

Abstract
We report 10 cases of a non‐neurocytic, purely neuronal tumor affecting adults. Situated in the cerebral hemispheres, with 7 of 10 confined to the temporal lobes, most presented with seizures as their principal clinical manifestations. On magnetic resosnance imaging (MRI), the tumors generally appeared solid and non‐contrast enhancing with minimal diffuse infiltration, edema, or mass effect. Six examples demonstrated internal nodularity. Microscopically, the tumor cells were largely distributed into discrete and coalescent nodules exhibiting varying degrees of matrix vacuolization, principally within the deep cortical ribbon and superficial subcortical white matter. Populating elements ranged from morphologically ambiguous to recognizably neuronal, with only two cases manifesting overt ganglion cell cytology. In all cases, tumor cells exhibited widespread nuclear immunolabeling for the HuC/HuD neuronal antigens, although expression of other neuronal markers, including synaptophysin, neurofilament and chromogranin was variable to absent. Tumor cells also failed to express GFAP, p53, IDH1 R132H, or CD34, although CD34‐labeling ramified neural elements were present in the adjoining cortex of seven cases. Molecular analysis in a subset of cases failed to reveal DNA copy number abnormalities or BRAF V600E mutation. Follow‐up data indicate that this unusual neuronal lesion behaves in benign, World Health Organization (WHO) grade I fashion and is amenable to surgical control.
Keywords: developmental tumor, eliptogenic tumor, gangliocytoma, HuC/HuD, multinodular, vacuolating neuronal tumor
Introduction
We present our experience with a group of purely neuronal tumors having distinctive morphologic attributes. Most became symptomatic in adulthood and all exhibited multinodular patterns of cerebral involvement on histopathologic assessment, multinodularity also being apparent in most cases on neuroradiologic study. Ambiguous cytologic features and an incompletely developed, “weakly” neuronal immunoprofile complicated the classification of these curious lesions, which evolved in benign clinical fashion.
Materials and Methods
Histopathology
Case materials were collected by the authors in the course of their hospital or consultative (MKR) practices. Formalin‐fixed and paraffin‐embedded tissues were employed for all immunohistochemical and molecular diagnostic assays. The streptavidin‐biotin peroxidase complex method was utilized for immunohistochemical studies with antibodies to: glial fibrillary acidic protein (GFAP; Biogenex, Fremont, CA, USA; polyclonal, 1:50), synaptophysin (SYN; Biogenex, monoclonal, 1:100), 160/200 kD neurofilament proteins (NFP; Zymed, Life Technologies, Grand Island, NY, USA; RMd0‐20 clone, monoclonal 1:200), neuronal nuclear protein (NeuN; Chemicon, Millipore, Billerica, MA, USA; monoclonal, 1:1000), neuronal protein HuC/HuD (HuC/HuD; Molecular Probes, Life Technologies; monoclonal, 1:50), chromogranin (CHR; Ventana Medical Systems, Tuscon, AZ, USA; monoclonal, prediluted), CD34 (Ventana, monoclonal, prediluted), p53 (Ventana, monoclonal, prediluted), Ki‐67 (MIB‐1; Ventana, monoclonal, prediluted), R132 IDH1 (Dianova, Hamburg, Germany; H09 clone, monoclonal, 1:50), and OLIG2 (Millipore; polyclonal, 1:100).
DNA copy number analysis
Array comparative genomic hybridization (aCGH), genomic DNA was extracted from unstained 5 uM sections in cases 5 and 8 using the Formapure kit (Agencourt, Beverly, MA, USA) using a modified version of the manufacturer's method in a semi‐automated fashion 12. Eluted double‐stranded DNA (dsDNA) was subsequently quantified [by Quant‐iT ™ PicoGreen ® reagent (Invitrogen, Carlsbad, CA, USA) the average yield was 373 ng of ds DNA] and subjected to whole genome amplification by Genome Plex (Sigma, St. Louis, MO, USA) following the manufacturer's instructions. Amplified genomic DNA was used for CGH on the Agilent 1M array platform employing whole genome amplified male genomic DNA (Promega, Madison, WI, USA) as normal. Regions of copy number variation were determined by RAE analysis as previously described 34.
Mutational genotyping
The Sequenom MassARRAY system was used in cases 1, 5, 6 and 8 to detect recognized somatic point mutations in the following eight genes: EGFR, ERBB2, KRAS, BRAF, NRAS, PIK3CA, MEK1 (MAP2K1) and AKT1. Mutation detection was based on the mass differences between the wild‐type and mutant polymerase chain reaction (PCR) products as resolved by matrix‐assisted laser desorption/ionization‐time‐of‐flight mass spectrometry 16.
Results
Detailed cases records are provided following a summation of major demographic, clinical and radiologic features (see also Table 1).
Table 1.
Clinical and demographic characteristics of patient cohort
| Case | Age | Sex | Location | Clinical manifestations (duration) | Treatment/follow‐up (duration) |
|---|---|---|---|---|---|
| 1 | 38 | M | R medial temporal | Dizziness, loss of attention (2 years) | Near‐total resection/no recurrence (8 months) |
| 2 | 54 | F | L amygdala | Dizziness, dysarthria, blurred vision, R numbness (one episode) | Gross total resection and radiation/no recurrence (6 years) |
| 3 | 38 | F | R parietal | Gran mal seizure (one episode) | Near‐total resection/no recurrence (16 months) |
| 4 | 35 | M | R medial temporal | Episodic confusion (14 months) | Subtotal resection/no regrowth (6 months) |
| 5 | 54 | M | R medial temporal | Partial complex and gran mal seizures (>40 years) | Gross total resection/no recurrence (11 months) |
| 6 | 31 | F | L temporal | Simple, complex, and gran mal seizures (2 years) | Subtotal resection/no regrowth (1 year) |
| 7 | 41 | M | R medial temporal | Confusion after motor vehicle accident (one episode) | Gross total resection/No recurrence (5 years) |
| 8 | 63 | F | R temporal | L numbness and tingling (1 year) | Gross total resection/no recurrence (3 years) |
| 9 | 64 | M | L temporal | Staring and mumbling (1 episode) | Biopsy/no follow‐up |
| 10 | 52 | F | L frontal | Episodic vertigo (2 years) | Gross total resection/no follow‐up |
F = female; L = left; M = male; R = right.
