

Abstract
Amyotrophic Lateral Sclerosis (ALS) is a heterogeneous disease in terms of progression rate and survival. This is probably one of the reasons for the failure of many clinical trials and the lack of effective therapies. Similar variability is also seen in SOD1G93A mouse models based on their genetic background. For example, when the SOD1G93A transgene is expressed in C57BL6 background the phenotype is mild with slower disease progression than in the 129Sv mice expressing the same amount of transgene but showing faster progression and shorter lifespan. This review summarizes and discusses data obtained from the analysis of these two mouse models under different aspects such as the motor phenotype, neuropathological alterations in the central nervous system (CNS) and peripheral nervous system (PNS) and the motor neuron autonomous and non‐cell autonomous mechanisms with the aim of finding elements to explain the different rates of disease progression. We also discuss the identification of promising prognostic biomarkers by comparative analysis of the two ALS mouse models. This analysis might possibly suggest new strategies for effective therapeutic intervention in ALS to slow significantly or even block the course of the disease.
Keywords: biomarkers, disease course variability, motor neuron, peripheral motor system, Transgenic SOD1G93A mice
Introduction
Amyotrophic Lateral Sclerosis (ALS) is the most common motor neuron disease first described by Jean‐Martin Charcot 150 years ago, for which there is no effective therapy. The neuropathology is characterized by progressive degeneration of motor neurons in the cortex, brainstem and spinal cord followed by extensive muscle wasting and atrophy leading to paralysis and death 3–5 years after disease onset. Around 10% of ALS cases are classified as familial (fALS) with a predominantly autosomal dominant pattern of inheritance whereas the remaining 90% of cases occur sporadically. Today, we know the genetic aetiology of two‐thirds of familial cases and about 11% of sporadic ALS cases 1, 88. The most frequent genetic causes in ALS population are related to the Superoxide dismutase 1 gene (SOD1, 20% of fALS and 2%–7% in sALS), with more than 180 mutations discovered (http://alsod.iop.kcl.ac.uk) since the first one found more than 20 years ago 91. More recently, mutations of the gene encoding C9ORF72 characterized by hexanucleotide repeat expansions in the intron 1 was found in about 40% of fALS and 10% of sALS 88. Mutations in other genes, including TAR DNA‐binding protein (TDP43)47, Fused in Sarcoma/Translocated in Liposarcoma (FUS/TLS) 51, 99, valosin‐containing protein (VCP) 100 ubiquilin 2 (UBQLN2) 24 and polymorphisms in sequestrosome 1 (SQSTM1, p62) 30, angiogenin (ANG) 37, alsin (ALS2) 103, vesicle associated membrane protein B (VAPB) 76, optineurin 66, Sigma 1 receptor 2, Charged multivesicular body protein 2B (CHMP2B)21 and TANK‐binding kinase (TBK1) 53, senataxin 105 and matrin 3 44, are together responsible for the 10% of familial cases and about 4%–9% of the sporadic ALS 88. All these genes encode proteins implicated in a wide range of cellular mechanisms such as oxidative stress, RNA processing, axonal transport, protein quality control and angiogenesis. This complexity indicates that the pathogenesis of ALS is rather composite with several different processes ultimately, leading to the selective motor neurodegeneration. Another level of complexity is related to the phenotypic variability of patients with ALS showing different rates of progression to death 8, 17, 64, 86.
Mouse models based on gene abnormalities associated with ALS have been instrumental in defining the biology of the disorder as well as in designing and testing potential targeted therapeutics.
Transgenic mice overexpressing the SOD1 mutation have been the first models to be developed soon after the discovery of a linkage between this gene mutation and ALS 40 and since then myriad of transgenic mutant SOD1‐overexpressing mice have been generated 46, 68, 84, 98. These various mouse lines invariably show progressive hind limb tremor and weakness, locomotor deficits and paralysis followed by premature death which recapitulates several core clinical features of ALS. These models have been widely used to dissect the pathogenic mechanisms of mutant SOD1 and to assess the efficacy of new therapeutic agents (see reviews,15, 68, 62, 98).
In the last decade the discovery of TDP‐43—positive cytoplasmic inclusions as major hallmarks of sporadic ALS and the identification of mutations of the gene coding for this protein in fALS has prompted several groups to create new genetic mouse models with the high expectations to mimic the human conditions 75. Most of these models have been amply described in recent reviews but in general, none of the TDP43 driven animals appears to replicate the progressive disease phenotype seen in ALS patients and SOD1 mutant mice 46, 68, 84.
More recently the discovery of the repeated expansion in the C9 or f72 gene as the most prevalent ALS‐causing gene identified, boosted several group to generate new mouse models. Given the complexity of the mutation, only few have been developed so far, however, none of them replicate the ALS pathology of human 50, 79, 82.
Thus, to date, the mutant SOD1 mouse is the model that best recapitulates several core clinical and neuropathological features of ALS. Several preclinical trials has been performed in this mouse model with positive effect in delaying the disease course 15, 62, 98. Unfortunately, none of these positive effects has been translated in successful clinical trial in ALS patients 9. This is in part due to an overestimation of the mouse studies, which are often inadequately powered and confounded by factors not properly considered such as variations in copy number, differential effects of sex and variability in mouse strain genetic backgrounds leading to nonreproducible preclinical results and potentially erroneous conclusions 81, 93. In attempt to overcome this problem guidelines for the use of SOD1 mutant mice have been recently issued 59 making comparative analysis more robust.
Another potential reason for the negative results of clinical trials is the phenotypic variability in patients, also present in genetic familial forms 70, 87. Controversy remains as to whether phenotypes can be linked to one cause, several potential causes, or the presence of modifying genes that affect the expression of ALS in a particular patient. With respect to this, there are growing evidence indicating that the severity of the disease may be remarkably influenced, also in mutant SOD1 mice, by their genetic background 42, 63, 65, 74. Therefore, discovering those genes that may be determinant for slowing the disease progression may lead to identify modifiers as potential druggable targets and staging‐disease biomarkers to be eventually translated in the clinical practice.
This review aims to draw the attention on this underestimated field in the preclinical research on ALS which may provide clues to understand the causes of different rates of disease progression. In particular, we illustrate the utility of mutant SOD1 mice in providing new insights in the cellular mechanisms underlying the variability in ALS disease phenotype.
Variability in the Disease Course of SOD1G93A Mice: Contribution of Genetic Background
When Gurney et al 40 generated the first SOD1G93A mouse model in 1994, it was clear that the disease expression in these mice closely depended on the number of transgene copies transferred to the mouse. In fact, Gurney's SOD1G93A mice carrying 18 copies of transgene became symptomatic at age 121 ± 23 days and moribund at 169 ± 16 days while mice carrying only 4 or less transgene copies which expressed normal levels of human mutant SOD1 did not show disease phenotype 40. However, the age of symptoms onset and death variably changed over the years not only because of the numbers of transgene copies 19, 98 but also for the change in the genetic background of mice breeded with the original SOD1G93A mice 42, 63, 83. The first observation in this respect came from Jackson laboratory that by transferring the mice to a C57B6/SJL background reduced the survival of the original Gurney's mice by 1 month (136 ± 15) 39. This corresponds to the mean survival reported later by the ALS‐TDI group from more than 4000 C57B6/SJL SOD1G93A mice analyzed over 4 years (134 ± 10) 93. However, among the different studies published over the last 15 years, the life span reported for these mice vary from 102 to 151 days 9, 15. Interestingly, such variability somehow reflects the variability in the clinical course of ALS patients 8, 17, 64, 86 including the familial cases. In fact, the patients carrying the SOD1 mutation in G93C has a survival that span from 62 to 243 months 87. Thus, understanding the mechanisms at the basis of such variability provides useful clues for the control of the disease severity in mouse models and eventually in patients.
Different groups, including ours, have clearly demonstrated the influence of the genetic background on the disease progression of SOD1G93A mice despite they expressed the same amount of mutant SOD1 42, 63, 65, 83. In particular, we observed that transgenic SOD1G93A mice on C57BL/6JOlaHsd genetic background (C57‐SOD1G93A), hereafter indicated as slow progressor, exhibited a delay in the onset of symptoms and a prolonged survival of about 2 and 8 weeks, respectively, compared with the SOD1G93A mice on 129SvHsd strain (129Sv‐SOD1G93A), hereafter indicated as fast progressor 65. The many differences between these two ALS mouse strains are summarized in Table 1. In terms of disease course, the 129Sv‐SOD1G93A mice develop a complete paralysis of the hind limbs soon after the first sign of motor weakness was detected by the impairment of hind limbs extension reflex. In less than 3 weeks, these mice completely lose their muscular strength and are no longer able to perform neither the hanging wire‐lid test or the rotarod test (Figure 1). At this stage their body weight is reduced by about 19% and drops rapidly until they have lost more than 25% of their original maximum weight at the time of euthanasia.
