

Abstract
Accumulation of beta‐amyloid (Aβ) in the brain has been implicated as a major contributor to the cellular pathology and cognitive impairment observed in Alzheimer's disease. Beta‐amyloid may exert its toxic effects by increasing reactive oxygen species and neuroinflammation in the brain. This study set out to investigate whether a genetically engineered derivative of the peroxisomal antioxidant enzyme catalase (CAT–SKL), is able to reduce the toxicity induced by intracerebroventricular injection of Aβ25–35 in the mature rat brain. Histopathological and immunohistochemical analyses were used to evaluate neuroinflammation, and neuronal loss. Spatial learning and reference memory was assessed using the Morris water maze. CAT–SKL treatment was able to reduce the pathology induced by Aβ25‐35 toxicity by significantly decreasing microglia activation in the basal forebrain and thalamus, and reducing cholinergic loss in the basal forebrain. Aβ25–35 animals showed deficits in long‐term reference memory in the Morris water maze, while Aβ25–35 animals treated with CAT–SKL did not demonstrate long‐term memory impairments. This preclinical data provides support for the use of CAT–SKL in reducing neuroinflammation and long‐term reference memory deficits induced by Aβ25–35.
Keywords: beta amyloid, catalase, neuroinflammation, oxidative stress
Introduction
Alzheimer's disease (AD) is a devastating age‐related neurodegenerative disease characterized by beta‐amyloid (Aβ) plaques, neurofibrillary tangles, neuronal degeneration and loss, and synaptic dysfunction and failure all of which contribute to progressive cognitive decline and dementia in afflicted patients 13, 24, 27. The exact pathogenesis of AD is not yet fully understood, however it is clear that accumulation of Aβ is an integral part of AD. Aβ is a peptide of 37–43 amino acids that is generated from proteolytic cleavage of the amyloid precursor protein (APP) by the action of β‐and γ‐secretases 11, 29. A number of studies have shown that Aβ exerts its toxicity in part by activating inflammatory pathways in the brain and facilitating the formation of reactive oxygen species (ROS) with a resultant increase in oxidative damage.
Prolonged activation of the inflammatory response results in a dysregulated process with neuroinflammatory cells releasing a variety of pro‐inflammatory mediators and potentially neurotoxic factors including cytokines, chemokines and complement pathway activation 1, 7, 24, 35. Added to this is the contribution of Aβ toxicity to oxidative stress in the brain. Reactive oxygen species are produced as a normal consequence of cellular activity and are usually maintained at low physiological levels by antioxidant enzymes 18, 31. These antioxidants are able to remove and or repair molecules that are oxidized, defending cells against free radical damage. When ROS levels exceed the antioxidant capabilities of the cell, such as is the case with Aβ toxicity and aging, oxidative damage results. Numerous studies have demonstrated oxidative damage in the brains of AD patients, including increased products of lipid peroxidation, protein oxidation and oxidative damage to nucleic acids 18, 32, 37. In considering the role of oxidative stress in the pathophysiology of AD, the balance between the generation and removal of ROS by antioxidant enzymes is of utmost importance. Modification of the activities of these antioxidant enzymes could be potential pharmacological targets for future AD therapy.
One such enzyme that plays a key role in maintaining oxidative balance in cells is catalase. Catalase is a heme‐containing tetrameric antioxidant enzyme, predominantly found in peroxisomes, that catalyzes the conversion of hydrogen peroxide (H2O2) to water and oxygen. Deficiencies in catalase activity, expression and peroxisomal localization are associated with oxidative stress, aging and human disease 16, 28, 33, 43. Aβ has been shown to inhibit catalase activity in cells, resulting in increased H2O2 levels 12, 20, 21. Moreover, addition of catalase to Aβ challenged cells has been shown to protect cells from Aβ toxicity by decreasing levels of H2O2 and reducing protein and lipid oxidation 4, 17, 25.
Catalase is targeted to peroxisomes by a type 1 peroxisomal targeting signal (PTS1), with the carboxy‐terminus residues, lysine‐alanine‐asparagine‐leucine (KANL). This targeting sequence is different from the classical PTS1 of other peroxisomal enzymes, which have a serine‐lysine‐leucine (SKL) consensus sequence 16, 28. The PTS1 KANL only poorly targets catalase to peroxisomes, and as cells age it has been shown that catalase is increasingly mislocalized to the cytosol 16. The poor targeting efficiency of the KANL sequence coupled with the mislocalization of catalase to peroxisomes as cells age is associated with accumulation of H2O2 in cells and resultant oxidative injury. In order to better target catalase to peroxisomes, a genetically engineered variant of the enzyme, CAT–SKL has been developed (United States patent 7,601,366 and 8,663,630 and several foreign patents). This recombinant enzyme is able to enter cells and traffic to organelles (mostly peroxisomes) where it is able to efficiently metabolize H2O2 16, 23, 36, 44. Use of CAT–SKL has been shown to restore catalase levels and oxidative equilibrium in a number of cellular settings 9, 10, 16, 36, 43, 44. CAT–SKL has also been shown to be protective against Aβ derived diffusible ligand (ADDL)‐induced cytotoxicity in rat primary cortical/hippocampal cultures via reduction of H2O2 levels 9.
Since Aβ results in increased H2O2 levels, and addition of catalase to cell culture alleviates this increase in H2O2, it is hypothesized that the use of CAT–SKL in vivo could be a targeted approach to reducing the toxicity induced by Aβ. Therefore, the aim of the present study was to evaluate whether the targeted antioxidant CAT–SKL is able to reduce the toxicity induced by Aβ25–35 administration in the mature rat brain. Previous work has demonstrated the toxicity induced by intracerebroventricular (icv) administration of Aβ25–35 in the rodent brain, with pathological changes including increased activation of inflammatory cells, loss of hippocampal and cholinergic neurons, and cognitive deficits being realized 2, 3, 5, 22, 40, 41, 42. Results from this study demonstrate the ability of CAT–SKL to reduce Aβ induced microglia and astrocyte activation, enhance cholinergic neuronal survival and attenuate long‐term reference memory deficits in rats.
