

Abstract
Clusterin, also known as apoJ, is a lipoprotein abundantly expressed within the CNS. It regulates Aβ fibril formation and toxicity and facilitates amyloid‐β (Aβ) transport across the blood‐brain barrier. Genome‐wide association studies have shown variations in the clusterin gene (CLU) to influence the risk of developing sporadic Alzheimer's disease (AD). To explore whether clusterin modulates the regional deposition of Aβ, we measured levels of soluble (NP40‐extracted) and insoluble (guanidine‐HCl‐extracted) clusterin, Aβ40 and Aβ42 by sandwich ELISA in brain regions with a predilection for amyloid pathology—mid‐frontal cortex (MF), cingulate cortex (CC), parahippocampal cortex (PH), and regions with little or no pathology—thalamus (TH) and white matter (WM). Clusterin level was highest in regions with plaque pathology (MF, CC, PH and PC), approximately mirroring the regional distribution of Aβ. It was significantly higher in AD than controls, and correlated positively with Aβ42 and insoluble Aβ40. Soluble clusterin level rose significantly with severity of cerebral amyloid angiopathy, and in MF and PC regions was highest in APOE ɛ4 homozygotes. In the TH and WM (areas with little amyloid pathology) clusterin was unaltered in AD and did not correlate with Aβ level. There was a significant positive correlation between the concentration of clusterin and the regional levels of insoluble Aβ42; however, the molar ratio of clusterin : Aβ42 declined with insoluble Aβ42 level in a region‐dependent manner, being lowest in regions with predilection for Aβ plaque pathology. Under physiological conditions, clusterin reduces aggregation and promotes clearance of Aβ. Our findings indicate that in AD, clusterin increases, particularly in regions with most abundant Aβ, but because the increase does not match the rising level of Aβ42, the molar ratio of clusterin : Aβ42 in those regions falls, probably contributing to Aβ deposition within the tissue.
Keywords: Alzheimer's disease, amyloid‐β, amyloid‐β clearance, apoJ, cerebral amyloid angiopathy, clusterin, plaque
Introduction
Alzheimer's disease (AD) is believed to be initiated by the accumulation and aggregation of amyloid‐β (Aβ) peptides [the so‐called amyloid cascade hypothesis 1]. The steady‐state level of Aβ reflects the balance between its production and removal from the brain 2. Aβ peptides are produced by sequential cleavage of Aβ protein precursor (APP) and mostly end at amino acid 40 or 42. Aβ42 is the more amyloidogenic form—relatively insoluble in the interstitial fluid and prone to parenchymal deposition. Aβ40 is more soluble, less prone to parenchymal deposition but more likely to accumulate in the walls of cortical and leptomeningeal blood vessels 3, 4. Most mutations in familial AD are associated with increased amyloidogenic processing of APP and elevated Aβ42 or an increase in the Aβ42 : Aβ40 ratio 5, 6, 7, 8. In sporadic AD, which accounts for most cases, the accumulation of Aβ is thought largely to reflect alterations in the pathways responsible for the removal of Aβ [reviewed in 2] or altered expression of chaperone proteins, such as apoE and clusterin (also known as apoJ) that regulate the structure, toxicity and clearance of Aβ [reviewed in 9].
Aβ peptides are produced throughout life 10, 11 but begin to accumulate and aggregate in the brain more than a decade before the onset of AD 12, 13. Risk factors for AD, such as ageing and APOE genotype 14, accelerate the parenchymal deposition of Aβ. The deposition of Aβ within the brain follows a hierarchical sequence first appearing in the neocortex and spreading to limbic areas, deep cerebral gray matter and brain stem regions and finally the cerebellum 15. The determinants of regional variability in the susceptibility of different brain regions to Aβ deposition remain unclear. No link was found between the distribution of plaque pathology and the regional distribution of enzymes involved in the amyloidogenic processing of APP (APP, APP‐CRFB, BACE‐1 and PS‐1; 16, 17, 18, 19). However, Shinohara et al 19 found a strong inverse relationship between apolipoprotein E (apoE) level and Aβ deposition in brain tissue from cognitively normal elderly people and those with mild cognitive impairment (MCI). The authors suggested that apoE had a role in preventing Aβ accumulation and was reduced in brain regions that would later develop significant plaque pathology.
Clusterin, also known as apolipoprotein‐J, is a 78‐80 kDa heterodimeric glycoprotein that is abundantly expressed in the CNS 20. Genome‐wide association studies have identified several single nucleotide polymorphisms within the clusterin gene (CLU) that are risk factors for AD 21, 22, 23, 24. Clusterin is up‐regulated in the brain in AD 25, 26 and is present in plaques 27, 28. In vitro studies suggest that clusterin influences Aβ fibril formation and neurotoxicity (reviewed in 29, 30) and can facilitate the transport of Aβ across the blood‐brain barrier 31. We have undertaken a comprehensive analysis of the regional distribution of clusterin, soluble and insoluble Aβ40 and Aβ42 in post‐mortem brain tissue across a number of brain regions that vary in their predilection to amyloid pathology. Our findings indicate that clusterin level rises with the accumulation of insoluble Aβ42 but the molar ratio of clusterin : Aβ42 falls, which probably influences the regional distribution of Aβ deposition.
