

Abstract
CADASIL and CARASIL are hereditary small vessel diseases leading to vascular dementia. CADASIL commonly begins with migraine followed by minor strokes in mid‐adulthood. Dominantly inherited CADASIL is caused by mutations (n > 230) in NOTCH 3 gene, which encodes Notch3 receptor expressed in vascular smooth muscle cells (VSMC). Notch3 extracellular domain (N3ECD) accumulates in arterial walls followed by VSMC degeneration and subsequent fibrosis and stenosis of arterioles, predominantly in cerebral white matter, where characteristic ischemic MRI changes and lacunar infarcts emerge. The likely pathogenesis of CADASIL is toxic gain of function related to mutation‐induced unpaired cysteine in N3ECD. Definite diagnosis is made by molecular genetics but is also possible by electron microscopic demonstration of pathognomonic granular osmiophilic material at VSMCs or by positive immunohistochemistry for N3ECD in dermal arteries.
In rare, recessively inherited CARASIL the clinical picture and white matter changes are similar as in CADASIL, but cognitive decline begins earlier. In addition, gait disturbance, low back pain and alopecia are characteristic features. CARASIL is caused by mutations (presently n = 10) in high‐temperature requirement. A serine peptidase 1 (HTRA 1) gene, which result in reduced function of HTRA1 as repressor of transforming growth factor‐β (TGF β) ‐signaling. Cerebral arteries show loss of VSMCs and marked hyalinosis, but not stenosis.
Keywords: CADASIL, CARASIL
Introduction
The acronyms CADASIL and CARASIL—two hereditary small vessel diseases (SVD)—differ from each other only by D vs. R (cerebral autosomal dominant vs. recessive arteriopathy with subcortical infarcts and leukoencephalopathy), which may confuse at least those who less actively follow the SVD literature. The acronyms imply a difference only in the mode of inheritance—dominant vs. recessive—however, their gene defects, clinical pictures, imaging findings and pathologies do differ from each other to such an extent that alert clinicians, radiologists and pathologists should be able to distinguish these two SVD entities, the definite diagnoses of which would then emerge at the latest in molecular genetic analyses. In this article, we describe first for CADASIL and thereafter for CARASIL the basic clinical findings, genetics, biochemistry, pathologies and suggested pathogeneses of these two entities with some similarities but at the same time major differences. Finally, the present situation of and prospects for evidence‐based therapeutics is discussed.
History
The first CADASIL family—later verified by genetic analysis of old tissue specimens—was reported by van Bogaert as “hereditary Binswanger's disease” in 1955 128. Thereafter, the disease entities resembling the present CADASIL received many different names (e.g. chronic familial vascular encephalopathy, Familiare zerebrale Arteriosklerose, Familiäre zerebrale Gefässerkrankung, familial disorder with subcortical ischemic strokes, dementia and leukoencephalopathy and familial Binswanger's syndrome). With the knowledge of that time, it is not surprising that CARASIL (also known as Maeda syndrome) in 1960 was described by Nemoto similarly as a form of Binswanger syndrome and then published by Maeda et al in 1976 81 as multifocal softenings in the cerebral white matter caused by cerebral angiitis (Binswanger's disease). The discovery of the causative gene defects then finally and definitely specified these two SVDs: For CADASIL, the linkage to chromosome 19 was identified in 1993 125 and the causative NOTCH3 gene on 19p13 was discovered in 1996 54. For CARASIL, the defective HTRA1 gene on chromosome 10q25 was determined in 2009 45.
Epidemiology
CADASIL
After the gene defect of CADASIL was established and doctors became better aware of this entity, the number of both reported and diagnosed cases increased rapidly. At present, CADASIL is the most common hereditary vascular dementia and it has been reported in all ethnic groups from Far West to Far East. However, exact surveys of its prevalence are few. In west Scotland, the prevalence of patients with definite diagnosis in 2005 was 1.98/100 000 and that of predicted mutation carriers was 4.14/100 000 105. In Northeast England, in an outbred Western European population, the minimum prevalence for definite cases in 2012 was 1.32/100 000 95. In geographic locations, where the population has a more limited genetic background, such as Finland and Gran Canaria island, ancestor phenomena and/or exceptionally high prevalences have been recorded. In Finland (population about 5 million), the dominant mutation p.Arg133Cys was introduced in the late 1600s or early 1700s 93, the prevalence being about 4/100 000. The ancestor effect is still more striking on the island of Gran Canaria, where the prevalence reaches 14/100 000 (121 patients with p.Arg207Cys mutation in a population of about 900 000; Drs. A Santana and J Delgado, pers. comm.). In Canada and United States, the prevalence numbers have been surprisingly low (in most Canadian provinces < 1/100 000, Dr. V Saly, pers. comm.) considering the high standard of neurology in those countries. Another approach by analyzing the number of CADASIL cases among patients with lacunar infarcts and leukoaraiosis gave frequencies of 11% (one out of nine) for patients under 50 years and 2% (one out of 48) for patients under 65 years of age 32. In a Korean study of 151 consecutive acute ischemic strokes (mean age 70 years), 4.0% (n = 6) were diagnosed to suffer from CADASIL 21. The percentages might be somewhat higher, as these two last mentioned studies used only a limited screening test for NOTCH3 mutations.
CARASIL
CARASIL is a very rare disease. Until recently, only somewhat over 50 cases have been reported 39, 97, 48 from Japan, three from China 19, 131 and one case each from Spain 83, Romania 9 and Turkey 8. With the growing awareness also outside the far eastern countries, more families are expected to be diagnosed and reported.
Genetics and Biochemistry
CADASIL
CADASIL is—as its name implies—autosomally dominantly inherited, and only a few patients with definite de novo mutations have been reported 55. The defective gene is NOTCH3 in 19p13.1–13.2. The 33 exons of NOTCH3 encode a single pass transmembrane heterodimer receptor protein Notch3 (N3) comprising 2321 amino acids. Similarly, as all Notch receptors, Notch3 is synthesized as a full‐length protein (Figure 1), which is subsequently proteolytically cleaved (S1 cleavage) by furin into two parts. The N‐terminal extracellular domain (N3ECD, 210 kDa) is composed of 34 epidermal growth factor‐like repeats (EGFr), each of which encompasses six cysteine residues, followed by three Notch/Lin12 repeats. At the heterodimerization domains (HD‐N and HD‐C), N3ECD is non‐covalently connected to the 97 kDa C‐terminal part, which comprises a transmembrane (N3TMD) and an intracellular domain (N3ICD; together N3TMIC). N3ICD contains seven ankyrin repeats (Figure 1).
Figure 1.
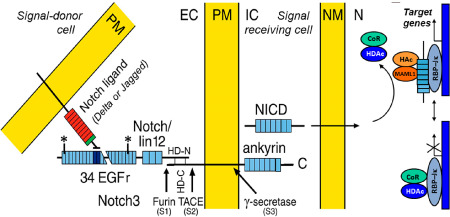
A schematic presentation of Notch signaling. Notch3 is constitutively cleaved by furin (S1) and the bipartite molecule is inserted to the plasma membrane (PM). The extracellular domain (N3ECD) consists of 34 epidermal growth factor (EGF)‐like repeats, followed by three notch/lin‐12 repeats, a transmembrane domain and an intracellular domain (NICD), which contains seven ankyrin repeats. The binding site of the ligand (in human Delta or Jagged) is at EGF repeats 10–11. Upon binding, N3ECD is cleaved external to the intramembranous domain (S2). Thereafter occurs the intramembranous S3 cleavage and NICD enters the nucleus to release the repressor molecules CoR and HDAc, and it binds to a transcription regulator of CSL family, RBP‐Jκ, and activates transcription. CADASIL mutations are in EGF repeats 1–32 (exons 2–23) between the asterisks. EC = extracellular space, IC = intracellular space, NM = nuclear membrane, N = nucleus, TACE = tumor necrosis factor α‐converting enzyme, HD = heterodimerization domain, CoR and HDAc = repressors, HAc and MAML1 = components of an activation complex.
Notch3 belongs to the Notch family (four members in mammals), whose signaling pathways are indispensable during development of most organs. Their importance is reflected by the presence of orthologous genes with high degree of homology from nematodes to man. Notch repertoire during organogenesis, including vasculogenesis, covers for example stem cell renewal, cell proliferation, determination of cell fate and differentiation and apoptosis. The exact function of Notch3 is not known in adult humans, in whom Notch3 is expressed predominantly in VSMCs 56, 103, the molecular phenotype and metabolic characteristics of which cells appear to be different in different vascular beds (for details see succeeding sections).
The Delta/Serrate/Lag‐2 (DSL) ligands of Notch receptors belong to the Delta‐like family (DLL1, 3 and 4) and Serrate family Jagged (Jag 1 and 2). They have similar molecular structure as Notch receptors, that is they are transmembrane proteins, the extracellular domain of which contains several EGFrs. Although both the ligands and Notch receptors are presented on the surface of vascular smooth muscle cells (VSMC), the ligands transactivate Notch receptor on the neighboring cells, whereas Notch receptors expressed on the same cell are cis‐inhibited 35. The ligand‐binding site in Notch3 is at EGFr 10–11. Upon binding of its ligand, N3ECD is non‐enzymatically dissociated from the heterodimeric Notch3 and the bound ligand and N3ECD are transendocytosed into the ligand‐expressing cell. Thereby the membrane‐bound C‐terminal part of Notch3 (composed of N3TMD and N3ICD) becomes exposed for cleavage by ADAM10 or by ADAM17 (also known as TNF‐alpha converting enzyme; TACE) just outside the plasma membrane (S2 cleavage), whereafter the final S3 cleavage by γ‐secretase (the same enzyme as in Alzheimer's disease) can occur within the membrane to detach N3ICD from N3TMD 11, 96. The transendocytosed non‐mutated N3ECD is degraded in the ligand‐presenting cell. N3ICD enters the nucleus of the receptor‐presenting cell and interacts with RBP‐Jκ and co‐activators to activate transcription of its target genes 37, 38 (Figure 1).
The pathogenic mutations—so far over 230 different reported—are located in exons 2–24 in the N3ECD, with prominent clustering in exons 3 and 4 encoding EGFr 2–5 (over 40% of mutations in over 70% of families occur in these exons). A great majority of mutations (>95%) are missense mutations, which cause either substitution of a wild type (wt) cysteine with another amino acid or vice versa. In addition, eight different deletions, two duplications 70, 82 and two splice site mutations 57, 112 have been reported. Remarkably, all these mutations result in an uneven number of cysteine residues in the affected EGFr, which situation appears to be crucial in the pathogenesis of CADASIL. However, at least 11 patients/families, whose clinical picture was considered compatible with CADASIL and who carried a non‐cysteine mutation in NOTCH3, have been reported. Besides, we have been consulted about several such patients. Whether the CADASIL‐like disease in these patients has a causal relationship with the non‐cysteine NOTCH3 mutation has been debated and remains to be investigated 23, 24, 88 (see also Pathogenesis section).
At present, five patients with homozygous cysteine mutations in NOTCH3 [Tuominen et al: p.Arg133Cys 126, Liem et al: p.Arg578Cys 78, Ragno et al: p.Gly528Cys 104, Soong et al: p.Arg544Cys 118 and Vinciguerra et al: p.Cys183Ser 130] have been reported.
CARASIL
Analogously, CARASIL is—as its name implies—autosomally recessively inherited and, thus, mutations on both alleles, either homozygous or compound heterozygous, are required to cause the disease.
The gene defect of CARASIL was discovered collaboratively by a large Japanese group of researchers directed by Professors O. Onodera and S. Tsuji, the majority of the participants being based at Niigata University 45. Five families with clinical CARASIL were enrolled and they were first analyzed by genome‐wide linkage and thereafter the defective gene was fine mapped to a 2.4‐Mb region on chromosome 10q. High‐temperature requirement A serine peptidase 1 (HTRA1) was selected as a candidate gene because it is expressed in the blood vessels, skin and bone. HTRA1 in human is composed of nine exons encoding a protein consisting of 480 residues and comprising four functional domains (Figure 2). The exons 3–6 encode the main domain of a trypsin‐like serine protease that represses signaling by transforming growth factor‐β (TGF‐β) family members 45.
Figure 2.
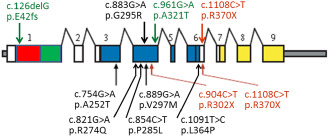
A schematic presentation of HTRA 1 mutations. The mutations reported in Far‐Eastern patients are presented below the gene and those in Caucasian patients are presented above the gene. Missense mutations are presented in black, nonsense mutations in red and the compound heterozygous mutation in green. [Domains of HTRA1. Red: Insulin‐like growth factor binding protein domain. Green: Kazai‐type serine protease inhibitor domain. Blue: Trypsin‐like serine protease domain. Yellow: PDZ domain. Scheme modified after Hara et al 45; for details see Table 1].
The sequence analysis by Hara et al 45 revealed in the serine protease domain of HTRA1 four homozygous mutations: two missense and two nonsense mutations (Table 1 and Figure 2), which were not discovered in 125 controls. Thereafter (at least), four novel missense mutations have been reported and among them the first Caucasian patient 83 (Table 1). Another Caucasian patient from Turkey 8 carried the same nonsense mutation as one of the original families of Hara et al 45. The first and only reported compound heterozygous patient was also Caucasian 9 (Table 1); this individual carried a classic missense mutation in exon 4 of HTRA1 paired with an exceptional single nucleotide deletion in exon 1 that causes a frameshift.
Table 1.