Materials and Methods
Animals and treatment groups
All experimental procedures were carried out in accordance with the guidelines of the Canadian Council on Animal Care and were approved by Western University Animal Use Subcommittee. Animals were carefully monitored and all efforts were made to minimize the number of animals used. Adult male Wistar rats (Charles River, Montreal QC, Canada) 6 months of age weighing 600–650 g at the beginning of the experiments were housed at a temperature of 22–24°C under a 12 h:12 h light:dark cycle. Rats were provided food and water ad libitum. Animals were randomly assigned to one of four groups. Group one received bilateral icv injections of the reverse Aβ35–25 peptide and ip saline injections (RP, n = 11), group two received bilateral icv injections of the reverse Aβ35‐25 peptide and ip CAT–SKL injections (RP + CAT–SKL, n = 12), group three received bilateral icv Aβ25–35 injections and ip saline injections (Aβ, n = 12) and group four received bilateral icv injections of Aβ25‐35, and ip CAT–SKL injections (Aβ + CAT–SKL, n = 12).
Aβ preparation and administration
Aβ25–35 or the reverse physiologically inactive, Aβ35–25 peptide (Bachem, Torrance, CA, USA) was prepared at a concentration of 21.2 μg/μL and administered via icv injection as previously described 22.
CAT–SKL treatment
The genetic reengineering of the catalase enzyme and purification of CAT–SKL is described elsewhere 16, 43, 44. Rats undergoing catalase‐SKL (CAT–SKL) treatment received a total of four catalase injections, once per week for 4 sequential weeks (Figure 1A). The decision to administer CAT–SKL once a week was based on a study that used a similar treatment paradigm of CAT–SKL administration in mice 8. Animals were weighed immediately prior to injection and were administered 1 mg/kg of CAT–SKL by ip injection. Those animals not receiving CAT–SKL injections received ip injections of an equivalent volume of saline. The recombinant enzyme CAT–SKL was acquired from Dr. Paul A. Walton and Dr. Stanley R. Terlecky (United States patent 7,601,366 and 8,663,630 and related international patents).
Figure 1.
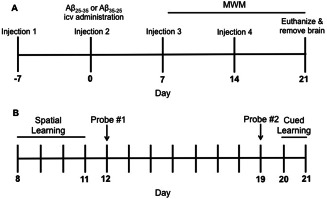
Treatment paradigm and time course for CAT–SKL injections, Aβ administration and behaviour testing.
(A) CAT–SKL or saline injections were administered ip once a week for four consecutive weeks. Aβ25–35 or the reverse peptide Aβ35–25 was injected icv on day 0 and animals were sacrificed on day 21. (B) Timeline for behavior testing in the Morris water maze (MWM) on days 8–21. Spatial learning took place from days 8 to 11. Rats underwent two probe trials, one on day 12 and the second probe trial on day 19. Cued learning occurred on days 20 and 21.
Behaviour testing: Morris water maze
Spatial learning and reference memory were tested in the Morris water maze (MWM) as described previously 22, 38. Briefly, animals underwent four days of spatial learning, followed by two probe trials (days 12 and 19) and two days of cued learning (day 20–day 21) (Figure 1B). Spatial learning took place from days 8 to 11 following icv injections of Aβ25–35 or Aβ35–25 and consisted of 16 training trials, with four trials per day for 4 days with an inter‐trial interval of 20 min. Twenty‐four hours following the last spatial acquisition trial, rats were subjected to a probe trial where the platform was removed from the pool and rats were allowed to swim freely in the water for 30 s. On day 19, rats received an additional probe trial for 30 s to determine long‐term memory retention. Rats were not trained during the time period between day 12 and day 19. The time spent and the distance travelled in the target quadrant was taken as an index of rats' memory capacity, with a value below 25% being considered chance. On days 20–21 rats were trained in a nonspatial cued version of the water maze. For the cued training rats received four trials per day for 2 days, with the location of the hidden platform and the rats start position varying with each trial. A cue directly attached to the platform was used to indicate the platforms position. Cued learning was used as a control procedure in order to determine if any differences observed in the MWM could be attributed to either a difference in motivation to escape the water, or an inability to use cues to locate the hidden platform. Animal behavior, including swimming speed, distance travelled and latency to find the platform were monitored using video‐tracking software for all MWM tasks (ANY‐maze®, Stoelting Co., Wood Dale, IL).
Tissue isolation/preparation
Twenty‐one days following Aβ25–35 or Aβ35–25 administration rats were weighed and then euthanized with an overdose of Euthanyl (pentobarbital sodium, 54.7 mg/mL). A subset of rats (Aβ + CAT–SKL n = 7; Aβ n = 6; RP + CAT–SKL n = 7; RP n = 6) were then perfused transaortically with 0.01 M phosphate buffer saline (PBS, pH 7.35) followed by 4% paraformaldehyde (PFA, pH 7.35, 300 mL). Brains were then removed and further fixed for 24 h in PFA at 4°C after which they were transferred to a 30% sucrose solution for 3 days. Thereafter, brains were frozen on dry ice and serially sliced into 35‐μm coronal sections (from 3.1 to −4.8 mm relative to bregma according to the atlas of Paxinos and Watson), using a Leica CM1850 Cryostat (Leica Biosystems, Concord, ON, Canada) in preparation for histochemical examination. Remaining animals (Aβ + CAT–SKL n = 5; Aβ n = 6; RP + CAT–SKL n = 5; RP n = 5) were sacrificed by decapitation and brain samples were isolated, flash frozen in dry ice, and subsequently stored at −80°C until used in biochemical assays.