Materials and Methods
Case selection
Brain tissue was obtained from the South West Dementia Brain Bank (SWDBB), University of Bristol, UK with local research ethics committee approval. All brains had been retrieved within 72 h of death. The right cerebral hemisphere had been fixed in 10% formalin for three weeks before the tissue was processed and paraffin blocks were taken for pathological assessment. The left cerebral hemisphere had been sliced and frozen at −80°C until used for biochemical assessment. According to the NIA‐AA guidelines AD neuropathological change was considered an adequate explanation for the dementia in all cases in the AD group 32. Controls were defined by an absence of clinical history of cognitive decline or other neurological disease and a lack of neuropathological abnormalities apart from sparse neuritic or diffuse plaques in some of the older cases, all of which were of Braak tangle stage III or lower. APOE genotyping and assessment of severity of cerebral amyloid angiopathy (CAA) had been performed as previously reported 33, 34. Demographic information, neuropathological findings and MRC identifiers for each case are shown in Supporting Information Tables S1 and S2.β
Brain tissue
Brain tissue (200 mg) was dissected from the midfrontal, cingulate, parahippocampal and medial parietal cortex, thalamus (pulvinar) and white matter (WM) underlying the parietal cortex. Brain tissue samples were prepared using a Precellys 24 homogenizer (Stretton Scientific, Derbyshire, UK) with 2.3 mm ceramic beads (Biospec, Stratech, Suffolk, UK) as previously described for Aβ measurements in human postmortem tissue 10, 11, 35, 36. Soluble and insoluble extracts were prepared sequentially following initial homogenization in 1% NP‐40 buffer containing 140 mM NaCl, 3 mM KCl, 25 mM TRIS, 5 mM ethylenediaminetetraacetic acid (EDTA) and 2 mM 1,10 phenanthroline). The homogenates were spun at 13 000 × g for 15 min at 4°C and the supernatant was removed and stored at −80°C. Insoluble extracts were prepared by homogenization of pelleted insoluble material in 6 M GuHCl and were left for 4 h at room temperature (RT) before storage at −80°C.
Measurement of clusterin levels by sandwich ELISA
Clusterin level was measured by sandwich ELISA (duoset kit # DY5874, R&D systems, Oxford, UK) according to the manufacturer's guidelines; 96‐well Maxisorp plates (R&D systems, Oxford, UK) were coated at RT overnight with mouse anti‐human clusterin. We washed the plates in phosphate‐buffered saline (PBS)/tween‐20 (0.01%), added 1% PBS/BSA at RT for 1 h to block nonspecific binding, then added recombinant human clusterin (62.5–4000 pg/mL) and tissue homogenates (2.5 μL supernatant diluted in 3.125 mL PBS, and 1.8 μL insoluble extract diluted in 10 mL PBS) for 2 h at RT. After a further wash, the plate was incubated for 2 h at RT with biotinylated mouse anti‐human clusterin. The plate was again washed and incubated with streptavidin‐horseradish peroxidase (HRP; 1:200 in 0.01% PBS : Tween‐20) for 20 minute at RT in the dark, washed, and incubated for 10 minute with chromogenic substrate (TMBS, R&D systems, Oxford, UK). Absorbance was read at 450 nM in a FLUOstar Optima plate reader (BMG Labtech, Ayelsbury, UK) after the addition of 50 μL of 2 N sulfuric acid. Measurements were repeated in duplicate and across two plates to ensure that there was minimal plate‐to‐plate variation.
Measurement of Aβ40 and Aβ42 by sandwich ELISA
We measured Aβ40 and Aβ42 level in both soluble (NP1‐40) and insoluble (guanidine‐HCl‐extracted) brain tissue fractions by sandwich ELISA as previously described 10, 11, 34, 35, 36, 37, 38, 39. For the Aβ40 ELISA, mouse monoclonal anti‐human Aβ (clone 6E10, raised against amino acids 1–16; Covance, Harrogate, UK), 2 µg/mL in PBS, was incubated overnight at RT, washed and then blocked with 300 µL protein‐free PBS blocking buffer (Thermo Fisher Scientific, Loughborough, UK) for 2 h at RT. Samples of brain homogenate (diluted 1:49 for guanidine extracts and 1:3 for soluble extracts) or recombinant human Aβ1‐40 (Sigma Aldrich, Dorset, UK) diluted in PBS containing 1% 1,10 phenanthroline (Sigma Aldrich, Dorset, UK) to prevent degradation of Aβ 40, were incubated for 2 h at RT on a rocking platform. After a further wash, the plates were incubated for 2 h at RT with mouse anti‐human Aβ1‐40 (1 µg/mL; 11A50‐B10; Covance, Harrogate, UK) that had been biotinylated using Lightning‐Link Biotinylation Kit (Innova Biosciences, Cambridge, UK) according to the manufacturer's guidelines. Streptavidin‐HRP (R&D Systems Europe, Abingdon, UK) diluted 1:200 was added to each well for 20 min at RT before they were washed and substrate solution (TMB; R&D Systems Europe, Abingdon, UK) was added for 30 min in the dark. The reaction was stopped with 2N sulfuric acid (R&D Systems Europe, Abingdon, UK) and the optical density of each well read at 450 nm in a FLUOstar plate reader (BMG Labtech, Aylesbury, UK).
For the Aβ42 ELISA, anti‐human Aβ1‐42 (12 F4, Covance) diluted 0.5 μg/mL in PBS was used as the capture antibody. Tissue samples (insoluble extracts diluted 1:9, soluble extracts diluted 1:3) were incubated at RT for 4 h. Biotinylated anti‐human Aβ (Thermo Fisher Scientific) diluted to 0.1 μg/mL in PBS was used for detection and left overnight at 4°C. After washing, streptavidin‐HRP was added for 1 h and chromogenic substrate for 20 min in the dark after a further wash. Aβ1‐42 concentration in brain tissue was interpolated from a standard curve generated by serial dilution (16 000 to 1.024 nM) of recombinant human Aβ1–42 (Sigma Aldrich). Each sample was assayed in duplicate. The Aβ1‐42 ELISA did not detect Aβ1‐40, and the Aβ1‐40 ELISA did not detect Aβ1‐42.
Statistical analysis
Unpaired two‐tailed t‐test or ANOVA with Dunnett's post hoc analysis was used for comparisons between groups, and Pearson's or Spearman's test was used to assess linear or rank order correlation, as appropriate, with the help of SPSS version 16 (SPSS, Chicago) and GraphPad Prism version 6 (GraphPad Software, La Jolla, CA). P‐values < 0.05 were considered statistically significant.