Demographic features
Cases were equally divided between men and women, patients ranging from 31 to 64 (mean = 47, median = 46) years of age at the time of tumor discovery.
Localization
All lesions were situated in the cerebral hemispheres and relatively superficial in position, six arising on the right and four on the left. Eight of 10 tumors involved the temporal lobes, seven being entirely intratemporal, five of these being medially positioned and four of these five being right‐sided. One lesion involved the left superior temporal gyrus and contiguous supramarginal gyrus, the two extratemporal cases arising in the medial left frontal and right parietal lobes.
Clinical manifestations
Seizures or seizure equivalents (eg, episodic confusion) were by far the most common neurologic complaints and were experienced by seven of 10 patients, an eighth suffering a motor vehicle accident that may have been seizure‐related as well. Focal and complex partial seizures (with secondary generalization in some cases), grand mal seizures and status epilepticus were all encountered in this series. Other complaints included numbness and tingling of the extremities and vertigo. One patient (see case 6 below) had a mother, brother and paternal aunt with chronic seizure disorders. Symptoms were of recent onset in five cases, while in five cases symptom duration prior to tumor discovery was 1 year or longer. Only one patient (number 5) had a relevant neurologic history (of pharmacoresistant epilepsy) dating back to childhood when he was found at 54 years of age to have a temporal lobe tumor.
Neuroradiologic features
Save for case 9, which exhibited a small cystic component, all lesions were solid on magnetic resonance imaging (MRI). Maximal dimensions ranged from 1.8 to 4.1 (mean = 2.7) centimeters. T1 iso/hypointensity, T2‐hyperintensity and increased signal in fluid attenuated inversion recovery (FLAIR) sequences characterized all lesions. Six cases demonstrated internal nodularity that was conspicuous in four and fainter in an additional two, this being best appreciated in FLAIR sequences but also demonstrable in T2‐weighted images (Figures 1 and 2). Eight of 10 lesions were devoid of contrast enhancement, two exhibiting focal and faint enhancement following administration of gadolinium. Involved regions of brain exhibited, at most, only minimal expansion and there was not evidence of edema or mass effect in any case. Additional small foci of non‐specific, increased T2 and FLAIR signal were observed in the cerebral hemispheric white matter of two patients. MR spectroscopy, performed in one case (number 8), demonstrated a relatively increased choline to NAA peak ratio. Four tumors manifested complete stability during pre‐operative MRI surveillance intervals ranging from 9 to 18 months, a fifth evidencing only questionable enlargement over a surveillance period of 2 years. Diagnoses entertained by reviewing neuroradiologists included glioma, cortical dysplasia, hamartoma, dysembryoplastic neuroepithelial tumor, focal cerebritis and contusion.
Figure 1.
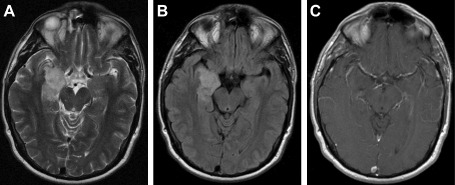
T2 –weighted (A), fluid attenuated inversion recovery (FLAIR B) and contrast‐enhanced (C) MR images from case 1. Note manner in which this solid, non‐enhancing lesion conforms to the contours of the right medial temporal lobe without significant tissue expansion or mass effect. T2/FLAIR‐ hyperintense, the lesion exhibits a subtle internal nodularity most evident in the FLAIR sequence.
Figure 2.
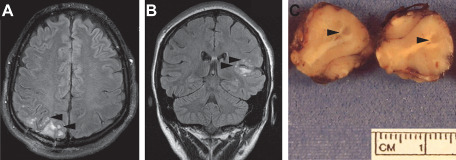
Conspicuous lesion multinodularity is apparent in fluid attenuated inversion recovery images from cases 3 (A) and 6 (B). Note the superficial location and manner in which underlying gyral conformations are preserved without mass effects. A tumor nodule spans the grey‐white junction in a gross photograph of the neurosurgical specimen from case 6 (at arrowheads, C).
Treatment and follow‐up
With the exception of one case (patient 10) in which only biopsies were performed, all patients underwent complete or substantial lesion excision. This was deemed gross total in five cases, near‐total in two and subtotal in two. Only one patient received adjuvant treatment (case 2; involved field radiotherapy). Post‐operative follow‐up information over intervals of 6 to 72 (mean = 27.5; median = 14) months was available for eight patients. All were free of recurrence regardless of extent of excision, the four patients with near‐total or subtotal resections having stable signal changes on MRI from 6 to 16 (mean = 10) months after surgery. Seven of the 8 patients for whom follow‐up information was available were neurologically asymptomatic, two on anticonvulsants and the remainder without medication, this group including patients with total, near‐total and subtotal resections. One patient (case 6) remained symptomatic after subtotal lesion removal, suffering seizures and cognitive disturbances despite continuing anticonvulsant therapy.
Case histories
Case 1. This 38‐year‐old man was evaluated in October 2011 for a 2‐year history of self‐described “dizzy spells” associated with loss of attention and balance, these being brief but increasing in frequency to several episodes daily by the time he consulted a physician. MRI revealed a 4.1 × 2.2 cm, non‐enhancing mass in the right medial temporal lobe that was centered in the hippocampal region with extension into the parahippocampal gyrus. This produced minimal expansion of the involved structures and demonstrated increased signal on T2‐weighted and FLAIR sequences with foci of subtle nodularity in its hippocampal component on coronal FLAIR imaging (Figure 1). A near‐total resection was performed in early January 2012. As of August 2012, the patient was neurologically asymptomatic and MRI demonstrated only a small focus of persistent T2/FLAIR signal abnormality in the right temporal lobe that was stable.