Histology (H&E staining; OX‐6, GFAP, ChAT immunolabeling)
For histology brain sections were stained with 0.1% Mayer's hematoxylin solution and 0.5% eosin Y solution, dehydrated, and coverslipped. Immunohistochemistry was performed on free‐floating serial coronal sections to visualize astrocytes, microglia and cholinergic neurons as previously described 1, 2, 22. The following primary antibodies were used: mouse monoclonal anti‐glial fibrillary acidic protein (GFAP; 1:1000; Sigma‐Aldrich, St. Louis MO, USA) to assess astrocyte activation, ramified microglia were detected using a mouse monoclonal antibody OX‐6 directed against the MHC II receptor (OX‐6; 1:1000; BD Pharmingen, Mississauga ON, Canada) and a monoclonal mouse anticholine acetyltransferase (ChAT; 1:500; Abcam, Cambridge MA, USA) was used to detect cholinergic neurons. After incubation with the primary antibody, sections were incubated with biotinylated antimouse secondary antibody (1:2000, Vector Laboratories, Burlingame, CA, USA) followed by incubation with avidin–biotinylated complex (Vector Laboratories, Burlingame, CA, USA) reagent and then visualized using 0.05% 3, 3′ diaminobenzidine tetrahydrochloride (Sigma‐Aldrich, St. Louis MO). Series representative of each treatment group were processed together to reduce variability between groups.
Catalase activity
Catalase activity was measured by its ability to degrade H2O2, as previously described 16, 30. For the determination of catalase activity levels, frontal cortex, hippocampal, thalamic and cerebellar brain tissue was isolated and homogenized in tissue protein extraction buffer (50 mM Tris–HCL, 150 mM NaCl, 0.1% Tween 20, dH20, protease cocktail inhibitor). Homogenized tissue was centrifuged (2× at 4°C 13,000 RPM for 20 min) and the supernatant was collected. Sample protein concentration was then measured using a Pierce BCA protein assay kit (Pierce, Rockford IL, USA) and 15 μg from each sample were taken for the catalase activity assay. Samples were added to a reaction mixture of 0.02 M imidazole buffer, 1 mg/mL BSA, 0.2% Triton‐X, and 0.01% hydrogen peroxide and incubated at room temperature. The reaction was stopped at 0, 4, 6, 8 or 10 min time points with titanium (IV) oxysulfate (TiOSO4) stop solution (TiOSO4 in 2N H2SO4) which reacts with remaining hydrogen peroxide in solution to produce a yellow peroxotitanium complex. Absorbance at 405 nm was then measured for sample and nonsample containing wells, whereby the difference yielded a rate of ΔOD405nm/min. Rates were then adjusted for protein concentration as determined by BCA protein assay, yielding a ΔOD405nm/min/mg total protein (Derived from Storrie and Madden, 30).
Imaging and quantification
Stained brain sections were photographed with a Leica DFC295 camera coupled to a Leica DM IRE2 microscope (Leica Microsystems, Concord, ON, Canada) with Leica Application Suite Version 4.1.0 image analysis software (Leica Microsystems). Analysis and quantification were carried out using ImageJ 1.45s software (Wayne Rasband, National Institute of Health, Bethesda, MD, USA).
Areas examined included the frontal cortex, striatum, hippocampus (CA1 and CA3), thalamus, medial septal nucleus (MSN), and vertical diagonal band (VDB) of the basal forebrain, and the corpus callosum. Further quantification was completed for the hippocampus (CA1 and CA3), thalamus (ventral posteromedial and ventral posterolateral thalamic nuclei), and MSN/VDB region of the basal forebrain. Microglia, astrocyte, and cholinergic neuronal analysis and quantification was completed as previously described 22. For H&E analysis photomicrographs were taken from the left and right CA1 and CA3 regions of the hippocampus. Two different observers counted the number of undamaged neurons in the CA1 and CA3 region of the hippocampus; the observers completed the counts independently from one another and were blinded to the experimental conditions. Only cells with a neuronal morphology were counted; undamaged neurons were those cells with intact cell membranes and full nucleus. The number of undamaged neurons per optical field (neuronal density) was determined in four tissue sections per rat.
Statistical analysis
Statistical analysis was performed using GraphPad Prisim 5.0 for MAC OSX. Data were analyzed by performing a one‐way analysis of variance (ANOVA) (F values). When ANOVA indicated significant treatment effects, means were separated using Tukey's multiple comparison test. Data are expressed as mean ± standard error of the mean (SEM), and a minimum of P < 0.05 was considered statistically significant. In some cases, statistical significance between treatment groups was indicated using a lettering system on graphs. Letters shared in common between or among groups indicated no significant differences.
Results
Catalase activity
Catalase activity was measured in the thalamus, frontal cortex, hippocampus, and cerebellum. CAT–SKL administration appeared to increase catalase activity levels in the cerebellum in RP and Aβ animals compared to their respective, non CAT–SKL administered controls. In the thalamus, catalase activity levels appeared higher in Aβ CAT–SKL administered animals compared to all other groups. However, these differences in catalase activity levels in the thalamus and cerebellum did not reach significance. Catalase activity levels in the frontal cortex and hippocampus appeared unaffected by treatment with CAT–SKL (Table 1).
Table 1.
Regional catalase activity levels
| RP | RP + CAT–SKL | Aβ | Aβ + CAT–SKL | |
|---|---|---|---|---|
| Cerebellum | 1.33 ± 0.04 | 1.38 ± 0.09 | 1.40 ± 0.08 | 1.47 ± 0.06 |
| Frontal Cortex | 0.55 ± 0.07 | 0.59 ± 0.01 | 0.42 ± 0.04 | 0.39 ± 0.13 |
| Hippocampus | 0.82 ± 0.14 | 0.78 ± 0.05 | 0.89 ± 0.07 | 0.85 ± 0.08 |
| Thalamus | 0.92 ± 0.09 | 0.86 ± 0.08 | 0.91 ± 0.06 | 1.06 ± 0.09 |
Catalase activity (ΔOD405nm/min/mg total protein) measured in the Cerebellum, Frontal Cortex, Hippocampus and Thalamus in rats subjected to icv injections of Aβ25–35 or reverse peptide Aβ35–25 with and without CAT–SKL treatment. Data are presented as mean ± SEM, n = 5–6 per group.