Table 1.
Phenotypic and molecular differences between ALS mouse models with a slow (C57SOD1G93A) vs. fast (129SvSOD1G93A) disease progression. In the table are reported only the parameters showing the major differences between the two SOD1G93A mouse strains at the disease onset. The onset of motor symptoms is defined when the mouse shows the first progressive impairment in grip strength. The survival time is evaluated when the mouse is unable to right itself within 10 s after being placed on each side and therefore is euthanised by deep anaesthesia. MN are counted from the lumbar spinal cord. NMJs denervation is evaluated in tibialis muscles. Abbreviations: MN = motor neuron; NMJ = neuromuscular junction; PPIA = cyclophilin A; CRYAB = alpha‐B‐crystallin; GFAP = glial fibrillary acidic protein; CLIC1 = Chloride intracellular channel protein 1; CCL2 = chemokine (C‐C motif) ligand 2; CC3 = Complement component 3; MHC‐I = major histocompatibility complex 1; β2m = beta‐2 microglobulin; LMP7 = immunoproteasome subunit; CD8+T = cytotoxic lymphocytes; NF200 = neurofilament 200Kd; TDP43‐P = phosphorylated TDP43; P‐ERK = phosphorylated extracellular signal‐regulated kinases; AChRγ = acetylcholine receptor gamma; Iba1 = Ionized calcium binding adaptor molecule 1; LY6Chigh = activated monocytes; Ntg = nontransgenic. 0 = no changes; + = increase; − = decrease.
| Parameters | C57SOD1G93A | 129SvSOD1G93A | |
|---|---|---|---|
| ONSET (days) | 122.5 ± 8.6 | 101.9 ± 3.9 | |
| SURVIVAL (days) | 180.9 ± 11.2 | 124.5 ± 5.6 | |
| MN loss at onset | −52% | −55% | |
| NMJs innervated at onset | −35% | −63% | |
| Alterations in spinal cord at onset | |||
| Protein aggregation | 0 | +++ | |
| Chaperones | PPIA | ++ | 0 |
| CRYAB | ‐ | ‐ ‐ ‐ | |
| Proteasome | 19S subunit | 0 | ‐ ‐ ‐ |
| β5 subunit | 0 | ‐ ‐ ‐ | |
| Mitochondrial activity | ‐ | ‐ ‐ ‐ | |
| Astrocytes | GFAP | + | + |
| Microglia | CD68 | ++ | +++ |
| CLIC1 | ‐ | + | |
| Immunity | CCL2 | ++ | 0 |
| CC3 | ++ | 0 | |
| MHCI | ++ | + | |
| β2m | + | 0 | |
| LMP7 | +++ | + | |
| CD8+T | ++ | 0 | |
| Biomarkers | Nitrotyrosine | 0 | + |
| Alterations in sciatic nerves at onset | |||
| Axonal changes | NF‐200 | 0 | ‐ |
| Acetyl‐Tubulin | 0 | ‐ ‐ | |
| TDP43‐P | + | 0 | |
| β‐Importin | 0 | ++ | |
| CCL2 | ++ | 0 | |
| MHCI | ++ | 0 | |
| CC3 | ++ | 0 (lower in Ntg) | |
| Schwann cells | GFAP | ++ | + (lower in Ntg) |
| P‐ERK | ++ | + (lower in Ntg) | |
| NMJs | AChRγ | + | +++ |
| Immune cells | CD8+T cells | +++ | ‐ ‐ |
| Iba1 | +++ | ++ | |
| Alterations in peripheral blood at onset | |||
| Monocytes | 0 (LY6Chigh) | 0 (CD45/CCR2) | |
| Lymphocytes | CD3+T | ‐ | 0 |
| PBMC | Nitroactin | 0 | +++ |
Figure 1.
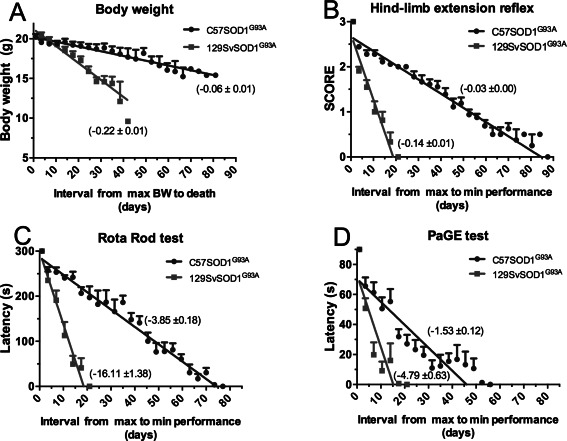
Rate of disease progression in 129Sv SOD1G93A and C57SOD1G93A mice monitored with four different tests. A. Body weight change from the maximum level to that reached at the time of death. B. Score of hind limbs extension reflex. Three and zero correspond, respectively, to the maximum or minimum opening of the hind limbs when the mouse is raised by tail for 5 s. C. Rotarod performance is evaluated on a rotating bar accelerated at a constant rate for a maximum of 5 min. The rate of disease progression is evaluated from the mouse age at the maximum latency to fall (300 s) to the age at which the mouse score 0. D. Paw grip endurance test is evaluated by placing the mouse on the wire‐lid of a housing cage which is then swiflty turned upside down. The rate of disease progression is evaluated from the mouse age at the maximum latency to fall (90 s) to the age at which the mouse score 0.
In slow progressors, the paralysis develops more steadily until they are totally paralyzed and unable to right themselves when laid on one side (Figure 1). Notheworthy, they become totally unable to execute the hanging wire‐lid test much earlier than the complete inability to stay on rotarod indicating a more rapid decline in muscular strength of all four limbs rather than in balance and motor coordination.
Thus, our and other studies clearly show that when the expression of the SOD1G93A transgene is in C57BL6 background the phenotype is milder than all the other inbred strains. It is to be emphasize, however, that there are C57BL/6 substrains that are genetically and phenotypically different (http://www.informatics.jax.org) 49, 106. For example, the survival of the SOD1G93A mice under C57B6OlaHsd substrain from Harlan, maintained in our colony for more than 15 generations, is prolonged by more than 10 days (180.9 ± 11.2 days) with respect to lifespan of congenic B6.Cg‐Tg(SOD1‐G93A) 1Gur/Jmice either reported by Heinemann‐Patterson's group (161 ± 10 days)42 and by AriSLA animal facility managed by our group (169.7 ± 12.9 days) (http://www.arisla.org/?page_id=247).
C57BL/6JOlaHsd mice carry gene deletion for the alpha‐synuclein (Snca) and for the multimerin 1, involved in the pathophysiology of the neuron and in platelet adhesion thrombus formation, respectively 96. Although we still ignore the effective contribution of these genetic alterations in the disease progression of ALS mice, it is important that investigators take into account this genetic polymorphisms when plan to breed knockout mice to identify ALS genetic modifiers and be as explicit as possible about which substrain is under study. In fact, it is clear that the influence of genetic background is far more effective in changing the course of the disease of SOD1 mutant mice than the effect of single genetic changes and therapeutic interventions tested so far in these mice 62, 68, 89.
Axonal Degeneration Rather than Motor Neuron Loss is the Cause of Disease Acceleration
A striking and unexpected difference between the C57‐SOD1G93A and the 129Sv‐SOD1G93A mouse models was found in the neuropathology of the central nervous system (CNS). In fact, the extent of motor neuron loss was similar in the lumbar spinal cord of these mice when measured either at the same stage of the disease, as expected, or at the same interval (2 weeks) after symptom onset corresponding, respectively, to the early or obvious symptomatic stage for the slow and fast progressing mice 65. These observations point out that the loss of motor neurons and survival are two unrelated events in line with that shown in other studies 25, 36, 92.
Even more unexpectedly, we found that in the brainstem the facial and motor trigeminal motor neurons displayed a later and milder vulnerability in fast‐progressing compared with slow‐progressing mice 14. Such difference was highlighted by the comparative longitudinal analysis of these cranial nuclei by Magnetic Resonance Imaging showing an increased T2 value associated with the formation of vacuoles in the motor neurons already at the presymptomatic stage in C57‐SOD1G93A mice, while this was present only at the advanced stage in 129Sv‐SOD1G93A progressing mice. One possible explanation for such difference is that the 129Sv‐SOD1G93A mice die so fast that the disease cannot propagate to the brainstem, however, this hypothesis needs to be verified.
