were deparaffinized and processed simultaneously using a DAKO Autostainer (DAKO Link 38; DAKO, Glostrup, Denmark) with heat‐induced antigen retrieval for 15 minutes at 95°C in Tris‐EDTA‐citrate buffer at pH 9.0 and application of primary antibody for 20 minutes. Bound antibody was visualized using the EnVision Kit (DAKO) according to the manufacturer's directions.
Antibodies used were rabbit polyclonal antibody against IgG (DAKO ready to use; DAKO) and mouse monoclonal antibody against IgG4 (1:100, REF MCO11, Clone: HP6025, Binding Site).
For analysis, total numbers of IgG‐ and IgG4‐positive cells were counted in up to three high power fields (HPF) in corresponding visual fields in serial sections and means were calculated for each case.
Results
Twenty‐nine cases previously diagnosed as primary hypophysitis spanning the years 2001–2016 were retrieved from the archives of the German pituitary adenoma registry and the Institutes of Neuropathology of the University Medical Center Hamburg‐Eppendorf and the University of Erlangen. Tentative diagnoses submitted by the surgeon were intra‐ or suprasellar mass, pituitary adenoma, hypophysitis, meningioma and tuberculosis. Two of the patients were in an early postpartum state (Table S1). Of these 29 cases, 15 (51.7%) had been diagnosed as lymphocytic hypophysitis, 6 (20.7%) as granulomatous and 8 (27.6%) as hypophysitis, not otherwise specified (NOS) (Table 1). The mean age at the time of the biopsy was 41.7 years, and female/male ratio was 2.2:1. All cases showed—at least focally—a dense lymphoplasmacytic infiltrate, the cardinal feature of IgG4‐related disease (Figure 1).
Table 1.
Data of cases included into the study.
| Patient | Sex | Age | Hypophysitis | IgG/HPF | IgG4/HPF | %IgG4/IgG | Fibrosis | Thrombosis |
|---|---|---|---|---|---|---|---|---|
| 1 | f | 75 | NOS | 48 | 46 | 95 | Yes | No |
| 2 | f | 37 | NOS | 41 | 35 | 85 | Yes | No |
| 3 | f | 60 | NOS | 117 | 84 | 71 | Yes | No |
| 4 | f | 19 | NOS | 23 | 15 | 64 | No | No |
| 5 | m | 33 | NOS | 168 | 21 | 12 | No | No |
| 6 | m | 37 | NOS | 39 | 0 | 0 | Yes | No |
| 7 | m | 36 | NOS | 31 | 0 | 0 | No | No |
| 8 | m | 44 | NOS | 0 | 0 | 0 | No | No |
| 9 | f | 23 | Lymphocytic | 52 | 38 | 74 | No | No |
| 10 | m | 28 | Lymphocytic | 17 | 13 | 74 | Yes | No |
| 11 | m | 53 | Lymphocytic | 127 | 93 | 73 | No | No |
| 12 | m | 62 | Lymphocytic | 99 | 60 | 61 | No | No |
| 13 | f | 28 | Lymphocytic | 69 | 41 | 60 | No | No |
| 14 | m | 40 | Lymphocytic | 95 | 51 | 53 | Yes | No |
| 15 | f | 65 | Lymphocytic | 58 | 29 | 51 | No | No |
| 16 | f | 26 | Lymphocytic | 65 | 31 | 47 | Yes | No |
| 17 | f | 62 | Lymphocytic | 70 | 17 | 24 | Yes | No |
| 18 | f | 39 | Lymphocytic | 42 | 9 | 22 | No | No |
| 19 | f | 45 | Lymphocytic | 73 | 3 | 4 | No | No |
| 20 | f | 48 | Lymphocytic | 70 | 1 | 1 | No | No |
| 21 | f | 35 | Lymphocytic | 75 | 0 | 0 | Yes | No |
| 22 | m | 37 | Lymphocytic | 12 | 0 | 0 | No | No |
| 23 | f | 40 | Lymphocytic | 0 | 0 | 0 | No | No |
| 24 | f | 40 | Granulomatous | 73 | 2 | 2 | No | No |
| 25 | f | 32 | Granulomatous | 76 | 1 | 1 | No | No |
| 26 | f | 41 | Granulomatous | 12 | 0 | 0 | No | No |
| 27 | f | 15 | Granulomatous | 39 | 0 | 0 | No | No |
| 28 | f | 50 | Granulomatous | 17 | 0 | 0 | No | No |
| 29 | f | 60 | Granulomatous | 0 | 0 | 0 | No | No |
Cases highlighted in italics fulfill the criteria for IgG‐related disease. NOS, not otherwise specified; IgG/HPF, IgG‐positive cells per high power field; IgG4/HPF, IgG4‐positive cells per high power field; %IgG4/IgG, percent IgG4‐positive cells per IgG‐positive cells; fibrosis, storiform fibrosis; thrombosis, venous thrombosis.
