

Abstract
Amyotrophic Lateral Sclerosis (ALS) is a complex multifactorial disorder, characterized by motor neuron loss with involvement of several other cell types, including astrocytes, oligodendrocytes and microglia. Studies in vivo and in in vitro models have highlighted that the contribution of non‐neuronal cells to the disease is a primary event and ALS pathogenesis is driven by both cell‐autonomous and non‐cell autonomous mechanisms. The advancements in genetics and in vitro modeling of the past 10 years have dramatically changed the way we investigate the pathogenic mechanisms involved in ALS. The identification of mutations in transactive response DNA‐binding protein gene (TARDBP), fused in sarcoma (FUS) and, more recently, a GGGGCC‐hexanucleotide repeat expansion in chromosome 9 open reading frame 72 (C9ORF72) and their link with familial ALS have provided new avenues of investigation and hypotheses on the pathophysiology of this devastating disease. In the same years, from 2007 to present, in vitro technologies to model neurological disorders have also undergone impressive developments. The advent of induced pluripotent stem cells (iPSCs) gave the field of ALS the opportunity to finally model in vitro not only familial, but also the larger part of ALS cases affected by sporadic disease. Since 2008, when the first human iPS‐derived motor neurons from patients were cultured in a petri dish, several different techniques have been developed to produce iPSC lines through genetic reprogramming and multiple direct conversion methods have been optimised. In this review, we will give an overview of how human in vitro models have been used so far, what discoveries they have led to since 2007, and how the recent advances in technology combined with the genetic discoveries, have tremendously widened the horizon of ALS research.
Keywords: astrocytes, cell models, iPSC, motor neurons
Introduction
The use of animal and in vitro models has highlighted that Amyotrophic Lateral Sclerosis (ALS) is a complex multifactorial disorder 16, where several different cell‐types, including astrocytes, microglia and oligodendrocytes, contribute to neuronal death 15, 25.
The advancements in genetics and in vitro modeling of the past 10 years have dramatically changed the way we investigate the pathogenic mechanisms involved in ALS.
Thorough pathological investigations 20, 39 led in 2008 to the genetic discovery that mutations in transactive response DNA‐binding protein gene (TARDBP) are linked to both familial and sporadic cases of ALS 27, 52. Shortly after, this finding led to the identification of a second RNA/DNA‐binding protein called fused in sarcoma (FUS) or translocated in liposarcoma (TLS) linked to the disease 30, 59. More recently, the field of ALS has seen a breakthrough with the discovery of the gene responsible for 35%–40% of familial cases and 5%–7% of sporadic cases, the GGGGCC‐hexanucleotide repeat expansion in chromosome 9 open reading frame 72 (C9ORF72) 11, 46.
These discoveries have relocated ALS within a spectrum of neurological disorders, ranging from pure motor neuron disease to frontotemporal dementia 3.
In the same years, from 2007 to present, in vitro technologies to model neurological disorders have also undergone impressive developments.
With the discovery that adult human fibroblasts could be reprogrammed to induced pluripotent stem cells (iPSCs) with the use of selected transcription factors 54, the field of ALS saw the opportunity to finally model not only the familial, but especially the sporadic disease in vitro. In fact, in 2008, the first human iPS‐derived motor neurons from patients were cultured in a petri dish 10. Since then, several different techniques have been developed to produce iPSC lines through genetic reprogramming of multiple somatic cell‐types, including adipocytes 55 and keratinocytes 1. Numerous lines have been made available to the research community (http://www.coriell.org/stem-cells).
Moreover, multiple direct conversion methods have been developed to produce induced neural progenitor cells (iNPCs) 35 and motor neurons 51 from ALS patients.
Neural progenitors cells (NPCs) have also been isolated from postmortem spinal cord samples of ALS patients and successfully cultured and differentiated into motor neurons, astrocytes and oligodendrocytes in vitro 23. This technology provided for the first time the possibility to model all forms of ALS in vitro without introducing major epigenetic alterations in the cells used. Only a few years later, in 2014, primary astrocytes were isolated from spinal cord and brain biopsies of ALS patients 45, providing one more valuable in vitro model to unravel the mechanisms involved in ALS pathophysiology.
Embryonic Stem Cells (ESCs) and Induced Pluripotent Stem Cells (iPSCS)
Stem cells are defined as a population of cells that maintains the ability to self‐renew and differentiate into several cell‐types of the adult body. In mammals, tissues such as muscle, brain and bone marrow, harbor subtypes of stem cells that can give rise to a relatively small variety of different cell‐types. These adult stem cells are committed to certain cell lineages and do not produce cells from other tissue types under normal conditions. Unlike adult stem cells, embryonic stem cells that can be isolated from the inner cell mass of early stage embryos are pluripotent and can, therefore, still differentiate into virtually any cell type of the human body. While the collection of embryonic stem cells from mice is a widely accepted approach used for disease modeling, the use of human embryonic stem cells is controversial and rises severe ethical concerns. With the discovery that adult human fibroblasts can be reprogrammed to an embryonic stem cell‐like state, new hope arose for stem cell based approaches in human research. Induced pluripotent stem cells (iPS) are usually generated by the introduction of 2–5 defined pluripotency transcription factors into somatic cells, ranging from fibroblasts 54, 65, to renal tubular cells present in urine 68. These transcription factors drastically alter gene expression in the target cells until some of them eventually become pluripotent and can then be isolated and amplified. Initially, the transcription factors were introduced by retroviral 54 or lentiviral 32, 65 constructs leading to the integration of the transgene into the target genome. As the random integration of additional genes can disrupt/alter the expression of endogenous genes, more recent approaches rely on less invasive techniques such as non‐integrating viral vectors, that is, adenoviruses 67, plasmids 42 or DNA‐free systems, like microRNAs (miRNAs) 37 and Sendai virus 19.