In contrast to the fact that the motor neurons were affected at the same extent or even more in slow progressing mice, the fast progressor showed earlier abnormalities in the axonal compartment as demonstrated by ex vivo diffusion tensor imaging and histopathological analysis of the white matter of the spinal cord 14. In fact, a decreased axial diffusivity was detected in the ventro‐lateral and dorsal white matter regions of 129Sv SOD1G93A mice with respect to nontransgenic mice, already at the onset of the disease while in C57 SOD1G93A mice this phenomenon was evident only at the advanced stage. In addition, in the gray matter of lumbar spinal cord the vacuolated axons appeared earlier in fast compared with the slow progressing mice 65.
Like in the CNS, the peripheral axons in the sciatic nerves and their innervation at the NMJ were more severely affected in 129Sv‐SOD1G93A compared with C57‐SOD1G93A mice at the onset of symptoms, predicting a faster disease progression in the first strain. We have examined the causes of this phenomenon, demonstrating an important role of mechanisms associated with the immune response mediated by the major histocompatibility complex I (MHCI) system at the axonal level (manuscript submitted).
Thus, the extent of axonal and neuromuscular dysfunction, rather than the motor neuron loss seems predictive of a more aggressive phenotype in the SOD1G93A mouse model. This partially agree with Mancuso's report of no differences in motor neuron loss between C57B6‐SOD1‐G93A and C57B6SJL‐SOD1‐G93A at the same age (16 weeks) despite of a large difference in their life‐span (153 ±1.7 vs. 138 ± 1.9 days, respectively) 63. In fact, despite both strains showed similar values of compound muscle action potential (CMAP) from the tibialis anterioris (TA) at 16 weeks of age, mice surviving less appeared to have faster muscle denervation once it was started 63.
Motor Neuron Autonomous Mechanisms Underlying Phenotype Variability
Our observation that the loss of spinal motor neurons was similar in mice with different rates of disease progression, prompt us to compare the intrinsic properties of these cells between the two strains. Using a transcriptome analysis of the laser captured lumbar spinal motor neurons from the two SOD1G93A mouse strains at the disease onset we identified marked differences in the transcriptional profile of these two mice providing the clues to explain the difference in the rate of the disease progression 74.
The cluster of genes more strikingly downregulated in the motor neurons of fast progressing mice at the disease onset are those related to the mitochondrial activity, protein catabolic process and axonal function impairment 74. Mitochondrial dysfunction and damage has major implications in the rapid decline of MN function not only because they are less efficient to produce energy necessary to the anterograde transport 22 but probably also because they can aberrantly accumulate along the axons, further altering the axonal transport 61.
Another gene cluster highly downregulated in motor neurons of fast rather than slow progressing mice at disease onset is the “protein catabolic process.” In fact, the accumulation of polyubiquitinated protein aggregates occurs already at the presymptomatic stages in MNs of 129Sv‐SOD1G93A mice, while it appears later in the C57‐SOD1G93A 65. This phenomenon is associated with a reduced expression of different chaperon proteins (CRYAB, PPIA, Hsp90, Hsc70) 7, 52, 65 as well as impairment of the ubiquitin‐proteasome system caused by a decreased expression of constitutive proteasome subunit (β5 and 19S) 11, 65, 74. In contrast, the autophagy‐lysosomal pathway is activated more in the spinal cord of fast progressing mice at the symptomatic stage 65. Interestingly, the activation of autophagosome has been proposed as a compensatory response of the surviving motor neurons to the proteasome impairment 23. This may explain why in 129Sv‐SOD1G93A mice the motor neurons loss is similar to C57‐SOD1G93A mice at the symptomatic stage. Thus, although the contribution of autophagy to the fALS linked pathogenesis is still debated, our data suggest that its impairment in the spinal cord does not contribute to the rapid disease progression. Autophagy may instead be involved in the earlier axonal damage of fast progressing mice. In fact, it has been demonstrated that an impaired autophagic removal of damaged mitochondria may result in their aberrant accumulation in axons, thus causing distal axons to become more vulnerable to dying back degeneration 101. Notably, in 129Sv‐SOD1G93A mice, at the presymptomatic stage, the myelinated axons of the ventral roots were massively vacuolated 65, a feature associated to the aberrant swelling of damaged mitochondria 10. Therefore, we cannot exclude that an aberrant accumulation of damaged mitochondria along the axons may result from an impaired mitophagy at this level.
Motor axon morphology and functionality in fast progressing mice are also negatively influenced by the downregulation of other gene clusters specifically related to neuron projection, microtubules, axonal/synapse stability and regeneration as well as to the anterograde axonal transport 74. In this context, special attention deserve the kinesin‐1 subunit, KiF5b, reported to be essential in regulating fast anterograde axonal transport of different cytoskeletal proteins as well as that of the proteasome complex 80 in sciatic nerve. KiF5b is 16 times less expressed in the MNs of fast with respect to slow progressing mice 74 underlying once again the key role of the axonal dysfunction in the rapid progression of the disease. Differently, in the motor neurons of slow progressing mice predominates the activation of specific biological processes related to the extracellular matrix, response to wounding and protein translation that may help underpin a more substantial response to stress which protect the axons in this mouse strain longer 74. In this respect, it should be emphasized that the motor neurons of C57BL6 mice express higher basal levels of the “stress transcription factor”ATF3, whose upregulation was reported to counteract protein aggregation and promote motor axon sprouting in the peripheral nervous system (PNS) 94. In addition, we found that the motor neurons of C57BL6 mice express higher levels of cytosolic ribosomal subunits (Rps9; Rps6; Rpl17) than 129Sv mice suggesting a higher rate of physiological protein translation in MNs of the slow progressing mice. Rps9 transcript levels, in particular, are 140 times higher in C57 vs. 129Sv MNs (Figure 2A). The levels of RSP9 protein in the spinal cord are also remarkably lower in 129Sv compared with the C57 mice and they decrease further in the MNs of 129Sv‐SOD1G93A mice at disease onset (Figure 2B–D). This suggests that an impairment of this ribosomal subunit and in general of the protein translation has an important role in accelerating the dysfunction of motor neurons particularly at the axonal level.
Figure 2.
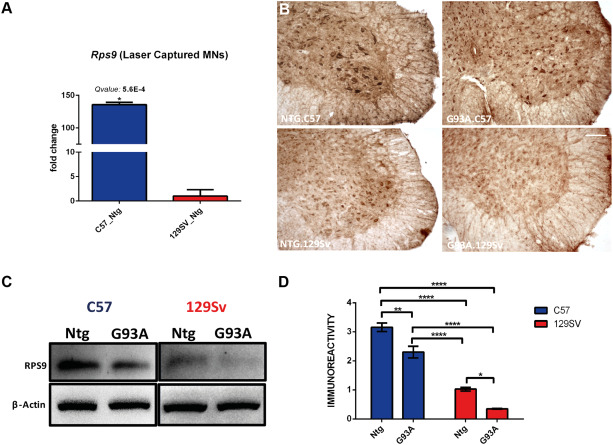
Rps9 expression in MNs distinguishes C57 from 129Sv mouse strains. A. Rps9 mRNA expression levels specifically increase in C57 Ntg MNs. mRNA levels are reported as mean fold change ratio (±} S.E.) between age matched C57 Ntg and 129Sv Ntg mice (n = 4 per group) at 60 days of age. Q‐values were generated from probability of positive log ratio (PPLR) using the following formula: Q‐val[i] = mean (1‐PPLR[1:i]); (*) Q‐value ≤0.01. B. Immunohistochemical comparison performed at the disease onset on lumbar spinal cord of C57 SOD1G93A, 129Sv SOD1G93A and Ntg littermates showing (i) the expression of RPS9 by MNs; (ii) lower levels of RPS9 in 129Sv Ntg and 129Sv SOD1G93A MNs (scale bar: 50 μm). C. RPS9 western blot analysis on longitudinally dissected lumbar ventral spinal cord protein extracts from C57 SOD1G93A, 129Sv SOD1G93A and Ntg littermates at disease onset. Immunoreactivity was normalized to Beta‐Actin. D. Densitometric analysis of RPS9 levels further validate immunohistochemical evidences. Data are reported as mean ±} S.E. of 4 mice per group. **P < 0.01; ****P < 0.0001 by Two‐way ANOVA with Tukey's postanalysis.
In line with this, Angiogenin, which is fundamental to stimulates the de novo biosynthesis of ribosomes, is eight fold more expressed in C57 than 129Sv MNs and it is remarkably downregulated in MNs of 129SvG93A mice at disease onset 74. Interestingly, missense mutation of ANG was found in a patient affected by sporadic ALS with the SOD1 G93D mutation that disclosed an unusual rapid disease progression 60. Taken together these results suggest that the motor neurons of the C57 mouse strain are better equipped to counteract the toxic effects caused by the overexpression of mutant SOD1 and therefore they can function better and live longer.
