

Abstract
The expression of subunits of mitochondrial respiratory complexes and components of the protein synthesis machinery from the nucleolus to the ribosome was analyzed in the mediodorsal thalamus in seven cases of fatal familial insomnia (FFI) compared with age‐matched controls. NDUFB8 (complex I subunit), SDHB (complex II subunit), UQCRC2 (complex III subunit), COX2 (complex IV subunit), and ATP50 (complex V subunit) expression levels, as revealed by western blotting, were reduced in FFI. Voltage‐dependent anion channel (VDAC) and ATP5H were also reduced due to the marked depopulation of neurons. In contrast, a marked increase in superoxide dismutase 2 (SOD2) was found in reactive astrocytes thus suggesting that astrocytes are key factors in oxidative stress responses. The histone‐binding chaperones nucleolin and nucleoplasmin 3, and histone H3 di‐methylated K9 were markedly reduced together with a decrease in the expression of protein transcription elongation factor eEF1A. These findings show severe impairment in the expression of crucial components of mitochondrial function and protein synthesis in parallel with neuron loss in mediodorsal thalamus at terminal stages of FFI. Therapeutic measures must be taken long before the appearance of clinical symptoms to prevent the devastating effects of FFI.
Keywords: fatal familial insomnia, mitochondria, mitochondrial respiratory chain, nucleolus, protein synthesis, ribosome
INTRODUCTION
Prion protein is encoded by PRNP located on the short arm of chromosome 20 in humans. The second exon of PRNP encodes a protein of 253 amino acids that is truncated post‐translationally to remove 22 amino acids at the amino‐terminal and 23 amino acids at the carboxyl‐terminal, being replaced at this site by the addition of a glycosylphosphatidylinositol (GPI) anchor. Additional putative glycosylation in two asparagine sites gives rise to three isoforms, fully di‐glycosylated, mono‐glycosylate, and nonglycosylated prion protein 3. Mutations in PRNP are causative of genetic prion diseases, some of which are manifested as familial Creutzfeldt‐Jakob disease (CJD), the phenotype of which depends on the site of PRNP mutation and also on the polymorphism of codon 129 in PRNP 3.
Fatal familial insomnia (FFI) is an autosomal dominant prion disease caused by a D178N mutation in PRNP in combination with methionine (Met) at codon 129 in the mutated allele of the same gene (D178N‐129M haplotype) 15, 23, 24, 25, 26, 30. Homozygosity, either methionine–methionine or valine–valine, at codon 129 in the normal allele of PRNP results in acceleration of clinical symptoms and shorter duration of the disease 5, 13, 28.
Clinically, FFI is principally manifested as sleep disturbances with insomnia, sleep fragmentation, and altered arousal and dreaming, accompanied by autonomic disturbances including increased salivation and sweating, tachycardia, hypertension, and impotence, as well as spontaneous and evoked myoclonus, among other neurological symptoms 5, 12, 14, 17, 20, 21, 28, 37.
Clinical symptoms reflect major atrophy of the anterior ventral and mediodorsal limbic nuclei of the thalamus that extends to the pulvinar, ventral anterior and ventral medial thalamic nuclei with disease progression. The inferior olives are the other major targets in FFI. Severe neuron loss and astrogliosis without spongiform change in these regions, in the context of familial disease, are the neuropathological characteristics of FFI. Moderate astrogliosis without neuron loss is common in the periaqueductal gray matter and hypothalamus. Involvement of the inner temporal cortex, including CA1 region of the hippocampus, cingular cortex and other areas of the neocortex, is variable and depends on the duration of the disease, which in turn largely depends on the codon 129 polymorphism 14, 21, 22, 26, 28.
