

Abstract
Models of ischemic brain injury in the nonhuman primate (NHP) are advantageous for investigating mechanisms of central nervous system (CNS) injuries and testing of new therapeutic strategies. However, issues of reproducibility and survivability persist in NHP models of CNS injuries. Furthermore, there are currently no pediatric NHP models of ischemic brain injury. Therefore, we have developed a NHP model of cortical focal ischemia that is highly reproducible throughout life to enable better understanding of downstream consequences of injury. Posterior cerebral arterial occlusion was induced through intracortical injections of endothelin‐1 in adult (n = 5) and neonatal (n = 3) marmosets, followed by magnetic resonance imaging (MRI), histology and immunohistochemistry. MRI revealed tissue hyperintensity at the lesion site at 1–7 days followed by isointensity at 14–21 days. Peripheral macrophage and serum albumin infiltration was detected at 1 day, persisting at 21 days. The proportional loss of total V1 as a result of infarction was consistent in adults and neonates. Minor hemorrhagic transformation was detected at 21 days at the lesion core, while neovascularization was detected in neonates, but not in adults. We have developed a highly reproducible and survivable model of focal ischemia in the adult and neonatal marmoset primary visual cortex, demonstrating similar downstream anatomical and cellular pathology to those observed in post‐ischemic humans.
Keywords: cerebral ischemia, endothelin‐1, MRI, neural degeneration, primary visual cortex, primate
Introduction
While the results from rodent models of central nervous system (CNS) injury such as stroke are very encouraging, the majority of experimental neuroprotective therapies discovered and trialed in rodents are not translated to clinical use 19. All too often, promising results from studies undertaken in rodent stroke models are not realized when applied clinically 53, 72. Investigating the consequential pathophysiology and testing of novel therapeutics in nonhuman primate (NHP) models of cerebral ischemia may provide better physiological and temporal representation to the human brain after CNS injuries.
The majority of transient and permanent arterial occlusions are performed on rodents and directed at the middle cerebral artery (MCA). In particular, the use of the vasoconstrictor endothelin‐1 (ET‐1) 57 was successfully used to induce MCA occlusion (MCAO) resulting in downstream ischemic injury in rodents. An advantage of this method is the minimal invasiveness of the technique and the potential for discrete ischemic lesions to be generated in neocortical targets 21. However, these rodent models of ET–1‐induced MCAO often result in relatively large zones of infarct, often invading subcortical and thalamic regions. The sequelae of these models often range from impaired sensorimotor capacity to death. Furthermore, the large infarct zones observed in most rodent and some NHP MCAO models are not representative of survivable strokes in humans, raising the issue of translatable clinical relevance. Focal MCAO, targeting distal branches in NHPs, has been attempted 30 with variable infarct size and severity. The high prevalence of pediatric strokes 33 has also prompted the development of animal models of peri‐/neonatal strokes in rodents 51, 67 for the purpose of identifying mechanisms underlying the greater capacity for functional recovery following early‐life injuries. However, NHP models of peri‐/neonatal ischemic strokes have yet to be developed.
While many models have focused on the motor neocortex, this study targeted the primary visual cortex (striate cortex; V1) of the NHP as the locus for the development of cortical ischemic injury. The primate's, including human's, visual cortex occupies 40%–50% of the neocortex and the circuitry of the primate visual system is also well characterized, and the topography is highly organized, unlike the rodent's. The visual cortex is also susceptible to cortical ischemia, through occlusion of the posterior cerebral artery (PCA), leading to cortical blindness in both neonates and adults 1, 22. These specific features afford the capacity to study the precise anatomical, connectional and functional consequences of focal injuries. For example, lesions of the visual cortex in NHP models have been used to precisely demonstrate both physiological and behavioral outcomes, as well as accurately map the extent of neuroplasticity following an injury (eg, 13, 14, 23, 34, 61, 70, 71, 80, 83), and can be linked to behavioral and functional outcomes such as those observed in the clinic. These specific features have certain advantages over lesions of the motor cortex, in which various NHPs have been extensively employed (eg, 28, 30, 47, 77, 81), where it can be difficult to determine transformations up‐ and downstream of the lesion site. The limited behavioral consequences associated with discrete lesions of V1, scotoma/“blind spot” in the contralateral visual field, ensure that the animal can still ambulate normally and does not require significant human intervention. This alone has specific ethical advantages.
The New‐World marmoset monkey (Callithrix jacchus) is a NHP with a visual system that has been extensively mapped in the in both adult and neonate 10, 59. The species shares similarities to humans in regards to its brain development, neocortical organization, retinotopy and connectivity (Figure 1A; 25, 59), and overall vascular anatomy 12, 27. These features provide the opportunity to define the effects of a lesion throughout the entire visual system (Figure 1A). The relatively small size of the species also makes it a popular model in the laboratory, with the ability to study larger cohorts required for preclinical testing of pharmacotherapeutics and better capacity for in vivo optical or magnetic resonance imaging (MRI) techniques in small‐bore, high Tesla systems.
Figure 1.

Intracortical injection of endothelin‐1 ( ET ‐1) proximal to the calcarine artery results in transient arterial occlusion. (A) Schematic illustration of the marmoset visual system. Visual representation in operculum V1 (red) corresponds to 0–3° (foveal) of the contralateral visual hemifield. (B) Schematic representation of the adult marmoset cranium in lateral and dorsal views. Craniotomies were created on the occipital pole of the cranium (shaded grey). The base of the craniotomy extends parallel and ∼2 mm lateral to the midline, from interaural −7 mm to −12 mm. The apex lies ∼7 mm lateral to the midline. (C) Red arrow denotes the calcarine artery (P4 branch of the posterior cerebral artery, PCAca) on the occipital pole of the marmoset brain. Dashed line outlines approximate operculum V1 area. (D–F). Time‐lapsed images demonstrating the PCAca before ET‐1 injections (D), ongoing arterial occlusion (E) and subsequent reperfusion (F). *Denotes injection sites. Empty arrows denote sites of occlusion and black arrows denote corresponding regions during reperfusion. (G) Anatomical overview of the occipital pole at 21 days post ischemia (DPI) after focal ischemia revealed a discrete lesion site localized to the occipital pole of the marmoset brain (outlined by dotted line) consistent with operculum V1. Orientation markers: D = dorsal; V = ventral; R = rostral; C = caudal; L = lateral; M = medial. Scale: (B) 5 mm, (C, G) 2.5 mm.
In this study, we developed a model in which focal ischemic injury was induced in V1 of the adult and neonate marmoset monkey through ET–1‐mediated vasoconstriction 9 of the calcarine artery (PCAca; Figure 1C). This paradigm induces significant occlusion of the targeted blood vessel. Previous studies with this vasoconstrictor have demonstrated that it accurately models the histopathological sequelae of stroke accurately in the rodent and primate. Our anatomical analyses of the neonate and adult post‐ischemic marmoset V1 demonstrate that the downstream pathologies bear similarities to pathophysiological consequences of focal stroke in humans.
Methods
Animals
Eight marmoset monkeys (C. jacchus) aged postnatal day (PD) 14 (n = 3) and adults (>18 months; n = 5) were used in this project. Gender was not a criterion in the selection of animals, and no siblings were used. Experiments were conducted according to the Australian Code of Practice for the Care and Use of Animals for Scientific Purposes and were approved by the Monash University Animal Ethics Committee. Animals were obtained and housed at the National Nonhuman Primate Breeding and Research Facility (Monash University).
Surgery and noninvasive imaging
Anesthesia was induced with alfaxalone (8 mg/kg) intramuscular (IM) (adults) or 2% inspired isoflurane (neonates) and maintained with inspired isoflurane (∼2%). The animal's head was shaved and swabbed with a topical antibiotic solution (Betadine; Sanofi, Victoria, Australia). Adults were administered a broad‐spectrum antibiotic (Norocilin; Norbrook, Lenexa, KS, USA; IM; 27 mg/kg, 0.1 mL) and dexamethasone (IM; 0.3 mg/kg) to prevent cerebral edema, then secured in a stereotaxic frame. Physiological conditions were constantly monitored using a pulse oximeter and temperature probe and kept within strict parameters. Following a skin incision along the midline of the cranium, a triangular craniotomy was created over the occipital pole using a burr drill with a 1‐mm ophthalmic drill bit. The base of the craniotomy (interaural: −7 to −12 mm) situated parallel and ∼2 mm lateral to the midline with the apex extending ∼5 mm laterally (Figure 1B). This was followed by dural thinning, with the aid of a diamond knife, to facilitate microsyringe penetration.
Once the target area was visualized, a 0.5‐mL capacity microsyringe (SGE; Analytical Sciences, Victoria, Australia) tipped with a custom‐pulled glass tip (∼80–90‐μm tip diameter) was positioned proximal to the calcarine artery (PCAca), originating from the P4 superficial segment of the posterior cerebral artery (PCA), to a depth of ≤200 μm (Figure 1C,D). Intracortical injections were performed in doses of 0.1 μL/30 s pulse at 30‐s intervals to administer ∼0.5 μL (neonates); ∼0.7 μL (adults) of ET‐1 1 μg/μL (Sigma‐Aldrich, St Louis, MO, USA). Post‐injection, the needle remained in situ for a further 2 minutes before withdrawal. Intracortical ET‐1 injections surrounding the PCAca were performed over four sites in neonates and ≤7 sites in adults. To determine the extent of ET–1‐induced ischemic injury, continuous video monitoring of cortical surface was performed before, during and after ET‐1 administration. Upon completion (1–2 h post‐injection), the exposed cortical surface was covered with a piece of soluble ophthalmic film (gelfilm; Pharmacia & Upjohn, Bridgewater, NJ, USA) presoaked in saline and the resected dura mater replaced over the film. The craniotomy was replaced and secured with tissue adhesive (vetbond; 3 M, Chelmsford, MA, USA) and the skin sutured closed.