Figure 1.
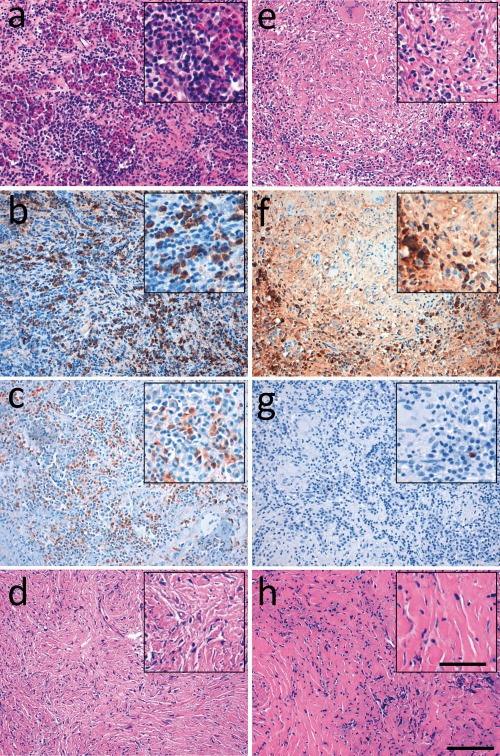
Histologic features of IgG4‐related hypophysitis. In the left panel, representative histological (H&E) and immunohistochemical pictures of a case of IgG4‐related hypophysitis that had previously been diagnosed as lymphocytic hypophysitis showing a dense lymphoplasmacytic infiltrate (a) with abundant IgG‐positive cells (b) and IgG4‐positive cells (c) and storiform fibrosis (d). In the right panel, a case of granulomatous hypophysitis showing a dense lymphoplasmacytic infiltrate (e) with abundant IgG‐positive cells (f) but only disseminated IgG4‐positive cells (g) and nonstoriform fibrosis (h). Scale bar = 100 µm.
To screen for IgG4‐related hypophysitis, immunohistochemical analyses with antibodies against IgG and IgG4 were performed and numbers of IgG‐positive and IgG4‐positive cells were counted as proposed in the consensus statement on the pathology of IgG4‐related disease 8. Cases were assumed to qualify as IgG4‐related hypophysitis if absolute numbers of IgG4‐positive cells exceeded 10 per high power field (HPF) and the ratio of IgG4‐ and IgG‐positive cells exceeded 40%, adhering to the minimum criteria defined in the consensus statement on the pathology of IgG4‐related disease 8.
Applying these histopathologic criteria, 12 cases (41.4%) qualified as IgG4‐related hypophysitis (Figure 2a). Interestingly, none of the cases previously diagnosed as granulomatous hypophysitis fulfilled the criteria for IgG4‐related hypophysitis, whereas eight (53.3%) cases previously diagnosed as lymphocytic hypophysitis and four (50%) cases previously diagnosed as hypophysitis NOS fulfilled these criteria (Figure 2b). The mean age of patients with IgG4‐related hypophysitis was 43 years, and female/male ratio was 2:1 and did not significantly differ from non‐IgG4‐related hypophysitis (Figure 2c,d). The hypophysitis of both women in early postpartum state fulfilled the criteria for IgG4‐related disease (Table S1).