Case 2. This 54‐year‐old woman was in her usual state of good health until March 2006 when she experienced a transient episode of dizziness, slurring of her speech, blurring of her vision and right‐sided numbness. Her family physician ordered a computed tomography (CT) scan, which proved to be unremarkable. A consulting neurologist subsequently ordered an MRI, this demonstrating a 1.7 × 1.8 cm region of increased T2 and FLAIR signal with internal nodularity in the left amygdala. There was not accompanying mass effect, edema or contrast enhancement. A presumptive diagnosis of low grade glioma was made. The patient was maintained on anti‐seizure medications and followed over the next 9 months, during which there was no change in serial MRIs. She desired surgery, however, and a gross total resection was performed in January 2007. As initial pathologic assessment suggested a potentially aggressive neoplasm, involved field radiotherapy was administered. The patient remained asymptomatic post‐treatment, follow‐up MRI in July 2012 demonstrating only a surgical cavity at the site of prior operation.
Case 3. This previously healthy 38‐year‐old woman suffered a grand mal seizure in October 2008. A CT scan proved negative, but on additional study the patient was found to have a 3.0 × 2.4 cm lesion in the posterior right parietal region that exhibited inhomogeneous signal characteristics on T1‐weighted MRI with multinodular T2 and FLAIR hyperintensity (Figure 2A). Faint enhancement was apparent in the deepest portion of the lesion, which produced mild expansion of the involved gyrus without mass effect or edema. The neuroradiologist suggested a diagnosis of dysembryoplastic neuroepithelial tumor. Additional subcortical T2 signal abnormalities of uncertain nature were noted in the right frontal and left frontotemporal areas. A biopsy was performed in December 2008 and near‐total resection accomplished in April 2009. The lesion did not exhibit any neuroradiologic changes during this interval. The patient was maintained on anticonvulsants and was seizure‐free with stable MRI as of August 2010.
Case 4. This 35‐year‐old man was well until early 2007, when he experienced several episodes of confusion. MRIs in March of that year demonstrated a 3.6 × 2.6 × 1.9 cm focus of increased T2 and FLAIR signal, without contrast enhancement or restricted diffusion, in the medial right temporal lobe. The affected region was mildly swollen without mass effect or edema. Surveillance MRIs in June 2007 and March 2008 were unchanged. A subtotal resection was performed in mid‐March 2008. An MRI in mid‐September of the same year (6 months postoperatively) demonstrated residual T2 signal abnormalities in the inferomedial right temporal lobe unchanged from prior studies. The patient was contacted repeatedly over the next 2 years but refused follow‐up examinations.
Case 5. This 54‐year‐old man (age at tumor discovery) had a history of complex partial epilepsy dating to childhood and had been on anticonvulsant therapy since age 18. At age 37, he began to experience secondary generalization of his seizures with urinary and fecal incontinence as well as tonic‐clonic convulsions. Over the next 17 years, seizure frequency increased gradually from one to two episodes per year to one episode every month or two. The patient also suffered progressive cognitive difficulties during this period that forced him to stop working. In May 2009, he suffered status epilepticus, requiring intubation and ventilatory support. MRI demonstrated a 1.7 × 2.6 × 1.4 cm lesion in the medial right temporal lobe that was isointense to grey matter on T1 and T2/FLAIR‐bright with vague internal nodularity. The lesion was non‐enhancing and without mass effect or edema. An anterior temporal lobectomy with gross total tumor excision was performed in July 2009. The patient was seizure‐free (on multiple anticonvulsants) and without MRI evidence of recurrence as of late June 2010.
Case 6. This 31‐year‐old woman with a 5‐year history of simple and complex partial epilepsy and a left temporal lobe abnormality demonstrated by MRI in 2007 experienced increased seizure frequency and severity culminating in a grand mal convulsion in January 2009. Of note is the fact that her mother, one brother and a paternal aunt suffered from chronic seizure disorders. MRI then demonstrated a 3.1 × 2.5 × 3.1 cm lesion conforming to the contour of the dorsal and superior aspects of the left superior temporal gyrus and following along the contour of the left supramarginal gyrus. This was iso‐to slightly hypointense on T1‐weighted study and T2/FLAIR‐hyperintense with a multinodular appearance in the latter weightings and foci of faint contrast enhancement (Figure 2B). There was question of slight lesional enlargement when compared to the appearance in MRI studies from February 2007. A diagnosis of cortical dysplasia was suggested with the possibility raised of a complicating low grade glioma. The left hippocampus exhibited increased T2 signal with an overall decrease in anterior hippocampal volume suggesting seizure‐related changes/early mesial temporal sclerosis. A subtotal resection was performed in August 2009. The patient continued to experience partial seizures with fugue episodes and cognitive disturbances despite anticonvulsant therapy, but there was no evidence of tumor regrowth on MRI 1 year after operation.
Case 7. This previously well 41‐year‐old man was involved in a minor motor vehicle accident in November 2006. A highway patrolman on the scene found him to be confused, though not obviously injured, and had the man taken to a local hospital where CT was unremarkable. A subsequent MRI, however, demonstrated a 2 cm focus of increased T2 signal in the right medial temporal/uncal region without contrast enhancement, mass effect or associated edema. Gross total resection was performed in January 2007. As of January 2012 the patient was asymptomatic and had no evidence of lesion regrowth on follow‐up MRI.
Case 8. This previously healthy 63‐year‐old woman experienced, over the course of 1 year, several brief episodes of numbness and tingling in the fingers of her left hand. In June 2008, an episode of transient left‐sided arm and body numbness prompted her to consult a physician, who noted a normal neurologic examination but ordered an MRI that disclosed a 2.2 cm lesion involving the right middle and inferior temporal gyri. This was characterized by T2 and FLAIR hyperintensity with a multinodular appearance. There was slight T1‐hypointensity. The lesion did not exhibit mass effect, contrast enhancement or associated edema. The patient was followed with serial MRIs, these exhibiting no interval changes through mid‐March 2009. MR spectroscopy at that time, however, demonstrated increased choline compared to NAA peaks and was read as consistent with a neoplastic process most likely representing an infiltrating glioma. A gross total resection was performed shortly after. The patient was asymptomatic and without MRI evidence of recurrence through April 2012.