Mouse embryonic stem cells (mESCs) and iPSCs
The use of stem cell technologies in ALS research started in 2007 when mouse embryonic stem cells (mESC) were established from the most commonly used model of this disease, the mouse carrying mutant G93A superoxide dismutase 1 (SOD1) 21. Interestingly, the expression of mutant SOD1 did not affect early motor neuron differentiation, however, after several weeks in culture, motor neurons derived from these mESCs developed SOD1 inclusions and accumulation of ubiquitinated proteins 21.
Similar findings were obtained when tail‐tip fibroblasts from SOD1 G93A mice were reprogrammed into iPSCs and subsequently differentiated into motor neurons 64. These cells, in fact, could be successfully differentiated into electrophysiologically active motor neurons, but they displayed significantly shorter neurites compared with their control counterpart 64.
To determine the similarities between iPSC‐derived motor neurons and ESC‐derived motor neurons, Toma et al 57 compared the protein expression profiles of these two cells‐types and showed that <4% of the proteins identified were differentially regulated. Like ESCs, mouse iPSCs treated with retinoic acid and a smoothened agonist differentiated into motor neurons expressing the LIM homeodomain protein Lhx3. On transplantation into the neural tube of developing chick embryos, iPSC‐derived motor neurons targeted hind limb muscles, as expected by Lhx3+ motor neurons. Also the electrophysiological properties of these neurons are similar, with iPSC‐derived motor neurons developing passive membrane and firing characteristic as expected from postnatal motor neurons after several weeks in culture.
Mouse ESC‐derived motor neurons from wild‐type and mutant SOD1 have also been recently utilized in promising small molecule screen studies 63. Kenpaullone, a GSK‐3 and HGK kinase inhibitor had a particularly impressive ability to prolong the healthy survival of both mouse ESC and patient‐derived iPSC motor neurons.
The advantages of using mouse ESCs are the availability of large numbers of neurons and the limited variability between cell preparations, thus providing reproducible results. Conversely, the results obtained with these cells are limited to only one gene, SOD1.
More challenging are the studies using human embryonic stem cells (hESC) and, even more, induced pluripotent stem cells (iPSCs). Their use, however, has greatly contributed to improve our understanding of the mechanisms involved in ALS pathophysiology.
Human ESCs and iPSCs
The first studies using human ESC‐derived neurons focused on elucidating the relationship between cell‐autonomous and non‐cell autonomous mechanisms in ALS. Studies in chimeric mice and transgenic mice expressing the Cre–Lox recombination system to exclude mutant SOD1 from motor neurons or non‐neuronal cells had already shown that both astrocytes and microglia play a key role in motor neuron death/survival 25, 62. However, in 2008 two independent studies showed that primary mouse astrocytes expressing mutant SOD1 can cause human ESC‐derived motor neuron death 22, 33. The study performed by Di Giorgio et al focused on the cell‐specific toxicity of astrocytes, specifically directed toward motor neurons as opposed to interneurons. The study by Marchetto et al focused on the causes of this toxicity, uncovering the important role of oxidative stress in motor neuron death. Human ESCs were also used to study the cell‐autonomous mechanisms involved in motor neuron degeneration. In 2009, a human embryonic stem cell line was used to generate motor neurons that were then transfected with different constructs containing SOD1 mutations 29. In line with the study performed in SOD1 G93A mESC‐derived motor neurons, the authors observed a reduction in neurite length and reduced cell survival. With the advent of iPSCs and the development and optimization of new protocols to increase yield and purity of human motor neuron preparations, hESCs are less frequently used in ALS research. Models based upon hESCs, in fact, represent only specific genetic cases carrying defined mutations, most often SOD1. Although limited in representing a variety of genetic causes of ALS, hESCs still represent a valuable tool for their limited genetic variability. Very recently Isobe et al 26 generated hESCs with identical genetic background, differing only for the SOD1 mutation they carried. Motor neurons derived from these stem cells displayed mutation‐dependent reduction in neurite length compared with controls and, more interesting, they displayed different response to drugs in a drug screen 26. This approach could be useful in identifying SOD1 mutation‐specific response to drugs and inform future drug discovery approaches and clinical trials.
With the development of iPS technology, huge efforts were put in the generation of patient‐specific iPS lines. Up to now, several hundred lines with various mutations have been generated and have become publicly available (http://www.coriell.org/stem-cells) thereby being accessible to a broad scientific community.
In 2008, Dimos et al reported the successful generation of motor neurons and glial cells from an ALS patient derived iPS line carrying a SOD1 mutation causing a mild disease phenotype 10. This characteristic could in part be explained by the patient's late onset and mild disease form.
Similarly, in 2011, iPSC‐derived motor neurons were generated from a patient harboring a mutation in the vesicle‐associated membrane protein‐associated protein‐B/C (VAPB/C) 40. These cells also did not display any phenotype, despite reduced levels of VAPB. This finding was in contrast with what was shown in transgenic animal models carrying mutant VAPB, where cytoplasmic aggregates represented a hallmark of the disease 36. Although it is impossible to draw general conclusions with only one patient line, this discrepancy might highlight one of the main advantages of using patient‐derived cells to model gene mutation‐related ALS: the absence of artifacts deriving from transgene overexpression.