Figure 2.
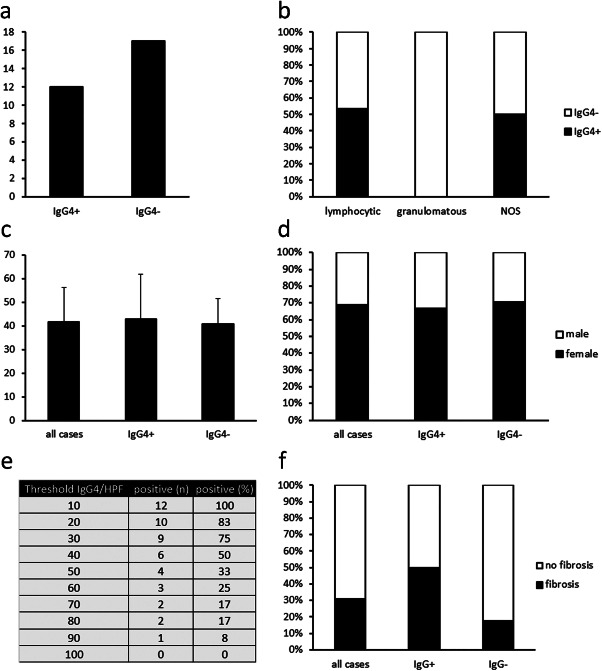
Statistical analysis of hypophysitis cases. (a) Absolute numbers of cases previously diagnosed as hypophysitis fulfilling the criteria for IgG4‐related hypophysitis (IgG4+) or failing to fulfill these criteria (IgG4−). (b) Percentages of IgG4+ and IgG4− hypophysitis among cases previously diagnosed as lymphocytic hypophysitis, granulomatous hypophysitis, or hypophysitis, not otherwise specified (NOS). (c) Mean ages ± standard deviation of all 29 cases analyzed and of IgG4+ and IgG4− hypophysitis. (d) Percentages of male and female patients among all cases and IgG4+ and IgG4− hypophysitis. (e) Sensitivity of different thresholds for the number of IgG4‐positive cells per high power field in the diagnosis of IgG4‐related hypophysitis. (f) Percentages of storiform fibrosis among all cases and IgG4+ and IgG4− hypophysitis.
A total of 10 IgG4‐positive cells per HPF constitute the minimum threshold for IgG4‐related disease but the appropriate cutoff point varies from organ to organ but has not been determined for the pituitary gland 8, and specificity increases with higher cutoff points at the expense of sensitivity. Therefore, we analyzed how higher cutoff points for absolute numbers of IgG4‐positive cells would influence the number of cases qualifying as IgG4‐related hypophysitis. As expected, the percentage of cases fulfilling the criteria dropped to 75% of initially identified cases of IgG4‐related hypophysitis when the cutoff point was raised to 30 IgG4‐positive cells per HPF, to 50%, when the cutoff point was raised to 40 IgG4‐positive cells per HPF and to 0%, when the cutoff point was raised to 100 IgG4‐positive cells per HPF (Figure 2e). Considering the potential therapeutic options, we propose a cutoff point of 10 IgG4‐positive cells per HPF to ensure sufficient sensitivity.
Storiform fibrosis is the second characteristic feature of IgG4‐related disease. Consequently, we analyzed the prevalence of storiform fibrosis in IgG4‐related hypophysitis. Six of the twelve cases (50%) identified as IgG4‐related hypophysitis and three of nineteen non‐IgG‐related cases (16%) of hypophysitis showed storiform fibrosis (Figure 2f). Thus, storiform fibrosis lacks sensitivity and specificity among IgG4‐related hypophysitis and should not be included as a necessary feature in the diagnostic process for IgG4‐related hypophysitis.