CARASIL mutations. Abbreviations: homoz = homozygous; heteroz = heterozygous; ms = missense; ns = nonsense; del = deletion
| E | Mutation type | Nucleotide | Protein | Ethnicity | Publication |
|---|---|---|---|---|---|
| Far‐Eastern patients | |||||
| 3 | homoz, ms | c.754G > A | p.Ala252Thr | Japanese | Hara et al N Engl J Med 2009; 360: 1729–1739. 45 |
| 4 | homoz, ms | c.889G > A | p.Val297Met | ||
| 4 | homoz, ns | c.904C > T | p.Arg302X | ||
| 6 | homoz, ns | c.1108C > T | p.Arg370X | ||
| 4 | homoz, ms | c.821G > A | p.Arg274Gln | Japanese | Nishimoto et al Neurology 2011; 76:1353–1355. |
| 4 | homoz, ms | c.854C > T | p.Pro285Leu | Chinese | Chen et al J Int Med Res 2013; 41:1445–1455. 19 |
| 6 | homoz, ms | c.1091T > C | p.Leu364Pro | Chinese | Wang et al CNS Neurosci Ther 2012; 18: 867–869. 131 |
| Caucasian patients | |||||
| 4 | homoz, ms | c.883G > A | p.Gly295Arg | Spanish | Mendioroz et al Neurology; 2010; 75: 2033–2035. 83 |
| 6 | homoz, ns | c.1108C > T | p.Arg370X | Turkish | Bayrakli et al Turk Neurosurg 2014; 24:67–69. 8 |
|
1 4 |
compound heteroz, del & ms |
c.126delG & c.961G > A |
p.Glu42fs & p.Ala321Thr |
Romanian | Bianchi et al Neurology 2014; 82:898–900. 9 |
HTRA1 belongs to the family of high‐temperature requirement A serine proteases, which in mammals participates in a variety of physiological processes, such as cell signaling and protein degradation, and is associated with development of the musculoskeletal system. For CARASIL, important repression of TGF‐β signaling occurs as follows 98, 116, 124: TGF‐β1 is synthesized as a pre‐pro‐protein. In the endoplasmic reticulum, proTGF‐β1 is dimerized and in trans‐Golgi network (TGN), proTGF‐β1 is then cleaved by furin convertase. The two cleaved products of proTGF‐β1, latency‐associated peptide (LAP) and the mature TGF‐β1 peptide, are connected non‐covalently to yield a small latent TGF‐β1 complex. This small latent TGF‐β1 complex is bound to LTBP1 by LAP forming the large latent TGF‐β1 complex, which complex is secreted and sequestered in the extracellular matrix (ECM). Different proteases/integrins in the ECM can cleave LAP to release the mature TGF‐β, which then can bind to its receptors. HTRA1 represses TGF‐β signaling by cleaving proTGF‐β1 in the endoplasmic reticulum (ER) and the cleaved products are degraded by ER‐associated protein degradation (ERAD), resulting in the reduction of mature TGF‐β1.
Hara et al 45 demonstrated that both missense mutations and one nonsense mutation (p.Arg302X) resulted in protein products with clearly lowered protease activity by 21%–50%, which was not enough to repress signaling by the TGF‐β family, whereas the other nonsense mutation (p.Arg370X) caused loss of HTRA1 protease activity by nonsense mediated decay of mRNA. Upregulated TGF‐β signaling is known to result in increased synthesis of ECM proteins with consequent vascular fibrosis 71. Both immunohistochemical analysis and in situ hybridization of the cerebral small arteries in CARASIL patients showed increased expression of fibronectin and versican in the thickened tunica intima and of TGF‐β1 in the tunica media.
Clinical Pictures
Even though the gene defects are known, the exact pathogenesis, that is the mechanisms by which the defective pathogenic genes cause these two SVDs are still to be discovered. In spite of that, we have now an effective arsenal of both knowledge and machinery to diagnose these patients.
Clinical picture of CADASIL
As the clinical picture of CADASIL has been described in detail in several excellent reviews 2, 18, 28, 61, 62, 101, only a short summary of the clinical findings—relevant to the theme of this mini‐symposium—is described. The wide variability of the clinical picture, also between family members with the same mutation and even between monozygotic twins 94, is repeatedly emphasized.
There is a relative lack of classic vascular risk factors, but for example arterial hypertension, hypercholesterolemia, smoking and diabetes mellitus are known to impact on CADASIL patients 2, 18, 28, 61, 62, 117. The four cardinal manifestations are:
-
(i)
Migraine with aura:
About 20%–40% of CADASIL patients manifest with this symptom, while up to 60% suffer from migrainous headache without aura. The average age of onset is around 30 years of age, although it may appear even before the age of 10 years, for example 18, 43, and besides, the aura may be exceptionally severe, for example 2, 18, 28, 43, 61, 129. Remarkably, in female patients, late pregnancy or puerperium often elicits their first attack of migraine with aura or aggravates such attacks 108.
-
(ii)
Ischemic attacks:
Ischemic strokes, often preceded by transient ischemic attacks (TIA, which are difficult to distinguish from severe migrainous aura) are the most common (up to 85% of symptomatic patients) manifestations. The age of first‐ever stroke varies vastly. The youngest reported patient with stroke was only 11 years of age 43, whereas some exceptional patients do not suffer their first‐ever stroke until they have reached their 70th birthday 133 (Miao et al unpublished). Such late‐onset cases may easily remain undiagnosed being considered “just common old age strokes”. The mean ages reported are around 40–50 years 2, 18, 28. The strokes are almost always subcortical and typically present as lacunar syndromes, such as pure motor or sensory deficits, dysarthria‐clumsy hand syndrome, expressive dysphasia or visual field defects. Major strokes because of larger infarcts in the supply territories of major cerebral arteries are rare, most likely coincidental, not related to CADASIL.
-
(iii)
Cognitive decline and dementia:
Impairment in executive function and working memory was recorded already before TIA and strokes. By testing for these two functions and for mental speed, non‐demented mutation carriers could be distinguished from controls. Episodic memory was relatively well preserved until late stage of the disease 3, 27, 101, 120, but eventually the patients develop a serious dementia of subcortical vascular type.
-
(iv)
Psychiatric manifestations:
20%–30% of CADASIL patients suffer from mood disturbances, the most common and serious being depression, which is expected in patients who have experienced the disease in their family and have an insight of their own disease. Apathy is a very common symptom; up to 41% of patients in a large French CADASIL cohort were apathetic. It appears to correlate with the patients' age, severity of cognitive impairment, other neuropsychiatric symptoms (often depression) and load of subcortical tissue lesions 106. Manic episodes are relatively uncommon (ca. 2%), although CADASIL may sometimes be mistaken for bipolar mood disorder before its true nature is discovered. Additional disturbances have been reported, including schizophrenia, psychosis, paranoia, aggression and dysthymia 14, 18, 28, 129.
Imaging in CADASIL
The imaging findings in SVDs including CADASIL are described in the article by Salamon in this mini‐symposium 113. Here, we present a few essential magnetic resonance imaging (MRI) findings [with reference to figures in the article 113] complemented by selected examinations applying special imaging methods.
White matter hyperintensities (WMHI) in temporopolar region and capsula externa on T2‐weighted (T2w) or Fluid‐Attenuated Inversion Recovery (FLAIR) MRI have been considered pathognomonic findings in CADASIL [figure 3B in 113]. These are complemented by periventricular, usually spotty leukoaraiosis, which gradually involves most of the cerebral WM [figure 4 in 113]. Interestingly, the prominence of the characteristic temporopolar WMHI may depend on the patients' ethnic background, being lesser in Chinese patients 72, 132. These MRI lesions are detectable already in the presymptomatic stage even before the age of 20 years and the youngest reported patient with subcortical foci of increased T2 hyperintensity was only 8 years old 48. The characteristic lacunar infarcts can be visualized in computerized tomography (CT) and in T1w or fluid‐attenuation inversion recovery (FLAIR) MRI as areas of decreased signal, predominantly in cerebral WM and deep grey matter (GM) [figure 3A in 113]. Lacunar infarct lesion load is the most important MRI parameter associated with cognitive dysfunction in CADASIL 77. Occurrence of simultaneous multiple acute infarcts, such as those usually caused by and ascribed to embolization, has been detected in CADASIL by diffusion‐weighted MRI 41.
Figure 3.
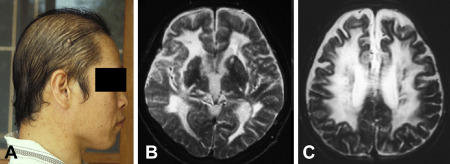
Loss of hair is clearly noticeable already at the age of 34 years in this male CARASIL patient (A). At the age of 33 years, T2‐weighted MRI disclosed diffuse leukoencephalopathy and lacunes (B and C).
Figure 4.
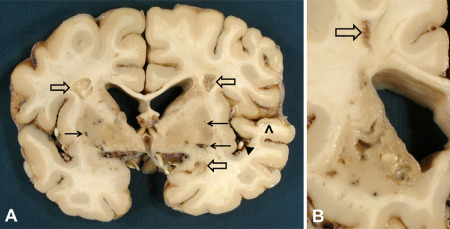
Three old infarcts of moderate size in cerebral white matter (open arrows) and smaller lacunar infarcts in putamen bilaterally (arrows) in a 65‐year‐old female CADASIL patient with p.Arg133Cys Notch3 mutation. Note the relatively spared cortex except for the ischemic lesion in temporal cortex (arrowhead) because of atherosclerotic occlusion of a middle cerebral artery branch [black triangle; (A)]. In this same patient's caudate nucleus, the lesions are more severe than in putamen. In the WM of the centrum semiovale, there is another small cystic infarct [open arrow; (B)]. Lateral ventricles in both (A) and (B) are widened because of ischemic tissue loss.
The above‐mentioned leukoaraiosis pattern implies a widely distributed diffuse leukoencephalopathy, which by diffusion tensor imaging (DTI) is detected as increased water diffusivity and loss of anisotropy, that is the diffusion of water can occur more freely in any direction. This indicates accumulation of fluid in enlarged extracellular space (vasogenic/interstitial brain edema) associated with microstructural pathology, myelin and axonal damage (see Histopathology post‐mortem section). Corresponding to such tissue damage, DTI results correlate better with the patients' clinical deterioration than T2w images 16. DTI alterations are also detectable in the normal‐appearing WM outside the T2w hyperintensities as a harbinger of incipient microstructural abnormalities.
In one‐third to two‐thirds of CADASIL patients, T2* (gradient echo) MRI discloses small hypointensities, microbleeds [figure 3A in 113], which represent perivascular accumulations of hemosiderin laden macrophages indicating focal extravasation of red blood cells (Figure 10). Microbleeds are clinically silent and their frequency was not associated with the severity of patient's cognitive dysfunction 73. Microbleeds are primarily located in the cerebral cortex, subcortical WM, basal ganglia and brainstem [figure 3a in 113; all outside ischemic lesions and 82% outside regions that are hyperintense in T2w MRI 29]. Their frequency increases with patients' age, blood pressure, volume of T2w lesions and antiaggregant therapy 29, 73. Microbleeds are not specific to CADASIL, but they also occur in other SVD of the central nervous system, such as arteriolosclerosis and amyloid angiopathy. They were considered to be largely an independent manifestation of the underlying angiopathy 29. Although direct association has not been reported, concerns that microbleeds may increase the risk of intracerebral hemorrhage have been expressed 73.
Figure 10.
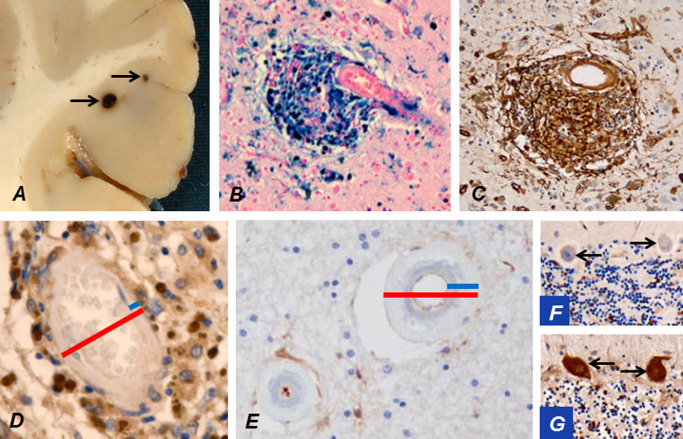
Occasionally microbleeds (arrows) in cerebral cortex may be visible already during brain cutting (A). An accumulation of hemosiderin around a cortical arteriole stains strongly positively for iron (B). The breached BBB at the microbleed leaks plasma fibrinogen (brown; C). The wall of a cortical arteriole (D) is markedly thinner than that of a WM arteriole (E; blue bars in D vs. E), while their outer diameters (red bars) are of approximately the same magnitude. This is a likely explanation for the predominant location of microbleeds in cortical and deep GM. The thick wall of a WM arteriole from a CADASIL patient at a quiescent stage of the disease has not allowed virtually any leakage of fibrinogen (E). Extravasated fibrinogen is normally taken up by Purkinje cells (PCs) from cerebrospinal fluid 51. Thus, negative fibrinogen staining of cerebellar PCs (arrows; F) verifies that significant leakage has not occurred in this patient at a quiescent stage, whereas in another CADASIL patient, who had suffered an infarct just before death, PCs (arrows) are strongly immunopositive for fibrinogen reflecting the breakdown of BBB (G).
As described in the succeeding sections, the brunt of brain pathology targets small arteries and arterioles. However, the arteries and arterioles in SVDs are too small to be specifically visualized by conventional MRI. The application of 7 tesla MRI 10 to in vivo visualization of the pathologies developing in small WM arteries in CADASIL and other SVDs will certainly provide interesting new data.