One year later, in 2012, the first report of an iPS line harboring a TDP‐43 mutation (TDP‐43 M337V) was published 7. The motor neurons generated from this line showed elevated TDP‐43 levels, but no change in localization or signs of aggregate formation. In addition, motor neurons from both control and TDP‐43 mutant were phenotypically and functionally similar despite an elevated sensitivity to PI3K signaling inhibition and mild elevated cell death in patient cells.
More encouraging in terms of disease modeling were the findings of Egawa et al 12. They found that ALS patient‐specific iPSC‐derived motor neurons formed cytosolic aggregates similar to those seen in postmortem tissue from ALS patients and exhibited shorter neurites. TDP‐43 motor neurons were characterized by increased mutant protein aggregates bound to a spliceosomal factor SNRPB2. Expression array analysis detected small increases in transcripts involved in RNA metabolism and decreases in transcripts encoding cytoskeletal proteins. iPSC‐derived motor neurons were then used to screen a small compound library, which led to the identification of a histone acetyltransferase inhibitor called anacardic acid. Treatment with this drug successfully rescued the abnormal ALS motor neuron phenotype, thus, suggesting that iPSC‐derived cells can be effectively used to identify new therapeutic compounds 12.
Interesting from a disease‐modeling point of view are the recent findings describing impaired TDP‐43 granule transport along the axons of both Drosophila motor neurons and iPSC‐derived motor neurons carrying mutations in TDP‐43 2.
Another breakthrough in the field of ALS was the association between C9ORF72 hexanucleotide‐repeat expansions and familial ALS and/or Frontotemporal Dementia (FTD) that identified the most common genetic cause for ALS known to date 11, 46. Due to the high frequency of this mutation, the availability of skin biopsies and other somatic cells allowed the production of several iPSC lines.
Since 2013, a large number of publications has reported that neurons and motor neurons derived from patients carrying C9orf72 repeat expansion display some of the neuropathological hallmarks of ALS/FTD.
Almeida et al reported the presence of RNA foci containing GGGGCC repeats in some iPSCs, iPSC‐derived human neurons and primary fibroblasts. Moreover, the authors identified repeat‐associated non‐ATG (RAN) translation products in neurons from patients, along with p62 staining. Interestingly, inhibition of autophagy, but not metabolic stress, ER stress or kinase inhibition, increased the amount of p62 aggregates, sign of cellular stress 4. This suggested a function for C9orf72 in endosomal trafficking and autophagy, as subsequently demonstrated using a neuronal cell model, the neuro2a cells, and SH‐SY5Y cells 14.
In the same year, Donnelly et al confirmed that neurons derived from patients carrying the C9orf72 repeat expansion indeed displayed nuclear foci and RAN translation peptides 9. Moreover, the authors showed that the repeat expansion has a large impact of gene expression through sequestration of the RNA binding protein ADARB2, but not only. Finally, neurons from patients also displayed increased susceptibility to glutamate excitotoxicity, resulting in a time dependent increase in cell death. Of importance under a therapeutic point of view, antisense oligonucleotide (ASO) approach successfully reduced the susceptibility of patients’ neurons to excitotoxicity.
A subsequent study identified hnRNPA1 and Pur‐α as binding partners of the hexanucleotide repeat expansion 49. The association with these two RNA binding proteins is very likely to interfere with physiological RNA metabolism. In fact, ALS motor neurons from C9orf72 patients showed altered expression of several genes, among which DPP6, a gene implicated in sporadic ALS in multiple independent GWA studies 13 and involved in membrane excitability. Consistently, C9orf72 neurons demonstrated a diminished capacity to fire continuous spikes on depolarization compared with control motor neurons. Similarly to what previously reported 9, ASO targeting the C9ORF72 transcript suppressed RNA foci formation and reversed gene expression alterations in C9orf72 ALS motor neurons 49.
Although there is some discrepancy between different studies, it seems that patient‐derived motor neurons, regardless of the specific mutation they carry, display clear electrophysiological abnormalities. Wainger et al, in fact, using multielectrode array and patch clamp recordings report that hyperexcitability is an intrinsic characteristic of iPSC‐derived motor neurons harbouring SOD1, C9orf72 and FUS mutations 60.
Very recently, a breakthrough has identified a new molecular mechanism that may contribute to C9orf72‐mediated pathogenic mechanism. Two independent groups, using a candidate‐based genetic screen in Drosophila expressing 30 G4C2 repeats 66 or an unbiased genetic screen in Drosophila expressing 8, 28 or 58 G4C2 repeat‐containing transcripts 18, sought genes that enhance or suppress the disease phenotype.
Zhang et al identified the gene encoding RanGAP, a key regulator of nucleocytoplasmic transport, and Freibaum et al identified various genes that encode components of the nuclear pore and the nucleocytoplasmic transport machinery. Thus, both studies identified deficits in nucleocytoplasmic transport in Drosophila cells expressing hexanucleotide repeats and in iPSC‐derived neurons from ALS patients. Zhang et al show that these defects can be rescued with antisense oligonucleotides or small molecules targeting the G‐quadruplexes 66.
Although most of the studies using iPSCs have focused on samples from patients carrying known genetic mutations linked to ALS, cell reprogramming is particularly important to mimic the larger portion of ALS cases with no family history of disease. More than 90% of all ALS cases are, in fact, sporadic.
So far, very few studies have focused on this prominent patient population that poses several challenges in terms of individual genetic variability, but also likely variability in terms of the pathogenic mechanisms they can unveil.