Results
Regional distribution of soluble and insoluble Aβ40 and Aβ42 in AD
We examined the regional distribution of soluble (NP‐40‐soluble) and insoluble (after guanidine‐HCl extraction) Aβ40 and Aβ42 in sequentially extracted brain homogenates from the following regions in AD and age‐matched controls: midfrontal cortex (MF), cingulate cortex (CC), parahippocampal cortex (PH), medial parietal cortex (PC), thalamus (TH) and parietal WM (Figure 1). In CC, MF, PH and PC, the concentrations of insoluble Aβ40 and Aβ42 were significantly higher in AD than age‐matched controls. The differences between AD and control brains in TH and WM did not reach significance.
Figure 1.
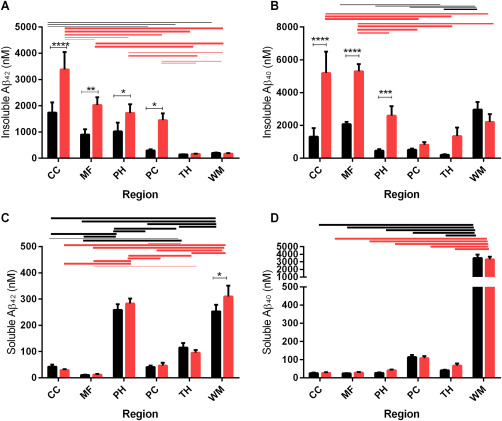
Bar charts showing regional levels of Aβ level in control (black bars) and AD brains (red bars), in MF, CC, PH, medial parietal cortex (PC), thalamus (TH) and parietal WM. Bars indicate the mean and SEM. *P < 0.05 **P < 0.01 ***P < 0.001 ****P < 0.0001. Lines indicate significant differences between regions, in the controls (black lines) and AD groups (red lines); the thickness of the line indicates the significance of the difference between the two regions, ranging from P < 0.01 to P < 0.0001.
In both control and AD groups, the concentration of insoluble Aβ40 and Aβ42 tended to decrease in the following order: CC > MF > PH > PC > TH and WM (Figure 1A,B). In AD, the concentration of insoluble Aβ40 and Aβ42 was significantly higher in neocortex (MF and CC) than in other regions (Figure 1A,B).
The regional distribution of soluble Aβ40 and Aβ42 differed substantially from that of insoluble Aβ40 and Aβ42. The concentration of the soluble forms of Aβ was lowest in MF and CC and tended to be higher in PC, PH, TH and WM (Figure 1C,D). Soluble Aβ level did not differ to a statistically significant extent between AD and controls, with the exception of increased Aβ42 in AD within WM. In general the concentration of Aβ in gray matter regions was much lower in the soluble than the insoluble tissue fractions. In contrast, Aβ40 and Aβ42 concentration was higher in the soluble than the insoluble tissue fractions of WM, and soluble Aβ level was several‐fold higher in WM than cortex. Soluble Aβ42 level was also relatively high in PH.
The relative contribution of soluble and insoluble Aβ40 and Aβ42 to “total” Aβ load in all regions studied is shown in Supporting Information Figure S1. “Total” Aβ was highest in MF and CC and tended to decrease in PH > PC > TH. Total Aβ in MF and CC consisted almost entirely of insoluble Aβ40 and Aβ42. In contrast, in WM, most of the Aβ consisted of soluble Aβ40 with a small amount of insoluble Aβ40 and negligible Aβ42.
Regional distribution of clusterin in AD
We examined the regional distribution of clusterin in the same soluble and insoluble brain fractions in AD and age‐matched controls that we had used to measure Aβ levels (Figure 2). In AD, clusterin level within both the soluble and insoluble extracts was highest in CC, MF and PH and PC and lowest in the TH and WM (approximately mirroring the regional distribution of “total” Aβ). The level of soluble clusterin was less variable between regions in the controls but was significantly higher in CC than PC or TH (Figure 2A). Clusterin level in the insoluble extract was significantly higher in all gray matter regions than in the WM (Figure 2A).
Figure 2.
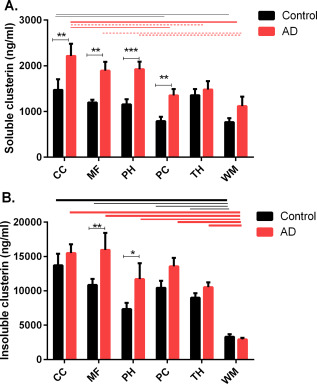
Bar chart showing regional levels of clusterin level in control (black bars) and AD brains (red bars), in the soluble and insoluble brain tissue fractions. Bars indicate the mean and SEM. *P < 0.05 **P < 0.01 ***P < 0.001. Lines indicate significant differences between regions, in the controls (black lines) and AD groups (red lines); the thickness of the solid lines indicates the significance of the difference between the two regions, ranging from P < 0.01 to P < 0.0001. The interrupted horizontal lines indicate differences significant at the P < 0.5 level.
The level of soluble clusterin was significantly higher in AD than controls in most regions (CC, MF, PH and PC; Figure 2A) and was increased in the insoluble extract in MF and PH (Figure 2B). Clusterin level within the soluble and insoluble extracts correlated significantly with soluble and insoluble Aβ42 and Aβ40 in MF (with the exception of insoluble Aβ42; Table 1). A similar trend was observed between clusterin and Aβ levels within the soluble extract in the PC but not the insoluble extract. A strong correlation was observed in the CC between clusterin and soluble and insoluble Aβ40. There was less correlation between clusterin and Aβ in the TH and WM (Table 1).
Table 1.