Case 9. This 64‐year‐old man suffered a transient attack of staring and unintelligible mumbling while at work in late April 2010. MRI revealed a non‐enhancing lesion in the left posteroinferior temporal lobe that was T1‐isointense relative to grey matter and T2/FLAIR‐hyperintense with a small cystic component. There was not restricted diffusion, mass effect or edema associated with this lesion. The patient underwent surgery in June 2010 at which time multiple biopsies were obtained. Further follow‐up information was unavailable.
Case 10. This 52‐year‐old woman with a 2‐year history of episodic vertigo was found on MRI in February 2012 to have a 2.5 × 1.4 × 2.0 cm lesion in the medial aspect of the left frontal lobe. This involved the cortex and subcortical white matter of the orbital and cingulate gyri, was solid, manifested increased signal on FLAIR and T2‐weighted images, did not enhance on contrast administration and was without mass effect or edema. Incidentally, noted were scattered small foci of increased FLAIR/T2 signal in the deeper white matter of both cerebral hemispheres and an arachnoid cyst in the left middle cranial fossa. Gross total excision was accomplished in May 2012.
Macroscopic findings
Gross descriptions of submitted neurosurgical materials noted specific abnormalities in two cases. The uncus included in the specimen from patient 4 was described as exhibiting grey discoloration and being abnormally firm. Gross examination of the specimen from patient 6 revealed discrete grey nodules measuring up to 1.5 mm in diameter scattered in the digitate white matter and along the cortical‐white matter junction (Figure 2C).
Histopathologic findings
Features observed in all cases and readily apparent on low power microscopic assessment included the distribution of tumor cells in discrete and coalescent nodules, these exhibiting conspicuous vacuolar alterations and being separated by apparently normal or astrogliotic neuroparenchyma (Figure 3A–D). In most cases, such nodules lay principally within the deep half of the cortical ribbon, along the grey‐white junction and within the digitate white matter. An entirely intracortical tumor topography was not encountered in any case. In addition to micronodular aggregates, tumor cells were also found in plaque‐like and more diffuse array in select cases.
Figure 3.
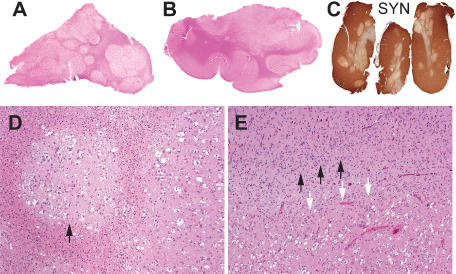
Multinodular tumor architecture is demonstrated in whole mount hematoxylin and eosin‐stained sections from cases 6 (A) and 5 (B) and in an anti‐synaptophysin immunohistochemical preparation from case 4 (C). Note distribution of nodules within cortex and at grey matter junction and greatly diminished intratumoral expression of synaptophysin in (C). Discrete tumor nodules surrounded by gliotic white matter are apparent at higher magnification (case 6; D). Note vacuolar alterations. Tumor (white arrows) abuts cortex (black arrows) exhibiting architectural disarray (case 6; E).
The abnormal neuroepithelial elements constituting the deposits described above ranged from small and astrocyte‐like forms having modest to moderate expanses of eosinophilic and, in some cases, granular cytoplasm to cells of more suggestively neuronal profile with vesicular nuclei, distinct nucleoli and amphophilic or lightly basophilic cytoplasm (Figure 4). Large, overtly ganglion cell‐like elements were encountered in only two cases (numbers 1 and 6), these including tumor cells with basophilic cytoplasmic stippling reminiscent of Nissl substance formation (Figure 4D). Within each case, constituent cells tended to a uniform size and cytomorphology. Whereas most lesions did not manifest any apparent architectural structuring, tumor cells in some cases exhibited focal clustering about small intracortical blood vessels. Multinucleation was not apparent, nor were the bizarre cytologic abnormalities displayed by some gangliogliomas in evidence. Mitotic figures were not found in any case. The vacuolar alterations apparent on scanning examination proved, on higher power view, to involve both tumor cell perikarya and the intercellular matrix, tumor cells also appearing to float within lacunar spaces. The tumor matrix generally manifested a delicate neuropil‐like appearance.
Figure 4.
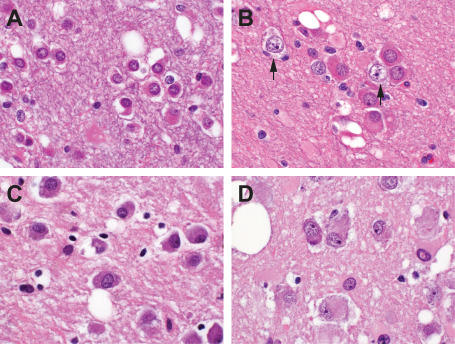
The cytologic variability of tumor cells is demonstrated in sections, all at the same magnification, from cases 5 (A), 4 (B), 1 (C) and 6 (D). These range from relatively small in size with eosinophilic cytoplasm (A) to large and overtly ganglion cell‐like (D). For comparative purposes, native neurons are included in B (arrows). Note vacuolar changes.
Conspicuous lymphoid infiltration, the formation of eosinophilic granular bodies or Rosenthal fibers, piloid or other neoplastic astroglial forms, myxoid/microcystic changes or microcalcifications were not observed in any case. Oligodendrocyte‐like cells organized in small aggregates (hamartia) were seen in the adjoining temporal cortex of patient 5, but not within the tumor micronodules of this or any other case. Absent from all cases were neurocytomatous components and anything resembling the “specific glioneuronal elements” of dysembryoplastic neuroepithelial tumor (DNT). Two cases (numbers 2 and 6) were associated with frankly dysplastic alterations of cerebral cortex included in the resection specimens, these demonstrating marked cytoarchitectural disarray with abnormal clustering of neurons and loss of normal lamination (Figure 3E).