Neuroinflammation: microglia and astrocyte activation
Immunohistochemical assessment showed an increase in the appearance of ramified microglia in the MSN/VDB (F 3,26 = 11.43, P < 0.0001) and thalamus (F 3,27 = 3.32, P < 0.03) of Aβ25–35 injected rats, which was reduced in animals treated with CAT–SKL. Microglia in the MSN/VDB of Aβ25–35 icv injected rats was significantly increased compared to the control RP (P < 0.001), and RP + CAT–SKL animals (P < 0.001). This microglia activation was effectively reduced by treatment with CAT–SKL with Aβ + CAT–SKL animals showing a significant decrease in microglia in the MSN/VDB when compared to Aβ25–35 animals (P < 0.05; Figure 2D). Microglia activation in the thalamus was significantly greater in Aβ25–35 administered animals compared to the control RP group (P < 0.05). CAT–SKL treatment combined with Aβ25–35 administration reduced microglia activation in the thalamus such that the microglia in the thalamus of Aβ + CAT–SKL animals was not significantly different from both control groups (RP or RP + CAT–SKL) (P > 0.05; Figure 2E).
Figure 2.
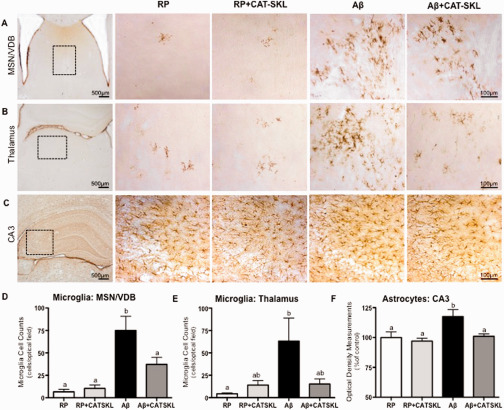
Neuroinflammation.
Representative photomicrographs of microglia immunolabeled with OX‐6 in (A) the medial septal nucleus/vertical diagonal band (MSN/VDB) of the basal forebrain (B) OX‐6 labeled activated microglia in the thalamus, and (C) astrocytes labeled with GFAP in the CA3 region of the hippocampus in RP, RP + CAT–SKL, Aβ, and Aβ + CAT–SKL rats, respectively. The number of OX‐6 positive microglia in the (D) MSN/VDB and (E) thalamus and (F) optical density measurements of GFAP immunopositive astrocytes in the CA3 region of the hippocampus shown as a percentage of the mean value of the RP group. Boxed areas in low magnification images in panel 1 illustrate the location of high magnification pictures in panels 2–4. Data presented as mean ± SEM, n = 6–7 animals per group, means with different letters are significantly different.
Optical density measurements of GFAP immunopositive astrocytes was taken as a measurement of astroglial reactivity in the hippocampus. One‐way ANOVA analysis revealed statistically significant differences between treatment groups (F 3,30 = 4.89, P < 0.007). Aβ25–35 administered animals showed a significant increase in astrocyte density in the CA3 region of the hippocampus compared to RP and RP + CAT–SKL treated animals (P < 0.05 vs. RP, P < 0.01 vs. RP + CAT–SKL). CAT–SKL treatment reversed this increase in astrocyte density induced by Aβ25–35 in the CA3 (P < 0.05 Aβ vs. Aβ + CAT–SKL) (Figure 2F). No differences in astrocyte density were detected in the CA1 region of the hippocampus between treatment groups (data not shown).
Cholinergic neurons in the MSN/VDB in CAT–SKL treated animals
Aβ25‐35 administered animals had a significantly reduced number of ChAT positive cholinergic neurons in the MSN/VDB compared to RP and RP + CAT–SKL treated rats (P < 0.05). With CAT–SKL treatment this significant reduction induced by Aβ25–35 administration was lost and there were no significant differences in cholinergic neuronal counts between Aβ + CAT–SKL treated rats and the control RP or RP + CAT–SKL treated animals (P > 0.05; Figure 3).
Figure 3.
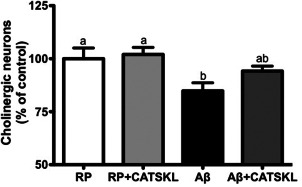
CAT‐SKL prevents cholinergic loss in the MSN/VDB.
The number of choline acetyltransferase (ChAT) immunolabeled cholinergic neurons shown as a percentage of the RP group in the medial septal nucleus/vertical diagonal band (MSN/VDB) of the basal forebrain in RP, RP + CAT–SKL, Aβ, and Aβ + CAT–SKL treated rats. Data are presented as mean ± SEM. Means with different letters signify significance.
Neuronal numbers in the hippocampus following Aβ25–35 administration and CAT–SKL treatment
Counts of H&E stained pyramidal neurons in the CA1 region of the hippocampus revealed no differences in neuronal numbers between treatment groups (Figure 4C). However, there was a significant reduction in neuronal numbers in the CA3 region of the hippocampus in Aβ25–35 treated rats compared to RP treated animals (P < 0.05). With CAT–SKL treatment this significant reduction in neuronal numbers was no longer observed. There were no significant differences in pyramidal cell numbers among Aβ, RP + CAT–SKL, and Aβ + CAT–SKL treated rats (Figure 4D).
Figure 4.