Correlation between concentrations of clusterin and Aβ in soluble and insoluble tissue fractions in different regions.
| Region | Insoluble Aβ42 | Soluble Aβ42 | Insoluble Aβ40 | Insoluble Aβ40 | |
|---|---|---|---|---|---|
| Mid‐frontal | Soluble clusterin | r = 0.359 | r = 0.507 | r = 0.496 | r = 0.354 |
| P = 0.048 | P = 0.008 | P = 0.005 | P = 0.090 | ||
| Insoluble clusterin | NS (not significant) | r = 0.68 | r = 0.462 | r = 0.374 | |
| P = 0.0001 | P = 0.009 | P = 0.072 | |||
| Cingulate | Soluble clusterin | NS | NS | r = 0.484 | NS |
| P = 0.080 | |||||
| Insoluble clusterin | NS | NS | r = 0.537 | NS | |
| P = 0.04 | |||||
| PH | Soluble clusterin | r = 0.428* | NS | NS | NS |
| P = 0.018 | |||||
| Insoluble clusterin | NS | NS | NS | NS | |
| Parietal cortex | Soluble clusterin | r = 0.640 | r = 0.236 | r = 0.521 | NS |
| P = 0.000002 | |||||
| P < 0.000001 | |||||
| P = 0.043 | |||||
| Insoluble clusterin | r = 0.549 | NS | NS | NS | |
| P = 0.002 | |||||
| TH | Soluble clusterin | NS | NS | NS | NS |
| Insoluble clusterin | r = 0.302 | NS | NS | NS | |
| P = 0.028 | |||||
| Parietal WM | Soluble clusterin | NS | r = 0.369 | NS | NS |
| P = 0.002 | |||||
| Insoluble clusterin | NS | NS | NS | NS |
*r = coefficient of correlation
Regional association of clusterin and aβ
To assess whether variations in clusterin concentration might influence the regional distribution of soluble and insoluble Aβ40 or Aβ42, we looked at the correlation between Aβ level and clusterin concentration across all regions. There was a significant correlation between the concentration of soluble clusterin and the level of insoluble Aβ42 within the AD cohort and a weaker, nonsignificant trend in the controls (Figure 3A,B). A trend approaching significance was observed between clusterin in the insoluble extract and insoluble Aβ42 in both controls and AD (Figure 3C,D). We did not find significant correlations between clusterin concentration and the level of soluble Aβ42, soluble Aβ40 or insoluble Aβ40.
Figure 3.
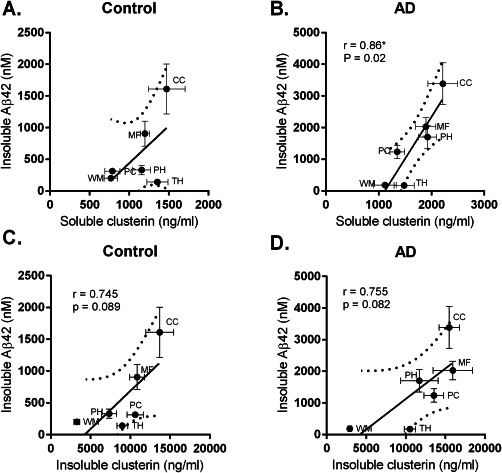
Regional association between clusterin and insoluble Aβ42 levels. The concentration of clusterin in soluble (A, B) and insoluble (C, D) brain tissue fractions was plotted against insoluble Aβ42 level in each region in controls and AD cases. The solid circles and thin bars indicate the mean values and SEM for clusterin (horizontal bars) and Aβ42 (vertical bars). The thick solid and dotted lines indicate the best‐fit linear regression and 95% confidence intervals.
To investigate further whether clusterin might promote the accumulation of insoluble Aβ42, we calculated the molar ratio of insoluble clusterin to insoluble Aβ42 in the different regions. In both controls and AD, the molar ratio of insoluble clusterin : insoluble Aβ42 was lowest in regions with the highest concentration of insoluble Aβ42 (Figure 4A,B) and vice versa.
Figure 4.
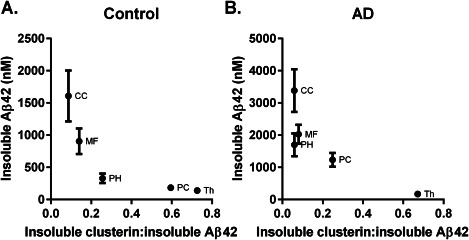
The ratio of clusterin : insoluble Aβ42 was lowest in regions with a predilection for Aβ42 deposition. The solid circles and bars indicate the mean values and SEM.
Clusterin levels influenced by APOE genotype
Clusterin level was highest in APOE ɛ4 homozygotes in MF and PC (Figure 5) and rose significantly with severity of CAA. Post‐hoc analysis revealed significantly higher clusterin level in ɛ4/4 than ɛ3/3 brains in PC (P < 0.05), and in ɛ4/4 than ɛ3/4 (P < 0.01) or ɛ3/3 (P < 0.05) in MF. Post‐hoc analysis also showed clusterin level in MF and PC to be significantly higher in brains with severe than absent CAA (P < 0.05 for both regions). Insoluble clusterin level did not vary significantly in relation to APOE genotype or CAA severity.
Figure 5.
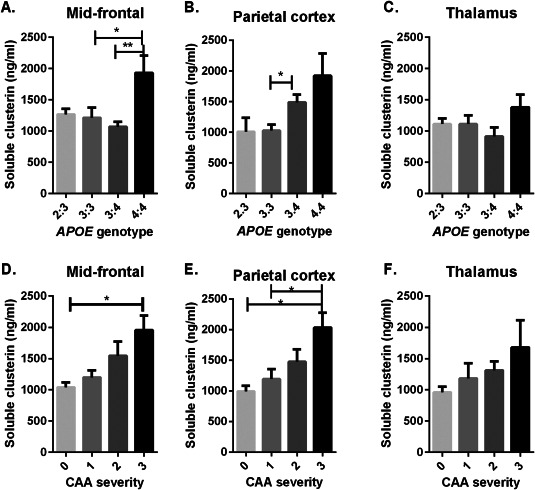
Bar charts showing clusterin level in relation to APOE genotype (A–C) and severity of CAA. Bars indicate the mean and SEM. *P < 0.05 **P < 0.01. Clusterin level was highest in APOE ɛ4 homozygotes in MF and PC and increased with severity of CAA.