The deposition of abnormal prion protein (PrPSc) varies from one region to another in FFI. Immunohistochemistry usually shows small granular deposits in the temporal neocortex in cases with relatively long duration, whereas PrPSc deposition in thalamus is very scanty, if present. However, western blotting identifies a particular pattern characterized by a weak band of proteinase‐resistant nonglycosylated PrPSc at 19 kDa (type 2 PrP) and relatively strong bands of mono‐glycosylated and di‐glycosylated PrPSc which disappear following PNGase digestion, giving rise to a robust nonglycosylated band 14, 28, 29, 30.
Recent studies of gene expression profiling have shown altered gene expression in the thalamus in FFI cases when compared with controls. The main changes correspond to the following biological processes: transcription, regulation of transcription, protein biosynthesis, protein folding, protein transport, RNA splicing, electron transport chain, oxidative phosphorylation, energy metabolism, transport, and oxidation reduction 36. Proteomics methods have also been employed to assess altered protein profiles in FFI. Proteins involved in protein export and oxidative phosphorylation, as well as proteins involved in other neurodegenerative diseases as Alzheimer's disease and Parkinson's disease have been identified in the cortex and cerebellum 34. Unfortunately, the thalamus was not examined.
Because of the limited knowledge about altered metabolic pathways in FFI, the present study was designed to learn about alterations of two major complex pathways, one regulating energy metabolism, and particularly mitochondrial function, and the other protein synthesis. The study was performed on postmortem samples of the thalamus of seven well‐characterized cases of FFI which were analyzed using combined qRT‐PCR (quantitative real‐time polymerase chain reaction), western blotting and immunohistochemistry.
MATERIALS AND METHODS
Human samples
All the cases were from the Basque Country in the north of Spain where FFI has a relative high incidence due to a founder effect in a historically small rural community with not uncommon endogamy 31. This study was focused on the mediodorsal thalamus from seven FFI cases and seven controls (Table 1).
Table 1.
Summary of the main clinical and pathologic features of cases used in this study. Abbreviations: PMD = postmortem delay; C = control; FFI = fatal familial insomnia; RIN = RNA integrity number; Thal = mediodorsal thalamus.
| Case | Sex | Age | PMD | Neuropathology | RIN Thal |
|---|---|---|---|---|---|
| 1 | M | 67 | 14 h 40 minutes | C | 6 |
| 2 | M | 61 | 4 h 30 minutes | C | 7.2 |
| 3 | M | 59 | 4 h 15 minutes | C | 6.6 |
| 4 | W | 66 | 12 h 10 minutes | C | 6.7 |
| 5 | M | 76 | 4 h 15 minutes | C | 6 |
| 6 | M | 81 | 11 h 40 minutes | C | 6 |
| 7 | M | 57 | 5 h | C | 6.9 |
| 8 | M | 53 | 12 h 10 minutes | FFI | 7.9 |
| 9 | M | 56 | 22 h 50 minutes | FFI | 5.3 |
| 10 | M | 36 | 17 h 30 minutes | FFI | 6.4 |
| 11 | M | 70 | 15 h 30 minutes | FFI | ‐ |
| 12 | M | 50 | 12 h 30 minutes | FFI | 6.8 |
| 13 | M | 47 | 3 h 30 minutes | FFI | 8.2 |
| 14 | M | 54 | 9 h 50 minutes | FFI | 7.5 |
All FFI cases were males, had the mutation D178N and were Met/Met homozygous at the codon 129 of PRNP. The most common age at onset was between 47 and 57 years (five cases) with the exception of one young person aged 36 and one old individual aged 70. The duration of the disease was between 6 and 16 months independently of the age at onset.