Clinical picture in CARASIL
The clinical features of CARASIL have been described in several articles 39, 40, 98, 136 (and many in Japanese), hence we present here again only an outline. Neurologic symptoms and signs are in many respects similar to those in CADASIL with lack of hypertension having been more explicitly emphasized in some reports on CARASIL 40. Half of the patients experience recurrent ischemic strokes, most often of the lacunar type. These lead to stepwise, progressive impairment of brain functions and finally to dementia, usually by the age of 30 to 40 years. Memory dysfunction in CARASIL appears to be more severe than in CADASIL. An interesting sign—visible to naked eye—is premature diffuse baldness, which predominantly concerns male patients (Figure 3A). Because the connective tissue is also affected in CARASIL, about 80% of the patients suffer from low back pain because of disk herniation and spondylosis deformans, usually between lower thoracic or upper lumbar levels. Cervical spondylosis is also often found.
Imaging in CARASIL
Brain MRI findings are in many respects similar to those in CADASIL 39, 98, 136. In T2w, MRI hyperintensities occur in periventricular and deep WM and sometimes even in anterior temporal WM and capsula externa, as is typical of CADASIL (Figure 3B and C). In CARASIL, the WM changes appear to develop more homogeneously than in CADASIL and the U‐fibers are relatively spared. Multiple lacunar infarct are also detected, predominantly in the basal ganglia and thalamus. On spinal MRI, cervical and lumbar spondylosis and disk degeneration become observable around the age of 30 years, often well after the onset of symptoms 98.
Circulatory Disturbances
CADASIL
The reduction of cerebral blood flow (CBF) in CADASIL has been found by applying several different imaging methods, such as positron emission tomography (PET) 15, 127, Doppler sonography 76 and MRI bolus tracking method 12, 17. Corresponding to the imaging findings and the sparing of cerebral cortex (see Pathology of CADASIL section), CBF is first and predominantly reduced in cerebral WM. This was detected by PET already at a presymptomatic stage, although it became more apparent at ages above 30 years, when also the strokes begin to appear. CBF in the cerebral cortex was not reduced until later ages and never to such an extent as in WM. Corresponding to the reduction in CBF, morphological studies have shown definite stenosis of WM arterioles (for details see Pathology of CADASIL section) 84. In a presymptomatic patient, the oxygen extraction fraction (OEF) was shown to increase indicating that the brain thereby attempts to compensate for the decreased CBF by increasing OEF 15. On the other hand, the cerebral metabolic rate of oxygen (CMRO2) 15 and glucose consumption (CMRgluc) 127 were not reduced until later stages parallel to the development of dementia and tissue loss, indicating that the early stage decrease in CBF is because of the arteriopathy instead of being just secondary to tissue loss.
Concordantly, by applying bolus tracking and Doppler methods, CBF and cerebral blood volume (CBV) were found to be reduced within T2w hyperintense areas in the WM, more severely in demented than in non‐demented patients 12, 17. Besides, in the normal‐appearing WM, a trend of reduced CBV was noticed 17. Furthermore, the hemodynamic reserve is reduced in CADASIL patients because acetazolamide induced a lesser increase in CBF in CADASIL patients' T2w hyperintense WM areas than in control subjects' WM 17. This is well understandable, as CADASIL patients' WM arteries have become stenosed and stiffened by fibrosis 84 (see Pathology of CADASIL section) and thus less responsive to acetazolamide. This may also be the cause of the prolonged arteriovenous cerebral transit time in both disabled and non‐disabled CADASIL patients as demonstrated by Liebetrau et al 76. Experimental studies have given similar results: transgenic mice expressing p.Arg90Cys‐mutant human NOTCH3 showed reduced responses to hypercapnia and acetazolamide, higher cerebrovascular resistance during hypertension and their lower limit of CBF autoregulation was shifted to higher blood pressures 69.
Retinal vasculature offers an exceptional and excellent in vivo view of arterioles. In CADASIL patients, retinal capillary blood flow was found to be mildly to moderately reduced 47. Furthermore, general arterial narrowing and arteriovenous nickings were common. Straightening of the retinal arterioles and a marked wall reflex occurred and the arterio‐venous (A/V) ratio was significantly lower than that in the controls. However, although CADASIL does induce a wide variety of ophthalmologic alterations, these resulted in only minimal functional disturbances and most often in no major ischemic injuries in the retina. Thus, the retina is relatively spared, which conclusion conforms with the results in the cerebral cortex, with which the retina and its arterioles are analogous 109.
As the arteriopathy in CADASIL is generalized, it is not surprising that vasoregulation is disturbed also in the systemic vasculature. Stenborg et al 119 showed in the forearm of CADASIL patients that endothelium‐dependent vasodilation is impaired in resistance arteries, but not in a large conduit artery, a. brachialis. This impairment results in reductions in both basal and stimulated blood flow. Similarly, Gobron et al 42 reported that after the release of forearm cuff occlusion and after nitroglycerin administration, the maximum changes in the diameter of the brachial artery did not differ between patients and controls, whereas skin circulation showed delayed post‐occlusive hyperemia response. They concluded that this modified kinetics of skin vasodilation was presumably caused by the structural changes in microvascular VSMCs. Experimental studies have yielded similar results: tail arteries from transgenic mice expressing p.Arg90Cys‐mutant human NOTCH3 showed in vitro significantly decreased flow‐induced dilation 33.
In spite of the above‐described systemic circulatory disturbances and autopsy verified structural involvement of arteries in most organs 110, hardly any clinical manifestations other than the neurological ones have been described. Myocardial infarctions have been reported in a Dutch CADASIL cohort 74, but not in some other studies, for example 14, 26, 28.
Circulatory disturbances in CARASIL
Very few studies on CBF in CARASIL have been published. Reduced CBF has been shown with single photon emission computed tomography (SPECT) and this reduction appeared to be relatively diffusely distributed 40, 115. Since at present there is no animal model for CARASIL, experimental studies on circulatory disturbances in HTRA1‐deficient animals have not been published.
Pathology of CADASIL
Autopsy findings
Macroscopic findings in CADASIL
In concordance with the imaging findings, the cerebral cortex is relatively spared, whereas multiple small lacunar infarcts are found predominantly in the cerebral WM and deep GM (Figure 4) as well as occasionally also in the brain stem. The severity of the lesions varies considerably between different, even neighboring anatomic regions. Stenoses in large cerebral arteries and rare larger, territorial infarcts 20, 85 have been reported, but they are most likely coincidental insults, not directly related to CADASIL. Besides, because CADASIL does not protect the patients from atherosclerosis, atherosclerotic infarcts may also occur, and these often impact also on cerebral cortex (Figure 4A).
T2*w MRI has shown that microbleeds are relatively common in CADASIL [see Imaging in CADASIL section and figure 3A in 113 and for pathology, Figure 10A–D] and they have been considered predictive for intracerebral hemorrhages (ICH). ICHs have been reported to be significantly associated with microbleeds among Chinese patients with p.Arg544Cys mutation, but on the other hand, these patients were also hypertensive 22. However, compared with the frequency of microbleeds, ICHs are relatively uncommon; just over 20 cases have been reported. Thus, microbleeds do not appear to impose a very serious threat of ICH. It appears that the fibrotic arterioles of CADASIL are less fragile than amyloid laden arteries in cerebral amyloid angiopathy (CAA). A few ICHs have been purely spontaneous, but most often they have occurred in patients, who have been hypertensive and/or treated with anticoagulants or antiaggregants. The majority of ICHs have been located in basal ganglia or thalamus with a few in cerebellum 75, 107.
Histopathology post‐mortem
Corresponding to MRI and macroscopic findings, the brunt of histopathology is seen in cerebral WM and deep GM, whereas cortical GM is involved only to a minor extent. Already in routine hematoxylin and eosin‐stained sections (Figure 5B), small arteries in cerebral WM appear thick‐walled and exceptionally rounded with basophilic (Figure 5B) and PAS‐positive (Figure 5C) staining of tunica media. Fibrous connective tissue accumulates in tunica adventitia, which may occasionally result in complete obliteration of arterioles (Figure 5D). Immunohistochemistry (IHC) discloses pathognomonic granular deposits of N3ECD immunopositivity in tunica media (Figure 5E). In parallel, α‐smooth muscle actin (α‐SMA) positivity decreases indicating degeneration of VSMCs (Figure 5F). In tunica adventitia, various types of collagen (Figure 5G), laminin, clusterin and other ECM proteins accumulate. Although cerebral cortex is relatively spared, N3ECD is also deposited in the tunica media of cortical arteries, but the adventitial thickening is markedly more prominent in WM arterioles (Figure 6A vs. B). Deposition of N3ECD expectedly begins already at the presymptomatic stage (Figure 6C). At advanced stages, N3ECD deposits are discernible also on veins and capillaries in both cerebral WM and GM (Figure 6D and E) and leptomeningeal arteries are also affected (Figure 6F).
Figure 5.
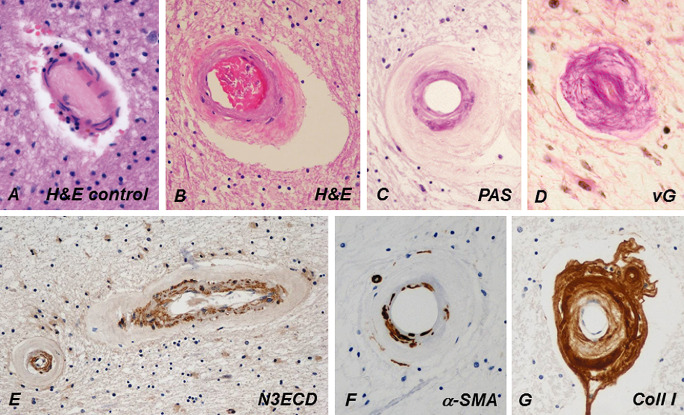
Compared with a control person's cerebral WM arteriole (A), the wall of a CADASIL patient's arteriole (B) is markedly thickened and fibrotic, and its rigidity renders the arteriole exceptionally circular (C). Tunica media is slightly basophilic in hematoxylin and eosin (H&E) (B) and prominently positive in periodic acid Schiff (PAS) staining (C). In advanced disease, the fibrosis has almost completely obliterated the lumen (D). Similarly, as in dermal arteries, NOTCH3 extracellular domain (N3ECD) immunopositivity accumulates in the tunica media of the thickened WM arterioles (E). The irregularly decreased immunoreactivity for α‐smooth muscle actin (α‐SMA) in the wall of a WM arteriole reflects the degeneration of VSMCs (G). The thickened adventitia harbors extracellular matrix proteins, here shown by collagen type I immunohistochemistry (F). vG = van Gieson.
Figure 6.
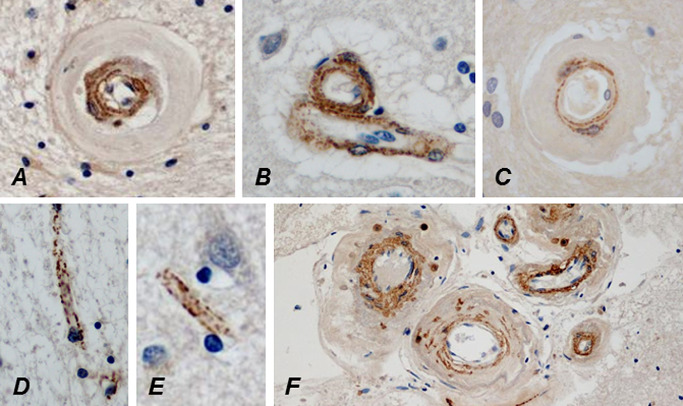
The lumina of a WM (A) and a cortical (B) arteriole are approximately as wide and approximately similar amount of N3ECD is deposited in their tunica media, but the wall of the WM arteriole is almost fourfold thicker than that of the cortical arteriole because of the marked fibrosis. N3ECD is also deposited in the wall of a probable vein beside the arteriole (B). Delicate N3ECD immunopositivity is already present in the wall of a markedly thickened arteriole in the cerebral WM of an only 32‐year‐old male CADASIL patient (C). In an elderly patient, N3ECD immunopositivity is seen on capillaries in both cerebral WM (D) and cortex (E), as well as in the walls of larger leptomeningeal arteries (F).
The predominant pathology—thickening of vascular walls and luminal stenosis—of arterioles, most prominent in cerebral WM and inflicting especially arterioles of smaller caliber, was explicitly demonstrated by Miao et al 84. This pathology is also clearly demonstrated in the elevation of the sclerotic index (SI) [defined and calculated as 1 − (internal diameter/external diameter)]. The SI of cerebral arterial vessels increased rapidly, when their lumina had narrowed to 20–30 μm and external diameter reached 130 μm. The mean SI of control persons' arterioles was about 0.42 in WM and 0.49 in GM, whereas in CADASIL patients' WM, the mean SI was 0.74 but in the relatively spared GM it increased only to 0.55 (Miao et al unpub. obs.). Interestingly, this increase in SI begins early, as in a 32‐year‐old CADASIL patient's WM arterioles the SI had reached the value of 0.64 85. This demonstrates the remarkable difference between WM and GM arterioles, the pathobiological background of which has not yet been fully clarified. This predominant stenosis in WM arterioles is also manifested in the reversal of the ratio between the mean luminal size in WM vs. cortical arterioles. In elderly controls, the mean luminal size of arterioles was found to be larger in WM than in cortex, that is 27.0 vs. 19.3 μm and the WM/GM ratio 1.40, whereas in elderly CADASIL patients, the disease had rendered the lumina of WM arterioles smaller than in cortical arterioles, that is 14.8 vs. 17.2 μm (WM/GM ratio 0.85) (Miao et al unpub. obs.).
The lacunar infarcts appear similar and follow the same sequence of cellular changes as any small ischemic infarct in WM, appearing at the subacute stage as a focal pannecrosis with accumulation of macrophages. For considerations on pathogenetic significance of the morphological findings, see the Pathogenetic significance of the morphological findings section.
Biopsy findings
Electron microscopy (EM) and granular osmiophilic material (GOM)
Baudrimont et al 7 were the first to examine a CADASIL patient's brain arteries by EM and they discovered granular electron‐dense extracellular material—to be later named GOM—located in the internal part of the media, reaching the basal lamina. No amyloid or intermediate filaments were visualized and wavelength dispersive EM showed that this material was not specific for mineral or metallic elements.