Immunohistochemical findings
All cases exhibited widespread immunolabeling of tumor cell nuclei (and, to a variable degree, perikarya) for the HuC/HuD neuronal antigens, although at noticeably lesser intensity than that manifested by native cortical neurons (Figure 5C,G,K,O). Three cases (numbers 1, 6 and 9) evidenced unequivocal, granular labeling of perikarya for synaptophysin, these including the two cases characterized by the most overt morphologic evidence of ganglion cell differentiation. Three cases were synaptophysin‐negative (numbers 4, 7 and 10), the remaining demonstrating focal and relatively weak, non‐granular perikaryal labeling that was difficult to interpret conclusively. Irregular, punctate synaptophysin reactivity in tumor matrices was apparent in cases with and without perikaryal labeling, but difficult to distinguish from background neuropil. Surface perikaryal synaptophysin expression of the sort often characterizing gangliogliomas was not observed in any case (Figure 5B,F,J,N). In a similar vein, tumor cells proved uniformly non‐reactive for chromogranin, NeuN (Figure 5D,H,L,P) and neurofilament proteins. Labeling for GFAP was restricted to a network of hyperplastic astrocytes within and around tumor micronodules (Figure 5A,E,I,M). All lesions studied for OLIG2 expression (cases 1, 2, 5, 6, 8, 9 and 10) manifested diffuse labeling of tumor cell nuclei (Figure 6). Save for one case (number 4) in which a minor population of tumor cells manifested weak granular labeling of their peripheral perikarya, immunoexpression of CD34 was restricted to process‐rich, ramified neural elements of the sort associated with gangliogliomas and pleomorphic xanthoastrocytomas (Figure 7). These elements were present in the cortices of seven cases (numbers 1, 2, 3, 5, 6, 9 and 10) and ranged in density from isolated examples (case 1, 2, 10) to dense aggregates, nodules and plaques (case 5 and 6). Five assessed cases (numbers 1, 3, 4, 6 and 10) were entirely p53‐negative and failed to demonstrate any immunoexpression of R132H mutant IDH1. MIB‐1/Ki‐67 labeling activity was restricted to only rare cells in each case and whether these represented tumor elements was difficult to determine.
Figure 5.
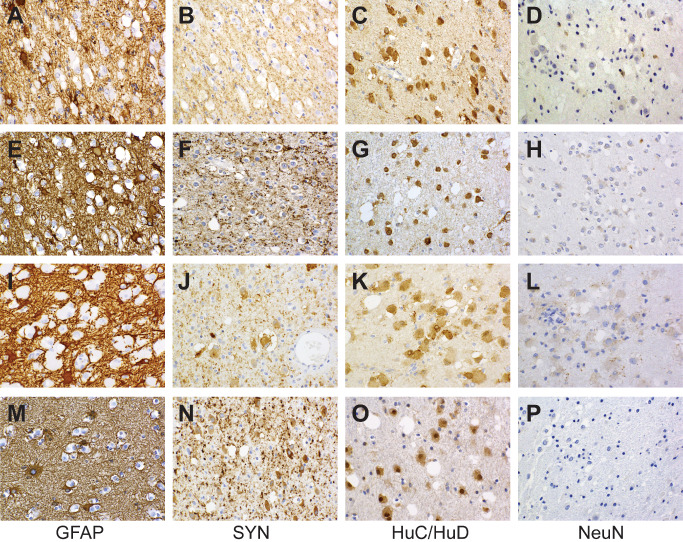
Immunohistochemical features of the cases illustrated in Figure 3 are shown (case 4: A–D; case 5: E–H; case 6: I–L; case 1: M–P). Regardless of cytologic presentation, shared immunophenotypic features include GFAP‐negativity (only the cell bodies and processes of reactive astrocytes are labeled) and nuclear as well as cytoplasmic expression of HuC/HuD in the absence of nuclear of NeuN labeling. Cytoplasmic reactivity for synaptophysin ranges from absent (B) to conspicuous (J, N) without surface perikaryal accentuation.
Figure 6.
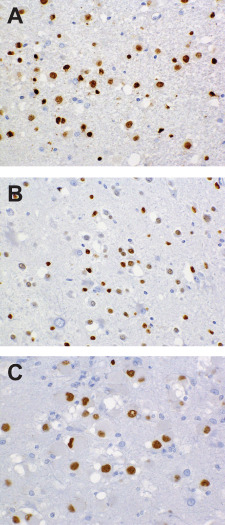
Nuclear OLIG2 expression, a feature of all examples studied, is depicted in fields from cases 1 (A), 5 (B) and 6 (C).
Figure 7.
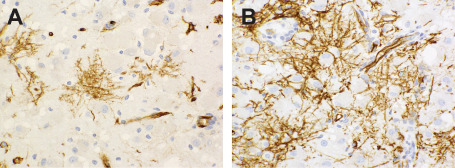
Ramified, CD34‐labeling neural elements are depicted from cases 5 (A) and 6 (B). Tumor cells are non‐reactive.
Molecular genetic findings
Array CGH analysis of cases 5 and 8 did not demonstrate obvious evidence of copy number variations. Sequenom‐based mutational analysis for the eight gene panel listed under Material and Methods demonstrated a point mutation involving MEK 1 (MAP 2K1) p. Q56P (c.167A>C) in the tumor sample of patient 5. BRAF v600E mutations were not detected in cases 1, 5, 6 or 8.
Discussion
The cases presented in this report are examples of a benign and sporadically occurring lesion that could be regarded as a form of gangliocytoma, but one that we have not found to be clearly delineated in the literature pertaining specifically to tumors of the ganglion cell group 2, 7, 11, 17, 24, 28, 36, 37, 39 or in major series detailing the surgical pathology of chronic seizure disorders 26, 27, 38. That these merit specific illustration and comment is underscored by the fact that most were collected from the consultation files of the senior author (MKR), having occasioned uncertainly as to their appropriate classification. The ganglion cell tumor series is heavily dominated by gangliogliomas, purely neuronal representatives being much the less common species. The cases collected here constitute the majority of benign, purely neuronal and non‐neurocytic tumors encountered in the senior author's consultation experience, which includes regular referrals from clinical practices specializing in the surgical management of epilepsy in children and adults.