Obliterative phlebitis, the third characteristic histologic feature of IgG4‐related disease, was not detected in any of our cases of IgG4‐related hypophysitis.
Discussion
The results of this study demonstrate that IgG4‐related hypophysitis is highly prevalent among histologically confirmed primary hypophysitis. In our study, 41.4% of all cases that had previously been diagnosed as primary hypophysitis fulfilled histologic criteria of IgG4‐related hypophysitis. Previous studies reported a lower prevalence between 5% and 30% 1, 17, 43, but these studies comprised only one 17, two 43 and seven 1 cases, respectively, and not all of these cases were histologically confirmed—in the study by Bando et al, only three of seven cases 1. Thus, the prevalence might have been under‐ or overestimated in these studies. IgG4‐related hypophysitis is a rare and new disease with the first histologically confirmed report in 2007 46. Before our study only fourteen histologically confirmed cases had been described in the English literature 1, 3, 13, 15, 26, 32, 35, 39, 43, 46 and another 25 cases without histological confirmation in pituitary specimen 2, 6, 11, 12, 18, 19, 20, 25, 26, 31, 33, 34, 36, 37, 41, 42, 44, 45, 47. The mean age of patients at biopsy with IgG4‐related hypophysitis (43 years) in our study was lower than the mean age at biopsy of previously published histologically confirmed cases (54 years) and significantly lower than the mean age at biopsy of all published cases including the cases that had not been histologically confirmed (66 years). The lower age in our patient cohort may be due to the novelty of IgG4‐related disease leading to a long history of the patient before the accurate diagnosis was made in cases with primary involvement of other organs when the diagnosis was made without histological confirmation in the pituitary. On the other hand, symptoms of primary involvement of the pituitary may have facilitated early diagnosis leading to a younger age at diagnosis in the histologically confirmed cases. This is supported by the fact that nine (65%) of the cases published before our study did not show involvement of other organs at the time of biopsy with a mean age of 49 years, whereas the remaining five (35%) cases that showed involvement of at least one more organ at the time of biopsy had a mean age of 62 years. Whether the cases without involvement of other organs comprise a clinical entity of isolated IgG4‐related hypophysitis or early manifestations of generalized IgG4‐related disease cannot be judged from the data available from previous published cases and in our study, no clinical data about other organ involvement was available but it is noticeable that previous studies reported pituitary involvement in IgG4‐related disease in only 0%–8% of patients 9, 27, 29, 30. It seems, thus, unlikely, that the cases of IgG4‐related hypophysitis without other organ involvement are early manifestations of generalized IgG4‐related disease. Furthermore, the female : male ratio in previously published cases without other organ involvement is 3:1, whereas this value is 0.25:1 in previously published cases with other organ involvement. Our study also showed a female preponderance (2:1). Thus, a subset of IgG4‐related hypophysitis might be a disease entity distinct from generalized IgG4‐related disease. Our cohort of 12 cases of histologically confirmed IgG4‐related hypophysitis is the largest cohort published to date with previous cohorts comprising three 1 and two 43 cases, respectively. In 2012, a consensus statement on the pathology of IgG4‐related