The deposition of GOM has proved to be an important alteration in CADASIL. Because the arteriopathy in CADASIL is systemic, GOM deposits can be detected in arteries of many different organs, including dermal arterioles. GOMs are actually best preserved and visualized in immediately fixed skin biopsies (see Biopsy findings section; Figure 7). GOM deposits commonly have a mushroom shape with the foot closely apposed to the VSMC plasma membrane and the head projecting to the ECM. Alternatively, GOMs are irregularly rounded bodies up to 1–2 μm in diameter, usually located in indentations of VSMCs, the plasma membrane beneath being decorated by pinocytotic vesicles (Figure 7B and C insets). GOMs are also often observed in the intercellular space apparently disconnected to VSMCs (Figure 7C). GOM itself is composed of strongly osmiophilic (thus electron‐dense) tiny granules Figure 7C inset).
Figure 7.
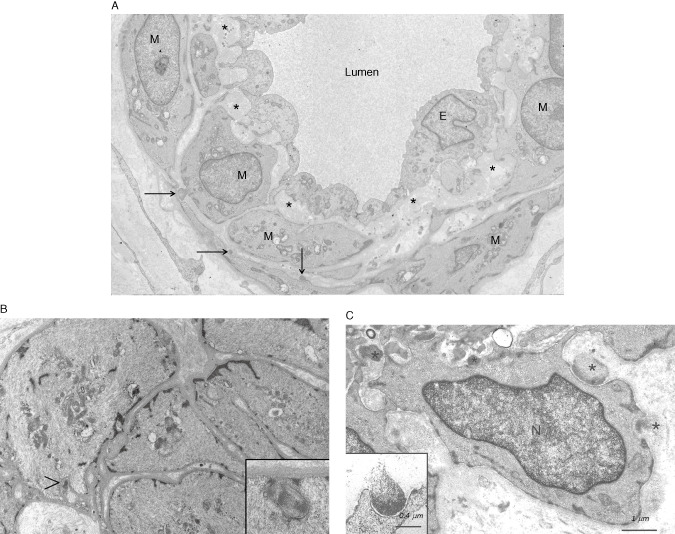
An electron micrograph (EM) of a small dermal artery from a 28‐year‐old male CADASIL patient with p.Arg133Cys NOTCH3 mutation. Three small deposits of GOM (arrows) are detectable (already at this age) on a few vascular smooth muscle cells (VSMC). The subendothelial space is widened with accumulation of extracellular matrix proteins (asterisks). E = endothelium, N = nucleus (A). An EM of a dermal arteriole from a 19‐year‐old CADASIL patient [younger brother of the patient in (A), both sons of a male patient homozygous for p.Arg133Cys mutation] shows the paucity of GOMs (arrowhead) and the inset shows that true GOMs do occur already at this young age (B). A higher magnification EM of a dermal arteriole from an elderly patient at a more advanced stage of the disease. GOMs are present both in indentations on a VSMC (black asterisks) and in the intercellular space apparently disconnected to the VSMC (white asterisk) (C). Note the pinocytotic vesicles in the VSMC beneath the GOM (B and C insets). N = nucleus. [Fig. 7A is reproduced from article (62) with permission]
Initial immuno‐EM analysis of human brain samples showed that N3ECD accumulates at the plasma membrane of VSMCs in close vicinity to the GOM deposits 56. Further immunogold EM studies from our and other laboratories, using a more sensitive methodology, revealed that N3ECD is actually a component of GOM deposits 52, 91, 135. It is conceivable that the distribution of N3ECD protein is heterogeneous within the GOM deposit, and taken together the observations suggest that the concentration of N3ECD might be highest at the foot of the GOM deposit.
Because GOM has not so far been reported in any other disease, CADASIL is an exceptional form of dementia: it can be diagnosed even with a simple skin biopsy. Because its arteriopathy also involves dermal arterioles, the deposition of the above‐described GOM is a specific diagnostic feature of CADASIL. In skin biopsies, GOM is detectable by EM in the walls of the diseased arterial vessels (Figure 7) as well as inconsistently of veins and rarely of capillaries. GOM appears to be present in all CADASIL patients with a pathogenic NOTCH3 mutation that causes an uneven number of cysteines in the affected EGFr of N3ECD 122 (for details see Diagnosis of CADASIL and CARASIL section). Thus, electron microscopists consider EM analysis a highly reliable diagnostic method 92, 122. The article by Tikka et al 122 presents a practical approach to the EM diagnostics of CADASIL, including directions for the light microscopic selection of representative small dermal arteries for EM thin sectioning as well as interpretation of EM findings to discriminate between fallacious and true GOM deposits.
IHC of N3ECD
However, because the number of laboratories available for performing reliable EM examinations is limited, immunohistochemical demonstration of N3ECD in skin biopsy was proffered as an alternative (Figure 8) 58. It is highly sensitive (85%–95%) and specific (95%–100%), although the inherent caveats of IHC must be kept in mind. Furthermore, its applicability at an early stage of CADASIL with minimal amount of N3ECD/GOM remains to be clarified (Figure 7B). With EM, we (HK) have detected GOM in a p.Arg133Cys patient already at the age of 19 years (Figure 7B inset), and positive IHC diagnosis (AJ) was also obtained already in a 28‐year‐old young adult patient 58. In confocal microscopy, N3ECD immunopositivity appears as delicate dots on VSMCs (Figure 8B), in concordance with the scattered appearance of GOM deposits in EM.
Figure 8.
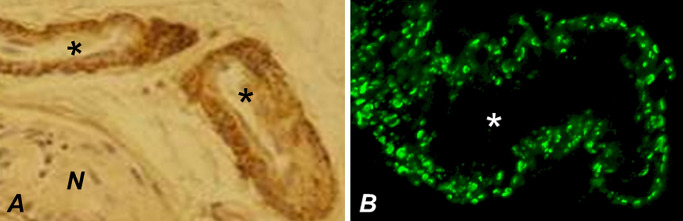
Immunohistochemical visualization of N3ECD deposition in the walls of dermal arteries (asterisks). The granular immunoreactivity is visualized either by using immunoperoxidase method (A) or—at a higher resolution—using confocal immunofluorescence, the punctate pattern being consistent with scattered distribution of GOMs (B). N = nerve.
As mentioned earlier a great majority of patients with unpaired cysteine in an EGFr of N3ECD appear to harbor GOM/N3ECD deposits and therefore the presence of such a cysteine residue has been suggested to be causally associated with the formation of GOMs. However, non‐cysteine mutations have been discovered in several patients with a SVD compatible with the CADASIL. Interestingly, in EM unequivocal, GOM has been reported only in two Japanese families with a non‐cysteine mutation (p.Arg75Pro), in whom despite extensive search cysteine involving Notch3 mutation was not detected 87. We have not detected GOM in any of the seven non‐cysteine cases that we have examined (Kalimo, unpub. obs.).
In addition to GOM deposits, EM also shows several degenerative alterations in the small arterioles: widening of subendothelial spaces, fragmentation of inner elastic lamina and presence of degenerating VSMCs with accumulation of cell debris in the extracellular space around them 122 (Figure 7A and C).
Pathology of CARASIL
The main cerebrovascular pathology in CARASIL falls on the same vascular bed as CADASIL, that is the small penetrating arteries, mainly in the cerebral WM and basal ganglia 6, 39. Histopathologically, the arteries show advanced arteriosclerosis (Figure 9B and C) without the light microscopic basophilic granularity of tunica media or electron microscopic GOM on VSMCs as in CADASIL. Neither is there deposition of amyloid. Intima shows fibrous thickening, internal elastic lamina is fragmented and media reveals loss of VSMCs with subsequent hyalinization. Arterial lumina are concentrically narrowed, but occasionally segmental dilation occurs. These vascular pathologies result in multifocal ischemic infarcts as well as diffuse ischemic changes, especially in cerebral WM (diffuse myelin loss with preservation of U‐fibers; Figure 9A) and consequent moderate atrophy. As there is no characteristic alteration in the diseased arteries, such as GOM in CADASIL, skin biopsies do not provide any diagnostic help, neither is the systemic nature of CARASIL similarly evident as it is in CADASIL. The vascular changes in other organs affected are less severe than those in cerebral arteries.
Figure 9.
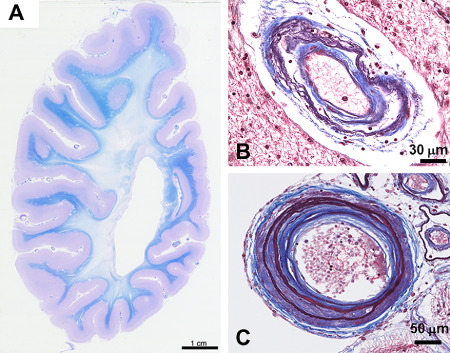
The cerebral white matter of a CARASIL patient who died at the age of 54 years shows diffuse widespread loss of myelin with preservation of U‐fibers (A). A small artery from a cerebral white matter shows irregular structure with double barreling of the thickened wall. Internal elastic lamina is multiplied (B). The wall of a medium‐sized leptomeningeal artery is markedly thickened with replacement of tunica muscularis by fibrous tissue, intimal proliferation and splitting of the internal elastic lamina. The smaller arteries beside show loose double barreling (C).
Diagnosis of CADASIL and CARASIL
The diagnosis of the patients, whose clinical picture is suggestive of CADASIL should be confirmed, both to inform the patient and to give correct counseling to the family members. Genetic testing is naturally the gold standard for the diagnosis of CADASIL. With the already wide knowledge of CADASIL, many genetic laboratories can provide even the full screening of the 21 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 18, 19, 20, 21, 22, 23, 24 exons that harbor the pathogenic NOTCH3 mutations. The specificity of mutations that lead to an uneven number of cysteines within any of the 34 EGFrs in N3ECD is 100% and the sensitivity of the analysis is close to 100% 14. However, in selected cases, neuropathology can be used to ascertain the diagnosis: (i) if reliable genetic screening is not readily available or (ii) if genetic screening gives an equivocal (especially non‐cysteine mutations), negative or previously unknown result (see Pathology of CADASIL section).
The clinical picture of premature alopecia, low back pain in addition to the neurological symptoms and presence of diffuse WM lesions in MRI should alert the clinician, especially in Far Eastern countries, to consider CARASIL as a possible, although a rare alternative. Similar patients among relatives indicating a hereditary disease is, of course, an important lead. The rarity of CARASIL in outside of Asia, of course, further increases the challenge in those countries. Thus, prompt molecular genetic analysis is warranted and demonstration of a pathogenic mutation in HTRA1 gives the definite diagnosis. As mentioned earlier, skin biopsy is not an auxiliary method, although it is helpful in differential diagnosis between CARASIL and CADASIL. The differential diagnostic alternatives for CARASIL have been presented in a comprehensive reviews by Fukutake 39 and Nozaki et al. 98.
Pathogenesis
CADASIL
Although the defective gene NOTCH3 that causes CADASIL has been identified almost 20 years ago and skilled research teams have striven to explain disease pathogenesis, the final goal has not yet been reached. The basic question whether the disease is caused by the loss‐of‐function or gain‐of‐function appears to become settled in favor of the latter alternative (see Neomorphic and toxic gain of function section).
Pathogenetic significance of the morphological findings
Vascular alterations
Stenosis of the affected arteries has been unequivocally demonstrated, but the decisive pathology—occlusion or thrombosis—of the feeding artery that leads to the lacunar infarct is rarely identified, although occasional completely obliterated arterioles may be detected (Figure 5D). In WM, where arteriolar stenosis is unequivocal, occlusion and/or thrombosis are most likely the predominant cause of the ischemic lesions. However, the arterioles with thickened and fibrotic walls, even if they are not significantly stenosed, do also bring on a risk of infarction, as demonstrated in deep GM (basal ganglia), where lacunar infarcts are common. Such arterioles have evidently lost their compliance and autoregulation, and thus the lacunar infarcts may also be caused by hemodynamic disturbances to which the rigid arterioles are not able to respond by dilatation, and critical ischemia occurs, although significant stenosis as such does not prevail 86, 119. On the other hand, experimental studies on transgenic mice expressing rat p.R169C mutant Notch3 (a preclinical mouse model of CADASIL) have shown cerebrovascular dysfunction with impaired neurovascular coupling and autoregulation of cerebral blood flow already prior to the appearance of fibrosis [and stenosis 60]. Thus, the problem may be also functional and not only mechanistic.
In CADASIL, the blood–brain barrier (BBB) is often multifocally breached as indicated by the common appearance of multiple microbleeds in T2*w MRI [see Imaging in CADASIL section and figure 3A in 113]. Rarely, small fresh cortical hemorrhages may be detected already during brain cutting (Figure 10A). Histopathologically, microbleeds appear as small perivascular hemosiderin or immunopositive plasma protein deposits around cortical/GM arterioles (Figure 10B and C), the walls of which are markedly thinner than in WM arterioles (Figure 10D vs. E), a likely reason for the predominant location of microbleeds in GM regions. IHC for plasma proteins shows that BBB is breached only locally at the sites of microbleeds or infarcts (Figure 10C–G), whereas the damage of the vascular walls does not appear to cause a more generalized breakdown of BBB that would result in widespread extravasation of plasma proteins and consequent vasogenic edema (Figure 10E–G). This suggests that the increased amount of free extracellular fluid detectable with DTI appears because of a CADASIL‐specific type of vasogenic edema.