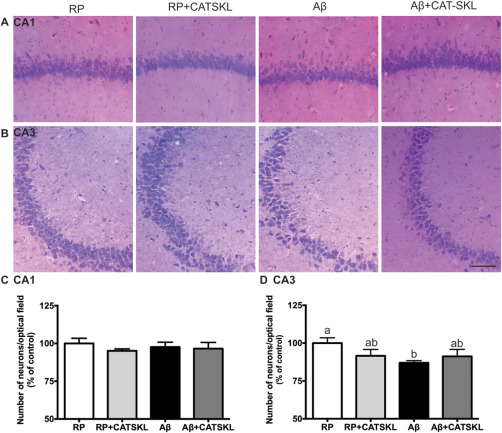
Neuronal numbers in the CA1 and CA3 regions of the hippocampus.
Representative photomicrographs of hematoxylin and eosin stained cells in (A) the CA1 and (B) the CA3 region of the hippocampus in RP, RP + CAT–SKL, Aβ, and Aβ + CAT–SKL treated rats respectively. (C,D) Pyramidal cell numbers in the CA1 and CA3 subfields of the hippocampus shown as a percentage of the control RP group. Data presented as mean ± SEM, n = 6–7 animals per group, different letters represent statistically significant differences.
Behavior testing: Morris water maze
Latency, path length and swimming speed during spatial learning
Latency and distance traveled to find the platform decreased significantly over the course of acquisition training for all treatment groups (P < 0.001 day 4 vs. day 1), indicative of successful learning of the task. There were no differences in latency or distance travelled to find the platform between treatment groups, suggesting Aβ25–35 toxicity did not impair spatial learning (Figure 5A,B). Average swimming speed was not significantly different across training days within a group and there were no differences in mean swimming speed between treatment groups across training days (data not shown).
Figure 5.
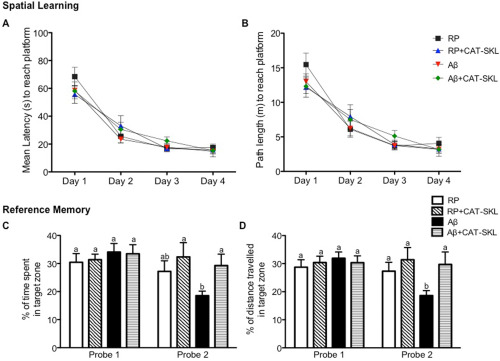
Spatial learning and reference memory in the Morris Water Maze (MWM).
(A) Mean latency and (B) path length to find the hidden platform in the MWM during four consecutive days of spatial learning for RP, RP + CAT–SKL, Aβ, and Aβ + CAT–SKL treatment groups. Spatial learning consisted of four training trials per day for four consecutive days. Reference memory was measured as (C) the percentage of total time spent and (D) percentage of total distance traveled in the quadrant where the platform was located 24 h following the last spatial learning trial (Probe 1) and 8 days after the last spatial training trial (Probe 2) for RP, RP + CAT–SKL, Aβ, and Aβ + CAT–SKL treatment groups. Data are presented as mean ± SEM, n = 8/9 animals per group, different letters represent statistically significant differences.
Probe trials: short and long‐term memory retention
On day 12, 24 h following the last spatial learning trial, all groups demonstrated memory of the platform location based on their preference for the target quadrant as indicated by the distance and time spent in that quadrant. No differences in percentage of time spent or distance travelled in the target zone was identified between treatment groups (Figure 5C,D). On day 19, 7 days after the first probe trial, rats were subjected to a second probe trial to assess long‐term reference memory retention. RP + CAT–SKL and Aβ + CAT–SKL treated animals spent a significantly greater percentage of time in the target zone than Aβ administered animals (P < 0.01 for RP + CAT–SKL vs. Aβ, P < 0.05 for Aβ + CAT–SKL vs. Aβ) (Figure 5C,D). RP, RP + CAT–SKL, and Aβ + CAT–SKL animals all travelled a greater distance in the target zone than Aβ only animals (P < 0.05 for RP vs. Aβ, P < 0.001 for RP + CAT–SKL vs. Aβ, P < 0.01 for Aβ + CAT–SKL vs Aβ).
Comparison between probe trial on D12 and D19
Aβ administered animals spent significantly less time and traveled significantly less distance in the target zone on day 19 compared to day 12 (P < 0.01). No significant differences in time spent or distance travelled in the target quadrant between day 12 and day 19 were found for RP, RP + CAT–SKL and Aβ + CAT–SKL treated rats (P > 0.05; Figure 5C,D). The reduction in time spent and distance traveled in the target quadrant on day 19 compared to day 12 for Aβ25–35 animals indicates long‐term reference memory deficits, which was effectively ameliorated in Aβ animals treated with CAT–SKL.
Cued learning
Animals across treatment groups showed no significant differences in the time it took them to locate the platform, or distance travelled to find the platform. Average swimming speed was not significantly different between treatment groups (data not shown). Thus, rats across treatment groups demonstrated similar motivation and abilities to escape the water.
Discussion
This study was the first to use the targeted antioxidant CAT–SKL to try to reduce the toxicity induced by Aβ25–35 in the mature rat brain. Previous work in animal models has shown infusion of Aβ increases H2O2 formation, reduces the activity of H2O2 degrading enzymes and increases the activity of H2O2‐generating enzymes in the rat brain 15. CAT–SKL is a genetically engineered derivative of the antioxidant enzyme catalase. The SKL targeting sequence enables catalase to be more effectively targeted to peroxisomes, where its main function is to metabolize H2O2 to oxygen and water. Metabolism of H2O2 is critical, because in addition to being a potentially toxic metabolite on its own, it can also react with Fe2+ to generate hydroxyl radicals, which are exquisitely reactive species capable of inducing protein, lipid and DNA damage 18, 19, 34.