disease defined histomorphological criteria for the diagnosis of IgG4‐related disease 8. In general, two out of the three major histomorphological features dense lymphoplasmacytic infiltrate, storiform fibrosis and obliterative phlebitis are a prerequisite for the diagnosis of IgG4‐related disease, but organ‐specific exceptions to this rule have been described. For example, storiform fibrosis is rare in manifestations of IgG4‐related disease in lymph nodes and lacrimal gland, and obliterative phlebitis is sometimes lacking in these organs 4, 5. In our cohort, storiform fibrosis was detectable in 50% of cases of IgG4‐related hypophysitis and also in 16% of cases of non‐IgG4‐related hypophysitis and, thus, lacked sensitivity and specificity, and obliterative phlebitis was not detectable in any case. Therefore, we propose that storiform fibrosis and obliterative phlebitis should not be a prerequisite for the diagnosis of IgG4‐related hypophysitis. The third major feature of IgG4‐related disease, a dense lymphoplasmacytic infiltrate, was met by all cases in our cohort. Thus, we consider this feature a prerequisite before testing the most important feature of IgG4‐related hypophysitis, the existence of IgG4‐positive cells. The consensus statement defines a threshold of 40% for the fraction of IgG4‐positive of all IgG‐positive cells, which is valid for all organ manifestations of IgG4‐related disease 8, a criterium that is met by all our cases of IgG4‐related hypophysitis. Furthermore, an organ‐specific minimum absolute number of IgG4‐positive cells per HPF are claimed ranging from 10 to 100. Our data show that the threshold value should not exceed 10 for the diagnosis of IgG4‐related hypophysitis in order to ensure a sufficient sensitivity. Applying these criteria, none of the included cases of granulomatous hypophysitis fulfilled the criteria for IgG4‐related disease. This is in line with previous reports describing that granulomatous inflammatory processes are relatively inconsistent with IgG4‐related disease 7, 40, 48. Adhering to these criteria, IgG4‐related hypophysitis can also be discriminated from Rosai Dorfman disease in most cases because although the disease is often associated with IgG4‐positive cells exceeding 10 per HPF because the fraction of IgG4‐positive cells of all IgG‐positive cells rarely exceeds 40% 28.
In summary, we could show that IgG4‐related hypophysitis is highly prevalent among histologically confirmed cases of hypophysitis and should always be considered in the differential diagnosis of nongranulomatous inflammatory lesions of the sella because IgG4‐related disease is treatable in early stages of the disease. Further studies will have to show whether a subset of IgG4‐related hypophysitis comprises an own disease entity independent of IgG4‐related disease.
Ethical Approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Declaration of Helsinki and its later amendments or comparable ethical standards. For this type of study, formal consent is not required.
Supporting information
Additional Supporting Information may be found in the online version of this article at the publisher's web‐site:
Table S1. Clinical data submitted by the surgeon.
Acknowledgments
The funding for the German Pituitary Tumor Registry to WS from Novartis Pharma GmbH (Nuremberg, Germany), Novo Nordisk Pharma GmbH (Mainz, Germany), Pfizer Pharma GmbH (Berlin, Germany), and Ipsen Pharma GmbH (Ettlingen, Germany) is gratefully acknowledged. We thank all colleagues for sending tumor material to the German Pituitary Tumor Registry.
References