Non‐Cell Autonomous Mechanisms Underlying Phenotype Variability: The Role of Glia and the Immune System
Despite similar number of spared motor neurons in the lumbar spinal cord of 129Sv‐SOD1G93A and C57‐SOD1G93A mice, we found that microglia was differently activated in this region in the two strains 65. In particular, the reactive microglia detected by immunostaining for CD11b and the expression level of CD68 mRNA, a marker of phagocytic microglia, were significantly elevated already at the presymptomatic stage and further increased during disease development in fast progressor 129SvG93A mice 65. In contrast, in C57G93A mice the levels of CD68 mRNA increased only from the symptom onset and increase further during the progression of the disease but more slowly than in fast progressing mice. As reported above the MNs of fast progressing mice, unlike those of slow progressors, show clear indices of dysfunction such as the accumulation of ubiquitinated protein aggregates already at the presymptomatic stage when the MNs are still alive. In addition, as previously discussed, although the number of remaining MNs is the same for both ALS models, the MNs from fast progressing mice show an earlier and more rapid muscle denervation than in slow progressors. An example of microglia activation in the spinal cord despite the absence of MNs loss, derives from the transgenic mice with muscle‐restricted expression of SOD1G93A gene. These mice develop progressive muscle atrophy, associated with a significant reduction in muscle strength without evident sign of motor neuron degeneration but with a clear activation of neuroinflammation in the spinal cord 27. Thus, the fact that the 129Sv‐SOD1G93A mice, showing very rapid decline in muscle strength, also express significantly higher levels of CD68 in the lumbar spinal cord, supports the hypothesis that toxic signals originating from skeletal muscle may contribute to microglia activation in the CNS. This might further compromise the functional connection between the MNs and the muscle, leading to a more severe disease. Unlike the microglia, the expression level of GFAP and its mRNA, markers of reactive astrocytosis, increased equally and constantly from the appearance of symptoms in both SOD1G93A mouse strains suggesting that the astroglial reactive response to the progressive motor neuron loss is not a determinant factor in the variability of the disease course.
We also found that the immune system made a major contribution to the variability in the disease course. In fact, from the transcriptomic comparative analysis of the two SOD1G93A mouse strains at the disease onset we observed that the complement and MHCI were upregulated either at mRNA and protein level in the MNs of C57‐SOD1G93A but not 129Sv‐SOD1G93A mice 74. In particular, we observed a high accumulation of MHCI and C3 in PNS of C57G93A mice, in line with previous observations 20, 43, 74, suggesting a specific role of these immune molecules at this level. A positive role of MHCI has been reported in the maintenance of the axonal function and axon regrowth when the MN is damaged by nerve injury 78, 97 or in case of spinal cord damage45. In addition, the activation of complement play an important role in neuronal survival 102 and consistently, the C1q or C3 depletion dramatically enhances the loss of synapses in spinal MNs of SOD1G93A mice 57. Taken together these findings point out that the upregulation of these immune molecules may be responsible of a more efficient axonal preservation. As regards the possible mechanism underlying this phenomenon, it is important to consider that MHCI and C3 in the peripheral nerves are responsible for the recruitment of hemathogenous CD8+ T cells and macrophages, respectively20, 48, whose coordinated activity may be essential to remove defective myelin (myelin debris) for preserving motor axon activity 38. In addition, while removing motor axon debris, these immune cells can release extracellular matrix molecules, cytokines and growth factors that further support axon regrowth. These processes are essential, particularly during Wallerian degeneration, when the distal portion of the axon degrades quickly, whereas the MN soma can often survive 33. Interestingly, in vivo imaging of SOD1G93A mice crossbred with transgenic mice carrying fluorescently labeled macrophages/microglia and neuronal projections, revealed distinct inflammatory activity of CNS microglia compared with PNS macrophages, with the latter showing no substantial morphological reaction during degeneration of peripheral motor axons, but a passive role in clearing debris, particularly lipid‐rich myelin 26. Barrette et al 6 reported that peripheral macrophages are essential to promote the production of neurotrophins in the regenerating PNS and macrophage depletion significantly slowed axon regeneration and functional recovery following sciatic nerve injury. At the same time, interactions between neurons and CD8+ T cells, mostly studied in vitro 51, 52 evidenced that activated CD8+ T cells establish stable contacts with axons in an antigen‐ and MHCI‐dependent manner 56. Further studies are needed to understand the mechanism underlying the effective contribution of immune cells in the PNS, however, the prominent activation of an immune response in the slowly progressing mice suggest that specific immune molecules are activated by MNs especially at the peripheral level, to preserve the activity of motor axons during the progression of ALS. Therefore, it appears that the contribution of the inflammatory response in PNS stands in stark contrast with that of the CNS, where the neuroinflammation tend to exacerbate the motor neuronal damage. This might explain the partial efficacy of anti‐inflammatory treatments in mutant SOD1 mice and their failure in ALS patients.
Altered Metabolism as a Cause of Disease Variability
The SOD1G93A mice are usually euthanised when they completely lose they righting reflex for more than 10 seconds if laid on either side or when their body weight falls below 20 to 30 % of their maximal weight 59. Therefore, we do not know the precise natural cause of death in ALS mice. Although the body weight loss can in part be the result of the mouse dysphagia due to the degeneration of nuclei trigeminus (V), facial (VII) and hypoglossal (XII) in C57SOD1G93A mice 55 this does not seem the case for the fast progressing mice that rapidly lose weight until they die even if the trigemini and facialis nuclei appeared normal during the disease progression 14.
Thus, a possible cause of the faster body weight loss in the fast progressing mice might be related to their hyperactive metabolism. In fact, the 129Sv mice have been reported to have a higher basal metabolic rate than the C57Bl6 mice measured as O2 consumption during feeding with either low or high fat diet 3. In addition, 129Sv mice have a remarkable lower circulating insulin levels compared with C57BL6 mice 71. The hypothesis of hypermetabolism as an additional contributor to ALS pathogenesis is supported by different studies from Loeffler's group 28. Noteworthy, the SOD1 mouse model used by this group, the transgenic mice expressing the murine G86R SOD1 mutation 90, also show very rapid disease progression (about 3 weeks) after the onset, similarly to the disease duration of the 129SvSOD1G93A mice. In addition, hyperactivity of metabolism has been reported for the B6SJL mouse strain as compared with the C57B6 mice potentially explaining why the B6SJLSOD1G93A have a shorter lifespan with respect to the C57B6 mice 83. Interestingly, an abnormal energy metabolism has been seen in ALS patients too weight loss and hypermetabolism being associated with poor prognosis 29.
Variability in Disease Course: Translational Biomarkers
Progression of the disease in ALS patients is inherently heterogeneous and influenced by clinical, demographic and genetic traits 95. Therefore, there is a pressing need of biomarkers to objectively quantify disease progression in longitudinal studies and eventually monitor response to therapy in clinical trials. In view of this fact, it would also be important to measure such biomarkers already in preclinical studies to be able to more easily translate the findings to the clinical context. However, reliable biomarkers are lacking and biomarkers in the animal models are not often measured in clinically accessible samples such as serum/plasma, PBMC and CSF. Few potential translational biomarkers, that is, molecules that have been measured in a clinical and preclinical setting and are similarly regulated, have been identified, such as neurofilament proteins and, in our laboratory, tyrosine‐nitrated actin (nitroactin), cyclophilin A (PPIA) and chloride intracellular channel protein 1 (CLIC1) 13, 58, 73. We found that high levels of nitroproteins, PPIA and CLIC1 are early biomarkers of disease in SOD1G93A animal models of ALS, both in spinal cord and PBMC 7, 16, 65, 67, 72, 73. We also confirmed that nitroactin, PPIA and CLIC1 were increased in PBMC of sporadic ALS patients compared with healthy individuals73. This indicates that there are some translational alterations in PBMC of animal models that may suggest common pathogenic pathways in the animal mutant SOD1‐linked and human sporadic forms and draws attention to PBMC analysis for mechanistic studies in patients. Nitroproteins are specific indicators of nitrative/oxidative stress 34. We measured nitroactin in PBMC of fast and slow progressors at presymptomatic, onset and symptomatic stages of the disease. Nitroactin started to rise in 129SvSOD1G93A mice compared with nontransgenic mice already at a presymptomatic stage of the disease, and increased steeply at the onset of the disease again compared to C57SOD1G93A mice (Figure 3), indicating that it could possibly be considered a biomarker of fast disease progression. Similar results were obtained in the lumbar spinal cord ventral horn, where at 14 weeks of age nitroactin increased in the fast progressing mice but did not change in the slow progressors (Table 1). PPIA is an enzyme acting as an acceleration factor in protein folding and assembly in nascent and mature proteins 31, 41. PPIA has a strong cytoprotective effect under oxidative stress conditions 12, mitigates toxicity induced by mutant SOD1 protein aggregates 54 and regulates key TDP‐43 functions, including the regulation of genes involved in clearance of protein aggregates 52. In the slow‐progressing mice PPIA is rapidly upregulated during disease progression in the lumbar spinal cord, but not in the fast‐progressors where it is instead more sequestered in the Triton‐insoluble aggregates, which may further reduce its protective function 65. This is in line with the accelerated time course of the disease in SOD1G93A mice knockout for PPIA 52.