Tissue alterations
The interpretation of the DTI findings earlier agrees well with the analysis of the microstructural changes in transgenic mice expressing rat wt or p.Arg169Cys‐mutant Notch3 25, 60. These mice are considered an animal model, in which the vascular and WM pathologies correspond to human preclinical CADASIL. It was demonstrated that in cerebral WM, myelin damage occurs first without loss of oligodendrocytes or major damage of axons (Figure 11). The axonal injury appears to be secondary to myelin degradation. These findings agree with the lack of an overt phenotype in these mice as well as with the fact that prominent MRI changes may be seen already in presymptomatic CADASIL patients' WM. The early WM lesions in these mice and hence in CADASIL were ascribed (linked?) to hypoperfusion‐induced energy deficiency, which causes failure of ATP‐dependent ion pumps and consequent impaired ion and water homeostasis. This leads to intramyelinic edema (Figure 11) 25, which in previous experimental studies has been shown to be a very early pathological change in ischemic WM 99. Chronic hypoperfusion in WM has been demonstrated both in the transgenic mouse model earlier 60 and in young CADASIL patients 127. Alternatively, impaired ion and water homeostasis could arise from a dysfunction of the astrocytic endfeet which cover the abluminal surface of pericytes and smooth muscle cells, where mutant Notch3 accumulates aberrantly 25.
Figure 11.
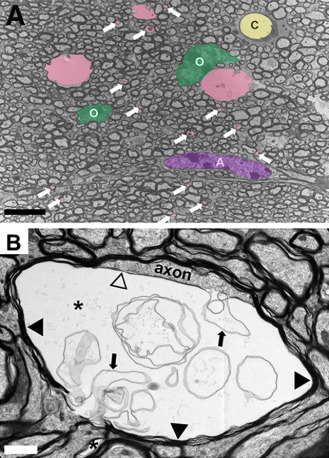
(A) Representative electron micrographs of the corpus callosum from a TgPAC‐Notch3R 169 C showing multiple membrane‐bound vacuoles (colored in pink) including numerous inframicrometric vacuoles (white arrows). O = oligodendrocytes (colored in green); A = astrocyte (colored in purple); C = capillary (colored in yellow). (B) Typical large vacuole (asterisk) in the innermost layer of the myelin sheath. The vacuole separates the axon from its myelin sheath (black arrowheads) and contains multiple aberrant myelin sheets (arrows). Notice the thin myelin sheet (open arrowhead) at the interface between the axon and the vacuole. Scale bar represents 5 μm in (A) and 1 μm in (B).
Processing and trafficking of mutated Notch3
The full‐length Notch3 is constitutively cleaved (site 1 = S1 cleavage) in the Golgi apparatus by furin 79 and targeted to the cell surface to form the heterodimer (N3ECD + N3TMIC, see Genetics and Biochemistry: CADASIL section and Figure 1) transmembrane receptor. It has been suggested that abnormal processing and trafficking of mutated Notch3 could be the cause of CADASIL. As almost all pathogenic mutations lead to an uneven number of cysteine residues in the mutated EGFr, the normal formation of sulphur bridges is prevented and thus misfolding of Notch3 most likely occurs. However, misfolded Notch3 proteins appear to escape the normal cellular quality control, ubiquitylation and proteosomal degradation, and are correctly processed and transported to cell surface in their partially misfolded state.
For example, in genuine patient‐derived VSMCs (with p.R133C mutation) cultured in vitro, the S1 cleavage appeared to occur correctly and intracellular aggregates were not more frequent than in control VSMCs. This suggests that in human VSMCs the microenvironment in Golgi and ER is able to prevent intracellular interreceptor cross‐linking and aggregation of the mutated endogenous Notch3 123. Thus, the characteristic aggregation of N3ECD does not appear to occur until the receptor has reached the cell surface and therefore the accumulation of N3ECD on VSMCs is most likely because of its local aggregation and/or its impaired clearance 34, 91.
In experimental studies, normal intracellular processing, trafficking and signaling were reported by Joutel et al 59 in cells transfected with human NOTCH3 harboring three different mutations and by Low et al 80 with NOTCH3 harboring four different mutations. However, in a study by Peters et al 100, cells transfected with mutated human NOTCH3 (p.R133C, p.C183R and p.C455R) showed reduced S1 (furin) cleavage. In another study, in cells transfected with mutated mouse mNotch3, a lesser amount of the overexpressed mutated than wt mNotch3 was transported to the cell surface. Besides, intracellular aggregates were formed, which may have resulted from increased accumulation or slowed transport in the secretory pathway 63. Abnormal S1 cleavage was also reported in cells expressing mutated rat Notch3, but presentation of this Notch3 on the cell surface was preserved 46. Yet, in the latter three studies, signaling by these mutated Notch3 receptors was not compromised. Thus, defective proteolytic processing or intracellular trafficking does not appear to be the key problem in CADASIL.
Impaired Notch3 signaling
Although many of the studies earlier have shown that mutated Notch3 retains its canonical signaling, loss of function has not been fully abandoned as a possible pathogenetic mechanism. Hypomorphic Notch3 function was suggested by Arboleda‐Velasquez et al 5 to be associated with CADASIL pathophysiology. They applied an in vivo assay, with which they showed that mutated (p.Cys455Arg or p.Arg1031Cys) NOTCH3 transgenes did not rescue the ischemic susceptibility phenotype of Notch3 knockout mice (evaluated by measuring the infarct size after standard filament occlusion of middle cerebral artery) as efficiently as wt NOTCH3. They also demonstrated decreased signaling activity in embryonic fibroblasts from transgenic mice expressing the above‐mentioned mutated NOTCH3 genes co‐cultured with ligand Delta‐like 1 expressing cells.
However, in some patients, CADASIL is caused by mutations at the ligand binding site, which abrogate ligand binding to Notch3 (Figure 1). Yet, the clinical pictures of these patients and of those with mutations outside the binding site are very similar. Thus, it is somewhat difficult to accept that the loss of function would be the sole/main cause of CADASIL. This view is supported for example by the experiments of Joutel et al 59 and Peters et al 100, who demonstrated in vitro that only Notch3 with specific mutations located in the ligand binding site in EGF repeats 10–11 (p.Cys428Ser and p.Cys455Arg) exhibited significantly reduced transcriptional activity via the RBP‐Jκ pathway, whereas the tested Notch3 receptors with mutations located outside the binding site had nearly normal signaling activity. In another set of in vivo experiments, the physiological levels of both wt human NOTCH3 and mutant p.Arg90Cys human NOTCH3 rescued the arterial defects of Notch3 knockout mice to similar degrees and mutant Notch3 exhibited normal level of Notch3/RBP‐Jκ activity in brain arteries 89. Furthermore, Notch3‐deficient (knockout) mice do not develop CADASIL pathognomonic GOM deposits or CADASIL pathology 23, 24, 65, 67, whereas they do appear in transgenic mice expressing mutant Notch3 5, 89, 111, but these deposits do not interfere with Notch3/RBP‐Jκ signaling activity 23, 24, 89. Finally, elimination of wt Notch3 did not influence the onset and burden of WM lesions in the TgPAC‐Notch3 R169C mice 23. Besides, the similar phenotype in the homozygous and heterozygous CADASIL patients 126 as well as the lack of truncating mutations in CADASIL 100 also favor a mechanism other than loss of the canonical Notch3 function.
Neomorphic and toxic gain of function
Thus, majority of CADASIL studies seem to favor a neomorphic (gain of novel function) pathogenetic mechanism rather than compromised canonical Notch3 function. The alteration that is common to all CADASIL mutations is the accumulation of N3ECD on the VSMC surface; unpaired cysteine residue with consequent partial misfolding and/or abnormal glycosylation of Notch3 seem to cause aberrant dimerization and higher order multimerization with cross‐linking by disulphide bonds, which results in the aggregation and deposition of N3ECD on VSMC surface 5, 34, 91. This aggregation phenomenon was also demonstrated in several transgenic mice expressing mutated (p.Arg90Cys or p.Cys428Ser) human Notch3 or (p.Arg169Cys) rat Notch3 and in a knocked‐in mouse model expressing the corresponding mouse p.Arg169Cys mutation, which did accumulate N3ECD on VSMCs like in CADASIL patients 23, 24, 89, 90, 111. Normally, N3ECD is transendocytosed into the ligand‐expressing cell and degraded through the endocytic‐lysosomal degradation pathway 44. The observation of abnormal accumulation of N3ECD in the vascular wall without accumulation of N3TMIC has raised the possibility that upon ligand binding, mutant Notch3 might be refractory to transendocytosis and further degradation 134. However, against this possibility speaks the observation that both transgenic mice and patients carrying the p.Cys428Ser mutation, which abolishes binding of the DSL ligands to Notch3, harbor vascular N3ECD deposits 59, 90. How mutated N3ECD may cause degeneration of VSMCs and the other con/subsequent pathogenic alterations has remained enigmatic.
Most studies have shown that neither most mutations in Notch3 nor accumulation of N3ECD compromise the canonical signaling, yet this accumulation appears to be pathogenic. The retention of misfolded N3ECD with an unpaired cysteine, could result in novel protein–protein interactions and induce (inactivate or aggravate) still unknown functions/dysfunctions that are toxic to VSMCs. A recent study showed that N3ECD deposits can make up a platform or act as a seed that promotes interactions with ECM proteins, such as tissue inhibitor of metalloproteinases 3 (TIMP3), that in turn can bind and recruit more and more proteins, such as vitronectin, generating new interaction surfaces and magnifying the toxic potential of aggregates in a snowball effect 91. Abnormal sequestration of proteins into aggregates usually results in the collapse of their biological function 97. TIMP3 and vitronectin have multiple biological activities that are directly relevant to CADASIL pathogenesis. For example, TIMP3 dysregulation could contribute to small vessel pathology through its well‐known metalloproteinase inhibitory activity. Also, by its ability to bind and stabilize plasminogen activator inhibitor‐1, vitronectin could modulate the balance of the fibrinolytic system, and consequently, the ECM homeostasis. Further studies are required to examine whether and how focal TIMP3 and vitronectin deposition can alter their biological activities and contribute to the disease process. Intriguingly, our preliminary studies showed an unexpected elevation of TIMP3 activity in the brain vessels of both CADASIL mouse models and patients with CADASIL.
A definite response is the degeneration of VSMCs with consequent impairment of vasomotor functions. This is reflected in the pathology of the key smooth muscle cell contractile protein α‐actin (interestingly, α‐actin is a direct target of canonical Notch3 signaling, yet disturbance in this pathway was by and large excluded). In cultured genuine human CADASIL (p.Arg133Cys) VSMCs, several proteins interacting with actin cytoskeleton were differentially expressed and the contractile capability of these VSMCs was impaired 50. In these cells, the actin cytoskeleton showed many derangements, the severity of which varied in VSMCs from different vascular beds being most severe in VSMCs derived from adult cerebral arteries. Again in slight disagreement with the above‐described lesser pathogenicity of hypomorphic Notch signaling, short hairpin RNA (shRNA) silencing of NOTCH3 in control cells did cause similar derangements of actin as in the p.Arg133Cys mutant human VSMCs 123. Furthermore, these cells disclosed also other features of hypomorphic Notch3 signaling by showing reduced upregulation of expression of Notch3 target genes PDGFR‐β, HES1 and HEY1 in response to ligand activation 53.
Degeneration of VSMCs is followed by increased production of ECM proteins resulting in marked thickening of the arterial walls, stenosis and decreased compliance [see above Pathology of CADASIL section and 84]. Whether this is a general response to degeneration of VSMCs or because of the activation of Notch‐specific fibrotic pathways is still to be examined (see discussion on TGF‐β signaling in CARASIL). By IHC, it has been shown that in CADASIL types I, II, IV and VI collagens are consistently increased in the walls of all caliber arterial vessels 31. These findings agree well with those in the study by Monet‐Leprêtre et al 91 in which the activity of TIMP3 was significantly elevated in both human patients and transgenic mouse model of CADASIL, as such an anticipated finding in vessel fibrosis reflecting reduced degradation of ECM components. Whether fibrosis also reflects transformation of VSMCs from contractile to productive mode like in atherosclerosis 68 is not known, although because VSMCs are degenerating, the most likely cell to produce ECM proteins is fibroblast.
CARASIL
Contrary to the complicated pathogenesis in CADASIL, the pathogenetic mechanisms in CARASIL appear relatively straightforward 45, 98, 116. As described in more detail in the section Genetics and Biochemistry, the function of HTRA1 as a repressor of TGF‐β signaling by cleaving proTGF‐β1 in the ER results in reduced amounts of mature TGF‐β1 in the ECM. HTRA1 mutations have been demonstrated to result in its decreased function and consequently in greater amounts of mature TGF‐β and upregulation of TGF‐β signaling. TGF‐β plays a critical role in ECM accumulation and vascular remodeling via upregulating the production of several agents including connective tissue growth factor (CTGF) and fibroblast growth factor. It has been shown to be the key mediator of vascular fibrosis 71. Concordantly, with the skeletal pathologies in CARASIL, TGF‐β also uniquely coordinates bone cell activity to maintain bone homeostasis. It regulates the differentiation and function of both osteoblasts and osteoclasts, from lineage recruitment to terminal differentiation, thereby balancing bone formation and resorption 121.