Brain tissue was obtained from the Brain Bank of the Araba University Hospital and Basque Biobank for Research (O + eHun) and the Institute of Neuropathology Biobank following the guidelines of the Spanish legislation on this matter and the approval of the local ethics committees. The postmortem interval between death and tissue processing was between 4 h 15 minutes and 22 h 50 minutes. One hemisphere was immediately cut in coronal sections, 1 cm thick, and selected areas of the encephalon were rapidly dissected, frozen on metal plates over dry ice, placed in individual airtight plastic bags, numbered with water‐resistant ink and stored at −80°C until use. The other hemisphere was fixed by immersion in 4% buffered formalin for 3 weeks. Neuropathological examination in all cases was routinely performed on 20 selected dewaxed paraffin sections comprising different regions of the cerebral cortex, diencephalon, thalamus, brain stem, and cerebellum which were stained with haematoxylin and eosin, Nissl staining, and for immunohistochemistry to microglia using antibodies Iba1 and CD68, glial fibrillary acidic protein (GFAP), β‐amyloid, phosphorylated tau (clone AT8), PrP (using the 3F4 antibody without and with pretreatment with proteinase K), α‐synuclein, TDP‐43, ubiquitin and p62. FFI cases were pretreated with formic acid. Age‐matched control cases had not suffered from neurologic, psychiatric, or metabolic diseases (including metabolic syndrome), and did not have abnormalities in the neuropathological examination excepting sporadic Alzheimer's disease‐related pathology stages I‐II/0 of Braak and Braak.
RNA extraction
RNA was obtained from about 100 mg of the dorsomedial thalamus of FFI cases and controls using RNeasy Lipid Tissue Mini Kit (Qiagen, Hilden, GE) following the protocol provided by the supplier. All samples were treated with RNase‐free DNase Set (Qiagen) for 15 minutes to eliminate genomic DNA contamination. The concentration of each sample was measured using a NanoDrop 2000 spectrophotometer (Thermo Scientific, Waltham, MA, USA) at 340 nm. RNA integrity was assessed with the RNA Integrity Number (RIN value) determined with the Agilent 2100 Bioanalyzer (Agilent, Santa Clara, CA, USA) 33. RIN values were similar in control and FFI cases (Table 1).
Retro‐transcription reaction
Retro‐transcription reaction of RNA samples was carried out with the High‐Capacity cDNA Archive kit (Applied Biosystems, Foster City, CA, USA) using 1000 ng of RNA for each sample following the protocol provided by the supplier using a Gene Amp 9700 PCR System thermos‐cycler (Applied Biosystems). RNA samples without reverse transcriptase were processed in parallel as controls of the reaction. No amplifications were obtained in any cases.
Real‐time polymerase chain reaction)
Quantitative RT‐PCR assays were performed in duplicate on cDNA samples obtained from the retro‐transcription reaction diluted 1:20 in 384‐well optical plates (Kisker Biotech, Steinfurt, GE) using the ABI Prism 7900 HT Sequence Detection System (Applied Biosystems). The reactions were carried out using 20xTaqMan Gene Expression Assays for genes involved in protein synthesis, energetic metabolism and purine metabolism, and 2xTaqMan Universal PCR Master Mix (Applied Biosystems). TaqMan probes used in the study are shown in Table 2. The reactions were conducted with the following parameters: 50°C for 2 minutes, 95°C for 10 minutes, 40 cycles at 95°C for 15 s and 60°C for 1 minute. The data were captured using the Sequence Detection Software (SDS version 2.2, Applied Biosystems). Threshold cycle (CT) data for each sample were analyzed with the Livak Method or double delta CT (ΔΔCT) method 19. First, delta CT (ΔCT) values were calculated as the normalized CT values for each target gene in relation to the endogenous control X‐prolyl aminopeptidase P1 (XPNPEP1) used as housekeeping gene 9. Second, ΔΔCT values were obtained from the ΔCT of each sample minus the mean ΔCT of control samples. The fold change was determined using the equation 2−ΔΔCT. Mean fold‐change values for every region between FFI cases and controls were analyzed with the Student's t‐test using GraphPad Prism 6 when the distribution was normal as revealed by the Kolmogorov–Smirnov test. If the distribution was not normal, mean fold‐changes were analyzed with the Mann–Whitney test. Differences between groups were considered statistically significant at *P < 0.05, **P < 0.01, and ***P < 0.001.