Discussion
We have examined the relationship between clusterin/apoJ level and the regional distribution of Aβ within the brain. Although the concentration of clusterin was elevated in AD and was highest in cortical regions with the most abundant Aβ deposition, the molar ratio of clusterin : Aβ was lowest in those regions and was highest in parts of the brain with little or no amyloid pathology, such as in the thalamus and WM. These findings in human brain tissue support experimental studies indicating that (i) clusterin level rises in association with increasing Aβ, (ii) within the physiological range of clusterin : Aβ, clusterin reduces aggregation and promotes clearance of Aβ, but (iii) when, despite a rise in clusterin, Aβ level increases to an extent that causes clusterin : Aβ to fall below the physiological range, Aβ‐clusterin complexes tend to aggregate and deposit within the brain parenchyma. We have also shown that clusterin concentration is influenced by APOE genotyope, being highest in brain tissue from ɛ4 homozygotes, and rises in relation to the severity of CAA.
Clusterin is highly expressed in the CNS, within which it is present at a similar concentration to that of apoE 20. Variations in the clusterin gene (CLU) are associated with sporadic AD 21, and previous studies showed that clusterin is increased in the CSF 41, 42 and brain tissue 25, 26 in AD. Clusterin is detectable immunohistochemically within plaques 27, 28 and increases in association with neuritic plaque density 43, 44, 45. In a transgenic APP/PS1 mouse model, clusterin level was elevated in plasma and brain tissue and found to co‐localize with amyloid plaques 42. Present findings show that the concentration of clusterin in human brain tissue in AD is highest in regions with the greatest concentration of Aβ. Within those regions, clusterin concentration correlates closely with Aβ level [as was also shown in transgenic mouse models of AD expressing mutant APP 46]. These findings are in keeping with clinical evidence of a correlation between raised plasma clusterin level and accelerated clinical progression of disease 41, 42, and imaging studies showing that increased plasma clusterin levels were a strong predictor of brain amyloid load in AD patients 42. Within subcortical regions that have a much lower level of Aβ, clusterin concentration does not differ significantly between AD and controls.
In vitro studies indicate that clusterin binds to Aβ and influences both fibril formation and toxicity 47, 48. Yerbury et al 49 reported that clusterin coprecipitates with Aβ as insoluble aggregates when Aβ is present in large molar excess. We have found the molar ratio of clusterin : Aβ42 to be lowest in regions that have the greatest accumulation of Aβ (almost entirely in the insoluble tissue fraction) even though clusterin levels are highest in those regions. In contrast, within the WM and thalamus, which had the highest ratio of clusterin : Aβ42, Aβ was almost all in a soluble form. It seems that when the clusterin : Aβ ratio falls low enough, clusterin actually promotes rather than simply fails to prevent the precipitation of Aβ, as evidenced by in vitro data 49. These data are consistent with experimental evidence in PDAPP mice (homozygous for the APPV717F transgene) showing that clusterin stimulates amyloid aggregation when Aβ is present in excess. PDAPP mice homozygous for knock‐out of the clusterin gene have significantly fewer fibrillar Aβ deposits and dystrophic neurites than PDAPP mice expressing clusterin 50. The rise in clusterin concentration that occurs with increasing Aβ seems likely to be a consequence of the latter. Thamsbietty et al 42 reported that plasma clusterin level was elevated almost 10 years in advance of fibrillary Aβ deposition, suggesting that clusterin production is raised at an early stage in the disease process, although we know from other studies that Aβ starts to accumulate even earlier 51, 52.
ApoE is also highly expressed within the CNS and has been implicated in the pathogenesis of AD. The APOE gene is a strong risk factor for sporadic AD and individuals possessing the ɛ4 allele have more abundant plaque and cerebrovascular deposition of Aβ and a higher level of this peptide 33, 34, 53. In vitro studies demonstrated that apoE interacts with and influences Aβ fibrillogenesis and clearance 54, 55, 56, 57, 58. However, apoE and clusterin play somewhat divergent roles in the progression of AD. In contrast to clusterin, apoE concentration shows a strong inverse correlation with regional Aβ load 19, and while fibril formation was reduced in clusterin‐deficient PDAPP mice 50 it was significantly increased in PDAPP mice deficient in both clusterin and apoE 59. It is of interest that no regional association was found between Aβ level and molecules involved in APP processing (APP, APP‐CTFβ, BACE‐1 or PS‐1) or enzymes involved in Aβ clearance (neprilysin and insulin‐degrading enzyme). Together, these data suggest that the regional distribution of Aβ is influenced to a greater extent by apoE and clusterin expression than by pathways involved in the production or enzymatic degradation of Aβ.
Clusterin has also been shown to facilitate the clearance of Aβ at the blood‐brain barrier 31 and is localized not only to plaques but also arterioles and capillaries within the brain 43, 44, 45. A recent immunohistochemical study showed that clusterin was associated with vascular Aβ, particularly Aβ40, in CAA 60. Co‐localization of clusterin with perivascular Aβ deposits and our finding of increased clusterin level in relation to CAA severity is supportive of a role in the perivascular drainage of Aβ, which is impaired in CAA 61, 62. Craggs et al 60 also reported increased clusterin in the frontal WM in cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL), suggesting that clusterin may also accumulate as a consequence of failed perivascular drainage of interstitial fluid.
In conclusion, we have shown that clusterin level is elevated in AD in regions with a predilection for plaque deposition. Yet despite that elevation, the molar ratio of clusterin : Aβ is lowest in those same regions, which is likely to influence the regional distribution of Aβ by promoting its aggregation and precipitation.