Figure 3.
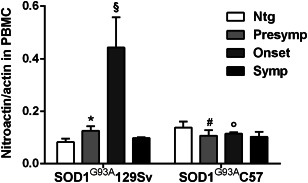
Analysis of nitroactin in PBMC from mice with fast (129SvSOD1G93A) and slow (C57SOD1G93A) disease progression. Nitroactin was measured by dot blot analysis using antinitrotyrosine antibody in‐house developed, as described 73, in 129SvSOD1G93A or C57SOD1G93A mice at presymptomatic (n = 6 per group, at 10 or 14 weeks of age for 129SvSOD1G93A and C57SOD1G93A, respectively), onset (n = 6 per group, at 14 or 16 weeks of age for 129Sv SOD1G93Aand C57SOD1G93A, respectively) and symptomatic stages of the disease (n = 6 per group, at 16 or 20 weeks of age for 129SvSOD1G93A and C57SOD1G93A, respectively) and in nontransgenic (Ntg) mice (n = 6). Data were normalized to the actual amount of protein loaded, by Red Ponceau staining, and to total actin immunoreactivity. *, significantly different from Ntg (P < 0.05); §, significantly different from Presymp and Symp (P < 0.05); ϒ, P < 0.05 vs. corresponding stage, #, P < 0.05 vs. corresponding age (two‐way ANOVA, Fisher's least significant difference test).
CLIC1 is considered a sensor and an effector of oxidative stress 4. In neurodegenerative conditions CLIC1 is up‐regulated and is mainly localized in the plasma membrane of activated microglia, where it functions as a chloride selective ion channel and is possibly involved in neuronal cell death 69, 77. CLIC1 was measured in the lumbar spinal cord ventral horn in slow‐ and fast‐progressing mice at 14 weeks of age, which corresponds to the presymptomatic and onset stages of the disease respectively. CLIC1 has an opposite behavior in the two mouse strains. In the slow‐progressing mice CLIC1 is lower than in the corresponding nontransgenic controls, while in the fast‐progressors has a tendency to increase compared to controls, suggesting an increased oxidative stress and/or microglial activation in 129SvSOD1G93A mice (Table 1).
Summarizing, all these data agree with the marked down‐regulation of specific pathways involved in mitochondrial function and protein quality control found in the fast‐progressing mice, 65, 74 that probably lead to increased nitrative/oxidative stress and a decreased ability to refold or degrade the oxidized/damaged proteins in the SOD1G93A129Sv mice. The analyses in the SOD1G93A mice with fast and slow disease progression suggested that nitroactin, PPIA and CLIC1 are promising prognostic biomarkers. Validation in patients with slow and fast disease progression is now called for.
Variability in Disease Course: Response to Treatments
The phenotypic variability of patients with ALS, in particular the different rate of disease progression, is considered one of the potential reasons for the negative results in randomized controlled clinical trials 70. As pointed out in the previous section, we still need biological markers of ALS progression that can be applied in routine clinical practice and clinical trials. It is likely that different phenotypes are linked to different pathogenic mechanisms associated to several potential causes or to the presence of modifying genes that affect the expression of ALS in a particular subgroup of patients. This implies that the response of randomized patients to a specific therapeutic intervention could differ depending on the phenotype and therefore the clinical trials need to be stratified to account for these different phenotypes.
Considering the fast and slow progressing mice, we observed different responses to specific treatments. For example, the treatment with lithium significantly anticipated the onset and reduced the survival in the fast progressing mice while it did not change the disease course of the slow progressors 85. This was confirmed by another group who found that lithium anticipated the disease onset of B6SJLTgN(SOD1‐G93A)1Gur) mice whose lifespan is similar to our fast‐progressors 35. Some ALS patients too treated with lithium showed in some cases a more rapid disease progression which was accompanied by an excess of serious adverse events 17. However, as the patients have not been stratified based on phenotype it is difficult to formulate any correlation.
On the contrary, treatment of either C57‐SOD1G93A and 129Sv‐SOD1G93A mice with Omega‐3 Fatty Acid Eicosapentaenoic Acid (EPA), before the onset of the symptoms, significantly accelerated the disease progression in slow but not in fast progressing mice suggesting that the long exposure to EPA may be detrimental 104. This unexpected effect was correlated with increased oxidation reflected in the higher level of 4‐HHE in the spinal cord, which may favor formation of small oligomeric SOD1 aggregates, potential precursors of the toxic forms 5. These data apparently contrast with the observation of a reduced risk of ALS associated with higher dietary intake of n‐3 PUFA in a large cohort study 32. However, the cohort study could not assess whether PUFA intake affected the risk of sporadic or familial ALS differently as no information is available on the genetic or family history or phenotype of these patients.
Conclusions
This review comparing SOD1G93A mouse strains with fast vs. slow progressing disease, highlights that: (1) genetic background largerly influences the course of the disease and lifespan more than each single genetic modifier or drug treatments. Therefore, the interpretation of results based on mixed genetic background, not properly balanced among experimental groups, can lead to erroneous conclusions. In addition, this underlines the need to target more than one gene or mechanism to significantly affect the disease progression; (2) motor neuron death is dissociated from motor neuron dysfunction underling a major role of the axonopathy and NMJ function in the severity of the disease. Thus, treatment aimed at only preserving the MN soma may be insufficient to counteract ALS disease progression; (3) the immune response has a protective rather than detrimental role in the ALS and this is particularly important for the preservation of the MNs at the peripheral level. This may explain the failure or the poor effect of various anti‐inflammatory and immuno‐suppressor treatments in patients and mouse models; (4) the disease course in these ALS mouse strains is subject to a multisystem control and the metabolic status may have a large role in the variability of their phenotype; (5) the detection of different biomarkers in these two ALS mouse models should help to identify prognostic factors for a proper stratification of patients in the clinical trials.
Acknowledgment
This work was supported by Motor Neuron Disease Association, UK, (grant 124695–1), European Community's Seventh Framework Programme (FP7/2007‐2013) under grant agreement EUROMOTOR(n°259867), Thierry Latran Foundation (F)‐project “MHCI and ALS”, Compagnia San Paolo‐Bando programma neuroscienze, associazione “Amici del Mario Negri” and Fondazione Italiana di Ricerca per la Sclerosi Laterale Amiotrofica (AriSLA)‐ project “IMMUNALS” and Animal Facility.