- 1. Bando H, Iguchi G, Fukuoka H, Taniguchi M, Yamamoto M, Matsumoto R et al (2014) The prevalence of IgG4‐related hypophysitis in 170 consecutive patients with hypopituitarism and/or central diabetes insipidus and review of the literature. Eur J Endocrinol 170:161–172. [DOI] [PubMed] [Google Scholar]
- 2. Batista RL, Ramos LS, Cescato VA, Musolino NR, Borba CG, Silva GO et al (2015) Thickened pituitary stalk associated with a mass in the sphenoidal sinus: an alarm to suspect hypophysitis by immunoglobulin G4? Int Arch Otorhinolaryngol 19:273–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Caputo C, Bazargan A, McKelvie PA, Sutherland T, Su CS, Inder WJ (2014) Hypophysitis due to IgG4‐related disease responding to treatment with azathioprine: an alternative to corticosteroid therapy. Pituitary 17:251–256. [DOI] [PubMed] [Google Scholar]
- 4. Cheuk W, Yuen HK, Chan JK (2007) Chronic sclerosing dacryoadenitis: part of the spectrum of IgG4‐related sclerosing disease? Am J Surg Pathol 31:643–645. [DOI] [PubMed] [Google Scholar]
- 5. Cheuk W, Yuen HK, Chu SY, Chiu EK, Lam LK, Chan JK (2008) Lymphadenopathy of IgG4‐related sclerosing disease. Am J Surg Pathol 32:671–681. [DOI] [PubMed] [Google Scholar]
- 6. Chiba K, Kamisawa T, Tabata T, Hara S, Kuruma S, Fujiwara T et al (2013) Clinical features of 10 patients with IgG4‐related retroperitoneal fibrosis. Intern Med 52:1545–1551. [DOI] [PubMed] [Google Scholar]
- 7. Deshpande V, Gupta R, Sainani N, Sahani DV, Virk R, Ferrone C et al (2011) Subclassification of autoimmune pancreatitis: a histologic classification with clinical significance. Am J Surg Pathol 35:26–35. [DOI] [PubMed] [Google Scholar]
- 8. Deshpande V, Zen Y, Chan JK, Yi EE, Sato Y, Yoshino T et al (2012) Consensus statement on the pathology of IgG4‐related disease. Mod Pathol 25:1181–1192. [DOI] [PubMed] [Google Scholar]
- 9. Ebbo M, Daniel L, Pavic M, Seve P, Hamidou M, Andres E et al (2012) IgG4‐related systemic disease: features and treatment response in a French cohort: results of a multicenter registry. Medicine 91:49–56. [DOI] [PubMed] [Google Scholar]
- 10. Hamano H, Kawa S, Horiuchi A, Unno H, Furuya N, Akamatsu T et al (2001) High serum IgG4 concentrations in patients with sclerosing pancreatitis. N Engl J Med 344:732–738. [DOI] [PubMed] [Google Scholar]
- 11. Haraguchi A, Era A, Yasui J, Ando T, Ueki I, Horie I et al (2010) Putative IgG4‐related pituitary disease with hypopituitarism and/or diabetes insipidus accompanied with elevated serum levels of IgG4. Endocr J 57:719–725. [DOI] [PubMed] [Google Scholar]
- 12. Harano Y, Honda K, Akiyama Y, Kotajima L, Arioka H (2015) A case of IgG4‐related hypophysitis presented with hypopituitarism and diabetes insipidus. Clin Med Insights Case Rep 8:23–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hattori Y, Tahara S, Ishii Y, Kitamura T, Inomoto C, Osamura RY et al (2013) A case of IgG4‐related hypophysitis without pituitary insufficiency. J Clin Endocrinol Metab 98:1808–1811. [DOI] [PubMed] [Google Scholar]
- 14. Hori M, Makita N, Andoh T, Takiyama H, Yajima Y, Sakatani T et al (2010) Long‐term clinical course of IgG4‐related systemic disease accompanied by hypophysitis. Endocr J 57:485–492. [DOI] [PubMed] [Google Scholar]
- 15. Hsing MT, Hsu HT, Cheng CY, Chen CM (2013) IgG4‐related hypophysitis presenting as a pituitary adenoma with systemic disease. Asian J Surg 36:93–97. [DOI] [PubMed] [Google Scholar]
- 16. Hunn BH, Martin WG, Simpson S, Jr , McLean CA (2014) Idiopathic granulomatous hypophysitis: a systematic review of 82 cases in the literature. Pituitary 17:357–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Imber BS, Lee HS, Kunwar S, Blevins LS, Aghi MK (2015) Hypophysitis: a single‐center case series. Pituitary 18:630–641. [DOI] [PubMed] [Google Scholar]