The study conducted by Burkhardt et al describes the reprogramming of 16 sporadic fibroblast cell lines and their differentiation into motor neurons 8. Due to the association between sporadic ALS and TDP‐43 protein aggregates in postmortem tissues, the authors characterised the iPSC‐derived motor neurons for TDP‐43 aggregates and found that only 3 out of the 16 lines displayed abnormal staining for this marker. Although variability in the cell model cannot be excluded, this finding could be associated to specific pathogenic pathways and be descriptive of a subgroup of sporadic patients. This pathogenic phenotype was then utilised to screen for compounds that can lower the amount of TDP‐43 aggregates in patients cells 8.
More recently, motor neurons differentiated from iPSCs derived from sporadic ALS patients were used in a gene expression profiling study. The sample numbers were low (two sporadic patients and two controls), however, transcriptome analysis identified mitochondrial dysfunction as one of the strongest dysregulated pathways in sporadic ALS motor neurons compared with controls 5.
Human Primary Cells and Direct Reprogramming Protocols
Human astrocytes
Reports implicating non‐neuronal cells and, in particular, astrocytes in the pathology of ALS have prompted a chain of studies into the effect of these cells on motor neuron survival. Experiments focusing on familial ALS and the SOD1 mutation have pointed out the increase in motor neuron mortality in the presence of mutant astrocytes 21, 38. To study the toxic effect of human astrocytes in ALS in vitro, numerous astrocyte and motor neuron coculture methods have been developed since. The first study to show the effect of human ALS astrocytes onto motor neurons, utilised neural progenitor cells isolated from postmortem samples of spinal cords of both familial and sporadic ALS patients 23. These were subsequently cultured and differentiated into astrocytes 23, and cocultured with wild‐type motor neurons derived from mouse embryonic stem cells. After 120 h in culture, motor neurons grown on mutant SOD1 and sporadic astrocytes showed 45%–70% decrease in survival in comparison with controls. This study was the first to demonstrate that human ALS astrocytes lead to a decrease in motor neuron survival. Results of this study have recently been confirmed using sporadic and SOD1 mutant primary astrocytes isolated from postmortem spinal cords as well as motor cortex 45. Furthermore, after comparing the toxicity of astrocytes from ALS, Alzheimer's disease and chronic obstructive respiratory disorder patients by coculturing them with human embryonic motor neurons, they observed a decrease in survival among cells grown on ALS astrocytes only, thus, suggesting that astrocyte toxicity is disease‐specific 45.
In spite of their effectiveness in in vitro models, conducting coculture experiments using postmortem samples poses challenges that are difficult to overcome. Not only is the availability of such samples limited, but they also represent the end‐stage of the disease only, hence potentially propagating the toxicity of an inflamed spinal cord environment. It is also argued that astrocytes produced from postmortem tissue‐derived neural precursors are immature and, thus, do not fully reproduce normal cell function 48. Therefore, the development of iPSC technology allowed for these challenges to be overcome and offered an opportunity to conduct in vitro investigations into different genetic subtypes of ALS.
A study conducted by Serio et al 50 describes the use of iPSC‐derived astrocytes expressing TDP‐43 mutation in coculture with iPSC‐derived motor neurons. Even though astrocytes have displayed all pivotal signs of TDP‐43 proteinopathy, such as decreased cell survival, increased level of protein and cytoplasmic mislocalisation of TDP‐43, they have not elicited any toxic effect on wild‐type motor neurons. This study 50 was the first to investigate the role of iPSC‐derived astrocytes onto motor neurons and describing a different outcome compared with the studies performed on astrocytes from SOD1 and sporadic cases 23, 45. It could be argued that this discrepancy stems either from a difference between ways in which iPSC‐derived and postmortem astrocytes behave in vitro, or from a distinct characteristics of different ALS mutations.
Indeed, the role of astrocytes carrying mutant TDP43 in motor neuron degeneration is controversial. Wild‐type mouse embryonic stem cell‐derived motor neurons cultured with astrocytes isolated from a mouse model of ALS overexpressing TDP‐43A315T, as well as knock‐outs, do not display increase in cell death 24. Conversely, another study showed opposing results when using conditioned medium from spinal cord astrocytes isolated from a mouse model carrying mutant TDP‐43A315T on primary wild‐type spinal cord motor neurons 47.
Generating iPSCs from patient fibroblasts is a lengthy process which can take up to 18 weeks 53. Moreover, further differentiation of iPSC‐derived neural progenitors into astrocytes can take further 6–8 weeks, with neuronal contamination remaining an issue for a considerable period of the process, though eventually faltering at 2% or less 48, 50. The breakthrough in the field of cell reprogramming in ALS research came with the development of a novel protocol for fibroblast reprogramming, which utilises the same defined factors as iPSC technology and manages to bypass the iPSC stage by converting fibroblast directly into induced neural progenitor cells (iNPCs) 35. The protocol lasts approximately 3–4 weeks, thus significantly shortening the reprogramming process. Moreover, a highly enriched astrocyte culture (∼95%) can be obtained after a week of further differentiation without a need for selection steps or purification 35. Skin biopsy samples used in this study have been obtained from sporadic patients and those carrying either a SOD1 mutation or expressing a C9orf72 repeat expansion. To determine the effect of astrocytes obtained from iNPCs on wild‐type mouse embryonic stem cell‐derived motor neurons, coculture assays were set up and the survival of cells was monitored over the course of 5 days. Motor neuron survival was significantly reduced in cocultures involving ALS astrocytes compared with controls, with all ALS subtypes exhibiting a similar level of toxicity. Astrocytes differentiated from iNPCs elicit the same toxic effect on motor neurons as observed in the postmortem samples, thus not only proving the validity of this method, but also highlighting the possibility of studying the disease progression in each individual by obtaining biopsy samples at different time points. Moreover, creating a reproducible and highly efficient coculture assay opens a window for the development of personalized drug screenings 35.