Dextran labeling of peripheral macrophages
Peripheral macrophage dextran loading was performed to allow downstream differentiation of infiltrating vs. local macrophage populations after focal ischemia. Peripheral macrophages were preloaded with blood–brain barrier (BBB)‐impermeable high molecular weight (hMW) tetramethylrhodamine‐labeled dextran (5 mg/mL; 1.66 mg/kg; 2 000 000 MW; Invitrogen, Carlsbad, CA, USA), which is taken up through endocytic processes. Animals undergoing dextran administration were anesthetized (as mentioned earlier) 24 h before surgery, and a bolus of hMW dextran diluted in 0.5 mL saline was injected into the left saphenous vein.
MRI
T2‐weighted scans were performed at the Monash Biomedical Imaging facility using an echo planar capable, 9.4‐T small animal MRI system (Agilent Technologies, Wokingham, UK) at 1, 7, 14 and 21 days post ischemia (DPI). Animals were anesthetized as outlined earlier (maintained with 1% inspired isoflurane) prior to imaging. Parameters of T2‐weighted imaging acquisition: thickness = 6 mm; repetition time = 3000 ms; echo time = 39.17 ms; echo train length = 8; averages = 8; flip angle = 90°.
Tissue preparation
At the end of the nominated recovery periods (1/21 DPI), animals were administered an overdose of pentobarbitone sodium (100 mg/kg; intraperitoneal). Following apnea, animals were transcardially perfused with warm 0.1 M heparinized phosphate buffer (PB, pH 7.2) containing 0.1% sodium nitrite (adults with heparinized saline), followed by 4% paraformaldehyde. Cerebral tissues were postfixed and cryoprotected as outlined in our previous study 79. Following separation of the hemispheres, each hemisphere was bisected coronally at the start of the caudal pole of the diencephalon. The occipital block, which encompassed V1, was cryosectioned in the parasagittal plane and the remaining block in the coronal plane (40 μm).
Immunohistochemistry (IHC)
Free‐floating sections were treated with 0.3% hydrogen peroxide and 50% methanol in 0.1 M PBS for 20 minutes to inactivate endogenous peroxidases. Sections were pre‐blocked in a solution of 10% normal goat serum in 0.1 M PBS, 0.3% Triton X‐100 (TX; Sigma‐Aldrich) before incubation with primary antibodies overnight at 4°C. Sections were rinsed in PBS‐Tween 0.1% (Sigma‐Aldrich) prior to incubation with biotinylated secondary antibodies for 1 h at room temperature. Sections were then treated with streptavidin‐horseradish peroxidase conjugate (GE Healthcare, Amersham, UK; 1:200) and visualized via a metal‐enhanced chromogen, 3,3′‐diaminobenzidine (Sigma‐Aldrich).
Immunofluorescent antigen detection was conducted with similar procedures, except steps including and after secondary antibody incubation, which were performed in the dark. Following pre‐incubation in blocking solution, sections were incubated overnight at 4°C with primary antibodies in blocking solution. Sections were then rinsed in PBS‐Tween, 0.1% before incubation with secondary antibodies (Table 1). Sections were also treated with Hoechst 333258 and 4′,6‐Diamidino‐2‐phenylindole (DAPI) nuclei stains.
Table 1.
List of primary and secondary antibodies used
| Host | Dilution | Source | |
|---|---|---|---|
| Antibodies (antigen) | |||
| NeuN (neuronal nuclear protein) | Ms | 1:1000 | Millipore |
| GFAP (glial fibrillary acidic protein) | Ms | 1:1000 | Millipore |
| Iba1 (ionized ca‐binding adapter protein1) | Rb | 1:1000 | Wako |
| hSA (human serum albumin) | Gt | 1:1000 | Bethyl |
| aCasp3 (activated caspase‐3) | Rb | 1:250 | Abcam |
| Ki‐67 (Ki‐67) | Rb | 1:500 | Abcam |
| IB4 (isolectin‐IB4) | Ms | 1:500 | Sigma |
| Secondary antibody | |||
| Anti‐Ms biotinylated | Rb | 1:500 | DAKO |
| Anti‐Rb biotinylated | Gt | 1:500 | DAKO |
| Anti‐Gt biotinylated | Rb | 1:500 | Sigma |
| Anti‐Rb Alexa Fluor 488 | Gt | 1:800 | Invitrogen |
| Anti‐Ms Alexa Fluor 594 | Gt | 1:800 | Invitrogen |
Ms = mouse; Gt = goat; Rb = rabbit.
Histology
For Nissl‐substance staining, a series representing the entire brain were mounted on glass slides, air‐dried, defatted overnight in equal‐volume absolute chloroform and ethanol and serially rehydrated in graded ethanol before treating with 0.1% cresyl violet.
For methyl green chromatin staining, air‐dried, slide‐mounted sections were rehydrated in distilled water before treating with 1% methyl green. Sections were dehydrated with 70% w/v ethanol and differentiated in acidified 100% ethanol.
For Perls' Prussian blue iron staining, air‐dried, slide‐mounted sections were similarly rehydrated before treatment with a solution containing equal‐volume 20% hydrochloric acid and 10% potassium ferrocyanide.
Qualitative and quantitative analysis
Standard brightfield and epifluorescent microscopic examination of processed sections were conducted using Zeiss SteREO discovery (V20) and Axio Imager Z1 microscopes (Zeiss, Jena, Germany). Images were obtained with a Zeiss Axiocam (HR rev3) using Axiovision software (V4.8.1.0, Zeiss). All photomicrographs were cropped and resized using Photoshop (CS4 v11.0, Adobe, San Jose, CA, USA) and GNU Image Manipulation Program (GIMP; V2.6.11, open source, www.gimp.org). Necessary manipulation (eg, brightness/contrast and image cropping) was performed to aid in analysis and for improved quality of final images.
Iba1+ and Iba1+/Dextran+ cell counts were performed using ImageJ. Photomicrographs of the infarct core and peri‐infarct area (∼600–800 μm from the core) obtained through a 10× objective were used for the counts (n = 2). Density counts of Iba1+ cells were made using a counting grid: 8 × 104 μm2 (282.85 μm × 282.85 μm) that was superimposed over each image. Counts were made over four grids per photomicrograph with three photomicrographs per area, three sections per animal. Results were expressed as mean cells per 8 × 104 μm2 on a histogram generated using Prism6 (GraphPad, La Jolla, CA, USA). Proportion of Iba1+/Dextran+ cells in each area was determined by counting double‐immunofluorescent positive cells and the results expressed as a percentage of total Iba1+ cell densities in each area, respectively.
V1 volume analyses were performed using ImageJ, utilizing the Cavalieri method 36, on full series Nissl‐stained sections (200‐μm separation) with a grid area of 0.64 mm2. Volume analyses were performed on even numbered sections (400 μm apart) with one replicate on odd numbered sections. Results were corrected for linear shrinkage and expressed as volume (mm3) and percent total of control V1. Statistical analyses were performed on Prism6 using the Mann–Whitney's U‐test (MW) and Welch's t‐test (Wt).
Results
Targeted intracortical injection of ET‐1 results in PCAca occlusion
Intracortical injections of ET‐1, proximal to the PCAca (Figure 1C,D), caused sustained vasoconstriction for up to 4 h, accompanied by vascular stasis and collapse immediately downstream of occlusion sites (Figure 1E) and visible pallor of the surrounding cortical surface. ET–1‐induced vasoconstriction also resulted in blood and fluid extravasation into the surrounding tissue causing localized cerebral edema. Reperfusion, characterized by visible vasodilation and restoration of blood flow usually occurred within 4 h of vessel occlusion (Figure 1F). Post‐perfusion images of whole brains (Figure 1G) revealed a clearly observable lesion core localized to the occipital pole of the ipsilateral marmoset neocortex consistent with operculum V1 (Figure 1A,C).
In vivo MRI post ischemia revealed temporal homology similar to the human neocortex
We profiled the spatiotemporal development of the ischemic core as a result of ET–1‐induced ischemia in the adult marmoset using T2‐weighted imaging at 1, 7, 14 and 21 DPI (Figure 2). The resultant data were compared against human post‐ischemic T2‐weighted MR data at similar time points 2. At 1 DPI, we observed development of an ischemic‐necrotic core at the injection site, characterized by a clear zone of hyperintense tissue relative to the contralateral hemisphere, indicative of cerebral edema. At 7 DPI, clearer demarcation of the necrotic core was observed, typified by a clearly visible void, with ongoing tissue hyperintensity. Between 14 and 21 DPI, a discrete core is discernible and T2 normalization was observed proximally. This was temporally consistent with transient T2 isointensity (“fogging;” 73) observed in humans at 2–3 weeks post ischemia 2, 15, 54, 73. These findings demonstrate that the development of the ischemic‐necrotic core following ET–1‐induced injury in the marmoset develops over approximately 14 days, accompanied by associated edema. Comparison with human T2‐weighted MR data revealed significant pathological time‐course homology between post‐ischemic marmoset and human neocortex.
Figure 2.