In this study, CAT–SKL was shown to reduce microglia activation in the MSN/VDB and thalamus of 6 months old Aβ25–35 administered rats. Reduction in microglia activation is likely a secondary consequence of the antioxidant properties of the CAT–SKL molecule. By decreasing ROS production, the toxicity induced by Aβ would be lessened thereby decreasing the activation and proliferation of inflammatory microglia and astrocytes. CAT–SKL may also have aided in reducing the production of inflammatory molecules. Previous studies in vitro have demonstrated the ability of CAT–SKL supplementation to reduce the expression of the inflammatory cytokine TNF‐α in a human cell model of psoriasis 44. Moreover, CAT–SKL has been shown to protect rat myocytes from hypoxia–reoxygeneation and ischemia reperfusion injury via reduction of oxidative stress in cell culture 36.
Treatment with CAT–SKL was also able to decrease cholinergic neuronal loss in the MSN/VDB of the basal forebrain, and promoted neuronal survival in the CA3 region of the hippocampus. Presumably this reduction in neuronal loss is related to the decreased inflammation seen following CAT–SKL treatment. Aβ, inflammation and ROS work in a self‐propagating cycle, with the result being excessive neuroinflammation and oxidative damage that can disrupt normal cellular functioning and ultimately lead to neuronal death. Stimulation of ROS production and activation of inflammatory molecules in culture has been shown to induce neuronal death. Moreover, Aβ has been shown to mediate cell death via its production of ROS 14. Therefore, the protective effect of CAT–SKL on neuronal functioning and survival could be via CAT–SKL mediated reduction in ROS and inflammation.
Presumably the protective effects of CAT–SKL are exerted in part by increasing catalase activity levels in the brain. However, the results from the catalase assay revealed no significant differences in catalase activity levels throughout the brain regions between groups. The lack of difference in catalase activity levels could be attributed to the time point at which catalase activity was measured. In this study we only examined catalase activity in brain tissue at a 21‐day time point. This is 7 days after the last CAT–SKL injection and thus this time frame may not have been optimal for detecting significant changes in catalase levels. This study did not evaluate levels of catalase protein expression in the brain. Although there is no direct evidence that CAT–SKL crossed the blood brain barrier and increased catalase levels, the impact on the brain was significant. This could be due to CAT–SKL directly in brain tissue or the systemic generation of other agents that did cross the blood brain barrier. In future studies analysis of protein levels is regions of the brain where the greatest changes in pathology are seen may help further our understanding of the mechanism of action of CAT–SKL especially when corroborated with data from catalase activity assays.
Cognitive performance was evaluated in a spatial learning and reference memory task in the MWM. All animals learned the task to the same degree, as shown in the spatial learning phase of the test. Upon evaluation of reference memory animals in all groups showed no differences in memory during the first probe trial. During the second probe trial, 19 days after Aβ25–35 injection, animals showed a significantly decreased preference for the target zone than animals from other treatment groups. Aβ25–35 injected animals also spent less time and traveled a shorter distance in the target zone during the second probe trial compared to the first probe trial. This decreased preference for the target zone during the second probe trial was not seen in Aβ25‐35 administered animals treated with CAT–SKL. Taken together this indicates that Aβ25‐35 icv administration induces long‐term reference memory deficits, and moreover treatment with CAT–SKL is able to attenuate Aβ25‐35 induced long‐term reference memory impairments.
Previous studies have investigated the role of catalase in maintaining oxidative equilibrium. Addition of catalase to neuronal cultures challenged with Aβ has been shown to reduce H2O2 levels and improve neuronal survival 4, 17, 45. Moreover, inhibition of catalase activity has been shown to enhance the cytotoxicity of Aβ in neuronal cultures (by increasing ROS levels), indicating an important role of this antioxidant enzyme in maintaining oxidative balance 4, 20. Work in a transgenic mouse model of AD has demonstrated the beneficial effects of using a superoxide dismutase/catalase mimetic, EUK‐207, to reduce Aβ pathology. EUK‐207 was shown to reduce oxidation of nucleic acids and lipid peroxidation, and was able to decrease Aβ and tau accumulation 6. Moreover, the impact of ROS, and in particular H2O2 levels on longevity has been examined in a transgenic mouse line overexpressing human catalase. The study demonstrated a significant enhancement in murine lifespan in mice overexpressing catalase when compared to wild type controls. This increased longevity was attributed in part to the reduction in H2O2 levels and oxidative stress 26. Taken together these studies provide evidence for the protective role of catalase in aging, and in reducing Aβ toxicity. The CAT–SKL molecule may be of further benefit due to its unique targeting signal that directs it to the organelle where it can carry out its function—the peroxisome.
Currently we have a limited understanding of the mechanism by which CAT–SKL supplementation reduces amyloid toxicity in vivo. Work in vitro has demonstrated CAT–SKL reduces ADDL toxicity in neuronal cultures via reduction of H2O2 levels and oxidative stress 9. Additionally, it has been shown in vitro that re‐establishing peroxisomal catalase has downstream protective effects on mitochondria via reestablishing redox balance 16, and that loss of catalase activity promotes oxidative damage in mitochondria 16, 39. However, future work investigating the mechanism of action of CAT–SKL in vivo is needed.
Finally, this study involved administration of CAT–SKL beginning one week prior to Aβ administration therefore the neuroprotective effects of CAT–SKL may be due in part to prevention rather than treatment of Aβ toxicity. Further studies are needed to elucidate whether CAT–SKL is able to reduce pre‐existing Aβ‐induced pathology, or whether it works primarily in a preventative manner, priming the brain and making it more resilient to neuroinflammation and the subsequent neurodegeneration that results from Aβ toxicity.