In spite of the discrepancy observed in vitro so far, with astrocytes carrying a TDP‐43 mutation not eliciting any toxicity in motor neurons and SOD1, C9orf72 as well as sporadic cases demonstrating an evident toxic effect, current in vitro models of ALS are showing great promise (Fig. 1). One of the major advantages of using patient skin biopsy‐derived astrocytes over the use of postmortem samples is a possibility of designing a personalised therapy for each patient while they are still alive. Moving toward a fully humanised disease model would allow for an effective set of treatments to be developed, hence improving prognosis for ALS patients.
Figure 1.
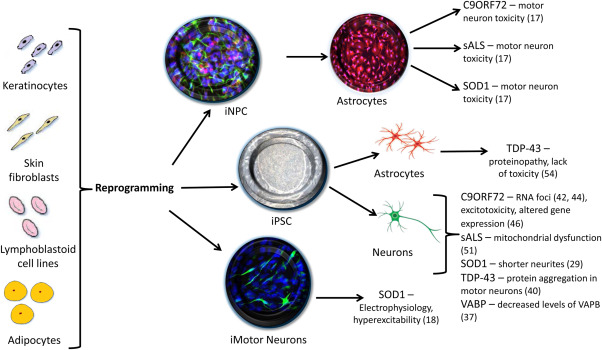
Schematic representation of the various direct and indirect reprogramming methods to obtain induced neural progenitor cells (iNPC), induced pluripotent stem cells (iPSC) and induced motor neurons (iMotor Neurons) from somatic cells. Studies conducted on cells derived from each protocol outlined above have successfully identified several pathological features of ALS across a number of genetic mutations (relevant references to each study in brackets).
The Promises and Limitations of Cell Reprogramming
Cell reprogramming holds great potential and promises for the future, from being an invaluable tool for in vitro disease modeling and identification of therapeutic targets to its potential for cell‐replacement therapies. Although iPSCs have already uncovered some important pathogenic mechanisms and potential therapeutic targets, they display several limitations.
One of the major challenges of cell reprogramming is the phenotypic inconsistency between stem cells 43 or between iPS clones differentiated from the same individual 56, a phenomenon called clonal variation. This kind of issue was reported in a study using iPS lines of ALS patients carrying TDP‐43 mutations. After differentiation of individual iPS clones from the same patient into motor neurons, the levels of TDP‐43 expression as well as aggregate formation and oxidative stress induced cell death showed substantial variation 12.
Despite the current discrepancies between the various methods used to generate patient‐derived iPSC lines, the observed clonal variations as well as the partial reflection of disease phenotypes, the field has advanced with tremendous speed if one considers that the first report describing the reprogramming of mouse fibroblasts was published only in 2006 53. Several steps have already been taken to make sure that protocols to obtain and differentiate iPSCs are optimized and standardized across the scientific community 6, 31, aiming to limit clonal variability.
One other important limitation of this in vitro model is that cells differentiated from induced pluripotent stem cells often reflect only certain aspects of disease pathophysiology and it often takes several weeks before such abnormalities can be recorded and/or the observed characteristics are very mild 10, 36. Observations from other neurological disorders, such as Alzheimer's 44 and Parkinson's disease 58, show a similar trend to what we have observed in ALS.
A very elegant recent study 34 has provided important information that might explain why iPSC‐derived neurons and glia might display mild signs of pathology 7, 8, 50, 60. RNA‐Sequencing analysis of fibroblasts, iPSCs, iPSC‐derived neurons, neurons reprogrammed directly from fibroblasts (iNeurons) and postmortem brain tissues revealed that iPSC‐derived neurons lose their aging signatures. This does not happen in iNeurons and directly converted cells 34. As ageing is the main risk factors for most neurodegenerative diseases, and in particular ALS, this characteristic might be of high relevance when modeling adult‐onset neurodegenerative diseases.
Although data on the ageing signature of induced neural progenitors (iNPCs) and derived astrocytes (iAstrocytes) 35 are not available, it might be possible that iAstrocytes from ALS patients successfully reproduce the toxicity observed in postmortem ALS astrocytes 23, 45 because of their faster reprogramming protocol compared to iPSC‐derived astrocytes 50. This crucial difference might enable iAstrocytes to retain their ageing signature and mimic the non‐cell autonomous mechanisms occurring in ALS more reliably than iPSC‐derived astrocytes.
One more important challenge to be faced in ALS, as well as other neurodegenerative disorders, will be the production of other glial cell‐types, including oligodendrocytes and microglia. These two cell‐types, in fact, have important implications in the pathogenesis of ALS and represent important therapeutic targets 17, 28.
While oligodendrocytes or their precursors have already been successfully differentiated from iPSCs for disease modeling 41 and therapy 61 in other fields of neurodegeneration, human microglia still remains a challenge.