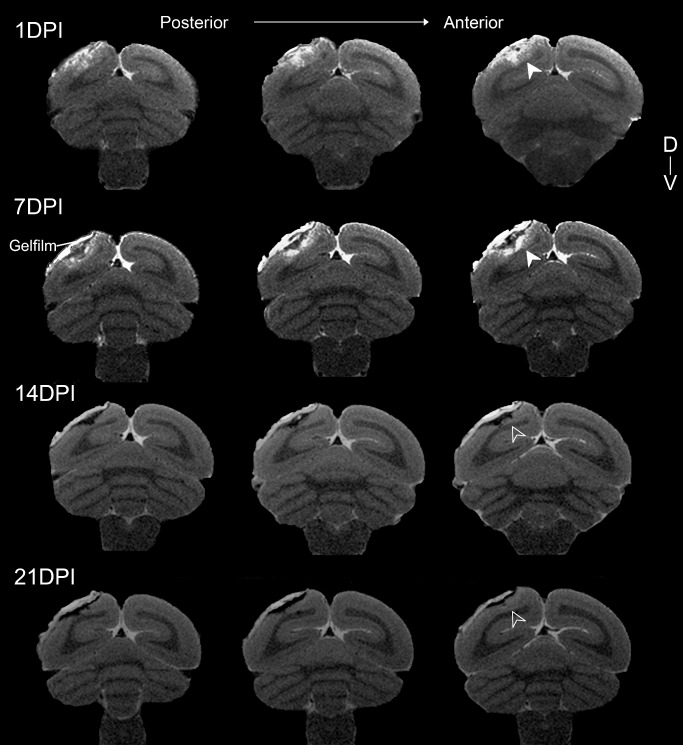
Establishing pathological time‐course of infarct development after focal ischemia through in vivo magnetic resonance imaging. Series of adjacent T2‐weighted images of the post‐ischemic marmoset brain proximal to the infarct site. Slices are 6 mm apart and obtained at 1, 7, 14 and 21 days post ischemia (DPI). T2 hyperintensity was observed at 1, 7 DPI (white arrowheads) indicating the location and extent of injury caused by arterial occlusion. Demarcation of the lesion core was most pronounced at 7 DPI, characterized by T2 hyperintensity, indicative of infarction and localized edema, and a clearly defined void most likely the result of necrotic tissue loss. T2 normalization in previously hyperintense regions was observed at 14 and 21 DPI (empty arrowheads).
BBB disruption and mononuclear immune cell infiltration
We examined BBB integrity in the adult marmoset following focal ischemia by analyzing serum albumin extravasation in the proximity of the lesion core at 1 and 21 DPI. IHC revealed elevated serum albumin immunoreactivity compared with controls at 1 DPI (Figure 3A) suggesting profound but localized vascular leakage. Elevated extravascular serum albumin persisted at 21 DPI (Figure 3A), indicating either delayed serum albumin clearing or sustained BBB pathology.
Figure 3.
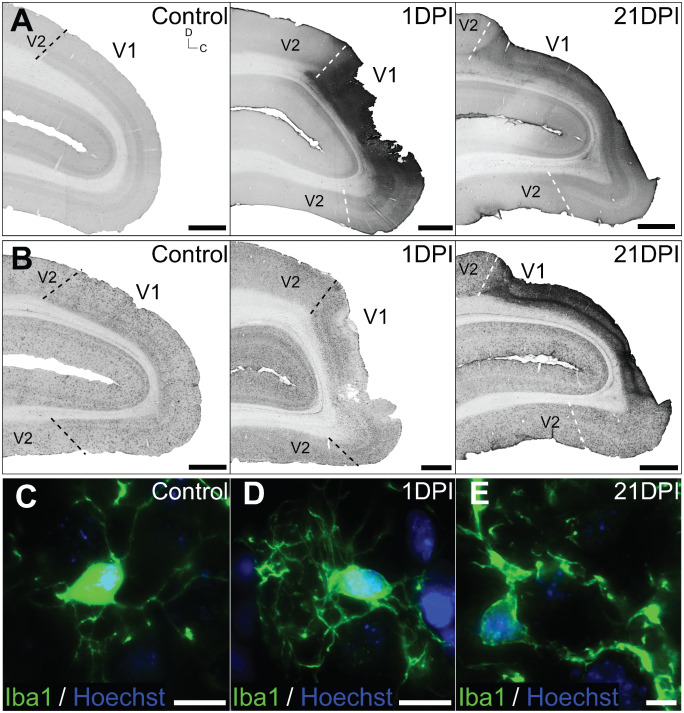
Inflammatory responses and blood–brain barrier ( BBB ) permeability after focal ischemia. Parasagittal sections of control, 1 and 21 days post ischemia (DPI) marmoset V1 immunolabeled for (A) serum albumin and (B) macrophage‐specific Iba1. (A) Serum albumin levels was markedly increased at 1 DPI, compared with controls, and remained elevated at 21 DPI. (B) No obvious difference in Iba1+ macrophage expression intensity or cellular distribution was detected at 1 DPI. However the density of Iba1+ immunolabeling was markedly more intense at 21 DPI, indicating ongoing microgliotic activity. Iba1+ macrophage morphology remained at resting state (C, D; ramified, long and thin processes) at 1 DPI, but was observed to be phagocytic (E; hypertrophic, thick processes) at 21 DPI. Dashed lines denote V1–V2 boundaries demarcated using adjacent Nissl‐stained sections. Scale: (A, B) 2 mm, (C–E) 20 μm.
At 1 DPI, macrophage populations proximal to the lesion site detected using the ionized calcium binding adapter protein1 (Iba1) revealed little to no upregulation (Figure 3B) or change from resting morphology defined by long, ramified processes (Figure 3C,D). At 21 DPI, an abundance of Iba1+ cells were detected adjacent to the ischemic core (Figure 3B) exhibiting hypertrophic, thick processes characteristic of activated (phagocytic) microglia/macrophages (Figure 3E).
We further investigated the acute infiltration of peripheral macrophages through detection of dextran‐loaded macrophages in the post‐ischemic region. hMW dextran does not cross the BBB in normal brains (35; Figure 4A), and is taken up by circulating macrophages through endocytic processes. This allowed for optical differentiation between Iba1+/dextran− resident microglia and Iba1+/dextran+ (dextran‐loaded) infiltrating macrophages subsequent to brain injury. Iba1+/dextran+ cells were detected in the ischemic core at 1 DPI (Figure 4B), indicating that a proportion of macrophages present were of peripheral blood origin. Quantitative data analyses were performed to determine the total macrophage population and proportion of infiltrating vs. local macrophage at 1 DPI. These data revealed a reduction in total macrophage density at the infarct site [∼7.9 cells per 8 × 104 μm2 ± 1.7 cells per 8 × 104 μm2 standard deviation (SD); Figure 4C] compared with controls (∼12.3 cells per 8 × 104 μm2 ± 2.2 cells per 8 × 104 μm2 SD; Figure 4C). Furthermore, we revealed that ∼71.65% of total macrophage present at the infarct core were peripheral infiltrates (Figure 4D). Total macrophage population in the peri‐infarct area remained close to control levels (∼11.8 cells per 8 × 104 μm2 ± 2.1 cells per 8 × 104 μm2 SD; Figure 4C), of which only ∼40.9% were of peripheral blood origin (Figure 4D). These results demonstrate the acute disruption of BBB integrity and infiltration of peripheral macrophage into the infarct and peri‐infarct area after focal ischemia.
Figure 4.
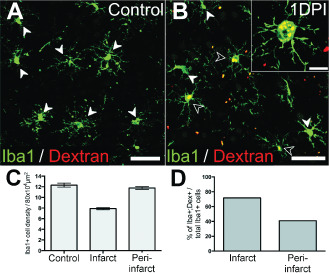
Infiltration of peripheral macrophages in the acute post‐ischemic period. Iba1+/dextran+ peripheral macrophages were detected in brain tissues proximal to lesion site at 1 days post ischemia (DPI) (B; enlarged in insert), but not in controls (A). White arrowheads denote Iba1+/dextran− macrophages. Empty arrowheads denote Iba1+/dextran+ macrophages. (C–D). Quantitative analysis of local and infiltrating macrophages at the infarct and peri‐infarct areas at 1 DPI. Total macrophage populations (Iba1+) was lower at the infarct site compared with peri‐infarct area (600–800 μm distal to core), which remained close to control levels. The percentage of infiltrating (Iba1+/dextran+) over total macrophage (Iba1+) population was greater in the infarct core compared with the peri‐infarct area. Scale: (A, B) 50 μm, (B insert) 20 μm.
Anatomical pathology is established rapidly and localized to operculum V1
To determine the extent of anatomical pathology following ET–1‐induced ischemia in adult marmosets, we performed volumetric analyses on brain tissue by measuring total (residual) V1 (calcarine and operculum; see Figure 1A) using Nissl–substance‐stained sections (Figure 5A) at 1 and 21 DPI. Volume analysis at 1 DPI revealed a statistically significant decrease (∼16%) in total V1 volume (Figures 5B and 7B; 178.16 ± 9.29 mm3 SD) compared with controls (209.92 mm3; P = 0.038; MW). At 21 DPI, no significant decrease in total ipsilateral V1 volume was observed compared with 1 DPI (Figures 5B and 7B; 179.2 mm3 ± 15.4 mm3 SD; P > 0.05; MW). Nissl‐substance histology also revealed that the ischemic injury was focal to operculum V1 only (Figure 5C), without affecting the juxtaposing secondary visual area (V2) and the underlying calcarine V1.
Figure 5.