Supporting information
Additional Supporting Information may be found in the online version of this article at the publisher's web‐site:
Figure S1. Stacked bar chart illustrating regional differences in the relative contributions of soluble and insoluble Aβ40 and Aβ42 to ‘total’ Aβ load in AD and control brains in MF, CC, PH, PC, TH, and WM.
Acknowledgements
This work was supported by Alzheimer's Research UK (ART‐PG2011‐1). The South West Dementia Brain Bank is part of the Brains for Dementia Research program, jointly funded by Alzheimer's Research UK and Alzheimer's Society, and is supported by BRACE (Bristol Research into Alzheimer's and Care of the Elderly) and the Medical Research Council.
References
- 1. Hardy J, Allsop D (1991) Amyloid deposition as the central event in the aetiology of Alzheimer's disease. Trends Pharmacol Sci 12:383–388. [DOI] [PubMed] [Google Scholar]
- 2. Miners JS, Baig S, Palmer J, Palmer LE, Kehoe PG, Love S (2008) Aβ‐degrading enzymes in Alzheimer's disease. Brain Pathol 18:240–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gravina SA, Ho L, Eckman CB, Long KE, Otvos L, Jr ., Younkin LH, et al (1995) Amyloid β protein (Aβ) in Alzheimer's disease brain. Biochemical and immunocytochemical analysis with antibodies specific for forms ending at Aβ40 or Aβ42(43). J Biol Chem 270:7013–7016. [DOI] [PubMed] [Google Scholar]
- 4. Iwatsubo T, Odaka A, Suzuki N, Mizusawa H, Nukina N, Ihara Y (1994) Visualization of Aβ42(43) and Aβ40 in senile plaques with end‐specific Aβ monoclonals: Evidence that an initially deposited species is Aβ42(43). Neuron 13:45–53. [DOI] [PubMed] [Google Scholar]
- 5. Price DL, Sisodia SS (1998) Mutant genes in familial Alzheimer's disease and transgenic models. Annu Rev Neurosci 21:479–505. [DOI] [PubMed] [Google Scholar]
- 6. Scheuner D, Eckman C, Jensen M, Song X, Citron M, Suzuki N, et al (1996) Secreted amyloid β‐protein similar to that in the senile plaques of Alzheimer's disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer's disease. Nat Med 2:864–870. [DOI] [PubMed] [Google Scholar]
- 7. Selkoe DJ (1998) The cell biology of β‐amyloid precursor protein and presenilin in Alzheimer's disease. Trends Cell Biol 8:447–453. [DOI] [PubMed] [Google Scholar]
- 8. Wolfe MS (2007) When loss is gain: Reduced presenilin proteolytic function leads to increased Aβ42/Aβ0. Talking Point on the role of presenilin mutations in Alzheimer disease. EMBO Rep 8:136–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Holtzman DM (2004) In vivo effects of ApoE and clusterin on amyloid‐β metabolism and neuropathology. J Mol Neurosci 23:247–254. [DOI] [PubMed] [Google Scholar]
- 10. van Helmond Z, Miners JS, Kehoe PG, Love S (2010) Higher soluble amyloid β concentration in frontal cortex of young adults than in normal elderly or Alzheimer's disease. Brain Pathol 20:787–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Miners JS, Jones R, Love S (2014) Differential changes in Aβ42 and Aβ40 with age. J Alzheimers Dis 40:727–735. [DOI] [PubMed] [Google Scholar]
- 12. Ballard C, Gauthier S, Corbett A, Brayne C, Aarsland D, Jones E (2011) Alzheimer's disease. Lancet 377:1019–1031. [DOI] [PubMed] [Google Scholar]
- 13. Jack CR, Jr. , Knopman DS, Jagust WJ, Shaw LM, Aisen PS, Weiner MW, et al (2010) Hypothetical model of dynamic biomarkers of the Alzheimer's pathological cascade. Lancet Neurol 9:119–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Morris JC, Roe CM, Xiong C, Fagan AM, Goate AM, Holtzman DM, Mintun MA (2010) APOE predicts amyloid‐β but not tau Alzheimer pathology in cognitively normal aging. Ann Neurol 67:122–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Thal DR, Rub U, Orantes M, Braak H (2002) Phases of Aβ‐deposition in the human brain and its relevance for the development of AD. Neurology 58:1791–1800. [DOI] [PubMed] [Google Scholar]
- 16. Benkovic SA, McGowan EM, Rothwell NJ, Hutton M, Morgan DG, Gordon MN (1997) Regional and cellular localization of presenilin‐2 RNA in rat and human brain. Exp Neurol 145:555–564. [DOI] [PubMed] [Google Scholar]
- 17. Page K, Hollister R, Tanzi RE, Hyman BT (1996) In situ hybridization analysis of presenilin 1 mRNA in Alzheimer disease and in lesioned rat brain. Proc Natl Acad Sci U S A 93:14020–14024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Takami K, Terai K, Matsuo A, Walker DG, McGeer PL (1997) Expression of presenilin‐1 and −2 mRNAs in rat and Alzheimer's disease brains. Brain Res 748:122–130. [DOI] [PubMed] [Google Scholar]
- 19. Shinohara M, Petersen RC, Dickson DW, Bu G (2013) Brain regional correlation of amyloid‐β with synapses and apolipoprotein E in non‐demented individuals: Potential mechanisms underlying regional vulnerability to amyloid‐β accumulation. Acta Neuropathol 125:535–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Roheim PS, Carey M, Forte T, Vega GL (1979) Apolipoproteins in human cerebrospinal fluid. Proc Natl Acad Sci U S A 76:4646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bertram L, McQueen MB, Mullin K, Blacker D, Tanzi RE (2007) Systematic meta‐analyses of Alzheimer disease genetic association studies: The AlzGene database. Nat Genet 39:17–23. [DOI] [PubMed] [Google Scholar]