References
- 1. Aasen T, Raya A, Barrero MJ, Garreta E, Consiglio A, Gonzalez F et al (2008) Efficient and rapid generation of induced pluripotent stem cells from human keratinocytes. Nat Biotechnol 26:1276–1284. [DOI] [PubMed] [Google Scholar]
- 2. Alami NH, Smith RB, Carrasco MA, Williams LA, Winborn CS, Han SSW et al (2014) Axonal transport of TDP‐43 mRNA granules is impaired by ALS‐causing mutations. Neuron 81:536–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Al‐Chalabi A, Jones A, Troakes C, King A, Al‐Sarraj S, van den Berg LH. (2012) The genetics and neuropathology of amyotrophic lateral sclerosis. Acta Neuropathol 124:339–352. [DOI] [PubMed] [Google Scholar]
- 4. Almeida S, Gascon E, Tran H, Chou HJ, Gendron TF, Degroot S et al (2013) Modeling key pathological features of frontotemporal dementia with C9ORF72 repeat expansion in iPSC‐derived human neurons. Acta Neuropathol 126:385–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Alves CJ, Dariolli R, Jorge FM, Monteiro MR, Maximino JR, Martins RS et al (2015) Gene expression profiling for human iPS‐derived motor neurons from sporadic ALS patients reveals a strong association between mitochondrial functions and neurodegeneration. Front Cell Neurosci 9:289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Andrews PW, Cavagnaro J, Cavanagro J, Deans R, Feigal E, Feigel E et al (2014) Harmonizing standards for producing clinical‐grade therapies from pluripotent stem cells. Nat Biotechnol 32:724–726. [DOI] [PubMed] [Google Scholar]
- 7. Bilican B, Serio A, Barmada SJ, Nishimura AL, Sullivan GJ, Carrasco M et al (2012) Mutant induced pluripotent stem cell lines recapitulate aspects of TDP‐43 proteinopathies and reveal cell‐specific vulnerability. Proc Natl Acad Sci USA 109:5803–5808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Burkhardt MF, Martinez FJ, Wright S, Ramos C, Volfson D, Mason M et al (2013) A cellular model for sporadic ALS using patient‐derived induced pluripotent stem cells. Mol Cell Neurosci 56:355–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Donnelly CJ, Zhang P‐W, Pham JT, Haeusler AR, Heusler AR, Mistry NA et al (2013) RNA toxicity from the ALS/FTD C9ORF72 expansion is mitigated by antisense intervention. Neuron 80:415–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dimos JT, Rodolfa KT, Niakan KK, Weisenthal LM, Mitsumoto H, Chung W et al (2008) Induced pluripotent stem cells generated from patients with ALS can be differentiated into motor neurons. Science 321:1218–1221. [DOI] [PubMed] [Google Scholar]
- 11. DeJesus‐Hernandez M, Mackenzie IR, Boeve BF, Boeve BF, Boxer AL, Baker M, Rutherford NJ et al (2011) Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p‐linked FTD and ALS. Neuron 72:245–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Egawa N, Kitaoka S, Tsukita K, Naitoh M, Takahashi K, Yamamoto T et al (2012) Drug screening for ALS using patient‐specific induced pluripotent stem cells. Sci Transl Med 4:145ra104. [DOI] [PubMed] [Google Scholar]
- 13. van Es MA, van Vught PWJ, Blauw HM, Franke L, Saris CGJ, Van den Bosch L et al (2008) Genetic variation in DPP6 is associated with susceptibility to amyotrophic lateral sclerosis. Nat Genet 40:29–31. [DOI] [PubMed] [Google Scholar]
- 14. Farg MA, Sundaramoorthy V, Sultana JM, Yang S, Atkinson RAK, Levina V et al (2014) C9ORF72, implicated in amytrophic lateral sclerosis and frontotemporal dementia, regulates endosomal trafficking. Hum Mol Genet 23:3579–3595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ferraiuolo L (2014) The non‐cell‐autonomous component of ALS: new in vitro models and future challenges. Biochem Soc Trans 42:1270–1274. [DOI] [PubMed] [Google Scholar]
- 16. Ferraiuolo L, Kirby J, Grierson AJ, Sendtner M, Shaw PJ (2011) Molecular pathways of motor neuron injury in amyotrophic lateral sclerosis. Nat Rev Neurol 7:616–630. [DOI] [PubMed] [Google Scholar]
- 17. Frakes AE, Ferraiuolo L, Haidet‐Phillips AM, Schmelzer L, Braun L, Miranda CJ et al (2014) Microglia induce motor neuron death via the classical NF‐κB pathway in amyotrophic lateral sclerosis. Neuron 81:1009–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Freibaum BD, Lu Y, Lopez‐Gonzalez R, Kim NC, Almeida S, Lee K‐H et al (2015) GGGGCC repeat expansion in C9orf72 compromises nucleocytoplasmic transport. Nature 525:129–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Fusaki N, Ban H, Nishiyama A, Saeki K, Hasegawa M (2009) Efficient induction of transgene‐free human pluripotent stem cells using a vector based on Sendai virus, an RNA virus that does not integrate into the host genome. Proc Jpn Acad Ser B Phys Biol Sci 85:348–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Geser F, Robinson JL, Malunda JA, Xie SX, Clark CM, Kwong LK et al (2010) Pathological 43‐kDa transactivation response DNA‐binding protein in older adults with and without severe mental illness. Arch Neurol 67:1238–1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Di Giorgio FP, Carrasco MA, Siao MC, Maniatis T, Eggan K (2007) Non‐cell autonomous effect of glia on motor neurons in an embryonic stem cell‐based ALS model. Nat Neurosci 10:608–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Di Giorgio FP, Boulting GL, Bobrowicz S, Eggan KC (2008) Human embryonic stem cell‐derived motor neurons are sensitive to the toxic effect of glial cells carrying an ALS‐causing mutation. Cell Stem Cell 3:637–648. [DOI] [PubMed] [Google Scholar]