The features complicating proper classification of these peculiar growths included their ambiguous cytologic profiles and the failure of those neuronal “immunomarkers” in widest employ to consistently label their constituent cellular elements. Gangliocytoma would be a misnomer as applied to the majority of these lesions inasmuch as only two manifested overtly ganglion cell‐type morphology, the remainder being composed of more modestly sized forms exhibiting a degree of cytoplasmic eosinophilia raising question of an astroglial neoplasm. Furthermore, nearly all examples became symptomatic in adulthood (unlike the great majority of conventional ganglion cell tumors) and none evidenced those commonly accompanying phenomena (eg, eosinophilic granular body formation, conspicuous lymphoplasmacellular infiltration and microcalcification) that necessarily prompt consideration of a ganglion cell tumor in differential diagnosis. Vacuolar alterations, on the other hand, are not uncommon in the neuron‐rich components of conventional ganglion cell tumors. As regards immunophenotypes, neither aberrant cell membrane labeling for synaptophysin nor diffuse cytoplasmic chromogranin expression—attributes facilitating the identification of neoplastic ganglion cells 15, 24 —were encountered in our material. In fact, synaptophysin expression was not a constant feature of the tumors in this series and we could not demonstrate reactivity for chromogranin, neurofilament proteins or NeuN in any of our assessed cases. Immunolabeling for these neuron‐associated proteins is not, however, a uniform attribute of ganglion cell tumors either 15, 29.
Of those antibodies that we employed to screen for evidence of neuronal differentiation, only the anti‐HuC/HuD reagent produced consistent labeling of tumor cells. Belonging to a family of highly conserved proteins that affect aspects of post‐transcriptional RNA metabolism from splicing to translation 14, HuC and HuD are relatively neuron‐restricted in their normal cellular distributions and play integral roles in neuronal differentiation and maintenance 8, 10, 25. The immunoexpression of HuC/HuD—associated epitopes has emerged from various studies as a sensitive and reasonably specific indicator of neuronal differentiation within tumors of the human central nervous system 13, 18, our findings clearly placing the lesions under discussion within the neuronal series. A state of maldevelopmental arrest may be reflected in the failure of these to regularly co‐express other antigens associated with neuronal maturation, animal studies indicating that Hu protein expression is a particularly early index of commitment to the neuronal lineage and a phenomenon predating the elaboration of neurofilament proteins and other neuron‐associated products 1, 22. In this connection, it is noteworthy that work in avians has documented the selective expression of these Hu family members by primitive cell populations specifically destined to neurogenesis 1, 21, 22. A developmental disturbance might also account for the consistent co‐expression of OLIG2 by cells exhibiting immunoreactivity for the neuron‐association Hu antigens and, in some cases, synaptophysin as well. A regulator of cell fate in the developing central nervous system, OLIG2 is normally expressed by progenitor elements in the ventral neuroectoderm that spawn both oligodendrocytic and select neuronal populations 19, 31. Particularly germane to our study may be the observation that OLIG2 is expressed in the human brain by ventricular and subventricular zone progenitor cells of the ganglionic eminences (and diencephalon) from which certain classes of interneurons, as well as oligodendroglia, derive 19. In the course of normal differentiation, however, OLIG2 expression persists only in oligodendrocytes, being rapidly downregulated in the neuronal progeny of these precursors and not detected in mature neurons 19. We should also note that some investigators have reported immunolabeling for OLIG2 in central neurocytomas, which are patently neuronal neoplasms, as well as medulloblastomas, tumors clearly disposed to neuronal differentiation 19.
Raising further question of dysregulated development playing a role in the pathogenesis of these unusual growths is their association with cortical disorganization, microhamartomas and, in six of nine cases, aberrantly CD34‐expressing neural elements of ramified type. The last, not visualized to date in the normal human brain, have been found in complex with glioneuronal hamartia and some forms of cortical dysplasia 3, 9, frequently colonize the cerebral cortex adjoining pediatric gangliogliomas 3, 4, 5, 9 and pleomorphic xanthoastrocytomas 9, 30, and have been detected by some observers in association with dysembryoplastic neuroepithelial tumors as well 35. The observation of transient CD34 expression in the embryonic murine neural tube 6, 20 has prompted speculation that these cells represent developmentally arrested or abnormally differentiated precursor elements that are, at the least, a testament to regional dysontogenesis if not the progenitors of the tumors with which they are associated. In the latter connection, we would point out that the demonstrably neuronal elements constituting the lesions we describe did not share the CD34 expression displayed by this ramified population. Furthermore, the decided over‐representation of adult‐onset cases encountered in this series would not be anticipated of a “developmental” tumor, although further studies could show children and adolescents to be more significantly represented among affected patients. Why CD34‐expressing cells of a type strongly associated with epileptogenic lesions of the young were apparent in a majority of our cases remains unclear. Whether the tumors included in this report represent bona fide neoplasms or are more dysplastic or hamartomatous in nature is debatable. We emphasize that the designation “tumor” is employed in this report in the noncommittal sense of a space‐occupying abnormality (as it was used, for example, in naming the dysembryoplastic neuroepithelial tumor) and is not meant to endorse a pathogenesis involving cellular transformation. The subset of cases specifically assessed for molecular genetic abnormalities did not manifest copy number variations or the BRAF v600E‐mutant profile that characterizes a minor group of conventional gangliogliomas 32.