References
- 1. Ajroud‐Driss S, Siddique T (2015) Sporadic and hereditary amyotrophic lateral sclerosis (ALS). Biochim Biophys Acta 1852:679–684. [DOI] [PubMed] [Google Scholar]
- 2. Al‐Saif A, Al‐Mohanna F, Bohlega S (2011) A mutation in sigma‐1 receptor causes juvenile amyotrophic lateral sclerosis. Ann Neurol 70:913–919. [DOI] [PubMed] [Google Scholar]
- 3. Almind K, Kahn CR (2004) Genetic determinants of energy expenditure and insulin resistance in diet‐induced obesity in mice. Diabetes 53:3274–3285. [DOI] [PubMed] [Google Scholar]
- 4. Averaimo S, Milton RH, Duchen MR, Mazzanti M (2010) Chloride intracellular channel 1 (CLIC1): sensor and effector during oxidative stress. FEBS Lett 584:2076–2084. [DOI] [PubMed] [Google Scholar]
- 5. Banci L, Bertini I, Boca M, Girotto S, Martinelli M, Valentine JS, Vieru M (2008) SOD1 and amyotrophic lateral sclerosis: mutations and oligomerization. PLoS One 3:e1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Barrette B, Hebert MA, Filali M, Lafortune K, Vallieres N, Gowing G et al (2008) Requirement of myeloid cells for axon regeneration. J Neurosci 28:9363–9376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Basso M, Samengo G, Nardo G, Massignan T, D'Alessandro G, Tartari S et al (2009) Characterization of detergent‐insoluble proteins in ALS indicates a causal link between nitrative stress and aggregation in pathogenesis. PLoS One 4:e8130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Beghi E, Chio A, Couratier P, Esteban J, Hardiman O, Logroscino G et al (2011) The epidemiology and treatment of ALS: focus on the heterogeneity of the disease and critical appraisal of therapeutic trials. Amyotroph Lateral Scler 12:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Benatar M (2007) Lost in translation: treatment trials in the SOD1 mouse and in human ALS. Neurobiol Dis 26:1–13. [DOI] [PubMed] [Google Scholar]
- 10. Bendotti C, Calvaresi N, Chiveri L, Prelle A, Moggio M, Braga M et al (2001) Early vacuolization and mitochondrial damage in motor neurons of FALS mice are not associated with apoptosis or with changes in cytochrome oxidase histochemical reactivity. J Neurol Sci 191:25–33. [DOI] [PubMed] [Google Scholar]
- 11. Bendotti C, Marino M, Cheroni C, Fontana E, Crippa V, Poletti A, De Biasi S (2012) Dysfunction of constitutive and inducible ubiquitin‐proteasome system in amyotrophic lateral sclerosis: implication for protein aggregation and immune response. Prog Neurobiol 97:101–126. [DOI] [PubMed] [Google Scholar]
- 12. Boulos S, Meloni BP, Arthur PG, Majda B, Bojarski C, Knuckey NW (2007) Evidence that intracellular cyclophilin A and cyclophilin A/CD147 receptor‐mediated ERK1/2 signalling can protect neurons against in vitro oxidative and ischemic injury. Neurobiol Dis 25:54–64. [DOI] [PubMed] [Google Scholar]
- 13. Boylan K, Yang C, Crook J, Overstreet K, Heckman M, Wang Y et al (2009) Immunoreactivity of the phosphorylated axonal neurofilament H subunit (pNF‐H) in blood of ALS model rodents and ALS patients: evaluation of blood pNF‐H as a potential ALS biomarker. J Neurochem 111:1182–1191. [DOI] [PubMed] [Google Scholar]
- 14. Caron I, Micotti E, Paladini A, Merlino G, Plebani L, Forloni G et al (2015) Comparative magnetic resonance imaging and histopathological correlates in two SOD1 transgenic mouse models of amyotrophic lateral sclerosis. PLoS One 10:e0132159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Carri MT, Grignaschi G, Bendotti C (2006) Targets in ALS: designing multidrug therapies. Trends Pharmacol Sci 27:267–273. [DOI] [PubMed] [Google Scholar]
- 16. Casoni F, Basso M, Massignan T, Gianazza E, Cheroni C, Salmona M et al (2005) Protein nitration in a mouse model of familial amyotrophic lateral sclerosis: possible multifunctional role in the pathogenesis. J Biol Chem 280(16):16295–16304. [DOI] [PubMed] [Google Scholar]
- 17. Chio A, Logroscino G, Hardiman O, Swingler R, Mitchell D, Beghi E, Traynor BG (2009) Prognostic factors in ALS: a critical review. Amyotroph Lateral Scler 10:310–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chio A, Borghero G, Calvo A, Capasso M, Caponnetto C, Corbo M et al (2010) Lithium carbonate in amyotrophic lateral sclerosis: lack of efficacy in a dose‐finding trial. Neurology 75:619–625. [DOI] [PubMed] [Google Scholar]
- 19. Chiu AY, Zhai P, Dal Canto MC, Peters TM, Kwon YW, Prattis SM, Gurney ME (1995) Age‐dependent penetrance of disease in a transgenic mouse model of familial amyotrophic lateral sclerosis. Mol Cell Neurosci 6:349–362. [DOI] [PubMed] [Google Scholar]
- 20. Chiu IM, Phatnani H, Kuligowski M, Tapia JC, Carrasco MA, Zhang M et al (2009) Activation of innate and humoral immunity in the peripheral nervous system of ALS transgenic mice. Proc Natl Acad Sci USA 106:20960–20965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cox LE, Ferraiuolo L, Goodall EF, Heath PR, Higginbottom A, Mortiboys H et al (2010) Mutations in CHMP2B in lower motor neuron predominant amyotrophic lateral sclerosis (ALS). PLoS One 5:e9872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cozzolino M, Ferri A, Valle C, Carri MT (2013) Mitochondria and ALS: implications from novel genes and pathways. Mol Cell Neurosci 55:44–49. [DOI] [PubMed] [Google Scholar]
- 23. Crippa V, Sau D, Rusmini P, Boncoraglio A, Onesto E, Bolzoni E et al (2010) The small heat shock protein B8 (HspB8) promotes autophagic removal of misfolded proteins involved in amyotrophic lateral sclerosis (ALS). Hum Mol Genet 19:3440–3456. [DOI] [PubMed] [Google Scholar]
- 24. Deng HX, Bigio EH, Zhai H, Fecto F, Ajroud K, Shi Y et al (2011) Differential involvement of optineurin in amyotrophic lateral sclerosis with or without SOD1 mutations. Arch Neurol 68:1057–1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Dewil M, dela Cruz VF, Van Den Bosch L, Robberecht W (2007) Inhibition of p38 mitogen activated protein kinase activation and mutant SOD1(G93A)‐induced motor neuron death. Neurobiol Dis 26:332–341. [DOI] [PubMed] [Google Scholar]
- 26. Dibaj P, Steffens H, Zschuntzsch J, Nadrigny F, Schomburg ED, Kirchhoff F, Neusch C (2011) In vivo imaging reveals distinct inflammatory activity of CNS microglia versus PNS macrophages in a mouse model for ALS. PLoS One 6:e17910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Dobrowolny G, Aucello M, Rizzuto E, Beccafico S, Mammucari C, Boncompagni S et al (2008) Skeletal muscle is a primary target of SOD1G93A‐mediated toxicity. Cell Metab 8:425–436. [DOI] [PubMed] [Google Scholar]
- 28. Dupuis L, Loeffler JP (2009) Neuromuscular junction destruction during amyotrophic lateral sclerosis: insights from transgenic models. Curr Opin Pharmacol 9:341–346. [DOI] [PubMed] [Google Scholar]
- 29. Dupuis L, Pradat PF, Ludolph AC, Loeffler JP (2011) Energy metabolism in amyotrophic lateral sclerosis. Lancet Neurol 10:75–82. [DOI] [PubMed] [Google Scholar]
- 30. Fecto F, Yan J, Vemula SP, Liu E, Yang Y, Chen W et al (2011) SQSTM1 mutations in familial and sporadic amyotrophic lateral sclerosis. Arch Neurol 68:1440–1446. [DOI] [PubMed] [Google Scholar]
- 31. Fischer G, Wittmann‐Liebold B, Lang K, Kiefhaber T, Schmid FX (1989) Cyclophilin and peptidyl‐prolyl cis‐trans isomerase are probably identical proteins. Nature 337:476–478. [DOI] [PubMed] [Google Scholar]
- 32. Fitzgerald KC, O'Reilly EJ, Falcone GJ, McCullough ML, Park Y, Kolonel LN, Ascherio A (2014) Dietary omega‐3 polyunsaturated fatty acid intake and risk for amyotrophic lateral sclerosis. JAMA Neurol 71:1102–1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Gaudet AD, Popovich PG, Ramer MS (2011) Wallerian degeneration: gaining perspective on inflammatory events after peripheral nerve injury. J Neuroinflammation 8:110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ghezzi P, Bonetto V (2003) Redox proteomics: identification of oxidatively modified proteins. Proteomics 3:1145–1153. [DOI] [PubMed] [Google Scholar]