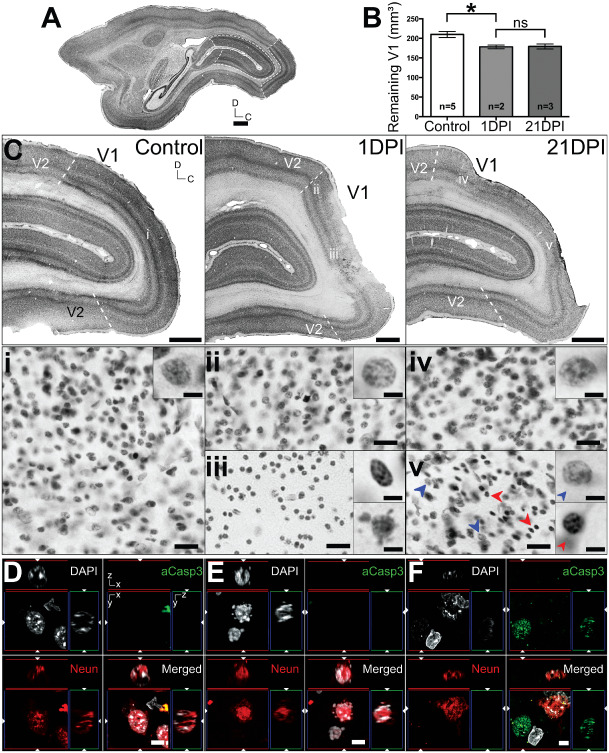
Analysis of lesion extent and neuronal injury after endothelin‐1 ( ET ‐1) induced PCA ca occlusion. (A) Parasagittal Nissl–substance‐stained section of adult marmoset brain. Dashed lines denote the borders of operculum and calcarine V1 used in volume measurement analysis. (B) Volume analysis ± standard error of the mean (SEM) of remaining V1 at 1 and 21 days post ischemia (DPI) compared with controls. *P = 0.038; not significant P > 0.05. (C). Enlarged parasagittal Nissl‐stained sections of control, 1 and 21 DPI adult V1 demonstrating the focal extent of the ischemic injury. Dashed lines in (C) denote the V1–V2 boundary. (i–v) denotes areas on accompanying enlarged regions (adjacent sections; 40 μm apart) visualized using methyl green nuclei histology. Pathological nuclei morphologies were observed in (iii) and (v), characterized by chromatin compaction or nuclear blebbing, compared with normal nuclei observed in (i, ii, iv). (D–F). Immunofluorescent detection of activated caspase‐3 (aCasp3) expression and neuronal nuclei (NeuN)+ neurons in control (D), at 1 DPI (E) and 21 DPI (F). Cellular nuclei visualized using DAPI. Scale: (A) 5 mm, (C) 2 mm, (i–v) 20 μm, (D–F) 5 μm.
Figure 7.
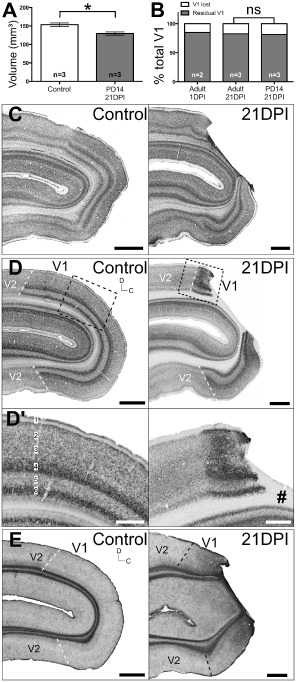
Reproducibility of V 1‐specific focal ischemia in the neonatal marmoset's visual cortex. (A) Volume analysis ± standard error of measurement (SEM) of neonatal V1 at 21 days post ischemia (DPI) compared with controls *P = 0.0152. (B) Proportion of V1 loss as a result of focal ischemia in adults compared with neonates (not significant, P > 0.05). Parasagittal Nissl‐stained sections (C) revealed a similar operculum V1‐specific localization of the lesion at 21 DPI, compared with adults, confirmed using adjacent sections immunolabelled for neuronal nuclei (NeuN) (D). Extent of neuronal degeneration was notably more severe in neonates compared with adults, affecting all six cortical layers indiscriminately (D′). Glial fibrillary acidic proteins (GFAP) immunohistochemistry revealed similar upregulation and cellular morphology at 21 DPI previously observed in adults. #denotes lesion core. Scale: (C, D, E) 2 mm, (D′) 1 mm.
Evidence for necrotic and apoptotic cell death in the lesion core during the acute and subacute periods post ischemia
Morphological evidence of cell death after focal ischemia in adult marmosets was revealed using methyl green histology for chromatin visualization. Presence of cellular pathology was characterized by chromatin compaction and nuclear blebbing at the ischemic core at 1 DPI (Figure 5Ciii) compared with standard nuclear morphology in the peri‐infarct area (Figure 5Cii) and control V1 (Figure 5Ci). At 21 DPI, normal nuclear morphology was detected in operculum V1 regions distal to the core (Figure 5Civ). However, cells exhibiting both normal and pathological nuclear morphologies were detected adjacent to the infarct core (Figure 5Cv). At 1 DPI, fluorescent co‐immunolabeling revealed activated caspase‐3 (aCasp3) independent neuronal death occurring proximal to the lesion core (Figure 5E), characterized by DAPI‐visualized nuclear pathologies (compared with control; Figure 5D) and absence of aCasp3 labeling. This result suggests that necrosis is most probably the primary cause of neuronal death at 1 DPI. At 21 DPI, no necrotic neurons were detected in residual V1. However, the presence of aCasp3+ NeuN− cells were detected proximal to the core (Figure 5F). Taken together, these results suggest that ongoing neuronal necrosis at 1 DPI is completed by 21 DPI. Furthermore, ongoing nonneuronal apoptotic cell death occurs the subacute period following focal ischemia.
Immunohistochemical characterization of neuronal and astrogliotic consequences of the post‐ischemic V1
NeuN IHC performed at 1 and 21 DPI in adult marmosets confirmed that the anatomical and neuronal pathology of ET–1‐induced PCAca occlusion was focal to operculum V1 only (Figure 6A). At 1 DPI, NeuN IHC revealed zones of reduced neuronal density compared with controls, consistent with an acute ischemic‐necrotic core (Figure 6A,A′), confirming previously described neuronal pathology. By 21 DPI, neuronal loss was observed to be most severe in the infarct core, affecting all six cortical layers. Affected V1 regions adjacent to the infarct core experienced less neuronal loss, involving only the supragranular cortical layers (layers 1–3) of operculum V1. Inter—(L4) and subgranular (L5–6) layers of operculum V1 outside the ischemic core remained mostly unaffected (Figure 6A′). This result confirmed the efficacy of using targeted PCAca occlusion to generate operculum V1‐specific focal ischemia.
Figure 6.
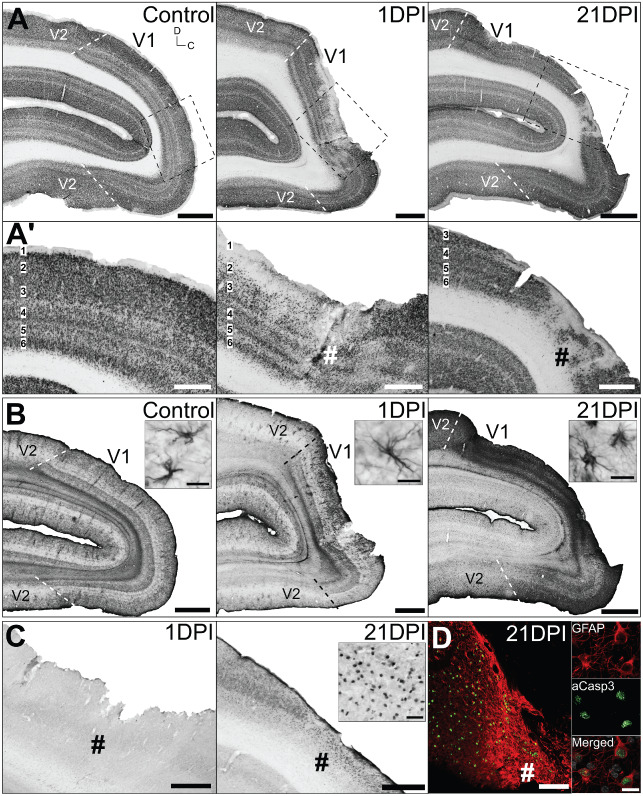
Neuronal and astroglial pathology after focal ischemia was localized to operculum V 1 only. Adjacent parasagittal sections of control, 1 and 21 days post ischemia (DPI) adult V1 immunolabelled for (A, A′) neuronal nuclei (NeuN), (B) astrocyte‐specific glial fibrillary acidic proteins (GFAP) and (C) activated caspase‐3 (aCasp3). Sections were selected where the lesion core was most apparent. Bounding boxes in (A) denotes areas enlarged in (A′, C, D). #denotes lesion core. (B inserts) Demonstrates astrocyte morphologies. (C insert) obtained from lesion core. (D) Co‐immunofluorescent labeling revealed that the majority of aCasp3+ cells detected were GFAP+ reactive astrocytes. Cellular nuclei (white) visualized using Hoechst stain. Scale: (A, B) 2 mm; (A′, C, D) 1 mm; (B insert, D insert) 20 μm.
Glial fibrillary acidic protein (GFAP) IHC for astrocyte detection revealed only slight upregulation proximal to the lesion site at 1 DPI, compared with controls, without obvious changes in overall morphology from resting state (small soma and long, thin processes; Figure 6B), indicating early‐stage astrocyte reactivity. However, profound upregulation of GFAP immunoreactivity within the lesion area was apparent by 21 DP1, most intense at the lesion core, exhibiting characteristic hypertrophic soma and short, thick processes (Figure 6B), indicating ongoing reactive astrocyte activities. Absence of any cellular aCasp3 immunoreactivity at 1 DPI confirmed that acute cell death was most likely necrotic and not apoptotic (Figure 6C). Cellular aCasp3 was detected within the infarct core and proximal peri‐infarct V1 at 21 DPI, confirming sustained apoptotic cell death in the subacute period post ischemia. Double labeling revealed that the majority of aCasp3+ cells detected were GFAP+ reactive astrocytes (Figure 6D). This result confirmed that apoptotic activity detected at 21 DPI was not of neuronal origin and most likely a result of either delayed astroglial death or reactive astrocyte turnover.