Conclusion
Substantial evidence exists implicating oxidative stress and neuroinflammation in the pathogenesis of AD. While it is unclear whether oxidative stress is an initiator of AD pathogenesis or a mediator of the disease process it is known that oxidative stress occurs during the early stages of the disease process, before the appearance of amyloid plaques and neurofibrillary tangles in both humans and in animals models of the disease 7, 37. Therapeutics aimed at restoring or maintaining the homeostatic balance between production and elimination of ROS, and thus reducing oxidative stress and inflammation during the early stages of the disease may help in slowing disease progression and may aide in the protection of at‐risk individuals from the development of AD. The antioxidant molecule, CAT–SKL, may therefore be a viable therapeutic approach for reducing oxidative stress and neuroinflammation during the beginning stages of AD pathogenesis.
Acknowledgments
This research was supported by an emerging team grant from the Canadian Institutes of Health Research (CIHR, R1478A47) to D.F.Cechetto. The recombinant enzyme CAT–SKL was acquired from P.A.Walton and S.R.Terlecky and is covered by United States patent 7,601,366 and 8,663,630 and related international patents. S.R. Terlecky and P.A. Walton have no commercial or equity interests in the CAT–SKL molecule at this time. Other authors state no conflict of interest. Finally, we would like to thank Lin Wang for her invaluable technical assistance on this research project.
References
- 1. Akiyama H, Barger S, Barnum S, Bradt B, Bauer J, Cole GM et al (2000) Inflammation and Alzheimer's disease. Neurobiol Aging 21:383–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Amtul Z, Nikolova S, Gao L, Keeley RJ, Bechberger JF, Fisher AL et al (2014) Comorbid Abeta toxicity and stroke: hippocampal atrophy, pathology, and cognitive deficit. Neurobiol Aging 35:1605–1614. [DOI] [PubMed] [Google Scholar]
- 3. Amtul Z, Whitehead S, Keeley R, Bechberger J, Fisher A, McDonald R et al (2015) Comorbid rat model of ischemia and beta‐amyloid toxicity: striatal and cortical degeneration. Brain Pathol 25:24–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Behl C, Davis JB, Lesley R, Schubert D (1994) Hydrogen peroxide mediates amyloid beta protein toxicity. Cell 77:817–827. [DOI] [PubMed] [Google Scholar]
- 5. Cheng G, Whitehead SN, Hachinski V, Cechetto DF (2006) Effects of pyrrolidine dithiocarbamate on beta‐amyloid (25–35)‐induced inflammatory responses and memory deficits in the rat. Neurobiol Dis 23:140–151. [DOI] [PubMed] [Google Scholar]
- 6. Clausen A, Xu X, Bi X, Baudry M (2012) Effects of the superoxide dismutase/catalase mimetic EUK‐207 in a mouse model of Alzheimer's disease: protection against and interruption of progression of amyloid and tau pathology and cognitive decline. J Alzheimers Dis 30:183–208. [DOI] [PubMed] [Google Scholar]
- 7. Dumont M, Beal MF (2011) Neuroprotective strategies involving ROS in Alzheimer disease. Free Radic Biol Med 51:1014–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Giordano CR, Roberts R, Krentz KA, Bissig D, Talreja D, Kumar A et al (2015) Catalase therapy corrects oxidative stress‐induced pathophysiology in incipient diabetic retinopathy. Invest Ophthalmol Vis Sci 56:3095–3102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Giordano CR, Terlecky LJ, Bollig‐Fischer A, Walton PA, Terlecky SR (2014) Amyloid‐beta neuroprotection mediated by a targeted antioxidant. Sci Rep 4:4983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Giordano CR, Terlecky SR (2012) Peroxisomes, cell senescence, and rates of aging. Biochim Biophys Acta 1822:1358–1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Haass C, Selkoe DJ (1993) Cellular processing of beta‐amyloid precursor protein and the genesis of amyloid beta‐peptide. Cell 75:1039–1042. [DOI] [PubMed] [Google Scholar]
- 12. Habib LK, Lee MT, Yang J (2010) Inhibitors of catalase‐amyloid interactions protect cells from beta‐amyloid‐induced oxidative stress and toxicity. J Biol Chem 285:38933–38943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hardy J, Selkoe DJ (2002) The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science 297:353–356. [DOI] [PubMed] [Google Scholar]
- 14. Kadowaki H, Nishitoh H, Urano F, Sadamitsu C, Matsuzawa A, Takeda K et al (2005) Amyloid beta induces neuronal cell death through ROS‐mediated ASK1 activation. Cell Death Differ 12:19–24. [DOI] [PubMed] [Google Scholar]
- 15. Kaminsky YG, Kosenko EA (2008) Effects of amyloid‐beta peptides on hydrogen peroxide‐metabolizing enzymes in rat brain in vivo . Free Radic Res 42:564–573. [DOI] [PubMed] [Google Scholar]
- 16. Koepke JI, Nakrieko KA, Wood CS, Boucher KK, Terlecky LJ, Walton PA et al (2007) Restoration of peroxisomal catalase import in a model of human cellular aging. Traffic 8:1590–1600. [DOI] [PubMed] [Google Scholar]
- 17. Manelli AM, Puttfarcken PS (1995) beta‐Amyloid‐induced toxicity in rat hippocampal cells: in vitro evidence for the involvement of free radicals. Brain Res Bull 38:569–576. [DOI] [PubMed] [Google Scholar]
- 18. Markesbery WR, Carney JM (1999) Oxidative alterations in Alzheimer's disease. Brain Pathol 9:133–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Milton NG (2004) Role of hydrogen peroxide in the aetiology of Alzheimer's disease: implications for treatment. Drugs Aging 21:81–100. [DOI] [PubMed] [Google Scholar]