Therapeutic Aspects
CADASIL
Because CADASIL is an autosomal dominant genetic disease, preventing the expression of the mutated pathogenic allele of NOTCH3 should—if successful—provide definite therapy. Based on the assumption that the decisive pathogenesis is the toxic gain of function, probably associated with the unpaired cysteine in the affected EGFr/EGFrs, there are two possible gene therapeutic approaches: (i) silencing the mutated gene allele with inhibitory RNA molecules and (ii) antisense‐mediated skipping of the NOTCH3 exon harboring the defective DNA sequence. Allele‐specific siRNAs have already been designed for several diseases, including familial forms of Alzheimer's disease, Parkinson's disease, amyotrophic lateral sclerosis and spinocerebellar ataxia 7 66, 114. Tikka et al 123 demonstrated efficient silencing of NOTCH3 in control VSMCs (from human umbilical artery) transfected with two different vectors (by 36% respectively 63%), which encode shRNAs targeting NOTCH3 gene. This predicts that the expression of the mutated NOTCH3 could also be silenced and thereby the assumed toxic effect could be prevented. Because of the great number of pathogenic mutations this approach would require a production of very many vectors to specifically silence only the pathogenic allele. A more inclusive possibility would be antisense‐mediated exon skipping, which is emerging as a promising gene therapy to combat Duchenne muscular dystrophy, a hereditary disease with a markedly larger number of different mutations than in CADASIL 1, 49. This approach would be advantageous also in CADASIL, because skipping of a few exons, for example exons 3 and 4 encoding EGFr 2–5, which harbor a large cluster of missense mutations [>40% of mutations in >70% of families occur in these exons 18], could—if successful—produce a truncated Notch3 in which the unpaired cysteine is eliminated, but at the same time it should still remain capable of implementing Notch3 functions. Thus, a limited number of skipping procedures could help many patients. The location of VSMCs just beyond the endothelium from circulating blood may well facilitate delivery of the antisense oligonucleotides to the target cell, VSMC.
Despite the promising gene therapeutic views, at present, only symptomatic therapy is available, beginning with the simple recommendation to refrain from smoking. If the migraneous headache requires medication, conventional “pain killers” are recommended, whereas treatment with classic migraine drugs, such as ergotamines or triptans is discouraged because of their vasoconstrictory effect 13. Exceptionally, acetazolamide has been applied to alleviate acute excruciating migraine attacks 36.
Anticoagulants and antiaggregants have been tried without definite positive effects; rather these medications may have caused a few fatal parenchymal brain hemorrhages. As the progressive stenosis and rigidity already impair blood flow in the diseased arterioles, increased blood viscosity, for example because of polycythemia or dehydration, appears to be harmful and should be avoided (Viitanen, unpub. obs.). On the same basis, medications which cause cerebral hypoperfusion, such as antihypertensive drugs, tricyclic antidepressants and neuroleptics, should be administered with caution. Because one‐half of the CADASIL patients have hypercholesterolemia, treatment with statins is indicated, although the role of statins in CADASIL per se is still unknown 102.
Cognitive decline begins already before ischemic attacks. Cholinergic deficits have been found in CADASIL patients in late middle age 64 and therefore cholinesterase inhibitors may alleviate cognitive decline. In a placebo controlled study with donepezil, executive functions were improved moderately but the clinical relevance of this treatment is still unknown 30.
CARASIL
For CARASIL, there is at present no effective treatment 39. The genetic defect causes a loss‐of‐function of HTRA1, which would necessitate inducing production of functional HTRA1 (or administration of another molecule capable of repressing TGF‐β signaling). Thus, similar approaches as suggested earlier for CADASIL, in which the toxic gain‐of‐function now seems to be the plausible pathogenic mechanism, are not possible. Shiga et al 116 and Nozaki et al. 98 proposed interesting approaches to treat patients carrying a nonsense mutation of HTRA1. Preventing/alleviating the nonsense‐mediated decay of mutated mRNA might be effective in increasing truncated p.Arg370X HTRA1, which has been shown to retain normal protease activity, since the truncated C‐terminal PDZ domain of HTRA1 was revealed to be dispensable (18, 98, 117). Alternatively a drug (e.g. aminoglycoside antibiotics) that suppresses the identification of the translation termination codon (induced by the mutation), may allow, sufficient production of active HTRA1 and thus repression of TGF‐β signaling could be rescued.
The treatment at present includes prevention of non‐CARASIL‐related ischemic stroke, genetic counseling, supportive care and medications for treating dementia. The roles of antithrombotic and anticoagulant drugs in the therapeutics are still unclear 39.
Because excessive TGF‐β signaling appears to be the major culprit in CARASIL, another approach—direct inhibition of TGF‐β signaling—has been suggested to be investigated as a therapeutic strategy 98. This suggestion is based on the fact that in Marfan syndrome, which is also caused by increased TGF‐β signaling, angiotensin I receptor antagonist (a drug in clinical use as antihypertensive) has been shown to inhibit TGF‐β signaling and ameliorate the progression of Marfan syndrome in both transgenic mice and patients. In human brain, angiotensin I receptor antagonist also inhibits TGF‐β signaling 98.
Acknowledgments
This study was supported by EVO research funds of the Helsinki University Hospital to the Finnish CADASIL team, Sigrid Juselius Foundation and Maud Kuistila Foundation, Finland to S.T., Fondation Leducq (Transatlantic Network of Excellence on the Pathogenesis of Small Vessel Disease of the Brain), France to A.J., Finnish Cultural Foundation, Varsinais‐Suomi regional fund, Finland to M.S. And we thank Prof. Osamu Onodera, Niigata University, Japan for the genetic analyses and Dr Shigeo Murayama, Departments of Neurology and Neuropathology, Tokyo Metropolitan Geriatric Hospital and Institute of Gerontology, Japan for the neuropathological examinations on CARASIL.
References
- 1. Aartsma‐Rus A, Fokkema I, Verschuuren J, Ginjaar I, van Deutekom J, van Ommen GJ, den Dunnen JT (2009) Theoretic applicability of antisense‐mediated exon skipping for Duchenne muscular dystrophy mutations. Hum Mutat 30:293–299. [DOI] [PubMed] [Google Scholar]
- 2. Adib‐Samii P, Brice G, Martin RJ, Markus HS (2010) Clinical spectrum of CADASIL and the effect of cardiovascular risk factors on phenotype. Study in 200 consecutively recruited individuals. Stroke 41:630–634. [DOI] [PubMed] [Google Scholar]
- 3. Amberla K, Wäljas M, Tuominen S, Almqvist O, Pöyhönen M, Tuisku S et al (2004) Insidious cognitive decline in CADASIL. Stroke 35:1598–1602. [DOI] [PubMed] [Google Scholar]
- 4. Arboleda‐Velasquez J, Rampal R, Fung E, Darland DC, Liu M, Martinez MC et al (2005) CADASIL mutations impair Notch3 glycosylation by Fringe. Hum Mol Genet 14:1631–1649. [DOI] [PubMed] [Google Scholar]
- 5. Arboleda‐Velasquez JF, Manent J, Lee JH, Tikka S, Ospina C, Vanderburg CR et al (2011) Hypomorphic Notch3 alleles link Notch signaling to ischemic cerebral small‐vessel disease. Proc Natl Acad Sci U S A 108:E128–E135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Arima K, Yanagawa S, Ito N, Ikeda S (2003) Cerebral arterial pathology of CADASIL and CARASIL (Maeda syndrome). Neuropathology 23:327–334. [DOI] [PubMed] [Google Scholar]
- 7. Baudrimont M, Dubas F, Joutel A, Tournier‐Lasserve E, Bousser MG (1993) Autosomal dominant leukoencephalopathy and subcortical ischemic stroke. A clinicopathological study. Stroke 24:122–125. [DOI] [PubMed] [Google Scholar]
- 8. Bayrakli F, Balaban H, Gurelik M, Hizmetli S, Topaktas S (2014) Mutation in the HTRA1 gene in a patient with degenerated spine as a component of CARASIL syndrome. Turk Neurosurg 24:67–69. [DOI] [PubMed] [Google Scholar]
- 9. Bianchi S, Di Palma C, Gallus GN, Taglia I, Poggiani A, Rosini F et al (2014) Two novel HTRA1 mutations in a European CARASIL patient. Neurology 82:898–900. [DOI] [PubMed] [Google Scholar]
- 10. Bouvy WH, Biessels GJ, Kuijf HJ, Kappelle LJ, Luijten PR, Zwanenburg JJ (2014) Visualization of perivascular spaces and perforating arteries with 7 T magnetic resonance imaging. Invest Radiol 49:307–313. [DOI] [PubMed] [Google Scholar]
- 11. Bozkulak EC, Weinmaster G (2009) Selective use of ADAM10 and ADAM17 in activation of Notch1 signaling. Mol Cell Biol 29:5679–5695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bruening R, Dichgans M, Berchtenbreiter C, Yousry T, Seelos KC, Wu RH et al (2001) Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy: decrease in regional cerebral blood volume in hyperintense subcortical lesions inversely correlates with disability and cognitive performance. AJNR Am J Neuroradiol 22:1268–1274. [PMC free article] [PubMed] [Google Scholar]
- 13. Chabriat H, Bousser MG (2008) Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy. Handb Clin Neurol 89:671–686. [DOI] [PubMed] [Google Scholar]
- 14. Chabriat H, Vahedi K, Iba‐Zizen MT, Joutel A, Nibbio A, Nagy TG et al (1995a) Clinical spectrum of CADASIL: a study of 7 families. Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy. Lancet 346:934–939. [DOI] [PubMed] [Google Scholar]
- 15. Chabriat H, Bousser M‐G, Pappata S (1995b) Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy: a positron emission tomography study in two affected family members. Stroke 26:1729–1730. [PubMed] [Google Scholar]
- 16. Chabriat H, Pappata S, Poupon C, Clark CA, Vahedi K, Poupon F et al (1999) Clinical severity in CADASIL related to ultrastructural damage in white matter: in vivo study with diffusion tensor MRI. Stroke 30:2637–2643. [DOI] [PubMed] [Google Scholar]
- 17. Chabriat H, Pappata S, Ostergaard L, Clark CA, Pachot‐Clouard M, Vahedi K et al (2000) Cerebral hemodynamics in CADASIL before and after acetazolamide challenge assessed with MRI bolus tracking. Stroke 31:1904–1912. [DOI] [PubMed] [Google Scholar]
- 18. Chabriat H, Joutel A, Dichgans M, Tournier‐Lasserve E, Bousser MG (2009) CADASIL. Lancet Neurol 8:643–653. [DOI] [PubMed] [Google Scholar]
- 19. Chen Y, He Z, Meng S, Li L, Yang H, Zhang X (2013) A novel mutation of the high‐temperature requirement A serine peptidase 1 (HTRA1) gene in a Chinese family with cerebral autosomal recessive arteriopathy with subcortical infarcts and leukoencephalopathy (CARASIL). J Int Med Res 41:1445–1455. [DOI] [PubMed] [Google Scholar]
- 20. Choi EJ, Choi CG, Kim JS (2005) Medium to large cerebral artery involvement in CADASIL. Neurology 65:1322–1324. [DOI] [PubMed] [Google Scholar]
- 21. Choi JC, Lee KH, Song SK, Lee JS, Kang SY, Kang JH (2013a) Screening for NOTCH3 gene mutations among 151 consecutive Korean patients with acute ischemic stroke. J Stroke Cerebrovasc Dis 22:608–614. [DOI] [PubMed] [Google Scholar]
- 22. Choi JC, Song SK, Lee JS, Kang SY, Kang JH (2013b) Diversity of stroke presentation in CADASIL: study from patients harboring the predominant Notch3 mutation R544C. J Stroke Cerebrovasc Dis 22:126–131. [DOI] [PubMed] [Google Scholar]