Table 2.
Abbreviated names of genes, their full names, and TaqMan probe references used for the study of mRNA expression of mitochondria, protein synthesis and purine metabolism enzymes including GUS‐β and XPNPEP1 used for normalization.
| Gene | Gene full name | Taqman probes |
|---|---|---|
| XPNPEP1 | X‐propylaminopeptidase 1 | Hs00958026_m1 |
| Mitochondria | ||
| NDUFA2 | NADH Dehydrogenase (ubiquinone) 1 apha subcomplex, 2, 8 kDa | Hs04187282_g1 |
| NDUFA7 | NADH dehydrogenase (ubiquinone) 1 alpha subcomplex, 7, 14.5 kDa | Hs01561430_m1 |
| NDUFA10 | NADH dehydrogenase (ubiquinone) 1 alpha subcomplex, 10, 42 kDa | Hs01071117_m1 |
| NDUFB3 | NADH Dehydrogenase (ubiquinone) 1 Beta subcomplex, 3, 12 kDa | Hs00991297_g1 |
| NDUFB7 | NADH Dehydrogenase (ubiquinone) 1 Beta subcomplex, 7, 18 kDa | Hs00188142_m1 |
| NDUFB10 | NADH dehydrogenase (ubiquinone) 1 beta subcomplex, 10, 22 kDa | Hs00605903_m1 |
| NDUFS7 | NADH dehydrogenase (ubiquinone) Fe‐S protein 7, 20 kDa (NADH‐coenzyme Q reductase) | Hs00257018_m1 |
| NDUFS8 | NADH dehydrogenase (ubiquinone) Fe‐S protein 8, 23 kDa (NADH‐coenzyme Q reductase) | Hs00159597_m1 |
| SDHB | Succinate dehydrogenase complex, subunit B, Iron Sulfur (Ip) | Hs01042482_m1 |
| UQCR11 | Ubiquinol‐cytochrome C reductase, complex III subunit XI | Hs00199138_m1 |
| UQCRB | Ubiquinol‐cytochrome C reductase binding protein | Hs01890823_s1 |
| COX7A2L | Cytochrome c oxidase subunit VIIa polypeptide 2 like | Hs00190880_m1 |
| COX7C | Cytochrome C oxidase subunit VIIc | Hs01595220_g1 |
| ATP5D | ATP Synthase, H+ transporting, mitochondrial F1 complex, Delta Subunit | Hs00961521_m1 |
| ATP5G2 | ATP synthase, H+ transporting, mitochondrial Fo complex, subunit C2 | Hs01086654_g1 |
| ATP5H | ATP Synthase, H+ transporting, mitochondrial Fo complex, subunit D | Hs01046892_gH |
| ATP5L | ATP Synthase, H+ transporting, mitochondrial Fo complex, subunit G | Hs00538946_g1 |
| ATP5O | ATP Synthase, H+ transporting, mitochondrial F1 complex, O subunit | Hs00426889_m1 |
| ATP6V0B | ATPase, H+ transporting, lysosomal 21 kDa, V0 subunit b | Hs01072388_m1 |
| ATP6V1H | ATPase, H+ transporting, lysosomal 50/57 kDa, V1 subunit H | Hs00977530_m1 |
| TOMM40 | Translocase of outer mitochondrial membrane 40 | Hs01587378_mH |
| Protein synthesis | ||
| NCL | Nucleolin | Hs01066668_m1 |
| NPM1 | Nucleophosmin/protein B23) | Hs02339479_g1 |
| UBTF | Eukaryotic upstream binding factor | Hs01115792_g1 |
| 18S rRNA | Eukaryotic 18S rRNA | Hs99999991_s1 |
| 28S rRNA | RNA, 28S ribosomal | Hs03654441_s1 |
| RPL5 | Ribosomal protein L5 | Hs03044958_g1 |
| RPL7 | Ribosomal protein L7 | Hs02596927_g1 |