- 22. Butler AW, Ng MY, Hamshere ML, Forabosco P, Wroe R, Al‐Chalabi A, et al (2009) Meta‐analysis of linkage studies for Alzheimer's disease–a web resource. Neurobiol Aging 30:1037–1047. [DOI] [PubMed] [Google Scholar]
- 23. Harold D, Abraham R, Hollingworth P, Sims R, Gerrish A, Hamshere ML, et al (2009) Genome‐wide association study identifies variants at CLU and PICALM associated with Alzheimer's disease. Nat Genet 41:1088–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lambert JC, Heath S, Even G, Campion D, Sleegers K, Hiltunen M, et al (2009) Genome‐wide association study identifies variants at CLU and CR1 associated with Alzheimer's disease. Nat Genet 41:1094–1099. [DOI] [PubMed] [Google Scholar]
- 25. Bertrand P, Poirier J, Oda T, Finch CE, Pasinetti GM (1995) Association of apolipoprotein E genotype with brain levels of apolipoprotein E and apolipoprotein J (clusterin) in Alzheimer disease. Brain Res Mol Brain Res 33:174–178. [DOI] [PubMed] [Google Scholar]
- 26. Lidstrom AM, Bogdanovic N, Hesse C, Volkman I, Davidsson P, Blennow K (1998) Clusterin (apolipoprotein J) protein levels are increased in hippocampus and in frontal cortex in Alzheimer's disease. Exp Neurol 154:511–521. [DOI] [PubMed] [Google Scholar]
- 27. Calero M, Rostagno A, Matsubara E, Zlokovic B, Frangione B, Ghiso J (2000) Apolipoprotein J (clusterin) and Alzheimer's disease. Microsc Res Tech 50:305–315. [DOI] [PubMed] [Google Scholar]
- 28. May PC, Lampert‐Etchells M, Johnson SA, Poirier J, Masters JN, Finch CE (1990) Dynamics of gene expression for a hippocampal glycoprotein elevated in Alzheimer's disease and in response to experimental lesions in rat. Neuron 5:831–839. [DOI] [PubMed] [Google Scholar]
- 29. Kanekiyo T, Xu H, Bu G (2014) ApoE and Aβ in Alzheimer's disease: Accidental encounters or partners? Neuron 81:740–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yu JT, Tan L (2012) The role of clusterin in Alzheimer's disease: Pathways, pathogenesis, and therapy. Mol Neurobiol 45:314–326. [DOI] [PubMed] [Google Scholar]
- 31. Zlokovic BV, Martel CL, Matsubara E, McComb JG, Zheng G, McCluskey RT, et al (1996) Glycoprotein 330/megalin: Probable role in receptor‐mediated transport of apolipoprotein J alone and in a complex with Alzheimer disease amyloid β at the blood‐brain and blood‐cerebrospinal fluid barriers. Proc Natl Acad Sci U S A 93:4229–4234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Montine TJ, Phelps CH, Beach TG, Bigio EH, Cairns NJ, Dickson DW, et al (2012) National Institute on Aging‐Alzheimer's Association guidelines for the neuropathologic assessment of Alzheimer's disease: A practical approach. Acta Neuropathol 123:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chalmers K, Wilcock GK, Love S (2003) APOE ɛ4 influences the pathological phenotype of Alzheimer's disease by favouring cerebrovascular over parenchymal accumulation of Aβ protein. Neuropathol Appl Neurobiol 29:231–238. [DOI] [PubMed] [Google Scholar]
- 34. van Helmond Z, Miners JS, Kehoe PG, Love S (2010) Oligomeric Aβ in Alzheimer's disease: Relationship to plaque and tangle pathology, APOE genotype and cerebral amyloid angiopathy. Brain Pathol 20:468–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Swirski M, Miners JS, de Silva R, Lashley T, Ling H, Holton J, et al (2014) Evaluating the relationship between amyloid‐β and α‐synuclein phosphorylated at Ser129 in dementia with Lewy bodies and Parkinson's disease. Alzheimers Res Ther 6:77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Thomas T, Miners S, Love S (2015) Post‐mortem assessment of hypoperfusion of cerebral cortex in Alzheimer's disease and vascular dementia. Brain 138:1059–1069. [DOI] [PubMed] [Google Scholar]
- 37. Ashby EL, Miners JS, Kumar S, Walter J, Love S, Kehoe PG (2015) Investigation of Aβ phosphorylated at serine 8 (pAβ) in Alzheimer's disease, dementia with Lewy bodies and vascular dementia. Neuropathol Appl Neurobiol 41:428–444. [DOI] [PubMed] [Google Scholar]
- 38. Barua NU, Miners JS, Bienemann AS, Wyatt MJ, Welser K, Tabor AB, et al (2012) Convection‐enhanced delivery of neprilysin: A novel amyloid‐β‐degrading therapeutic strategy. J Alzheimers Dis 32:43–56. [DOI] [PubMed] [Google Scholar]
- 39. Miners JS, Palmer JC, Love S. (in press) Pathophysiology of hypoperfusion of the precuneus in early Alzheimer's disease. Brain Pathol. doi: 10.1111/bpa.12331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Qiu WQ, Ye Z, Kholodenko D, Seubert P, Selkoe DJ (1997) Degradation of amyloid β‐protein by a metalloprotease secreted by microglia and other neural and non‐neural cells. J Biol Chem 272:6641–6646. [DOI] [PubMed] [Google Scholar]
- 41. Schrijvers EM, Koudstaal PJ, Hofman A, Breteler MM (2011) Plasma clusterin and the risk of Alzheimer disease. JAMA 305:1322–1326. [DOI] [PubMed] [Google Scholar]
- 42. Thambisetty M, Simmons A, Velayudhan L, Hye A, Campbell J, Zhang Y, et al (2010) Association of plasma clusterin concentration with severity, pathology, and progression in Alzheimer disease. Arch Gen Psychiatry 67:739–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Giannakopoulos P, Kovari E, French LE, Viard I, Hof PR, Bouras C (1998) Possible neuroprotective role of clusterin in Alzheimer's disease: A quantitative immunocytochemical study. Acta Neuropathol 95:387–394. [DOI] [PubMed] [Google Scholar]