- 35. Gill A, Kidd J, Vieira F, Thompson K, Perrin S (2009) No benefit from chronic lithium dosing in a sibling‐matched, gender balanced, investigator‐blinded trial using a standard mouse model of familial ALS. PLoS One 4:e6489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gould TW, Buss RR, Vinsant S, Prevette D, Sun W, Knudson CM et al (2006) Complete dissociation of motor neuron death from motor dysfunction by Bax deletion in a mouse model of ALS. J Neurosci 26:8774–8786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Greenway MJ, Andersen PM, Russ C, Ennis S, Cashman S, Donaghy C et al (2006) ANG mutations segregate with familial and 'sporadic' amyotrophic lateral sclerosis. Nat Genet 38:411–413. [DOI] [PubMed] [Google Scholar]
- 38. Gurlo T, von Grafenstein H (2003) Antigen‐independent cross‐talk between macrophages and CD8+ T cells facilitates their cooperation during target destruction. Int Immunol 15:1063–1071. [DOI] [PubMed] [Google Scholar]
- 39. Gurney ME (1997) The use of transgenic mouse models of amyotrophic lateral sclerosis in preclinical drug studies. J Neurol Sci 152(Suppl. 1):S67–73. [DOI] [PubMed] [Google Scholar]
- 40. Gurney ME, Pu H, Chiu AY, Dal Canto MC, Polchow CY, Alexander DD et al (1994) Motor neuron degeneration in mice that express a human Cu, Zn superoxide dismutase mutation. Science 264:1772–1775. [DOI] [PubMed] [Google Scholar]
- 41. Hanes SD (2015) Prolyl isomerases in gene transcription. Biochim Biophys Acta 1850:2017–2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Heiman‐Patterson TD, Sher RB, Blankenhorn EA, Alexander G, Deitch JS, Kunst CB et al (2011) Effect of genetic background on phenotype variability in transgenic mouse models of amyotrophic lateral sclerosis: a window of opportunity in the search for genetic modifiers. Amyotroph Lateral Scler 12:79–86. [DOI] [PubMed] [Google Scholar]
- 43. Heurich B, El Idrissi NB, Donev RM, Petri S, Claus P, Neal J et al (2011) Complement upregulation and activation on motor neurons and neuromuscular junction in the SOD1 G93A mouse model of familial amyotrophic lateral sclerosis. J Neuroimmunol 235:104–109. [DOI] [PubMed] [Google Scholar]
- 44. Johnson JO, Pioro EP, Boehringer A, Chia R, Feit H, Renton AE et al (2014) Mutations in the Matrin 3 gene cause familial amyotrophic lateral sclerosis. Nat Neurosci 17:664–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Joseph MS, Bilousova T, Zdunowski S, Wu ZP, Middleton B, Boudzinskaia M et al (2011) Transgenic mice with enhanced neuronal major histocompatibility complex class I expression recover locomotor function better after spinal cord injury. J Neurosci Res 89:365–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Joyce PI, Fratta P, Fisher EM, Acevedo‐Arozena A (2011) SOD1 and TDP‐43 animal models of amyotrophic lateral sclerosis: recent advances in understanding disease toward the development of clinical treatments. Mamm Genome 22:420–448. [DOI] [PubMed] [Google Scholar]
- 47. Kabashi E, Valdmanis PN, Dion P, Spiegelman D, McConkey BJ, Vande Velde C et al (2008) TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat Genet 40:572–574. [DOI] [PubMed] [Google Scholar]
- 48. Kiefer R, Kieseier BC, Stoll G, Hartung HP (2001) The role of macrophages in immune‐mediated damage to the peripheral nervous system. Prog Neurobiol 64:109–127. [DOI] [PubMed] [Google Scholar]
- 49. Kiselycznyk C, Holmes A (2011) All (C57BL/6) mice are not created equal. Front Neurosci 5:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Koppers M, Blokhuis AM, Westeneng HJ, Terpstra ML, Zundel CA, Vieira de Sa R et al (2015) C9orf72 ablation in mice does not cause motor neuron degeneration or motor deficits. Ann Neurol 78:426–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kwiatkowski TJ Jr, Bosco DA, Leclerc AL, Tamrazian E, Vanderburg CR, Russ C et al (2009) Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 323:1205–1208. [DOI] [PubMed] [Google Scholar]
- 52. Lauranzano E, Pozzi S, Pasetto L, Stucchi R, Massignan T, Paolella K et al (2015) Peptidylprolyl isomerase A governs TARDBP function and assembly in heterogeneous nuclear ribonucleoprotein complexes. Brain 138(Pt 4):974–991. [DOI] [PubMed] [Google Scholar]
- 53. Le Ber I, De Septenville A, Millecamps S, Camuzat A, Caroppo P, Couratier P et al (2015) TBK1 mutation frequencies in French frontotemporal dementia and amyotrophic lateral sclerosis cohorts. Neurobiol Aging 36:3116 e5–8. [DOI] [PubMed] [Google Scholar]
- 54. Lee JP, Palfrey HC, Bindokas VP, Ghadge GD, Ma L, Miller RJ, Roos RP (1999) The role of immunophilins in mutant superoxide dismutase‐1linked familial amyotrophic lateral sclerosis. Proc Natl Acad Sci USA 96:3251–3256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Lever TE, Gorsek A, Cox KT, O'Brien KF, Capra NF, Hough MS, Murashov AK (2009) An animal model of oral dysphagia in amyotrophic lateral sclerosis. Dysphagia 24:180–195. [DOI] [PubMed] [Google Scholar]
- 56. Liblau RS, Gonzalez‐Dunia D, Wiendl H, Zipp F (2013) Neurons as targets for T cells in the nervous system. Trends Neurosci 36:315–324. [DOI] [PubMed] [Google Scholar]
- 57. Lobsiger CS, Boillee S, Pozniak C, Khan AM, McAlonis‐Downes M, Lewcock JW, Cleveland DW (2013) C1q induction and global complement pathway activation do not contribute to ALS toxicity in mutant SOD1 mice. Proc Natl Acad Sci USA 110:E4385–4392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Lu CH, Petzold A, Kalmar B, Dick J, Malaspina A, Greensmith L (2012) Plasma neurofilament heavy chain levels correlate to markers of late stage disease progression and treatment response in SOD1(G93A) mice that model ALS. PLoS One 7:e40998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Ludolph AC, Bendotti C, Blaugrund E, Chio A, Greensmith L, Loeffler JP et al (2010) Guidelines for preclinical animal research in ALS/MND: a consensus meeting. Amyotroph Lateral Scler 11:38–45. [DOI] [PubMed] [Google Scholar]
- 60. Luigetti M, Lattante S, Zollino M, Conte A, Marangi G, Del Grande A, Sabatelli M (2011) SOD1 G93D sporadic amyotrophic lateral sclerosis (SALS) patient with rapid progression and concomitant novel ANG variant. Neurobiol Aging 32:1924 e15–8. [DOI] [PubMed] [Google Scholar]
- 61. Magrane J, Manfredi G (2009) Mitochondrial function, morphology, and axonal transport in amyotrophic lateral sclerosis. Antioxid Redox Signal 11:1615–1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Mancuso R, Navarro X (2015) Amyotrophic lateral sclerosis: current perspectives from basic research to the clinic. Prog Neurobiol 133:1–26. [DOI] [PubMed] [Google Scholar]
- 63. Mancuso R, Olivan S, Mancera P, Pasten‐Zamorano A, Manzano R, Casas C et al (2012) Effect of genetic background on onset and disease progression in the SOD1‐G93A model of amyotrophic lateral sclerosis. Amyotroph Lateral Scler 13:302–310. [DOI] [PubMed] [Google Scholar]
- 64. Marin B, Couratier P, Arcuti S, Copetti M, Fontana A, Nicol M et al (2016) Stratification of ALS patients' survival: a population‐based study. J Neurol 263: 100–111. [DOI] [PubMed] [Google Scholar]
- 65. Marino M, Papa S, Crippa V, Nardo G, Peviani M, Cheroni C et al (2015) Differences in protein quality control correlate with phenotype variability in 2 mouse models of familial amyotrophic lateral sclerosis. Neurobiol Aging 36:492–504. [DOI] [PubMed] [Google Scholar]
- 66. Maruyama H, Morino H, Ito H, Izumi Y, Kato H, Watanabe Y et al (2010) Mutations of optineurin in amyotrophic lateral sclerosis. Nature 465:223–226. [DOI] [PubMed] [Google Scholar]
- 67. Massignan T, Casoni F, Basso M, Stefanazzi P, Biasini E, Tortarolo M et al (2007) Proteomic analysis of spinal cord of presymptomatic amyotrophic lateral sclerosis G93A SOD1 mouse. Biochem Biophys Res Commun 353:719–725. [DOI] [PubMed] [Google Scholar]
- 68. McGoldrick P, Joyce PI, Fisher EM, Greensmith L (2013) Rodent models of amyotrophic lateral sclerosis. Biochim Biophys Acta 1832:1421–1436. [DOI] [PubMed] [Google Scholar]
- 69. Milton RH, Abeti R, Averaimo S, DeBiasi S, Vitellaro L, Jiang L et al (2008) CLIC1 function is required for beta‐amyloid‐induced generation of reactive oxygen species by microglia. J Neurosci 28:11488–11499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Mitsumoto H, Brooks BR, Silani V (2014) Clinical trials in amyotrophic lateral sclerosis: why so many negative trials and how can trials be improved? Lancet Neurol 13:1127–1138. [DOI] [PubMed] [Google Scholar]