We conclude with additional remarks pertaining to the neuroradiologic identification, differential diagnosis, and clinical management of the unusual entity addressed in this report. While definitive diagnosis requires tissue examination, particularly suggestive on MRI (albeit inconstant) is a multinodular pattern of increased signal, best appreciated in FLAIR sequences but potentially apparent in T2‐weighted studies, within a superficially positioned and non‐enhancing cerebral lesion producing, at most, minimal expansion of the involved neuroparenchyma. This profile, foreign to the large run of conventional gliomas and gangliogliomas, may be shared, however, by the dysembryoplastic neuroepithelial tumor (which also exhibits a predilection for the temporal lobes but typically presents in childhood or adolescence). Distinction of these entities at the light microscopic level should not be problematic. Again (see Histologic Findings), the “specific glioneuronal element” that characterizes the dysembryoplastic neuroepithelial tumor was not identified as a component of any of the lesions reported here. Specifically, oligodendrocyte‐like clear cells in columnar (or, for that matter, any other) array, accumulations of myxoid matrix material containing “floating” neurons, and patterned, alveolar nodules were uniformly absent. Conversely, we have not encountered within dysembryoplastic neuroepithelial tumors vacuolated micronodules populated by cells having the cytologic and immunohistochemical profiles on display in our cases. Nodular neuronal heterotopias, another potential consideration, typically exhibit the MRI signal characteristics of normal grey matter, but can occasionally appear T2/FLAIR‐hyperintense 33. These, however, are composed of highly differentiated neuronal elements that closely resemble their counterparts in the native cortex and that retain, in further contrast to the abnormal cells described in this report, the NeuN expression characterizing cortical neurons of all classes 23. As regards treatment, our experience to date indicates that the neuronal tumors annotated in this study are benign and that gross total or substantial resection not only suffices for disease control, but is likely to alleviate lesion‐associated neurologic manifestations.
Disclosures
The authors have no disclosures or conflicts of interest.
Acknowledgments
We would like to thank Miriam Fayad for her technical assistance.
References
- 1. Barami K, Iversen K, Furneaux H, Goldman SA (1995) Hu protein as an early marker of neuronal phenotypic differentiation by subependymal zone cells of the adult songbird forebrain. J Neurobiol 28:82–101. [DOI] [PubMed] [Google Scholar]
- 2. Becker AJ, Wiestler OD, Figarella‐Branger D, Blumcke I (2007) Ganglioglioma and gangliocytoma. In: WHO Classification of Tumours of the Central Nervous System, Louis DN, Ohgaki H, Wiestler OD, Cavenee WK (eds), pp. 103–105. IARC: Lyon. [Google Scholar]
- 3. Blumcke I, Giencke K, Wardelmann E, Beyenburg S, Kral T, Sarioglu N et al (1999) The CD34 epitope is expressed in neoplastic and malformative lesions associated with chronic, focal epilepsies. Acta Neuropathol 97:481–490. [DOI] [PubMed] [Google Scholar]
- 4. Blumcke I, Lobach M, Wolf HK, Wiestler OD (1999) Evidence for developmental precursor lesions in epilepsy‐associated glioneuronal tumors. Microsc Res Tech 46:53–58. [DOI] [PubMed] [Google Scholar]
- 5. Blumcke I, Wiestler OD (2002) Gangliogliomas: an intriguing tumor entity associated with focal epilepsies. J Neuropathol Exp Neurol 61:575–584. [DOI] [PubMed] [Google Scholar]
- 6. Brown J, Greaves MF, Molgaard HV (1991) The gene encoding the stem cell antigen, CD34, is conserved in mouse and expressed in haemopoietic progenitor cell lines, brain, and embryonic fibroblasts. Int Immunol 3:175–184. [DOI] [PubMed] [Google Scholar]
- 7. Burger PC, Scheithauer BW (2007) Tumors of the central nervous system. AFIP atlas of tumor pathology, fourth series, fascicle 7. Am Regist Pathol 209–218. [Google Scholar]
- 8. Dalmau J, Furneaux HM, Cordon‐Cardo C, Posner JB (1992) The expression of the Hu (paraneoplastic encephalomyelitis/sensory neuronopathy) antigen in human normal and tumor tissues. Am J Pathol 141:881–886. [PMC free article] [PubMed] [Google Scholar]
- 9. Deb P, Sharma MC, Tripathi M, Sarat Chandra P, Gupta A, Sarkar C (2006) Expression of CD34 as a novel marker for glioneuronal lesions associated with chronic intractable epilepsy. Neuropathol Appl Neurobiol 32:461–468. [DOI] [PubMed] [Google Scholar]
- 10. Deschenes‐Furry J, Perrone‐Bizzozero N, Jasmin BJ (2006) The RNA‐binding protein HuD: a regulator of neuronal differentiation, maintenance and plasticity. BioEssays 28:822–833. [DOI] [PubMed] [Google Scholar]
- 11. Felix I, Bilbao JM, Asa SL, Tyndel F, Kovacs K, Becker LE (1994) Cerebral and cerebellar gangliocytomas: a morphological study of nine cases. Acta Neuropathol 88:246–251. [DOI] [PubMed] [Google Scholar]
- 12. Gilbert MT, Haselkorn T, Bunce M, Sanchez JJ, Lucas SB, Jewell LD et al (2007) The isolation of nucleic acids from fixed, paraffin‐embedded tissues‐which methods are useful when? PLoS ONE 2:e537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gultekin SH, Dalmau J, Graus Y, Posner JB, Rosenblum MK (1998) Anti‐Hu immunolabeling as an index of neuronal differentiation in human brain tumors: a study of 112 central neuroepithelial neoplasms. Am J Surg Pathol 22:195–200. [DOI] [PubMed] [Google Scholar]
- 14. Hinman MN, Lou H (2008) Diverse molecular functions of Hu proteins. Cell Mol Life Sci 65:3168–3181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hirose T, Scheithauer BW, Lopes MB, Gerber HA, Altermatt HJ, Vandenberg SR (1997) Ganglioglioma: an ultrastructural and immunohistochemical study. Cancer 79:989–1003. [PubMed] [Google Scholar]