- 23. Cognat E, Hervé D, Joutel A (2014a) Response to letter regarding article, “Archetypal Arg169Cys mutation in Notch3 does not drive the pathogenesis in cerebral autosomal dominant arteriopathy with subcortical infarcts and leucoencephalopathy via a loss‐of‐function mechanism”. Stroke 45:e129. [DOI] [PubMed] [Google Scholar]
- 24. Cognat E, Baron‐Menguy C, Domenga‐Denier V, Cleophax S, Fouillade C, Monet‐Leprêtre M et al (2014b) Archetypal Arg169Cys mutation in Notch3 does not drive the pathogenesis in cerebral autosomal dominant arteriopathy with subcortical infarcts and leucoencephalopathy via a loss‐of‐function mechanism. Stroke 45:842–849. [DOI] [PubMed] [Google Scholar]
- 25. Cognat E, Cleophax S, Domenga‐Denier V, Joutel A (2014c) Early white matter changes in CADASIL: evidence of segmental intramyelinic oedema in a pre‐clinical mouse model. Acta Neuropathol Commun. 2:49. doi: 10.1186/2051-5960-2-49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cumurciuc R, Henry P, Gobron C, Vicaut E, Bousser MG, Chabriat H, Vahedi K (2006) Electrocardiogram in cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy patients without any clinical evidence of coronary artery disease: a case‐control study. Stroke 37:1100–1102. [DOI] [PubMed] [Google Scholar]
- 27. Dichgans M (2009) Cognition in CADASIL. Stroke 40:S45–S47. [DOI] [PubMed] [Google Scholar]
- 28. Dichgans M, Mayer M, Uttner I et al (1998) The phenotypic spectrum of CADASIL: clinical findings in 102 cases. Ann Neurol 44:731–739. [DOI] [PubMed] [Google Scholar]
- 29. Dichgans M, Holtmannspotter M, Herzog J, Peters N, Bergmann M, Yousry TA (2002) Cerebral microbleeds in CADASIL: a gradient‐echo magnetic resonance imaging and autopsy study. Stroke 33:67–71. [DOI] [PubMed] [Google Scholar]
- 30. Dichgans M, Markus HS, Salloway S, Verkkoniemi A, Moline M, Wang Q et al (2008) Donepezil in patients with subcortical vascular cognitive impairment: a randomised double‐blind trial in CADASIL. Lancet Neurol 7:310–318. [DOI] [PubMed] [Google Scholar]
- 31. Dong H, Blaivas M, Wang MM (2012) Bidirectional encroachment of collagen into the tunica media in cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy. Brain Res 1456:64–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Dong Y, Hassan A, Zhang Z, Huber D, Dalageorgou C, Markus HS (2003) Yield of screening for CADASIL mutations in lacunar stroke and leukoaraiosis. Stroke 34:203–205. [DOI] [PubMed] [Google Scholar]
- 33. Dubroca C, Lacombe P, Domenga V, Maciazek J, Levy B, Tournier‐Lasserve E et al (2005) Impaired vascular mechanotransduction in a transgenic mouse model of CADASIL arteriopathy. Stroke 36:113–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Duering M, Karpinska A, Rosner S, Hopfner F, Zechmeister M, Peters N et al (2011) Co‐aggregate formation of CADASIL‐mutant Notch3: a single‐particle analysis. Hum Mol Genet 20:3256–3265. [DOI] [PubMed] [Google Scholar]
- 35. Fleming RJ, Hori K, Sen A, Filloramo GV, Langer JM, Obar RA et al (2013) An extracellular region of Serrate is essential for ligand‐induced cis‐inhibition of Notch signaling. Development 140:2039–2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Forteza AM, Brozman B, Rabinstein AA, Romano JG, Bradley WG (2001) Acetazolamide for the treatment of migraine with aura in CADASIL. Neurology 57:2144–2145. [DOI] [PubMed] [Google Scholar]
- 37. Fouillade C, Monet‐Leprêtre M, Baron‐Menguy C, Joutel A (2012) Notch signalling in smooth muscle cells during development and disease. Cardiovasc Res 95:138–146. [DOI] [PubMed] [Google Scholar]
- 38. Fouillade C, Baron‐Menguy C, Domenga‐Denier V, Thibault C, Takamiya K, Huganir R, Joutel A (2013) Transcriptome analysis for Notch3 target genes identifies Grip2 as a novel regulator of myogenic response in the cerebrovasculature. Arterioscler Thromb Vasc Biol 33:76–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Fukutake T (2011) Cerebral autosomal recessive arteriopathy with subcortical infarcts and leukoencephalopathy (CARASIL): from discovery to gene identification. J Stroke Cerebrovasc Dis 20:85–93. [DOI] [PubMed] [Google Scholar]
- 40. Fukutake T, Hirayama K (1995) Familial young adult–onset arteriosclerotic leukoencephalopathy with alopecia and lumbago without arterial hypertension. Eur Neurol 35:69–79. [DOI] [PubMed] [Google Scholar]
- 41. Gobron C, Viswanathan A, Bousser MG, Chabriat H (2006) Multiple simultaneous cerebral infarctions in cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy. Cerebrovasc Dis 22:445–446. [DOI] [PubMed] [Google Scholar]
- 42. Gobron C, Vahedi K, Vicaut E, Stucker O, Laemmel E, Baudry N et al (2007) Characteristic features of in vivo skin microvascular reactivity in CADASIL. J Cereb Blood Flow Metab 27:250–257. [DOI] [PubMed] [Google Scholar]
- 43. Granild‐Jensen J, Jensen UB, Schwartz M, Hansen US (2009) Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy resulting in stroke in an 11‐year‐old male. Dev Med Child Neurol 51:754–757. [DOI] [PubMed] [Google Scholar]
- 44. Hansson EM, Lanner F, Das D, Mutvei A, Marklund U, Ericson J et al (2010) Control of Notch‐ligand endocytosis by ligand‐receptor interaction. J Cell Sci 123:2931–2942. [DOI] [PubMed] [Google Scholar]
- 45. Hara K, Shiga A, Fukutake T, Nozaki H, Miyashita A, Yokoseki A et al (2009) Association of HTRA1 mutations and familial ischemic cerebral small‐vessel disease. N Engl J Med 360:1729–1739. [DOI] [PubMed] [Google Scholar]
- 46. Haritunians T, Chow T, De Lange RP, Nichols JT, Ghavimi D, Dorrani N et al (2005) Functional analysis of a recurrent missense mutation in Notch3 in CADASIL. J Neurol Neurosurg Psychiatry 76:1242–1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Harju M, Tuominen S, Summanen P, Viitanen M, Pöyhönen M, Nikoskelainen E et al (2004) Scanning laser Doppler flowmetry shows reduced retinal capillary blood flow in CADASIL. Stroke 35:2449–2452. [DOI] [PubMed] [Google Scholar]
- 48. Hartley J, Westmacott R, Decker J, Shroff M, Yoon G (2010) Childhood‐onset CADASIL: clinical, imaging, and neurocognitive features. J Child Neurol 25:623–627. [DOI] [PubMed] [Google Scholar]
- 49. Hoffman EP, McNally EM (2014) Exon‐skipping therapy: a roadblock, detour, or bump in the road? Sci Transl Med. 6(230):230fs14. doi: 10.1126/scitranslmed.3008873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ihalainen S, Soliymani R, Iivanainen E, Mykkänen K, Sainio A, Pöyhönen M et al (2007) Proteome analysis of cultivated vascular smooth muscle cells from a CADASIL patient. Mol Med 13:305–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Ikegaya H, Heino J, Laaksonen H, Toivonen S, Kalimo H, Saukko P (2004) Accumulation of plasma proteins in Purkinje cells as an indicator of blood‐brain barrier breakdown. Forensic Sci Int 146:121–124. [DOI] [PubMed] [Google Scholar]
- 52. Ishiko A, Shimizu A, Nagata E, Takahashi K, Tabira T, Suzuki N (2006) Notch3 ectodomain is a major component of granular osmiophilic material (GOM) in CADASIL. Acta Neuropathol 112:333–339. [DOI] [PubMed] [Google Scholar]
- 53. Jin S, Hansson EM, Tikka S, Lanner F, Sahlgren C, Farnebo F et al (2008) Notch signaling regulates platelet‐derived growth factor receptor‐beta expression in vascular smooth muscle cells. Circ Res 102:1483–1491. [DOI] [PubMed] [Google Scholar]
- 54. Joutel A, Corpechot C, Ducros A, Vahedi K, Chabriat H, Mouton P et al (1996) Notch3 mutations in CADASIL, a hereditary late‐onset condition causing ischaemic stroke and dementia. Nature 383:707–710. [DOI] [PubMed] [Google Scholar]
- 55. Joutel A, Dodick DD, Parisi JE, Cecillon M, Tournier‐Lasserve E, Bousser MG (2000a) De novo mutation in the NOTCH3 gene causing CADASIL. Ann Neurol 47:388–391. [PubMed] [Google Scholar]
- 56. Joutel A, Andreux F, Gaulis S, Domenga V, Cecillon M, Battail N et al (2000b) The ectodomain of the Notch3 receptor accumulates within the cerebrovasculature of CADASIL patients. J Clin Invest 105:597–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Joutel A, Chabriat H, Vahedi K, Domenga V, Vayssière C, Ruchoux MM et al (2000c) Splice site mutation causing a seven amino acid Notch3 in‐frame deletion in CADASIL. Neurology 54:1874–1875. [DOI] [PubMed] [Google Scholar]
- 58. Joutel A, Favrole P, Labauge P, Chabriat H, Lescoat C, Andreux F et al (2001) Skin biopsy immunostaining with a Notch3 monoclonal antibody for CADASIL diagnosis. Lancet 358:2049–2051. [DOI] [PubMed] [Google Scholar]
- 59. Joutel A, Monet M, Domenga V, Riant F, Tournier‐Lasserve E (2004) Pathogenic mutations associated with cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy differently affect Jagged1 binding and Notch3 activity via the RBP/Jκ signaling pathway. Am J Hum Genet 74:338–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Joutel A, Monet‐Leprêtre M, Gosele C, Baron‐Menguy C, Hammes A, Schmidt S et al (2010) Cerebrovascular dysfunction and microcirculation rarefaction precede white matter lesions in a mouse genetic model of cerebral ischemic small vessel disease. J Clin Invest 120:433–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Kalimo H, Ruchoux MM, Viitanen M, Kalaria RN (2002) CADASIL a common form of hereditary arteriopathy causing brain infarcts and dementia. Brain Pathol 12:371–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Kalimo H, Miao Q, Tikka S, Mykkänen K, Junna M, Roine S et al (2008) CADASIL, the most common hereditary vascular subcortical dementia. Future Neurol 3:683–704. [Google Scholar]
- 63. Karlström H, Beatus P, Dennaeus K, Chapman G, Lendahl U, Lundqvist J (2002) A CADASIL mutated receptor exhibits impaired intracellular trafficking and maturation but normal ligand‐induced signaling. Proc Natl Acad Sci U S A 99:17119–17124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Keverne JS, Low WC, Ziabreva I, Court JA, Oakley AE, Kalaria RN (2007) Cholinergic neuronal deficits in CADASIL. Stroke 38:188–191. [DOI] [PubMed] [Google Scholar]
- 65. Kitamoto T, Takahashi K, Takimoto H, Tomizuka K, Hayasaka M, Tabira T, Hanaoka K (2005) Functional redundancy of the Notch gene family during mouse embryogenesis: analysis of Notch gene expression in Notch3‐deficient mice. Biochem Biophys Res Commun 331:1154–1162. [DOI] [PubMed] [Google Scholar]
- 66. Koutsilieri E, Rethwilm A, Scheller C (2007) The therapeutic potential of siRNA in gene therapy of neurodegenerative disorders. J Neural Transm Suppl 72:43–49. [DOI] [PubMed] [Google Scholar]
- 67. Krebs LT, Xue Y, Norton CR, Sundberg JP, Beatus P, Lendahl U et al (2003) Characterization of Notch3‐deficient mice: normal embryonic development and absence of genetic interactions with Notch 1 mutation. Genesis 37:139–143. [DOI] [PubMed] [Google Scholar]
- 68. Lacolley P, Regnault V, Nicoletti A, Li Z, Michel JB (2012) The vascular smooth muscle cell in arterial pathology: a cell that can take on multiple roles. Cardiovasc Res 95:194–204. [DOI] [PubMed] [Google Scholar]
- 69. Lacombe P, Oligo C, Domenga V, Tournier‐Lasserve E, Joutel A (2005) Impaired cerebral vasoreactivity in a transgenic mouse model of cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy arteriopathy. Stroke 36:1053–1058. [DOI] [PubMed] [Google Scholar]
- 70. Laitinen V, Siitonen M, Pasanen P (2008) Previously unpublished data. DNA Diagnostic Laboratory, Department of Medical Genetics, University of Turku, Turku, Finland.