- 44. Howlett DR, Hortobagyi T, Francis PT (2013) Clusterin associates specifically with Aβ40 in Alzheimer's disease brain tissue. Brain Pathol 23:623–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. McGeer PL, Kawamata T, Walker DG (1992) Distribution of clusterin in Alzheimer brain tissue. Brain Res 579:337–341. [DOI] [PubMed] [Google Scholar]
- 46. Matarin M, Salih DA, Yasvoina M, Cummings DM, Guelfi S, Liu W, et al (2015) A genome‐wide gene‐expression analysis and database in transgenic mice during development of amyloid or tau pathology. Cell Rep 10:633–644. [DOI] [PubMed] [Google Scholar]
- 47. Matsubara E, Soto C, Governale S, Frangione B, Ghiso J (1996) Apolipoprotein J and Alzheimer's amyloid β solubility. Biochem J 316:671–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Oda T, Wals P, Osterburg HH, Johnson SA, Pasinetti GM, Morgan TE, et al (1995) Clusterin (apoJ) alters the aggregation of amyloid β‐peptide (Aβ1‐42) and forms slowly sedimenting Aβ complexes that cause oxidative stress. Exp Neurol 136:22–31. [DOI] [PubMed] [Google Scholar]
- 49. Yerbury JJ, Poon S, Meehan S, Thompson B, Kumita JR, Dobson CM, Wilson MR (2007) The extracellular chaperone clusterin influences amyloid formation and toxicity by interacting with prefibrillar structures. Faseb J 21:2312–2322. [DOI] [PubMed] [Google Scholar]
- 50. DeMattos RB, O'Dell MA, Parsadanian M, Taylor JW, Harmony JA, Bales KR, et al (2002) Clusterin promotes amyloid plaque formation and is critical for neuritic toxicity in a mouse model of Alzheimer's disease. Proc Natl Acad Sci U S A 99:10843–10848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Benzinger TL, Blazey T, Jack CR, Jr. , Koeppe RA, Su Y, Xiong C, et al (2013) Regional variability of imaging biomarkers in autosomal dominant Alzheimer's disease. Proc Natl Acad Sci U S A 110:E4502–E4509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Binnewijzend MA, Kuijer JP, Benedictus MR, van der Flier WM, Wink AM, Wattjes MP, et al (2013) Cerebral blood flow measured with 3D pseudocontinuous arterial spin‐labeling MR imaging in Alzheimer disease and mild cognitive impairment: A marker for disease severity. Radiology 267:221–230. [DOI] [PubMed] [Google Scholar]
- 53. Yamaguchi H, Sugihara S, Ogawa A, Oshima N, Ihara Y (2001) Alzheimer β amyloid deposition enhanced by apoE ϵ4 gene precedes neurofibrillary pathology in the frontal association cortex of nondemented senior subjects. J Neuropathol Exp Neurol 60:731–739. [DOI] [PubMed] [Google Scholar]
- 54. Castano EM, Prelli F, Wisniewski T, Golabek A, Kumar RA, Soto C, Frangione B (1995) Fibrillogenesis in Alzheimer's disease of amyloid β peptides and apolipoprotein E. Biochem J 306:599–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. LaDu MJ, Falduto MT, Manelli AM, Reardon CA, Getz GS, Frail DE (1994) Isoform‐specific binding of apolipoprotein E to β‐amyloid. J Biol Chem 269:23403–23406. [PubMed] [Google Scholar]
- 56. Ma J, Yee A, Brewer HB, Jr. , Das S, Potter H (1994) Amyloid‐associated proteins alpha 1‐antichymotrypsin and apolipoprotein E promote assembly of Alzheimer β‐protein into filaments. Nature 372:92–94. [DOI] [PubMed] [Google Scholar]
- 57. Sanan DA, Weisgraber KH, Russell SJ, Mahley RW, Huang D, Saunders A, et al (1994) Apolipoprotein E associates with β amyloid peptide of Alzheimer's disease to form novel monofibrils. Isoform apoE4 associates more efficiently than apoE3. J Clin Invest 94:860–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Strittmatter WJ, Weisgraber KH, Huang DY, Dong LM, Salvesen GS, Pericak‐Vance M, et al (1993) Binding of human apolipoprotein E to synthetic amyloid β peptide: Isoform‐specific effects and implications for late‐onset Alzheimer disease. Proc Natl Acad Sci U S A 90:8098–8102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. DeMattos RB, Cirrito JR, Parsadanian M, May PC, O'Dell MA, Taylor JW, et al (2004) ApoE and clusterin cooperatively suppress Aβ levels and deposition: Evidence that ApoE regulates extracellular Abeta metabolism in vivo. Neuron 41:193–202. [DOI] [PubMed] [Google Scholar]
- 60. Craggs L, Taylor J, Slade JY, Chen A, Hagel C, Kuhlenbaeumer G, et al (2016) Clusterin/Apolipoprotein J immunoreactivity is associated with white matter damage in cerebral small vessel diseases. Neuropathol Appl Neurobiol 42:194–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Hawkes CA, Jayakody N, Johnston DA, Bechmann I, Carare RO (2014) Failure of perivascular drainage of β‐amyloid in cerebral amyloid angiopathy. Brain Pathol 24:396–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Weller RO, Subash M, Preston SD, Mazanti I, Carare RO (2008) Perivascular drainage of amyloid‐beta peptides from the brain and its failure in cerebral amyloid angiopathy and Alzheimer's disease. Brain Pathol 18:253–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting Information may be found in the online version of this article at the publisher's web‐site:
Figure S1. Stacked bar chart illustrating regional differences in the relative contributions of soluble and insoluble Aβ40 and Aβ42 to ‘total’ Aβ load in AD and control brains in MF, CC, PH, PC, TH, and WM.