- 16. Jurinke C, van den Oeth P, Boom D (2004) MALDI‐TOF mass spectrometry: a versatile tool for high‐performance DNA analysis. Mol Biotechnol 26:147–164. [DOI] [PubMed] [Google Scholar]
- 17. Krouwer HG, Davis RL, McDermott MW, Hoshino T, Prados MD (1993) Gangliogliomas: a clinicopathological study of 25 cases and review of the literature. J Neurooncol 17:139–154. [DOI] [PubMed] [Google Scholar]
- 18. Laeng RH, Scheithauer BW, Altermatt HJ (1998) Anti‐neuronal nuclear autoantibodies, types 1 and 2: their utility in the study of tumors of the nervous system. Acta Neuropathol 96:329–339. [DOI] [PubMed] [Google Scholar]
- 19. Ligon KL, Alberta JA, Kho AT, Weiss J, Kwaan MR, Nutt CL et al (2004) The oligodendroglial lineage marker OLIG2 is universally expressed in diffuse gliomas. J Neuropath Exp Neurol 63:499–509. [DOI] [PubMed] [Google Scholar]
- 20. Lin G, Finger E, Gutierrez‐Ramos JC (1995) Expression of CD34 in endothelial cells, hematopoietic progenitors and nervous cells in fetal and adult mouse tissues. Eur J Immunol 25:1508–1516. [DOI] [PubMed] [Google Scholar]
- 21. Marusich MF, Furneaux HM, Henion PD, Weston JA (1994) Hu neuronal proteins are expressed in proliferating neurogenic cells. J Neurobiol 25:143–155. [DOI] [PubMed] [Google Scholar]
- 22. Marusich MF, Weston JA (1992) Identification of early neurogenic cells in the neural crest lineage. Dev Biol 149:295–306. [DOI] [PubMed] [Google Scholar]
- 23. Meroni A, Galli C, Bramerio M, Tassi L, Colombo N, Cossu M et al (2009) Nodular heterotopia: a neuropathological study of 24 patients undergoing surgery for drug‐resistant epilepsy. Epilepsia 50:116–124. [DOI] [PubMed] [Google Scholar]
- 24. Miller DC, Lang FF, Epstein FJ (1993) Central nervous system gangliogliomas. Part 1: pathology. J Neurosurg 79:859–866. [DOI] [PubMed] [Google Scholar]
- 25. Pascale A, Amadio M, Quattrone A (2008) Defining a neuron: neuronal ELAV proteins. Cell Mol Life Sci 65:128–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Pasquier B, Peoc HM, Fabre‐Bocquentin B, Bensaadi L, Pasquier D, Hoffmann D et al (2002) Surgical pathology of drug‐resistant partial epilepsy. A 10‐year‐experience with a series of 327 consecutive resections. Epileptic Disord 4:99–119. [PubMed] [Google Scholar]
- 27. Plate KH, Wieser HG, Yasargil MG, Wiestler OD (1993) Neuropathological findings in 224 patients with temporal lobe epilepsy. Acta Neuropathol 86:433–438. [DOI] [PubMed] [Google Scholar]
- 28. Prayson RA (2006) Ganglion cell tumors: gangliocytoma, ganglioglioma and anaplastic ganglioglioma. In: Russell and Rubinstein's Pathology of Tumors of the Nervous System, 7th edn. McLendon RE, Rosenblum MK, Bigner DD (eds), pp. 305–320. Hodder Arnold: London. [Google Scholar]
- 29. Preusser M, Laggner U, Haberler C, Heinzl H, Budka H, Hainfellner JA (2006) Comparative analysis of NeuN immunoreactivity in primary brain tumours: conclusions for rational use in diagnostic histopathology. Histopathology 48:438–444. [DOI] [PubMed] [Google Scholar]
- 30. Reifenberger G, Kaulich K, Wiestler OD, Blumcke I (2003) Expression of the CD34 antigen in pleomorphic xanthoastrocytomas. Acta Neuropathol 105:358–364. [DOI] [PubMed] [Google Scholar]
- 31. Rowitch DH, Lu R, Kessaris N, Richardson WD (2002) An “oligarchy” rules neural development. Trends Neurosci 25:417–422. [DOI] [PubMed] [Google Scholar]
- 32. Schindler G, Capper D, Meyer J, Janzarik W, Omran H, Herold‐Mende C et al (2011) Analysis of BRAF V600E mutation in 1,320 nervous system tumors reveals high mutation frequencies in pleomorphic xanthoastrocytoma, ganglioglioma and extra‐cerebellar pilocytic astrocytoma. Acta Neuropathol 121:397–405. [DOI] [PubMed] [Google Scholar]
- 33. Tassi L, Colombo N, Cossu M, Mai R, Francione S, Lo Russo G et al (2005) Electroclinical, MRI and neuropathological study of 10 patients with nodular heterotopia, with surgical outcomes. Brain 128:321–337. [DOI] [PubMed] [Google Scholar]
- 34. Taylor BS, Barretina J, Socci ND, DeCarolis P, Ladanyi M, Meyerson M et al (2008) Functional copy‐number alterations in cancer. Plos ONE 3:e3179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Thom M, Toma A, An S, Martinian L, Hadjivassiliou G, Ratilal B et al (2011) One hundred and one dysembryoplastic neuroepithelial tumors: an adult epilepsy series with immunohistochemical, molecular genetic, and clinical correlations and a review of the literature. J Neuropathol Exp Neurol 70:859–878. [DOI] [PubMed] [Google Scholar]
- 36. Wolf HK, Campos MG, Zentner J, Hufnagel A, Schramn J, Elger CE, Wiestler OD (1993) Surgical pathology of temporal lobe epilepsy. Experience with 216 cases. J Neuropathol Exp Neurol 52:499–506. [DOI] [PubMed] [Google Scholar]
- 37. Wolf HK, Muller MB, Spanle M, Zentner J, Schramm J, Wiestler OD (1994) Ganglioglioma: a detailed histopathological and immunohistochemical analysis of 61 cases. Acta Neuropathol 88:166–173. [DOI] [PubMed] [Google Scholar]
- 38. Wolf HK, Zentner J, Hufnagel A, Campos MG, Schramm J, Elger CE, Wiestler OD (1993) Surgical pathology of chronic epileptic seizure disorders: experience with 63 specimens from extratemporal corticectomies, lobectomies and functional hemispherectomies. Acta Neuropathol 86:466–472. [DOI] [PubMed] [Google Scholar]
- 39. Zentner J, Wolf HK, Ostertun B, Hufnagel A, Campos MG, Solymosi L, Schramm J (1994) Gangliogliomas: clinical, radiological, and histopathological findings in 51 patients. J Neurol Neurosurg Psychiatry 57:1497–1502. [DOI] [PMC free article] [PubMed] [Google Scholar]