ET–1‐induced PCAca occlusion is reproducible in neonatal marmosets and produces an identical injury to adults
The ischemic injury model was recapitulated in neonate marmosets because of a lack of NHP models of peri‐/neonatal strokes, despite the ongoing prevalence in humans. To demonstrate the reproducibility of ischemic injury in the neonatal marmoset V1, we performed identical PCAca occlusion surgeries on PD 14 marmoset monkeys. PD 14 animals were selected because of the relative maturity of V1 at this age 11, which is comparable with 3–5 months in human 4. A 21 DPI recovery period was adopted to allow for time‐matched comparison with adults. Live monitoring ensured an equivalent level of occlusion was achieved in neonates. Volume analysis revealed a statistically significant ∼18% loss of total V1 volume (Figure 7A; 129.71 mm3 ± 10.79 mm3 SD) compared with controls (153.60 mm3 ± 11.43 mm3 SD; P = 0.0152; MW). Furthermore, analysis revealed that neonates experienced a similar loss in total V1 volume compared with adults (Figure 7B; P > 0.05; Wt). Nissl‐substance histology and NeuN IHC revealed similarities in injury extent with adults, affecting only operculum V1, with adjacent V2 and underlying calcarine V1 spared completely (Figure 7C,D). Compared with adults, the extent of neuronal loss was more profound at the infarct core and peri‐infarct area, indiscriminately affecting all six layers of the neocortex (Figure 7D′). With respect to GFAP immunoreactivity, this was more intense at the lesion site compared with controls at 21 DPI in both adults and neonates (Figures 6B and 7E), although the intensity was weaker in neonates compared with adults at 21 DPI. This suggests that the reactive astroglial activity post‐injury may be more potent in adults compared with neonates.
Hemorrhagic transformation after ET‐1‐induced focal ischemia in the marmoset V1
To determine if secondary hemorrhagic transformation is a subacute complication post ischemia in our model, Perls' Prussian blue histology for ferric iron detection was performed on control and post‐ischemic adult and neonatal V1 sections (Figure 8A,B). An adult rat brain that underwent a simple stab wound injury to the cortex was used as a positive control (Figure 8C). No hemorrhage was detected in the adult at 1 DPI (Figure 7A), suggesting that blood extravasation observed during the lesion surgery (Figure 1F) was most probably superficial and not intracerebral. Ferric iron was detected in cerebral tissue in the infarct core at 21 DPI in adults and neonates, suggesting that secondary hemorrhagic transformation occurred subacutely. However, the extent of hemorrhage was small and localized to a restricted region within the central infarct core at both ages (Figure 8A,B), compared with the more widespread and intense ferric iron staining observed in the stab wound‐positive control (Figure 8C). This result suggests that both adult and neonates experience minor secondary punctate hemorrhages in the subacute period post ischemia.
Figure 8.
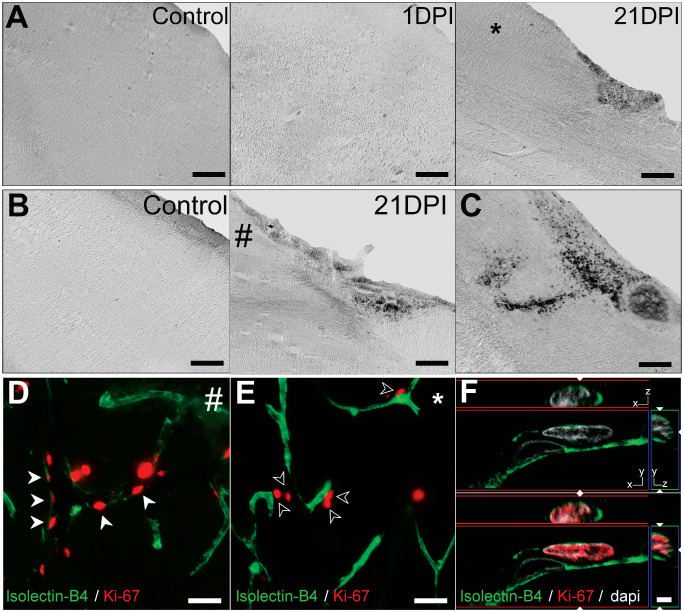
Hemorrhagic transformation and neovascularization after neonatal and adult focal ischemia. (A, B) Differential interference contrast (DIC) photomicrographs of Perls' Prussian blue stained sections corresponding the lesion core shown in Figures 5A′ and 6D′. No ferric iron staining was apparent in adult and neonatal control or at 1 days post ischemia (DPI). At 21 DPI, small punctate‐like regions of ferric iron staining was detected, localized to the central lesion core in both neonates and adults. (C) Positive control of a rat stab wound lesion demonstrating large region of intracerebral hemorrhage. # and * denotes regions in (D) and (E), respectively. Isolectin‐B4 immunolabeled vascular endothelial cells positive for the proliferation marker Ki‐67 were observed 21 days after neonatal (D; red arrowheads) but not adult (E; empty arrowheads) focal ischemia. Enlarged example of IB4+/Ki‐67 cells with orthogonal views shown in (F). Scale: (A, B) 0.5 mm, (D, E) 25 μm, (F) 2.5 μm.
Neovascularization after focal ischemia in the neonate and adult marmoset V1
We further investigated the possibility of subacute neovascularization in the adult and neonate residual V1. Fluorescent co‐immunolabeling was performed using vascular endothelial cell marker isolectin‐B4 (IB4) and Ki‐67 antigen detections. Large populations of Ki‐67+/IB4+ vascular endothelial cells were detected in the neonatal residual V1 at 21 DPI (Figure 8D,F). No Ki‐67+/IB4+ cells were detected in the post‐ischemic adult residual V1 at 21 DPI (Figure 8E), although a population Ki67+ cells were observed adjacent to IB4+ blood vessels (Figure 8E) in the adult post‐ischemic residual V1. Our results support the capacity for post‐ischemic neovascularization following early‐life, but not adult‐life strokes.
Transneuronal retrograde degeneration (TRD) following V1 focal ischemia in adult and neonate marmosets
We investigated the extent of TRD at 21 DPI in the lateral geniculate nucleus (LGN; primary thalamic visual relay nucleus in primates) from retrograde degeneration of the optic radiation after V1 focal ischemia. NeuN IHC revealed no obvious changes in the laminar structure of the adult LGN at 21 DPI (Figure 9A,B). However, a clear zone of neuronal degeneration was observed in the neonatal LGN (Figure 9A′,B′), within the foveal representation (central 0°–3°) of the contralateral visual hemifield 82, consistent with the corresponding retinotopy of the marmoset operculum V1 (Figure 1A; 59). The extent of neuronal degeneration was more obvious in the dorsal (parvocellular) layers compared with deep (magnocellular) layers. No cellular aCasp3 immunoreactivity was detected in the adult or neonatal LGN at 21 DPI (Figure 9C,C′). This result suggests that TRD‐associated neuronal apoptosis in the LGN is not yet apparent in the adults but could be complete in neonates by 21 DPI, consistent with previous NHP data 38, 80, 83.
Figure 9.
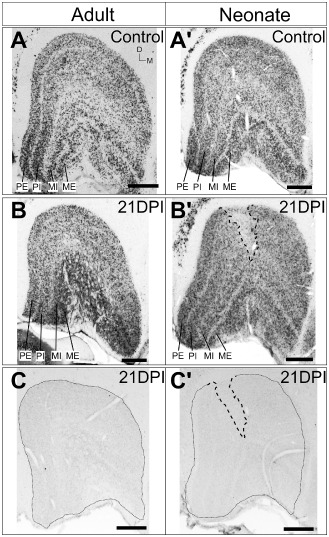
Transneuronal retrograde degeneration in the adult and neonatal LGN at 21 days post ischemia ( DPI ) after focal ischemia. Coronal sections of control adult (A) and neonatal (A′) LGN and after focal ischemia at 21 DPI (B, B′) immunolabaled for neuronal nuclei (NeuN). No obvious neuronal degeneration was detected in the adult LGN at 21 DPI but the neonatal LGN revealed a clear zone devoid of neurones. Dashed lines denote neuronal degeneration zone. PE = parvocellular external; PI = parvocellular internal; MI = magnocellular internal; ME = magnocellular external. (C, C′) no cellular aCasp3 expression was detected in both adult and neonatal LGN at 21 DPI. Scale: 500 μm.
Discussion
In this study, we sought to develop a model of focal ischemia in the NHP visual cortex and characterize the anatomical and cellular sequelae during the acute and subacute period post ischemia. This is also the first demonstration of a model of neonatal/pediatric stroke in a NHP. The approach provided a 100% survival rate with no unexpected postsurgical complications in either adults or neonates. Animals were able to ambulate and feed normally after recovery from anesthesia, followed by successful reintegration into family groups.