- 71. Mori MA, Liu M, Bezy O, Almind K, Shapiro H, Kasif S, Kahn CR (2010) A systems biology approach identifies inflammatory abnormalities between mouse strains prior to development of metabolic disease. Diabetes 59:2960–2971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Nardo G, Pozzi S, Mantovani S, Garbelli S, Marinou K, Basso M et al (2009) Nitroproteomics of peripheral blood mononuclear cells from patients and a rat model of ALS. Antioxid Redox Signal 11:1559–1567. [DOI] [PubMed] [Google Scholar]
- 73. Nardo G, Pozzi S, Pignataro M, Lauranzano E, Spano G, Garbelli S et al (2011) Amyotrophic lateral sclerosis multiprotein biomarkers in peripheral blood mononuclear cells. PLoS One 6:e25545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Nardo G, Iennaco R, Fusi N, Heath PR, Marino M, Trolese MC et al (2013) Transcriptomic indices of fast and slow disease progression in two mouse models of amyotrophic lateral sclerosis. Brain 136(Pt 11):3305–3332. [DOI] [PubMed] [Google Scholar]
- 75. Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT et al (2006) Ubiquitinated TDP‐43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314:130–133. [DOI] [PubMed] [Google Scholar]
- 76. Nishimura AL, Mitne‐Neto M, Silva HC, Richieri‐Costa A, Middleton S, Cascio D et al (2004) A mutation in the vesicle‐trafficking protein VAPB causes late‐onset spinal muscular atrophy and amyotrophic lateral sclerosis. Am J Hum Genet 75:822–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Novarino G, Fabrizi C, Tonini R, Denti MA, Malchiodi‐Albedi F, Lauro GM et al (2004) Involvement of the intracellular ion channel CLIC1 in microglia‐mediated beta‐amyloid‐induced neurotoxicity. J Neurosci 24:5322–5330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Oliveira AL, Thams S, Lidman O, Piehl F, Hokfelt T, Karre K et al (2004) A role for MHC class I molecules in synaptic plasticity and regeneration of neurons after axotomy. Proc Natl Acad Sci USA 101:17843–17848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. O'Rourke JG, Bogdanik L, Muhammad AK, Gendron TF, Kim KJ, Austin A et al (2015) C9orf72 BAC transgenic mice display typical pathologic features of ALS/FTD. Neuron 88:892–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Otero MG, Alloatti M, Cromberg LE, Almenar‐Queralt A, Encalada SE, Pozo Devoto VM et al (2014) Fast axonal transport of the proteasome complex depends on membrane interaction and molecular motor function. J Cell Sci 127(Pt 7):1537–1549. [DOI] [PubMed] [Google Scholar]
- 81. Perrin S (2014) Preclinical research: make mouse studies work. Nature 507:423–425. [DOI] [PubMed] [Google Scholar]
- 82. Peters OM, Cabrera GT, Tran H, Gendron TF, McKeon JE, Metterville J et al (2015) Human C9ORF72 hexanucleotide expansion reproduces RNA foci and dipeptide repeat proteins but not neurodegeneration in BAC transgenic mice. Neuron 88:902–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Pfohl SR, Halicek MT, Mitchell CS (2015) Characterization of the contribution of genetic background and gender to disease progression in the SOD1 G93A mouse model of amyotrophic lateral sclerosis: a meta‐analysis. J Neuromuscul Dis 2:137–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Philips T, Rothstein JD (2015) Rodent models of amyotrophic lateral sclerosis. Curr Protoc Pharmacol 69:5 67 1–5 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Pizzasegola C, Caron I, Daleno C, Ronchi A, Minoia C, Carri MT, Bendotti C (2009) Treatment with lithium carbonate does not improve disease progression in two different strains of SOD1 mutant mice. Amyotroph Lateral Scler 10:221–228. [DOI] [PubMed] [Google Scholar]
- 86. Pupillo E, Messina P, Logroscino G, Beghi E (2014) Long‐term survival in amyotrophic lateral sclerosis: a population‐based study. Ann Neurol 75:287–297. [DOI] [PubMed] [Google Scholar]
- 87. Regal L, Vanopdenbosch L, Tilkin P, Van den Bosch L, Thijs V, Sciot R, Robberecht W (2006) The G93C mutation in superoxide dismutase 1: clinicopathologic phenotype and prognosis. Arch Neurol 63:262–267. [DOI] [PubMed] [Google Scholar]
- 88. Renton AE, Chio A, Traynor BJ (2014) State of play in amyotrophic lateral sclerosis genetics. Nat Neurosci 17:17–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Riboldi G, Nizzardo M, Simone C, Falcone M, Bresolin N, Comi GP, Corti S (2011) ALS genetic modifiers that increase survival of SOD1 mice and are suitable for therapeutic development. Prog Neurobiol 95:133–148. [DOI] [PubMed] [Google Scholar]
- 90. Ripps ME, Huntley GW, Hof PR, Morrison JH, Gordon JW (1995) Transgenic mice expressing an altered murine superoxide dismutase gene provide an animal model of amyotrophic lateral sclerosis. Proc Natl Acad Sci USA 92:689–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A et al (1993) Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 362:59–62. [DOI] [PubMed] [Google Scholar]
- 92. Rouaux C, Panteleeva I, Rene F, Gonzalez de Aguilar JL, Echaniz‐Laguna A et al (2007) Sodium valproate exerts neuroprotective effects in vivo through CREB‐binding protein‐dependent mechanisms but does not improve survival in an amyotrophic lateral sclerosis mouse model. J Neurosci 27:5535–5545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Scott S, Kranz JE, Cole J, Lincecum JM, Thompson K, Kelly N et al (2008) Design, power, and interpretation of studies in the standard murine model of ALS. Amyotroph Lateral Scler 9:4–15. [DOI] [PubMed] [Google Scholar]
- 94. Seijffers R, Zhang J, Matthews JC, Chen A, Tamrazian E, Babaniyi O et al (2014) ATF3 expression improves motor function in the ALS mouse model by promoting motor neuron survival and retaining muscle innervation. Proc Natl Acad Sci USA 111:1622–1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Simon NG, Turner MR, Vucic S, Al‐Chalabi A, Shefner J, Lomen‐Hoerth C, Kiernan MC (2014) Quantifying disease progression in amyotrophic lateral sclerosis. Ann Neurol 76:643–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Specht CG, Schoepfer R (2004) Deletion of multimerin‐1 in alpha‐synuclein‐deficient mice. Genomics 83:1176–1178. [DOI] [PubMed] [Google Scholar]
- 97. Thams S, Brodin P, Plantman S, Saxelin R, Karre K, Cullheim S (2009) Classical major histocompatibility complex class I molecules in motoneurons: new actors at the neuromuscular junction. J Neurosci 29:13503–13515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Turner BJ, Talbot K (2008) Transgenics, toxicity and therapeutics in rodent models of mutant SOD1‐mediated familial ALS. Prog Neurobiol 85:94–134. [DOI] [PubMed] [Google Scholar]
- 99. Vance C, Rogelj B, Hortobagyi T, De Vos KJ, Nishimura AL, Sreedharan J et al (2009) Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 323:1208–1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Williams KL, Solski JA, Nicholson GA, Blair IP (2012) Mutation analysis of VCP in familial and sporadic amyotrophic lateral sclerosis. Neurobiol Aging 33:1488 e15–16. [DOI] [PubMed] [Google Scholar]
- 101. Xie Y, Zhou B, Lin MY, Wang S, Foust KD, Sheng ZH (2015) Endolysosomal deficits augment mitochondria pathology in spinal motor neurons of asymptomatic fALS Mice. Neuron 87:355–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Yanamadala V, Friedlander RM (2010) Complement in neuroprotection and neurodegeneration. Trends Mol Med 16:69–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Yang Y, Hentati A, Deng HX, Dabbagh O, Sasaki T, Hirano M et al (2001) The gene encoding alsin, a protein with three guanine‐nucleotide exchange factor domains, is mutated in a form of recessive amyotrophic lateral sclerosis. Nat Genet 29:160–165. [DOI] [PubMed] [Google Scholar]
- 104. Yip PK, Pizzasegola C, Gladman S, Biggio ML, Marino M, Jayasinghe M et al (2013) The omega‐3 fatty acid eicosapentaenoic acid accelerates disease progression in a model of amyotrophic lateral sclerosis. PLoS One 8:e61626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Zhao ZH, Chen WZ, Wu ZY, Wang N, Zhao GX, Chen WJ, Murong SX (2009) A novel mutation in the senataxin gene identified in a Chinese patient with sporadic amyotrophic lateral sclerosis. Amyotroph Lateral Scler 10:118–122. [DOI] [PubMed] [Google Scholar]
- 106. Zurita E, Chagoyen M, Cantero M, Alonso R, Gonzalez‐Neira A, Lopez‐Jimenez A et al (2011) Genetic polymorphisms among C57BL/6 mouse inbred strains. Transgenic Res 20:481–489. [DOI] [PubMed] [Google Scholar]