- 23. Haidet‐Phillips AM, Hester ME, Meyer K, Braun L, Frakes A et al (2011) Astrocytes from familial and sporadic ALS patients are toxic to motor neurons. Nat Biotechnol 29:824–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Haidet‐Phillips AM, Gross SK, Williams T, Miranda CJ, Tuteja A, Sherman A, Ko M et al (2013) Altered astrocytic expression of TDP‐43 does not influence motor neuron survival. Exp Neurol 250C:250–9. [DOI] [PubMed] [Google Scholar]
- 25. Ilieva H, Polymenidou M, Cleveland DW (2009) Non‐cell autonomous toxicity in neurodegenerative disorders: ALS and beyond. J Cell Biol 187:761–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Isobe T, Tooi N, Nakatsuji N, Aiba K (2015) Amyotrophic lateral sclerosis models derived from human embryonic stem cells with different superoxide dismutase 1 mutations exhibit differential drug responses. Stem Cell Res 15:459–468. [DOI] [PubMed] [Google Scholar]
- 27. Kabashi E, Valdmanis PN, Dion P, Spiegelman D, McConkey BJ, Vande VC et al (2008) TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat Genet 40:572–574. [DOI] [PubMed] [Google Scholar]
- 28. Kang SH, Li Y, Fukaya M, Lorenzini I, Cleveland DW, Ostrow LW et al (2013) Degeneration and impaired regeneration of gray matter oligodendrocytes in amyotrophic lateral sclerosis. Nat Neurosci 16:571–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Karumbayaram S, Kelly TK, Paucar AA, Roe AJT, Umbach JA, Charles A et al (2015) Human embryonic stem cell‐derived motor neurons expressing SOD1 mutants exhibit typical signs of motor neuron degeneration linked to ALS. Dis Model Mech 2:189–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kwiatkowski TJ Jr, Bosco DA, Leclerc AL, Tamrazian E, Vanderburg CR, Russ C et al (2009) Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 323:1205–1208. [DOI] [PubMed] [Google Scholar]
- 31. Maherali N, Hochedlinger K (2008) Guidelines and techniques for the generation of induced pluripotent stem cells. Cell Stem Cell 3:595–605. [DOI] [PubMed] [Google Scholar]
- 32. Maherali N, Ahfeldt T, Rigamonti A, Utikal J, Cowan C, Hochedlinger K (2008) A high‐efficiency system for the generation and study of human induced pluripotent stem cells. Cell Stem Cell 3:340–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Marchetto MCN, Muotri AR, Mu Y, Smith AM, Cezar GG, Gage FH (2008) Non‐cell‐autonomous effect of human SOD1 G37R astrocytes on motor neurons derived from human embryonic stem cells. Cell Stem Cell 3:649–57. [DOI] [PubMed] [Google Scholar]
- 34. Mertens J, Paquola ACM, Ku M, Hatch E, Böhnke L, Ladjevardi S et al (2015) Directly reprogrammed human neurons retain aging‐associated transcriptomic signatures and reveal age‐related nucleocytoplasmic defects. Cell Stem Cell 17:705–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Meyer K, Ferraiuolo L, Miranda CJ, Likhite S, McElroy S, Renusch S et al (2014) Direct conversion of patient fibroblasts demonstrates non‐cell autonomous toxicity of astrocytes to motor neurons in familial and sporadic ALS. Proc Natl Acad Sci USA 111:829–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Mitne‐Neto M, Machado‐Costa M, Marchetto MCN, Bengtson MH, Joazeiro CA, Tsuda H et al (2011) Downregulation of VAPB expression in motor neurons derived from induced pluripotent stem cells of ALS8 patients. Hum Mol Genet 20:3642–3652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Miyoshi N, Ishii H, Nagano H, Haraguchi N, Dewi DL, Kano Y et al (2011) Reprogramming of mouse and human cells to pluripotency using mature microRNAs. Cell Stem Cell 8:633–638. [DOI] [PubMed] [Google Scholar]
- 38. Nagai M, Re DB, Nagata T, Chalazonitis A, Chalazonitis A, Jessell TM, Wichterle H et al (2007) Astrocytes expressing ALS‐linked mutated SOD1 release factors selectively toxic to motor neurons. Nat Neurosci 10:615–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT et al (2006) Ubiquitinated TDP‐43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314:130–133. [DOI] [PubMed] [Google Scholar]
- 40. Nishimura AL, Mitne‐Neto M, Silva HCA, Richieri‐Costa A, Middleton S, Cascio D et al (2004) A mutation in the vesicle‐trafficking protein VAPB causes late‐onset spinal muscular atrophy and amyotrophic lateral sclerosis. Am J Hum Genet 75:822–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Numasawa‐Kuroiwa Y, Okada Y, Shibata S, Kishi N, Akamatsu W, Shoji M et al (2014) Involvement of ER stress in dysmyelination of Pelizaeus‐Merzbacher disease with PLP1 missense mutations shown by iPSC‐derived oligodendrocytes. Stem Cell Reports 2:648–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Okita K, Nakagawa M, Hyenjong H, Ichisaka T, Yamanaka S (2008) Generation of mouse induced pluripotent stem cells without viral vectors. Science 322:949–953. [DOI] [PubMed] [Google Scholar]
- 43. Osafune K, Caron L, Borowiak M, Martinez RJ, Fitz‐Gerald CS, Sato Y et al (2008) Marked differences in differentiation propensity among human embryonic stem cell lines. Nat Biotechnol 26:313–315. [DOI] [PubMed] [Google Scholar]