This study demonstrates the reproducibility of selective distal‐arterial occlusion to produce area‐specific focal ischemia in the adult and neonatal NHP. Previous studies in rodents, which involved ET–1‐induced occlusion of large arteries (main/proximal branch; eg, MCAOs; 6) or intracortical injections into the cortex or striatum (eg, 39) resulted in inconsistent spatial profiles and large infarct sizes, affecting subcortical and/or thalamic structures. However, by selectively occluding a distal branch of the PCA, we have achieved a precise and focal ischemic injury in the marmoset monkey V1, which is highly reproducible in neonates and adults alike, without affecting extrastriate/association cortices. This approach, which can potentially be applied to other sensory cortical areas, confers reduced variability in infarct size, providing better control and ability to analyze up‐ and downstream changes, ultimately reducing the cohort of animals required for studies. These features are necessary to allow more precise research to better define the long‐term pathophysiological and functional consequences following cerebral ischemia in a complex brain. Therefore, this model provides an excellent approach for future investigation into the functional and connectional changes as a consequence of focal ischemia in V1. Examples of common methods that have been employed include electrophysiological (eg, 31, 58, 64) and functional MRI (eg, 62, 63) analyses of visual responses in extrastriate areas after V1 injury. Recent advancements have also highlighted the potential for functional MRI experiments to be performed on unanesthetized, awake marmosets, a strategy previously only employed in rodent studies 17, 18, 20, 48. Awake brain imaging of NHPs will be crucial to generate more accurate maps of brain circuitry during resting state 5 or in response to stimuli 45, 46, reducing the risk of anesthetic‐related physiological artifacts. Moreover, the lissencephalic neocortex of the marmoset monkey and isotropic growth during development 49 is advantageous over the gyrencephalic brains of other NHPs used in research (eg, macaque monkey), enabling more stringent control of lesion induction and subsequent comparative evaluation in both young and old animals. Another advantage of the lissencephalic brain is the position of foveal V1 on the outer surface of the brain and not within the calcarine sulcus, which provides uncomplicated and minimally invasive access to the PCAca throughout life.
Our experimental model utilized an isoflurane‐sustained anesthetized setup for both the arterial occlusion and repeated MRI procedures. Previous reports, especially in rodents, have demonstrated isofluorane's ability to afford a level of neuroprotection; that is improving survivability, reducing edema and inflammatory injury and increasing cerebral blood flow during MCAO 8, with paradoxically detrimental effects on the neonatal/developing rodent brain (for a comprehensive review, see 60). However, there is a lack of evidence of significant long‐term isoflurane‐related neuroprotection available in NHP models or the clinic 44. While investigating anesthetic‐related differences in the development of the ischemic‐necrotic core in our model is beyond the scope of our study, we argue that this model remains enormously useful for the study of the cellular and molecular sequelae of neocortical focal ischemia as well as the resulting long‐term connectional changes. Comparative analysis of the injury at 21 DPI between the animals that underwent repeated anesthetic induction for MRI with animals that did not, revealed no obvious cellular differences in infarct or peri‐infarct area or size of areas. However, it should be noted that, given the current lack of information on anesthetic‐related differences, without proper controls or modifications to the anesthetic regime, this model may not be suitable for studies relating to pharmacological or cellular therapies for neuroprotection.
With respect to the time‐course of neuropathology, we reveal striking similarities to previous T2 MRI data from the post‐ischemic human cortex 2. Tissue hyperintensity at 1 DPI is consistent with fluid extravasation and edema observed immediately after PCAca occlusion. This is supported by validation of BBB disruption/increased vascular permeability at the lesion site, characterized by extravasation of serum proteins and infiltration of dextran pre‐labeled peripheral macrophages. Quantitative analysis of infiltrating macrophages post ischemia indicates that peripheral infiltrates, although present in both the infarct and peri‐infarct areas, were present in a higher proportion in the infarct core. The lower density of total immune cells in the infarct core may be explained by the ischemic‐necrotic microenvironment at the infarct site, which has been demonstrated to result in microglia death in rodents 40 during the acute post‐ischemic period. The distribution of peripheral macrophages was consistent with the spatial profile of serum albumin extravasation, validating BBB disruption.
T2 normalization in previously hyperintense regions at 14–21 DPI is consistent with transient T2 isointensity following ischemic strokes observed in human. This phenomenon, known as “fogging,” occurs in ∼50% of T2‐weighted MR images during subacute stages (2–3 weeks) after injury 15, 52 followed by a secondary T2 increase. Although not fully understood, it has been proposed that the transient isointensity is caused by displacement of free water at the site through hemorrhagic transformations or cellular activity including angiogenesis, tissue clearing, accumulation of infiltrating and local macrophages and reactive astrogliosis 78. Although some measure of secondary hemorrhagic transformation did occur, the extent of the bleed was small and punctate like, clustered superficially within the central core of the infarct at 21 DPI. We suggest that the extent of hemorrhage observed is most likely not a contributing factor to T2 fogging. Although evidence of neovascularization was present in the post‐ischemic neonatal peri‐infarct areas, similar evidence was not observed in the adults at 21 DPI. This result precludes angiogenesis as a contributing factor to the T2 fogging. The presence of Ki‐67+ cells adjacent to blood vessels may be consistent with the post‐ischemic proliferation of perivascular astrocytes in adults but not in neonates. Furthermore, our data demonstrate an accumulation of large GFAP+ reactive astrocyte and Iba1+ macrophage populations proximal to the lesion site at 21 DPI. This suggests that the T2 fogging observed in our model is most likely the result of accumulating reactive astrocytes and immune cells proximal to the infarct and peri‐infarct regions. Although this event has parallels with rodent models of focal ischemia, the decrease in T2 hyperintensity and T2‐derived apparent lesion volumes in post‐ischemic rodents occurs earlier, between 3 DPI and 9 DPI 16, 50, consistent with the peak of microglial accumulation at ∼5–7 DPI in rodents 66.
The morphology of neuronal nuclei proximal to the infarct core and absence of neuronal aCasp3 immunoreactivity at 1 DPI implicates necrosis as the primary mechanism of acute post‐ischemic cell death. This result corresponds with human data demonstrating apoptotic neuronal morphology detected only after 24 h post ischemia 55, despite the earlier upregulation of the pre‐cleaved caspase3 proenzyme 69. This is contrary to the hyperacute activation of apoptosis in rodents at ∼4 h 75. These data provide corroborating evidence that the neuropathological timeline in NHP models of CNS injury are more characteristic of what is observed in the human post‐ischemic cortex than the rodent.
Despite ongoing research, the mechanisms by which neonatal plasticity are thought to confer improved functional recovery after early life vs. adult brain insults are still debated and remains poorly understood 3, 32, 37. Therefore, the need for comparable early‐ and late‐life brain injury models is crucial for future research in this area. The similar ischemic core localization between neonatal and adult marmoset V1 that we have developed presents a robust tool for this purpose, that is, to elucidate differences in pathophysiology and capacity for functional recovery after neonatal compared with adulthood injuries. In this study, we demonstrate that selective occlusion of the PCAca results in an operculum V1‐specific focal ischemic injury that is anatomically and volumetrically consistent between neonates and adults. This comparative model of early‐ and late‐life focal ischemia provides evidence of potentially less potent reactive astroglial response from neonatal brain insults compared with injuries in adulthood, an observation that will require subsequent validation. The extent of neuronal degeneration observed in the neonates was more pronounced compared with the adult, supporting previous reports of greater neuronal susceptibility to injury during early life compared with adults 7. The degeneration occurring in all cortical layers, an observation not always present in cases of neonatal stroke is most likely dependent on the position and portion of the occluded artery and the downstream territory. Previously published reports exists describing similar extent of complete cortical degeneration in human perinatal stroke 42, following main‐branch MCAO 24, 26, 29, 43, 56, 68 vs. lenticulostriate branch occlusion, which affects deeper structures and sparing superficial cortical layers. These data are consistent with our own following superficial PCAca occlusion. Moreover, angiogenic responses present in the post‐ischemic neonatal, but not in the adult brain provides substantiating proof of differential cellular and physiological events underlying the response to injury that may be a contributing factor to post‐injury neonatal plasticity. The V1‐specificity of the ischemic core at both ages will also prove valuable in studies of the processes underlying TRD in relation to long‐term functional visual outcomes following pediatric vs. adult PCA strokes in humans 65, 76, in which we demonstrate that the neonate undergoes a more rapid process of degeneration compared with the adult, similar to that seen in the human 41, 74 and other models of brain injury in the marmoset 38. Better understanding of these mechanisms is vital, as potential therapies should be directed to prevent degeneration of afferent inputs to sensorimotor systems, which may otherwise be lost post‐injury.
In conclusion, while this model may not directly mimic physiological etiologies observed in the clinic, the goal of this model was to generate the consequential anatomical and cellular effects of ischemic injury observed in the clinic using a minimally invasive and simple surgical technique that significantly improves focality and reproducibility. We believe vasoconstrictor‐induced vascular occlusion provided the ischemic events required to generate these downstream effects. This model minimized variability of affected cortical territories and infarct size with no post‐ischemic animal mortality. We have demonstrated that the temporal profile of pathology as well as cellular events during the acute and subacute period mimics that of previously published data observed in the post‐ischemic human as evidence that the post‐ischemic sequelae is clinically relevant.
The pathophysiological and temporal homology between our model and the post‐ischemic human brain provides new opportunities for translational research into the neuropathological, physiological and behavioral consequences of early‐life vs. adult strokes. More importantly, the highly focal nature of the ischemic injury, as well as its reproducibility throughout life, offers better prospect for the development and preclinical testing of new, translatable therapeutic strategies in a clinically relevant model of pediatric and adult ischemic stroke.
Disclosures/Conflict of interests
None.
Acknowledgments
The authors wish to acknowledge the technical assistance and contributions of J. Homman‐Ludiye, K. Burman, J. Godwin, A. Samson, Q. Wu, R. Vreys, J. Pearson and A. Pinto. We would like to thank C. Sobey for his useful comments on earlier versions of the article. LT is supported by a South Melbourne Alliance for Research and Technology scholarship. National Health and Medical Research Council Project Grants to JAB and a National Stroke Foundation grant to LT supported this work. The Australian Regenerative Medicine Institute is supported by grants from the State Government of Victoria and the Australian Government.