| RPL21 | Ribosomal protein L21 | Hs00823333_s1 |
| RPL22 | Ribosomal protein L22 | Hs01865331_s1 |
| RPL23A | Ribosomal protein L23A | Hs01921329_g1 |
| RPL26 | Ribosomal protein L26 | Hs00864008_m1 |
| RPL27 | Ribosomal protein L27 | Hs03044961_g1 |
| RPL30 | Ribosomal protein L30 | Hs00265497_m1 |
| RPL31 | Ribosomal protein L31 | Hs01015497_g1 |
| RPS3A | Ribosomal protein S3A | Hs00832893_sH |
| RPS5 | Ribosomal protein S5 | Hs00734849_g1 |
| RPS6 | Ribosomal protein S6 | Hs04195024_g1 |
| RPS10 | Ribosomal protein S10 | Hs01652370_gH |
| RPS13 | Ribosomal protein S13 | Hs01011487_g1 |
| RPS16 | Ribosomal protein S16 | Hs01598516_g1 |
| RPS17 | Ribosomal protein S17 | Hs00734303_g1 |
| RPS20 | Ribosomal protein S20 | Hs00828752_gH |
Protein extraction and western blotting
Thalamus samples (0.1 g) were homogenized using lysis buffer composed of 100 mM Tris pH 7, 100 mM NaCl, 10 mM EDTA, 0.5% NP‐40 and 0.5% sodium deoxycholate plus protease and phosphatase inhibitors (Roche Molecular Systems, Pleasanton, CA, USA). Samples were centrifuged at 4°C for 5 minutes at 10,000 g USING Eppendorf centrifuge 5417R (Eppendorf®, Hamburg, Germany) and the supernatants obtained were stored at −80°C. Proteins were separated in sodium dodecylsulfate‐polyacrylamide gel electrophoresis (SDS‐PAGE). Control of protein loading was carried out with Coomassie blue staining. Proteins were then electrophoretically transferred to nitrocellulose membranes using the Trans‐Blot® SD Semi‐Dry Transfer Cell (Bio‐Rad, Hercules, CA, USA) at 60 mA/membrane for 90 minutes). Nonspecific bindings were blocked by incubation in 5% milk in Tris‐buffered saline (TBS) containing 0.2% Tween for 1 h at room temperature. After washing, the membranes were incubated at 4°C overnight with one of the primary antibodies listed in Table 3 in TBS containing 5% albumin and 0.2% Tween. Afterwards, the membranes were incubated for 1 h with the appropriate HRP‐conjugated secondary antibody (1:1000, Dako, Glostrup, Denmark, Northern Europe), and the immune complexes were visualized with a chemiluminescence reagent (ECL, Amersham, GE Healthcare, Buckinghamshire, UK). Densitometry of western blot bands was assessed with the TotalLab program (TotalLab Quant, Newcastle, UK) and analyzed with Graphpad Prism using Student's t‐test when the distribution was normal and Mann–Whitney test when the distribution was not normal, as assessed with the Kolmogorov–Smirnov normality test. Differences were considered statistically significant at *P < 0.05, **P < 0.01, and ***P < 0.001.
Table 3.
List of antibodies used for western blotting and immunohistochemistry.