- 44. Ovchinnikov DA, Wolvetang EJ (2014) Opportunities and limitations of modelling Alzheimer's disease with induced pluripotent stem cells. J Clin Med 3:1357–1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Re DB, Le Verche V, Yu C, Amoroso MW, Politi KA, Phani S et al (2014) Necroptosis drives motor neuron death in models of both sporadic and familial ALS. Neuron 81:1001–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Renton AE, Majounie E, Waite A, Simon‐Sanchez J, Rollinson S, Gibbs JR et al (2011) A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21‐linked ALS‐FTD. Neuron 72:257–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Rojas F, Cortes N, Abarzua S, Dyrda A, van Zundert B (2014) Astrocytes expressing mutant SOD1 and TDP43 trigger motoneuron death that is mediated via sodium channels and nitroxidative stress. Front Cell Neurosci 8:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Roybon L, Lamas NJ, Garcia‐Diaz A, Yang EJ, Sattler R, Jackson‐Lewis V et al (2013) Human stem cell‐derived spinal cord astrocytes with defined mature or reactive phenotypes. Cell Rep 4:1035–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Sareen D, O'Rourke JG, Meera P, Muhammad AKMG, Grant S, Simpkinson M et al (2013) Targeting RNA foci in iPSC‐derived motor neurons from ALS patients with a C9ORF72 repeat expansion. Sci Transl Med 5:208ra149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Serio A, Bilican B, Barmada SJ, Ando DM, Zhao C, Siller R et al (2013) Astrocyte pathology and the absence of non‐cell autonomy in an induced pluripotent stem cell model of TDP‐43 proteinopathy. Proc Natl Acad Sci USA 110:4697–4702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Son EY, Ichida JK, Wainger BJ, Toma JS, Rafuse VF, Woolf CJ et al (2011) Conversion of mouse and human fibroblasts into functional spinal motor neurons. Cell Stem Cell 9:205–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Sreedharan J, Blair IP, Tripathi VB, Hu X, Vance C, Rogelj B et al (2008) TDP‐43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 319:1668–1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Takahashi K, Yamanaka S (2006) Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126:663–676. [DOI] [PubMed] [Google Scholar]
- 54. Takahashi K, Okita K, Nakagawa M, Yamanaka S (2007) Induction of pluripotent stem cells from fibroblast cultures. Nat Protoc 2:3081–3089. [DOI] [PubMed] [Google Scholar]
- 55. Taura D, Noguchi M, Sone M, Hosoda K, Mori E, Okada Y et al (2009) Adipogenic differentiation of human induced pluripotent stem cells: comparison with that of human embryonic stem cells. FEBS Lett 583:1029–1033. [DOI] [PubMed] [Google Scholar]
- 56. Thatava T, Kudva YC, Edukulla R, Squillace K, De Lamo JG, Khan YK et al (2013) Intrapatient variations in type 1 diabetes‐specific iPS cell differentiation into insulin‐producing cells. Mol Ther 21:228–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Toma JS, Shettar BC, Chipman PH, Pinto DM, Borowska JP, Ichida JK et al (2015) Motoneurons derived from induced pluripotent stem cells develop mature phenotypes typical of endogenous spinal motoneurons. J Neurosci 35:1291–1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Torrent R, De Angelis RF, Dell'Era P, Memo M, Raya A, Consiglio A (2015) Using iPS cells toward the understanding of Parkinson's disease. J Clin Med 4:548–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Vance C, Rogelj B, Hortobagyi T, De Vos KJ, Nishimura AL, Sreedharan J et al (2009) Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 323:1208–1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Wainger BJ, Kiskinis E, Mellin C, Wiskow O, Han SSW, Sandoe J et al (2014) Intrinsic membrane hyperexcitability of amyotrophic lateral sclerosis patient‐derived motor neurons. Cell Rep 7:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Wang S, Bates J, Li X, Schanz S, Chandler‐Militello D, Levine C et al (2013) Human iPSC‐derived oligodendrocyte progenitor cells can myelinate and rescue a mouse model of congenital hypomyelination. Cell Stem Cell 12:252–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Yamanaka K, Chun SJ, Boillee S, Fujimori‐Tonou N, Yamashita H, Gutmann DH et al (2008) Astrocytes as determinants of disease progression in inherited amyotrophic lateral sclerosis. Nat Neurosci 11:251–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Yang YM, Gupta SK, Kim KJ, Powers BE, Cerqueira A, Wainger BJ et al (2013) A small molecule screen in stem‐cell‐derived motor neurons identifies a kinase inhibitor as a candidate therapeutic for ALS. Cell Stem Cell 12:713–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Yao X‐L, Ye C‐H, Liu Q, Wan J, Zhen J, Xiang AP et al (2013) Motoneuron differentiation of induced pluripotent stem cells from SOD1G93A mice. PLoS One 8:e64720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Yu J, Vodyanik MA, Smuga‐Otto K, Antosiewicz‐Bourget J, Frane JL, Tian S et al (2007) Induced pluripotent stem cell lines derived from human somatic cells. Science 318:1917–1920. [DOI] [PubMed] [Google Scholar]
- 66. Zhang K, Donnelly CJ, Haeusler AR, Grima JC, Machamer JB, Steinwald P et al (2015) The C9orf72 repeat expansion disrupts nucleocytoplasmic transport. Nature 525:56–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Zhou W, Freed CR (2009) Adenoviral gene delivery can reprogram human fibroblasts to induced pluripotent stem cells. Stem Cells 27:2667–2674. [DOI] [PubMed] [Google Scholar]
- 68. Zhou T, Benda C, Dunzinger S, Huang Y, Ho JC, Yang J et al (2012) Generation of human induced pluripotent stem cells from urine samples. Nat Protoc 7:2080–2089. [DOI] [PubMed] [Google Scholar]