References
- 1. Aldrich MS, Alessi AG, Beck RW, Gilman S (1987) Cortical blindness: etiology, diagnosis, and prognosis. Ann Neurol 21:149–158. [DOI] [PubMed] [Google Scholar]
- 2. Allen LM, Hasso AN, Handwerker J, Farid H (2012) Sequence‐specific MR imaging findings that are useful in dating ischemic stroke. Radiographics 32:1285–1297, discussion 97–9. [DOI] [PubMed] [Google Scholar]
- 3. Anderson V, Spencer‐Smith M, Wood A (2011) Do children really recover better? Neurobehavioural plasticity after early brain insult. Brain 134(Pt 8):2197–2221. [DOI] [PubMed] [Google Scholar]
- 4. Ang LC, Munoz DG, Shul D, George DH (1991) SMI‐32 immunoreactivity in human striate cortex during postnatal development. Brain Res Dev Brain Res 61:103–109. [DOI] [PubMed] [Google Scholar]
- 5. Belcher AM, Yen CC, Stepp H, Gu H, Lu H, Yang Y et al (2013) Large‐scale brain networks in the awake, truly resting marmoset monkey. J Neurosci 33:16796–16804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Biernaskie J, Corbett D, Peeling J, Wells J, Lei H (2001) A serial MR study of cerebral blood flow changes and lesion development following endothelin‐1‐induced ischemia in rats. Magn Reson Med 46:827–830. [DOI] [PubMed] [Google Scholar]
- 7. Bittigau P, Sifringer M, Pohl D, Stadthaus D, Ishimaru M, Shimizu H et al (1999) Apoptotic neurodegeneration following trauma is markedly enhanced in the immature brain. Ann Neurol 45:724–735. [DOI] [PubMed] [Google Scholar]
- 8. Bleilevens C, Roehl AB, Goetzenich A, Zoremba N, Kipp M, Dang J et al (2013) Effect of anesthesia and cerebral blood flow on neuronal injury in a rat middle cerebral artery occlusion (MCAO) model. Exp Brain Res 224:155–164. [DOI] [PubMed] [Google Scholar]
- 9. Bouallegue A, Daou GB, Srivastava AK (2007) Endothelin‐1‐induced signaling pathways in vascular smooth muscle cells. Curr Vasc Pharmacol 5:45–52. [DOI] [PubMed] [Google Scholar]
- 10. Bourne JA (2010) Unravelling the development of the visual cortex: implications for plasticity and repair. J Anat 217:449–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bourne JA, Warner CE, Rosa MG (2005) Topographic and laminar maturation of striate cortex in early postnatal marmoset monkeys, as revealed by neurofilament immunohistochemistry. Cereb Cortex 15:740–748. [DOI] [PubMed] [Google Scholar]
- 12. Brandt T, Steinke W, Thie A, Pessin MS, Caplan LR (2000) Posterior cerebral artery territory infarcts: clinical features, infarct topography, causes and outcome. Multicenter results and a review of the literature. Cerebrovasc Dis 10:170–182. [DOI] [PubMed] [Google Scholar]
- 13. Bridge H, Thomas O, Jbabdi S, Cowey A (2008) Changes in connectivity after visual cortical brain damage underlie altered visual function. Brain 131 (Pt 6):1433–1444. [DOI] [PubMed] [Google Scholar]
- 14. Burke MW, Kupers R, Ptito M (2012) Adaptive neuroplastic responses in early and late hemispherectomized monkeys. Neural Plast 2012:852423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chalela JA, Kasner SE (2000) The fogging effect. Neurology 55:315. [DOI] [PubMed] [Google Scholar]
- 16. Chen F, Suzuki Y, Nagai N, Jin L, Yu J, Wang H et al (2007) Rodent stroke induced by photochemical occlusion of proximal middle cerebral artery: evolution monitored with MR imaging and histopathology. Eur J Radiol 63:68–75. [DOI] [PubMed] [Google Scholar]
- 17. Chin CL, Pauly JR, Surber BW, Skoubis PD, McGaraughty S, Hradil VP et al (2008) Pharmacological MRI in awake rats predicts selective binding of alpha4beta2 nicotinic receptors. Synapse 62:159–168. [DOI] [PubMed] [Google Scholar]
- 18. Desai M, Kahn I, Knoblich U, Bernstein J, Atallah H, Yang A et al (2011) Mapping brain networks in awake mice using combined optical neural control and fMRI. J Neurophysiol 105:1393–1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Dirnagl U (2006) Bench to bedside: the quest for quality in experimental stroke research. J Cereb Blood Flow Metab 26:1465–1478. [DOI] [PubMed] [Google Scholar]
- 20. Duong TQ (2007) Cerebral blood flow and BOLD fMRI responses to hypoxia in awake and anesthetized rats. Brain Res 1135:186–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Durukan A, Tatlisumak T (2007) Acute ischemic stroke: overview of major experimental rodent models, pathophysiology, and therapy of focal cerebral ischemia. Pharmacol Biochem Behav 87:179–197. [DOI] [PubMed] [Google Scholar]
- 22. Edmond JC, Foroozan R (2006) Cortical visual impairment in children. Curr Opin Ophthalmol 17:509–512. [DOI] [PubMed] [Google Scholar]
- 23. El‐Shamayleh Y, Kiorpes L, Kohn A, Movshon JA (2010) Visual motion processing by neurons in area MT of macaque monkeys with experimental amblyopia. J Neurosci 30:12198–12209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Estan J, Hope P (1997) Unilateral neonatal cerebral infarction in full term infants. Arch Dis Child Fetal Neonatal Ed 76:F88–F93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Felleman DJ, Van Essen DC (1991) Distributed hierarchical processing in the primate cerebral cortex. Cereb Cortex 1:1–47. [DOI] [PubMed] [Google Scholar]
- 26. Filipek PA, Krishnamoorthy KS, Davis KR, Kuehnle K (1987) Focal cerebral infarction in the newborn: a distinct entity. Pediatr Neurol 3:141–147. [DOI] [PubMed] [Google Scholar]
- 27. Fonta C, Imbert M (2002) Vascularization in the primate visual cortex during development. Cereb Cortex 12:199–211. [DOI] [PubMed] [Google Scholar]
- 28. Freret T, Bouet V, Toutain J, Saulnier R, Pro‐Sistiaga P, Bihel E et al (2008) Intraluminal thread model of focal stroke in the non‐human primate. J Cereb Blood Flow Metab 28:786–796. [DOI] [PubMed] [Google Scholar]
- 29. Fujimoto S, Yokochi K, Togari H, Nishimura Y, Inukai K, Futamura M et al (1992) Neonatal cerebral infarction: symptoms, CT findings and prognosis. Brain Dev 14:48–52. [DOI] [PubMed] [Google Scholar]
- 30. Fukuda S, del Zoppo G (2003) Models of focal cerebral ischemia in the nonhuman primate. ILAR J 44:96–104. [DOI] [PubMed] [Google Scholar]
- 31. Girard P, Salin PA, Bullier J (1992) Response selectivity of neurons in area MT of the macaque monkey during reversible inactivation of area V1. J Neurophysiol 67:1437–1446. [DOI] [PubMed] [Google Scholar]
- 32. Giza CC, Prins ML (2006) Is being plastic fantastic? Mechanisms of altered plasticity after developmental traumatic brain injury. Dev Neurosci 28:364–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Go AS, Mozaffarian D, Roger VL, Benjamin EJ, Berry JD, Borden WB et al (2013) Heart disease and stroke statistics––2013 update: a report from the American Heart Association. Circulation 127:e6–e245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Goldshmit Y, Bourne J (2010) Upregulation of EphA4 on astrocytes potentially mediates astrocytic gliosis after cortical lesion in the marmoset monkey. J Neurotrauma 27:1321–1332. [DOI] [PubMed] [Google Scholar]
- 35. Guerin CJ, Nolan CC, Mavroudis G, Lister T, Davidson GM, Holton JL, Ray DE (2001) The dynamics of blood–brain barrier breakdown in an experimental model of glial cell degeneration. Neuroscience 103:873–883. [DOI] [PubMed] [Google Scholar]
- 36. Gundersen HJ, Jensen EB (1987) The efficiency of systematic sampling in stereology and its prediction. J Microsc 147(Pt 3):229–263. [DOI] [PubMed] [Google Scholar]
- 37. Guzzetta A, D'Acunto G, Rose S, Tinelli F, Boyd R, Cioni G (2010) Plasticity of the visual system after early brain damage. Dev Med Child Neurol 52:891–900. [DOI] [PubMed] [Google Scholar]
- 38. Hendrickson A, Warner CE, Possin D, Huang J, Kwan WC, Bourne JA (2013) Retrograde transneuronal degeneration in the retina and lateral geniculate nucleus of the v1 lesioned marmoset monkey. Brain Struct Funct. doi: 10.1007/s00429-013-0659-7. [DOI] [PubMed] [Google Scholar]
- 39. Horie N, Maag AL, Hamilton SA, Shichinohe H, Bliss TM, Steinberg GK (2008) Mouse model of focal cerebral ischemia using endothelin‐1. J Neurosci Methods 173:286–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ji KA, Yang MS, Jeong HK, Min KJ, Kang SH, Jou I, Joe EH (2007) Resident microglia die and infiltrated neutrophils and monocytes become major inflammatory cells in lipopolysaccharide‐injected brain. Glia 55:1577–1588. [DOI] [PubMed] [Google Scholar]
- 41. Jindahra P, Petrie A, Plant GT (2012) The time course of retrograde trans‐synaptic degeneration following occipital lobe damage in humans. Brain 135(Pt 2):534–541. [DOI] [PubMed] [Google Scholar]
- 42. Kirton A, deVeber G (2009) Advances in perinatal ischemic stroke. Pediatr Neurol 40:205–214. [DOI] [PubMed] [Google Scholar]