- 20. Milton NG (2001) Inhibition of catalase activity with 3‐amino‐triazole enhances the cytotoxicity of the Alzheimer's amyloid‐beta peptide. Neurotoxicology 22:767–774. [DOI] [PubMed] [Google Scholar]
- 21. Milton NG (1999) Amyloid‐beta binds catalase with high affinity and inhibits hydrogen peroxide breakdown. Biochem J 344 (Pt 2 ):293–296. [PMC free article] [PubMed] [Google Scholar]
- 22. Nell HJ, Whitehead SN, Cechetto DF (2014) Age‐dependent effect of beta‐amyloid toxicity on basal forebrain cholinergic neurons and inflammation in the rat brain. Brain Pathol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Price M, Terlecky SR, Kessel D (2009) A role for hydrogen peroxide in the pro‐apoptotic effects of photodynamic therapy. Photochem Photobiol 85:1491–1496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Querfurth HW, LaFerla FM (2010) Alzheimer's disease. N Engl J Med 362:329–344. [DOI] [PubMed] [Google Scholar]
- 25. Sagara Y, Dargusch R, Klier FG, Schubert D, Behl C (1996) Increased antioxidant enzyme activity in amyloid beta protein‐resistant cells. J Neurosci 16:497–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Schriner SE, Linford NJ, Martin GM, Treuting P, Ogburn CE, Emond M et al (2005) Extension of murine life span by overexpression of catalase targeted to mitochondria. Science 308:1909–1911. [DOI] [PubMed] [Google Scholar]
- 27. Selkoe DJ (2001) Alzheimer's disease: genes, proteins, and therapy. Physiol Rev 81:741–766. [DOI] [PubMed] [Google Scholar]
- 28. Sheikh FG, Pahan K, Khan M, Barbosa E, Singh I (1998) Abnormality in catalase import into peroxisomes leads to severe neurological disorder. Proc Natl Acad Sci USA 95:2961–2966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Shoji M, Golde TE, Ghiso J, Cheung TT, Estus S, Shaffer LM et al (1992) Production of the Alzheimer amyloid beta protein by normal proteolytic processing. Science 258:126–129. [DOI] [PubMed] [Google Scholar]
- 30. Storrie B, Madden EA (1990) Isolation of subcellular organelles. Methods Enzymol 182:203–225. [DOI] [PubMed] [Google Scholar]
- 31. Su B, Wang X, Nunomura A, Moreira PI, Lee HG, Perry G et al (2008) Oxidative stress signaling in Alzheimer's disease. Curr Alzheimer Res 5:525–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Tabner BJ, El‐Agnaf OM, German MJ, Fullwood NJ, Allsop D (2005) Protein aggregation, metals and oxidative stress in neurodegenerative diseases. Biochem Soc Trans 33:1082–1086. [DOI] [PubMed] [Google Scholar]
- 33. Terlecky SR, Koepke JI, Walton PA (2006) Peroxisomes and aging. Biochim Biophys Acta 1763:1749–1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Trippier PC, Jansen Labby K, Hawker DD, Mataka JJ, Silverman RB (2013) Target‐ and mechanism‐based therapeutics for neurodegenerative diseases: strength in numbers. J Med Chem 56:3121–3147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Tuppo EE, Arias HR (2005) The role of inflammation in Alzheimer's disease. Int J Biochem Cell Biol 37:289–305. [DOI] [PubMed] [Google Scholar]
- 36. Undyala V, Terlecky SR, Vander Heide RS (2011) Targeted intracellular catalase delivery protects neonatal rat myocytes from hypoxia–reoxygenation and ischemia–reperfusion injury. Cardiovasc. Pathol. 20:272–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Venkateshappa C, Harish G, Mahadevan A, Srinivas Bharath MM, Shankar SK (2012) Elevated oxidative stress and decreased antioxidant function in the human hippocampus and frontal cortex with increasing age: implications for neurodegeneration in Alzheimer's disease. Neurochem. Res. 37:1601–1614. [DOI] [PubMed] [Google Scholar]
- 38. Vorhees CV, Williams MT (2006) Morris water maze: procedures for assessing spatial and related forms of learning and memory. Nat Protoc 1:848–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Walton PA, Pizzitelli M (2012) Effects of peroxisomal catalase inhibition on mitochondrial function. Front Physiol 3:108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Whitehead S, Cheng G, Hachinski V, Cechetto DF (2005) Interaction between a rat model of cerebral ischemia and beta‐amyloid toxicity: II. Effects of triflusal. Stroke 36:1782–1789. [DOI] [PubMed] [Google Scholar]
- 41. Whitehead SN, Cheng G, Hachinski VC, Cechetto DF (2007) Progressive increase in infarct size, neuroinflammation, and cognitive deficits in the presence of high levels of amyloid. Stroke 38:3245–3250. [DOI] [PubMed] [Google Scholar]
- 42. Whitehead SN, Hachinski VC, Cechetto DF (2005) Interaction between a rat model of cerebral ischemia and beta‐amyloid toxicity: inflammatory responses. Stroke 36:107–112. [DOI] [PubMed] [Google Scholar]
- 43. Wood CS, Koepke JI, Teng H, Boucher KK, Katz S, Chang P et al (2006) Hypocatalasemic fibroblasts accumulate hydrogen peroxide and display age‐associated pathologies. Traffic 7:97–107. [DOI] [PubMed] [Google Scholar]
- 44. Young CN, Koepke JI, Terlecky LJ, Borkin MS, Boyd Savoy L, Terlecky SR (2008) Reactive oxygen species in tumor necrosis factor‐alpha‐activated primary human keratinocytes: implications for psoriasis and inflammatory skin disease. J Invest Dermatol 128:2606–2614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Zhang Z, Rydel RE, Drzewiecki GJ, Fuson K, Wright S, Wogulis M et al (1996) Amyloid beta‐mediated oxidative and metabolic stress in rat cortical neurons: no direct evidence for a role for H2O2 generation. J Neurochem 67:1595–1606. [DOI] [PubMed] [Google Scholar]