- 18. Isaka Y, Yoshioka K, Nishio M, Yamagami K, Konishi Y, Inoue T et al (2008) A case of IgG4‐related multifocal fibrosclerosis complicated by central diabetes insipidus. Endocr J 55:723–728. [DOI] [PubMed] [Google Scholar]
- 19. Iseda I, Hida K, Tone A, Tenta M, Shibata Y, Matsuo K et al (2014) Prednisolone markedly reduced serum IgG4 levels along with the improvement of pituitary mass and anterior pituitary function in a patient with IgG4‐related infundibulo‐hypophysitis. Endocr J 61:195–203. [DOI] [PubMed] [Google Scholar]
- 20. Joshi D, Jager R, Hurel S, Pereira SP, Johnson GJ, Chapman M et al (2015) Cerebral involvement in IgG4‐related disease. Clin Med (Lond) 15:130–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kamisawa T, Egawa N, Nakajima H (2003) Autoimmune pancreatitis is a systemic autoimmune disease. Am J Gastroenterol 98:2811–2812. [DOI] [PubMed] [Google Scholar]
- 22. Kamisawa T, Funata N, Hayashi Y, Eishi Y, Koike M, Tsuruta K et al (2003) A new clinicopathological entity of IgG4‐related autoimmune disease. J Gastroenterol 38:982–984. [DOI] [PubMed] [Google Scholar]
- 23. Kamisawa T, Zen Y, Pillai S, Stone JH (2015) IgG4‐related disease. Lancet 385:1460–1471. [DOI] [PubMed] [Google Scholar]
- 24. Kanoke A, Ogawa Y, Watanabe M, Kumabe T, Tominaga T (2013) Autoimmune hypophysitis presenting with intracranial multi‐organ involvement: three case reports and review of the literature. BMC Res Notes 6:560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Khong P, Enno A, Darwish B (2014) Lymphoplasmacytic hypophysitis associated with immunoglobulin G4. J Clin Neurosci 21:342–344. [DOI] [PubMed] [Google Scholar]
- 26. Leporati P, Landek‐Salgado MA, Lupi I, Chiovato L, Caturegli P (2011) IgG4‐related hypophysitis: a new addition to the hypophysitis spectrum. J Clin Endocrinol Metab 96:1971–1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lin W, Lu S, Chen H, Wu Q, Fei Y, Li M et al (2015) Clinical characteristics of immunoglobulin G4‐related disease: a prospective study of 118 Chinese patients. Rheumatology 54:1982–1990. [DOI] [PubMed] [Google Scholar]
- 28. Liu L, Perry AM, Cao W, Smith LM, Hsi ED, Liu X et al (2013) Relationship between Rosai‐Dorfman disease and IgG4‐related disease: study of 32 cases. Am J Clin Pathol 140:395–402. [DOI] [PubMed] [Google Scholar]
- 29. Masaki Y, Dong L, Kurose N, Kitagawa K, Morikawa Y, Yamamoto M et al (2009) Proposal for a new clinical entity, IgG4‐positive multiorgan lymphoproliferative syndrome: analysis of 64 cases of IgG4‐related disorders. Ann Rheum Dis 68:1310–1315. [DOI] [PubMed] [Google Scholar]
- 30. Masaki Y, Kurose N, Yamamoto M, Takahashi H, Saeki T, Azumi A et al (2012) Cutoff values of serum IgG4 and histopathological IgG4+ plasma cells for diagnosis of patients with IgG4‐related disease. Int J Rheumatol 2012:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Nakasone Y, Oguchi K, Sato Y, Okubo Y, Yamauchi K, Aizawa T (2015) Rapid conversion of autoimmune hypophysitis to an empty sella with immediate lowering of the serum IgG4 level. Case report. Neuro Endocrinol Lett 36:112–114. [PubMed] [Google Scholar]