- 71. Lan TH, Huang XQ, Tan HM (2013) Vascular fibrosis in atherosclerosis. Cardiovasc Pathol 22:401–407. [DOI] [PubMed] [Google Scholar]
- 72. Lee YC, Liu CS, Chang MH, Lin KP, Fuh JL, Lu YC et al (2009) Population‐specific spectrum of Notch3 mutations, MRI features and founder effect of CADASIL in Chinese. J Neurol 256:249–255. [DOI] [PubMed] [Google Scholar]
- 73. Lesnik Oberstein SA, van den Boom R, van Buchem MA, van Houwelingen HC, Bakker E, Vollebregt E et al (2001) Cerebral microbleeds in CADASIL. Neurology 57:1066–1070. [DOI] [PubMed] [Google Scholar]
- 74. Lesnik Oberstein SA, Jukema JW, van Duinen SG, Macfarlane PW, van Houwelingen HC, Breuning MH et al (2003) Myocardial infarction in cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL). Medicine 82:251–256. [DOI] [PubMed] [Google Scholar]
- 75. Lian L, Li D, Xue Z, Liang Q, Xu F, Kang H et al (2013) Spontaneous intracerebral hemorrhage in CADASIL. J Headache Pain 14:98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Liebetrau M, Herzog J, Kloss CU, Hamann GF, Dichgans M (2002) Prolonged cerebral transit time in CADASIL: a transcranial ultrasound study. Stroke 33:509–512. [DOI] [PubMed] [Google Scholar]
- 77. Liem MK, van der Grond J, Haan J, van den Boom R, Ferrari MD, Knaap YM et al (2007) Lacunar infarcts are the main correlate with cognitive dysfunction in CADASIL. Stroke 38:923–928. [DOI] [PubMed] [Google Scholar]
- 78. Liem MK, Lesnik Oberstein SA, Vollebregt MJ, Middelkoop HA, van der Grond J, Helderman‐van den Enden AT (2008) Homozygosity for a Notch3 mutation in a 65‐year‐old CADASIL patient with mild symptoms: a family report. J Neurol 255:1978–1980. [DOI] [PubMed] [Google Scholar]
- 79. Logeat F, Bessia C, Brou C, LeBail O, Jarriault S, Seidah NG, Israel A (1998) The Notch1 receptor is cleaved constitutively by a furin‐like convertase. Proc Natl Acad Sci U S A 95:8108–8112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Low WC, Santa Y, Takahashi K, Tabira T, Kalaria RN (2006) CADASIL‐causing mutations do not alter Notch3 receptor processing and activation. Neuroreport 17:945–949. [DOI] [PubMed] [Google Scholar]
- 81. Maeda S, Nakayama H, Isaka K, Aihara Y, Nemoto S (1976) Familial unusual encephalopathy of Binswanger's type without hypertension. Folia Psychiatr Neurol Jpn 30:165–177. [DOI] [PubMed] [Google Scholar]
- 82. Mazzei R, Guidetti D, Ungaro C, Conforti FL, Muglia M, Cenacchi G et al (2008) First evidence of a pathogenic insertion in the NOTCH3 gene causing CADASIL. J Neurol Neurosurg Psychiatry 79:108–110. [DOI] [PubMed] [Google Scholar]
- 83. Mendioroz M, Fernández‐Cadenas I, Del Río‐Espinola A, Rovira A, Solé E, Fernández‐Figueras MT et al (2010) A missense HTRA1 mutation expands CARASIL syndrome to the Caucasian population. Neurology 75:2033–2035. [DOI] [PubMed] [Google Scholar]
- 84. Miao Q, Paloneva T, Tuominen S, Pöyhönen M, Tuisku S, Viitanen M et al (2004) Fibrosis and stenosis of the long penetrating cerebral arteries: the cause of the white matter pathology in cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy. Brain Pathol 14:358–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Miao Q, Kalimo H, Bogdanovic N, Kostulas K, Börjesson‐Hanson A, Viitanen M (2006) Cerebral arteriolar pathology in a 32‐year‐old patient with CADASIL. Neuropathol Appl Neurobiol 32:455–458. [DOI] [PubMed] [Google Scholar]
- 86. Miao Q, Paloneva T, Tuisku S, Roine S, Pöyhönen M, Viitanen M, Kalimo H (2006) Arterioles of the lenticular nucleus in CADASIL. Stroke 37:2242–2247. [DOI] [PubMed] [Google Scholar]
- 87. Mizuno T, Muranishi M, Torugun T, Tango H, Nagakane Y, Kudeken T et al (2008) Two Japanese CADASIL families exhibiting Notch3 mutation R75P not involving cysteine residue. Intern Med 47:2067–2072. [DOI] [PubMed] [Google Scholar]
- 88. Moccia M, Penco S, Barone P (2014) Letter by Moccia et al regarding article “Archetypal Arg169Cys mutation in Notch3 does not drive the pathogenesis in cerebral autosomal dominant arteriopathy with subcortical infarcts and leucoencephalopathy via a loss‐of‐function mechanism”. Stroke 45:e128. [DOI] [PubMed] [Google Scholar]
- 89. Monet M, Domenga V, Lemaire B, Souilhol C, Langa F, Babinet C et al (2007) The archetypal R90C CADASIL‐NOTCH3 mutation retains Notch3 function in vivo. Hum Mol Genet 16:982–992. [DOI] [PubMed] [Google Scholar]
- 90. Monet‐Leprêtre M, Bardot B, Lemaire B, Domenga V, Godin O, Dichgans M et al (2009) Distinct phenotypic and functional features of CADASIL mutations in the Notch3 ligand binding domain. Brain 132:1601–1612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Monet‐Leprêtre M, Haddad I, Baron‐Menguy C, Fouillot‐Panchal M, Riani M, Domenga‐Denier V et al (2013) Abnormal recruitment of extracellular matrix proteins by excess Notch3 ECD: a new pathomechanism in CADASIL. Brain 136:1830–1845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Morroni M, Marzioni D, Ragno M, Di Bella P, Cartechini E, Pianese L et al (2013) Role of electron microscopy in the diagnosis of CADASIL syndrome: a study of 32 patients. PLoS ONE 8:e65482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Mykkänen K, Savontaus ML, Juvonen V, Sistonen P, Tuisku S, Tuominen S et al (2004) Detection of the founder effect in Finnish CADASIL families. Eur J Hum Genet 12:813–819. [DOI] [PubMed] [Google Scholar]
- 94. Mykkänen K, Junna M, Amberla K, Bronge L, Kääriäinen H, Pöyhönen M et al (2009) Different clinical phenotypes in monozygotic CADASIL twins with a novel NOTCH3 mutation. Stroke 40:2215–2218. [DOI] [PubMed] [Google Scholar]
- 95. Narayan SK, Gorman G, Kalaria RN, Ford GA, Chinnery PF (2012) The minimum prevalence of CADASIL in northeast England. Neurology 78:1025–1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Nichols JT, Miyamoto A, Olsen SL, D'Souza B, Yao C, Weinmaster G (2007) DSL ligand endocytosis physically dissociates Notch1 heterodimers before activating proteolysis can occur. J Cell Biol 176:445–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Nozaki H, Nishizawa M, Onodera O (2014) Features of Cerebral Autosomal Recessive Arteriopathy With Subcortical Infarcts and Leukoencephalopathy. Stroke Aug 12. pii: STROKEAHA.114.004236. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 98. Olzscha H, Schermann SM, Woerner AC, Pinkert S, Hecht MH, Tartaglia GG et al (2011) Amyloid‐like aggregates sequester numerous metastable proteins with essential cellular functions. Cell 144:67–78. [DOI] [PubMed] [Google Scholar]
- 99. Pantoni L, Garcia JH, Gutierrez JA (1996) Cerebral white matter is highly vulnerable to ischemia. Stroke 27:1641–1646. [DOI] [PubMed] [Google Scholar]
- 100. Peters N, Opherk C, Zacherle S, Capell A, Gempel P, Dichgans M (2004) CADASIL‐associated Notch3 mutations have differential effects both on ligand binding and ligand‐induced Notch3 receptor signaling through RBP‐Jκ. Exp Cell Res 299:454–464. [DOI] [PubMed] [Google Scholar]
- 101. Peters N, Opherk C, Danek A, Ballard C, Herzog J, Dichgans M (2005) The pattern of cognitive performance in CADASIL: a monogenic condition leading to subcortical ischemic vascular dementia. Am J Psychiatry 162:2078–2085. [DOI] [PubMed] [Google Scholar]
- 102. Peters N, Freilinger T, Opherk C, Pfefferkorn T, Dichgans M (2007) Effects of short term atorvastatin treatment on cerebral hemodynamics in CADASIL. J Neurol Sci 260:100–105. [DOI] [PubMed] [Google Scholar]
- 103. Prakash N, Hansson E, Betsholtz C, Mitsiadis T, Lendahl U (2002) Mouse Notch3 expression in the pre‐ and postnatal brain: relationship to the stroke and dementia syndrome CADASIL. Exp Cell Res 278:31–44. [DOI] [PubMed] [Google Scholar]
- 104. Ragno M, Pianese L, Morroni M, Cacchiò G, Manca A, Di Marzio F et al (2013) CADASIL coma in an Italian homozygous CADASIL patient: comparison with clinical and MRI findings in age‐matched heterozygous patients with the same G528C Notch3 mutation. Neurol Sci 34:1947–1953. [DOI] [PubMed] [Google Scholar]
- 105. Razvi SS, Davidson R, Bone I, Muir KW (2005) The prevalence of cerebral autosomal dominant arteriopathy with subcortical infarcts and leucoencephalopathy (CADASIL) in the west of Scotland. J Neurol Neurosurg Psychiatry 76:739–741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Reyes S, Viswanathan A, Godin O, Dufouil C, Benisty S, Hernandez K (2009) Apathy: a major symptom in CADASIL. Neurology 72:905–910. [DOI] [PubMed] [Google Scholar]
- 107. Rinnoci V, Nannucci S, Valenti R, Donnini I, Bianchi S, Pescini F et al (2013) Cerebral hemorrhages in CADASIL: report of four cases and a brief review. J Neurol Sci 330:45–51. [DOI] [PubMed] [Google Scholar]
- 108. Roine S, Pöyhönen M, Timonen S, Tuisku S, Marttila R, Sulkava R et al (2005) Neurologic symptoms are common during gestation and puerperium in CADASIL. Neurology 64:1441–1443. [DOI] [PubMed] [Google Scholar]
- 109. Roine S, Harju M, Kivelä TT, Pöyhönen M, Nikoskelainen E, Tuisku S et al (2006) Ophthalmologic findings in cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy: a cross‐sectional study. Ophthalmology 113:1411–1417. [DOI] [PubMed] [Google Scholar]
- 110. Ruchoux MM, Guerouaou D, Vandenhaute B, Pruvo JP, Vermersch P, Leys D (1995) Systemic vascular smooth muscle cell impairment in cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy. Acta Neuropathol 89:500–512. [DOI] [PubMed] [Google Scholar]
- 111. Ruchoux MM, Domenga V, Brulin P, Maciaze J, Limol S, Tournier‐Lasserve E et al (2003) Transgenic mice expressing mutant Notch3 develop vascular alterations characteristic of cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy. Am J Pathol 162:329–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Saiki S, Sakai K, Saiki M, Kitagawa Y, Umemori T, Murata K et al (2006) Varicose veins associated with CADASIL result from a novel mutation in the NOTCH3 gene. Neurology 67:337–339. [DOI] [PubMed] [Google Scholar]
- 113. Salomon N (2014) Neuroimaging of small vessel disease. Brain Pathol. [Google Scholar]
- 114. Scholefield J, Watson L, Smith D, Greenberg J, Wood MJ (2014) Allele‐specific silencing of mutant Ataxin‐7 in SCA7 patient‐derived fibroblasts. Eur J Hum Genet doi: 10.1038/ejhg.2014.39; Mar 26, [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Shibata M (2012) [Clinical manifestations and neuroradiological findings of CARASIL with a novel mutation]. Rinsho Shinkeigaku 52:1363–1364, (Article in Japanese). [DOI] [PubMed] [Google Scholar]
- 116. Shiga A, Nozaki H, Yokoseki A, Nihonmatsu M, Kawata H, Kato T et al (2011) Cerebral small‐vessel disease protein HTRA1 controls the amount of TGF‐β1 via cleavage of proTGF‐β1. Hum Mol Genet 20:1800–1810. [DOI] [PubMed] [Google Scholar]
- 117. Singhal S, Bevan S, Barrick T, Rich P, Markus HS (2004) The influence of genetic and cardiovascular risk factors on the CADASIL phenotype. Brain 127:2031–2038. [DOI] [PubMed] [Google Scholar]
- 118. Soong BW, Liao YC, Tu PH, Tsai PC, Lee IH, Chung CP, Lee YC (2013) A homozygous Notch3 mutation p.R544C and a heterozygous TREX1 variant p.C99MfsX3 in a family with hereditary small vessel disease of the brain. J Chin Med Assoc 76:319–324. [DOI] [PubMed] [Google Scholar]
- 119. Stenborg A, Kalimo H, Viitanen M, Terent A, Lind L (2007) Impaired endothelial function of forearm resistance arteries in CADASIL patients. Stroke 38:2692–2697. [DOI] [PubMed] [Google Scholar]
- 120. Taillia H, Chabriat H, Kurtz A, Verin M, Levy C, Vahedi K et al (1998) Cognitive alterations in non‐demented CADASIL patients. Cerebrovasc Dis 8:97–101. [DOI] [PubMed] [Google Scholar]
- 121. Tang SY, Alliston T (2013) Regulation of postnatal bone homeostasis by TGFβ. Bonekey Rep 2:255. doi: 10.1038/bonekey.2012.255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Tikka S, Mykkänen K, Ruchoux MM, Bergholm R, Junna M, Pöyhönen M et al (2009) Congruence between NOTCH3 mutations and GOM in 131 CADASIL patients. Brain 132:933–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Tikka S, Ng YP, Di Maio G, Mykkänen K, Siitonen M, Lepikhova T et al (2012) CADASIL mutations and shRNA silencing of Notch3 affect actin organization in cultured vascular smooth muscle cells. J Cereb Blood Flow Metab 32:2171–2180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Todorovic V, Rifkin DB (2012) LTBPs, more than just an escort service. J Cell Biochem 113:410–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Tournier‐Lasserve E, Joutel A, Melki J et al (1993) Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy maps to chromosome 19q12. Nat Genet 3:256–259. [DOI] [PubMed] [Google Scholar]
- 126. Tuominen S, Juvonen V, Amberla K, Jolma T, Rinne JO, Tuisku S et al (2001) Phenotype of a homozygous CADASIL patient in comparison to 9 age‐matched heterozygous patients with the same R133C Notch3 mutation. Stroke 32:1767–1774. [DOI] [PubMed] [Google Scholar]
- 127. Tuominen S, Miao Q, Kurki T, Tuisku S, Pöyhönen M, Kalimo H et al (2004) Positron emission tomography examination of cerebral blood flow and glucose metabolism in young CADASIL patients. Stroke 35:1063–1067. [DOI] [PubMed] [Google Scholar]
- 128. van Bogaert L (1955) Encephalopathie sous‐corticale progressive (Binswanger) a evolution rapide sous deuz soeurs. Med Hellen 24:961–972. [Google Scholar]
- 129. Verin M, Rolland Y, Landgraf F, Chabriat H, Bompais B, Michel A et al (1995) New phenotype of the cerebral autosomal dominant arteriopathy mapped to chromosome 19: migraine as the prominent clinical feature. J Neurol Neurosurg Psychiatry 59:579–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Vinciguerra C, Rufa A, Bianchi S, Sperduto A, De Santis M, Malandrini A et al (2014) Homozygosity and severity of phenotypic presentation in a CADASIL family. Neurol Sci 35:91–93. [DOI] [PubMed] [Google Scholar]
- 131. Wang XL, Li CF, Guo HW, Cao BZ (2012) A novel mutation in the HTRA1 gene identified in Chinese CARASIL pedigree. CNS Neurosci Ther 18:867–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Wang Z, Yuan Y, Zhang W, Lv H, Hong D, Chen B et al (2011) Notch3 mutations and clinical features in 33 mainland Chinese families with CADASIL. J Neurol Neurosurg Psychiatry 82:534–539. [DOI] [PubMed] [Google Scholar]
- 133. Watanabe M, Adachi Y, Jackson M, Yamamoto‐Watanabe Y, Wakasaya Y et al (2012) An unusual case of elderly onset cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL) with multiple cerebrovascular risk factors. J Stroke Cerebrovasc Dis 21:143–145. [DOI] [PubMed] [Google Scholar]
- 134. Watanabe‐Hosomi A, Watanabe Y, Tanaka M, Nakagawa M, Mizuno T (2012) Transendocytosis is impaired in CADASIL‐mutant Notch3. Exp Neurol 233:303–311. [DOI] [PubMed] [Google Scholar]
- 135. Yamamoto Y, Craggs LJ, Watanabe A, Booth T, Attems J, Low RW et al (2013) Brain microvascular accumulation and distribution of the Notch3 ectodomain and granular osmiophilic material in CADASIL. J Neuropathol Exp Neurol 72:416–431. [DOI] [PubMed] [Google Scholar]
- 136. Yanagawa S, Ito N, Arima K, Ikeda S (2002) Cerebral autosomal recessive arteriopathy with subcortical infarcts and leukoencephalopathy. Neurology 58:817–820. [DOI] [PubMed] [Google Scholar]