- 43. Koelfen W, Freund M, König S, Varnholt V, Rohr H, Schultze C (1993) Results of parenchymal and angiographic magnetic resonance imaging and neuropsychological testing of children after stroke as neonates. Eur J Pediatr 152:1030–1035. [DOI] [PubMed] [Google Scholar]
- 44. Koerner IP, Brambrink AM (2006) Brain protection by anesthetic agents. Curr Opin Anaesthesiol 19:481–486. [DOI] [PubMed] [Google Scholar]
- 45. Leite FP, Tsao D, Vanduffel W, Fize D, Sasaki Y, Wald LL et al (2002) Repeated fMRI using iron oxide contrast agent in awake, behaving macaques at 3 Tesla. Neuroimage 16:283–294. [DOI] [PubMed] [Google Scholar]
- 46. Liu JV, Hirano Y, Nascimento GC, Stefanovic B, Leopold DA, Silva AC (2013) fMRI in the awake marmoset: somatosensory‐evoked responses, functional connectivity, and comparison with propofol anesthesia. Neuroimage 78:186–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Marshall JW, Ridley RM (1996) Assessment of functional impairment following permanent middle cerebral artery occlusion in a non‐human primate species. Neurodegeneration 5:275–286. [DOI] [PubMed] [Google Scholar]
- 48. Martin C, Martindale J, Berwick J, Mayhew J (2006) Investigating neural‐hemodynamic coupling and the hemodynamic response function in the awake rat. Neuroimage 32:33–48. [DOI] [PubMed] [Google Scholar]
- 49. Missler M, Eins S, Merker HJ, Rothe H, Wolff JR (1993) Pre‐ and postnatal development of the primary visual cortex of the common marmoset. I. A changing space for synaptogenesis. J Comp Neurol 333:41–52. [DOI] [PubMed] [Google Scholar]
- 50. Neumann‐Haefelin T, Kastrup A, de Crespigny A, Yenari MA, Ringer T, Sun GH, Moseley ME (2000) Serial MRI after transient focal cerebral ischemia in rats: dynamics of tissue injury, blood–brain barrier damage, and edema formation. Stroke 31:1965–1972, discussion 72–3. [DOI] [PubMed] [Google Scholar]
- 51. Northington FJ (2006) Brief update on animal models of hypoxic‐ischemic encephalopathy and neonatal stroke. ILAR J 47:32–38. [DOI] [PubMed] [Google Scholar]
- 52. O'Brien P, Sellar RJ, Wardlaw JM (2004) Fogging on T2‐weighted MR after acute ischaemic stroke: how often might this occur and what are the implications? Neuroradiology 46:635–641. [DOI] [PubMed] [Google Scholar]
- 53. O'Collins VE, Macleod MR, Donnan GA, Horky LL, van der Worp BH, Howells DW (2006) 1026 experimental treatments in acute stroke. Ann Neurol 59:467–477. [DOI] [PubMed] [Google Scholar]
- 54. Pereira AC, Doyle VL, Clifton A, Howe FA, Griffiths JR, Brown MM (2000) The transient disappearance of cerebral infarction on T2weighted MRI: case reports. Clin Radiol 55:725–727. [DOI] [PubMed] [Google Scholar]
- 55. Qi JP, Wu AP, Wang DS, Wang LF, Li SX, Xu FL (2004) Correlation between neuronal injury and caspase‐3 after focal ischemia in human hippocampus. Chin Med J 117:1507–1512. [PubMed] [Google Scholar]
- 56. Ricci D, Mercuri E (2012) Neonatal arterial stroke. Chapter 139. In: Neonatology. Buonocore G, Bracci R, Weindling M (eds), pp. 1192–1198. Springer: Milan. [Google Scholar]
- 57. Robinson MJ, Macrae IM, Todd M, Reid JL, McCulloch J (1990) Reduction of local cerebral blood flow to pathological levels by endothelin‐1 applied to the middle cerebral artery in the rat. Neurosci Lett 118:269–272. [DOI] [PubMed] [Google Scholar]
- 58. Rodman HR, Gross CG, Albright TD (1989) Afferent basis of visual response properties in area MT of the macaque. I. Effects of striate cortex removal. J Neurosci 9:2033–2050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Rosa MG (2002) Visual maps in the adult primate cerebral cortex: some implications for brain development and evolution. Braz J Med Biol Res 35:1485–1498. [DOI] [PubMed] [Google Scholar]
- 60. Schifilliti D, Grasso G, Conti A, Fodale V (2010) Anaesthetic‐related neuroprotection: intravenous or inhalational agents? CNS Drugs 24:893–907. [DOI] [PubMed] [Google Scholar]
- 61. Schiller PH, Tehovnik EJ (2008) Visual prosthesis. Perception 37:1529–1559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Schmid MC, Mrowka SW, Turchi J, Saunders RC, Wilke M, Peters AJ et al (2010) Blindsight depends on the lateral geniculate nucleus. Nature 466:373–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Schmid MC, Panagiotaropoulos T, Augath MA, Logothetis NK, Smirnakis SM (2009) Visually driven activation in macaque areas V2 and V3 without input from the primary visual cortex. PLoS ONE 4:e5527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Schmid MC, Schmiedt JT, Peters AJ, Saunders RC, Maier A, Leopold DA (2013) Motion‐sensitive responses in visual area v4 in the absence of primary visual cortex. J Neurosci 33:18740–18745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Seghier ML, Lazeyras F, Zimine S, Saudan‐Frei S, Safran AB, Huppi PS (2005) Visual recovery after perinatal stroke evidenced by functional and diffusion MRI: case report. BMC Neurol 5:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Skriver EB, Olsen TS (1981) Transient disappearance of cerebral infarcts on CT scan, the so‐called fogging effect. Neuroradiology 22:61–65. [DOI] [PubMed] [Google Scholar]
- 67. Sola A, Peng H, Rogido M, Wen TC (2008) Animal models of neonatal stroke and response to erythropoietin and cardiotrophin‐1. Int J Dev Neurosci 26:27–35. [DOI] [PubMed] [Google Scholar]
- 68. Sran SK, Baumann RJ (1988) Outcome of neonatal strokes. Am J Dis Child 142:1086–1088. [DOI] [PubMed] [Google Scholar]
- 69. Taylor RC, Cullen SP, Martin SJ (2008) Apoptosis: controlled demolition at the cellular level. Nat Rev Mol Cell Biol 9:231–241. [DOI] [PubMed] [Google Scholar]
- 70. Tehovnik EJ, Slocum WM (2005) Microstimulation of V1 affects the detection of visual targets: manipulation of target contrast. Exp Brain Res 165:305–314. [DOI] [PubMed] [Google Scholar]
- 71. Tehovnik EJ, Slocum WM, Smirnakis SM, Tolias AS (2009) Microstimulation of visual cortex to restore vision. Prog Brain Res 175:347–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Teo L, Rosenfeld J, Bourne J (2012) Models of CNS injury in the nonhuman primate: a new era for treatment strategies. Transl Neurosci 3:181–195. [Google Scholar]
- 73. Uchino A, Miyoshi T, Ohno M (1990) Fogging effect and MR imaging: a case report of pontine infarction. Radiat Med 8:99–102. [PubMed] [Google Scholar]
- 74. Uggetti C, Egitto MG, Fazzi E, Bianchi PE, Zappoli F, Martelli A, Lanzi G (1997) Transsynaptic degeneration of lateral geniculate bodies in blind children: in vivo MR demonstration. AJNR Am J Neuroradiol 18:233–238. [PMC free article] [PubMed] [Google Scholar]
- 75. Unal‐Cevik I, Kilinc M, Can A, Gursoy‐Ozdemir Y, Dalkara T (2004) Apoptotic and necrotic death mechanisms are concomitantly activated in the same cell after cerebral ischemia. Stroke 35:2189–2194. [DOI] [PubMed] [Google Scholar]
- 76. van der Aa NE, Dudink J, Benders MJNL, Govaert P, van Straaten HLM, Porro GL et al (2013) Neonatal posterior cerebral artery stroke: clinical presentation, MRI findings, and outcome. Dev Med Child Neurol 55:283–290. [DOI] [PubMed] [Google Scholar]
- 77. Virley D, Hadingham SJ, Roberts JC, Farnfield B, Elliott H, Whelan G et al (2004) A new primate model of focal stroke: endothelin–1‐induced middle cerebral artery occlusion and reperfusion in the common marmoset. J Cereb Blood Flow Metab 24:24–41. [DOI] [PubMed] [Google Scholar]
- 78. Wagner DC, Deten A, Hartig W, Boltze J, Kranz A (2012) Changes in T2 relaxation time after stroke reflect clearing processes. Neuroimage 61:780–785. [DOI] [PubMed] [Google Scholar]
- 79. Warner CE, Goldshmit Y, Bourne JA (2010) Retinal afferents synapse with relay cells targeting the middle temporal area in the pulvinar and lateral geniculate nuclei. Front Neuroanat 4:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Weller RE, Kaas JH (1989) Parameters affecting the loss of ganglion cells of the retina following ablations of striate cortex in primates. Vis Neurosci 3:327–349. [DOI] [PubMed] [Google Scholar]
- 81. West GA, Golshani KJ, Doyle KP, Lessov NS, Hobbs TR, Kohama SG et al (2009) A new model of cortical stroke in the rhesus macaque. J Cereb Blood Flow Metab 29:1175–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. White AJ, Wilder HD, Goodchild AK, Sefton AJ, Martin PR (1998) Segregation of receptive field properties in the lateral geniculate nucleus of a New‐World monkey, the marmoset Callithrix jacchus . J Neurophysiol 80:2063–2076. [DOI] [PubMed] [Google Scholar]
- 83. Wong‐Riley MT (1972) Changes in the dorsal lateral geniculate nucleus of the squirrel monkey after unilateral ablation of the visual cortex. J Comp Neurol 146:519–548. [DOI] [PubMed] [Google Scholar]