- 32. Ngaosuwan K, Trongwongsa T, Shuangshoti S (2015) Clinical course of IgG4‐related hypophysitis presenting with focal seizure and relapsing lymphocytic hypophysitis. BMC Endocr Disord 15:64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Nishina N, Kaneko Y, Kuwana M, Hanaoka H, Kameda H, Mikami S, Takeuchi T (2012) IgG4‐related disease without overexpression of IgG4: pathogenesis implications. Case Rep Rheumatol 2012:754935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ohkubo Y, Sekido T, Takeshige K, Ishi H, Takei M, Nishio S et al (2014) Occurrence of IgG4‐related hypophysitis lacking IgG4‐bearing plasma cell infiltration during steroid therapy. Intern Med 53:753–757. [DOI] [PubMed] [Google Scholar]
- 35. Osawa S, Ogawa Y, Watanabe M, Tominaga T (2009) Hypophysitis presenting with atypical rapid deterioration: with special reference to immunoglobulin G4‐related disease‐case report. Neurol Med Chir 49:622–625. [DOI] [PubMed] [Google Scholar]
- 36. Patel SM, Szostek JH (2011) IgG4‐related systemic disease in a Native American man. Intern Med 50:931–934. [DOI] [PubMed] [Google Scholar]
- 37. Ralli S, Lin J, Farrell J (2007) Autoimmune pancreatitis. N Engl J Med 356:1586, author reply 7. [DOI] [PubMed] [Google Scholar]
- 38. Rivera JA (2006) Lymphocytic hypophysitis: disease spectrum and approach to diagnosis and therapy. Pituitary 9:35–45. [DOI] [PubMed] [Google Scholar]
- 39. Sosa GA, Bell S, Christiansen SB, Pietrani M, Glerean M, Loto M et al (2014) Histologically confirmed isolated IgG4‐related hypophysitis: two case reports in young women. Endocrinol Diabetes Metab Case Rep 2014:140062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Stone JH, Zen Y, Deshpande V (2012) IgG4‐related disease. N Engl J Med 366:539–551. [DOI] [PubMed] [Google Scholar]
- 41. Tanabe T, Tsushima K, Yasuo M, Urushihata K, Hanaoka M, Koizumi T et al (2006) IgG4‐associated multifocal systemic fibrosis complicating sclerosing sialadenitis, hypophysitis, and retroperitoneal fibrosis, but lacking pancreatic involvement. Intern Med 45:1243–1247. [DOI] [PubMed] [Google Scholar]
- 42. Taniguchi T, Kobayashi H, Fukui S, Ogura K, Saiga T, Okamoto M (2006) A case of multifocal fibrosclerosis involving posterior mediastinal fibrosis, retroperitoneal fibrosis, and a left seminal vesicle with elevated serum IgG4. Hum Pathol 37:1237–1239, author reply 9. [DOI] [PubMed] [Google Scholar]
- 43. Tauziede‐Espariat A, Polivka M, Bouazza S, Decq P, Robert G, Laloi‐Michelin M, Adle‐Biassette H (2015) The prevalence of IgG4‐positive plasma cells in hypophysitis: a possible relationship to IgG4‐related disease. Clin Neuropathol 34:181–192. [DOI] [PubMed] [Google Scholar]
- 44. Tsuboi H, Inokuma S, Setoguchi K, Shuji S, Hagino N, Tanaka Y et al (2008) Inflammatory pseudotumors in multiple organs associated with elevated serum IgG4 level: recovery by only a small replacement dose of steroid. Intern Med 47:1139–1142. [DOI] [PubMed] [Google Scholar]
- 45. van der Vliet HJ, Perenboom RM (2004) Multiple pseudotumors in IgG4‐associated multifocal systemic fibrosis. Ann Intern Med 141:896–897. [DOI] [PubMed] [Google Scholar]
- 46. Wong S, Lam WY, Wong WK, Lee KC (2007) Hypophysitis presented as inflammatory pseudotumor in immunoglobulin G4‐related systemic disease. Hum Pathol 38:1720–1723. [DOI] [PubMed] [Google Scholar]
- 47. Yamamoto M, Takahashi H, Ohara M, Suzuki C, Naishiro Y, Yamamoto H et al (2006) A case of Mikulicz's disease (IgG4‐related plasmacytic disease) complicated by autoimmune hypophysitis. Scand J Rheumatol 35:410–411. [DOI] [PubMed] [Google Scholar]
- 48. Zen Y, Nakanuma Y (2010) IgG4‐related disease: a cross‐sectional study of 114 cases. Am J Surg Pathol 34:1812–1819. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting Information may be found in the online version of this article at the publisher's web‐site:
Table S1. Clinical data submitted by the surgeon.