| Antibody | Reference | Supplier | Host | wb dilution | ihq dilution |
|---|---|---|---|---|---|
| Nucleolin | Ab22758 | Abcam (Cambridge, UK) | rb | 1/100 | 1/1000 |
| Anti‐eukaryotic translation initiation factor 2 (eIF2‐α) | 5A5 | Thermo Scientific (Waltham, MA, USA) | m | 1/50 | |
| Anti‐phospho‐eIF2‐alpha pSer51 (p‐EIF2‐α) | S.674.5 | Thermo Scientific (Waltham, MA, USA) | rb | 1/50 | |
| Anti‐eukaryotic translation initiation factor 5 (eIF5) | sc‐282 | Santa Cruz (Dallas, TX, USA) | rb | 1/400 | |
| Anti‐eukaryotic elongation factor 1A (eEF1A) | 2551 | Cell signaling (Danvers, MA, USA) | rb | 1/100 | |
| Anti‐eukaryotic elongation factor 2 (eEF2) | 2332 | Cell signaling (Danvers, MA, USA) | rb | 1/500 | |
| Total OXPHOS Antibody Cocktail | ab110411 | Mitosciences, Abcam (Cambridge, UK) | m | 1/1000 | |
| ATP5O (OSCP antibody) | 10994‐1‐AP | Proteintech (Chicago, IL, USA) | rb | 1/1000 | |
| UQCRB | 10756‐1‐AP | Proteintech (Chicago, IL, USA) | rb | 1/250 | |
| VDAC1 | Ab15895 | Abcam (Cambridge, UK) | rb | 1/400 | 1/100 |
| β‐actin | A5316 | Sigma‐Aldrich (St Louis, MO, USA) | m | 1/5000 | |
| ATP5H | 17589‐1‐AP | Proteintech (Chicago, IL, USA) | rb | 1/100 | |
| Anti‐manganese superoxide dismutaseSOD2 | SOD‐110 | Stressgen (San Diego, CA, USA) | rb | 1/100 | |
| Anti‐histone H3K9me2 | Ab1220 | Abcam (Cambridge, UK) | m | 1/90 |
Immunohistochemistry
Immunohistochemical study was performed in 4‐µm thick dewaxed paraffin sections of the thalamus. Endogenous peroxidases were blocked with peroxidase (Dako, Glostrup) followed by 10% normal goat serum. The primary antibodies are shown in Table 3. Following incubation with the primary antibody at room temperature overnight, the sections were incubated with EnVision+ system peroxidase (Dako, Barcelona, Spain) at room temperature for 15 minutes. The peroxidise reaction was visualized with diaminobenzidine (DAB) and H2O2. No antigenic peptides were available to carry out preadsorption studies of primary antibodies. However, the omission of the primary antibody in some sections was used as a control of the immunostaining; no signal was obtained after incubation with only the secondary antibody. Sections were slightly counterstained with haematoxylin. Quantification of Nissl‐stained and immunoreactive cells identified with antibodies against glial fibrillary acidic protein (GFAP), CD68, voltage‐dependent anion channel (VDAC), nucleolin (NCL), nucleoplasmin 3 (NPM3), histone H3K9me2, ATP5H protein and superoxide dismutase 2 (SOD2) was carried out in the mediodorsal thalamus in five controls and five FFI cases (two fields per case) at a magnification of ×200 which corresponds to an area of 450 × 280 μm2. Results were expressed as mean values ± standard deviation (SD).
RESULTS
General neuropathological findings
Severe neuronal loss and marked astrocytic gliosis was observed in every case in the mediodorsal and anterior nuclei of the thalamus, and inferior olives; the ventral nuclei of the thalamus and the entorhinal cortex were variably affected (five and six of seven cases, respectively). Spongiform change was not seen in the mediodorsal thalamus and only in the entorhinal cortex when affected PrP. Discrete astrogliosis was also found in the periaqueductal gray matter. Representative changes in the mediodorsal thalamus and entorhinal cortex compared with corresponding regions in age‐matched controls are shown in Figure 1. Neuron loss in the mediodorsal thalamus was severe accounting for about 80%–90%; only isolated small neurons can be seen in some cases. Microglial response was analyzed using the antibodies Iba1 which identifies microglia and CD68 which recognizes active microglia. Increased numbers of Iba1‐ and CD68‐immunoreactive cells was found in the mediodorsal thalamus, particularly in two of seven cases, and entorhinal cortex in the six cases in which this region was affected. Details of cell types are recognized at higher magnification (Figure 1 inserts)
Figure 1.
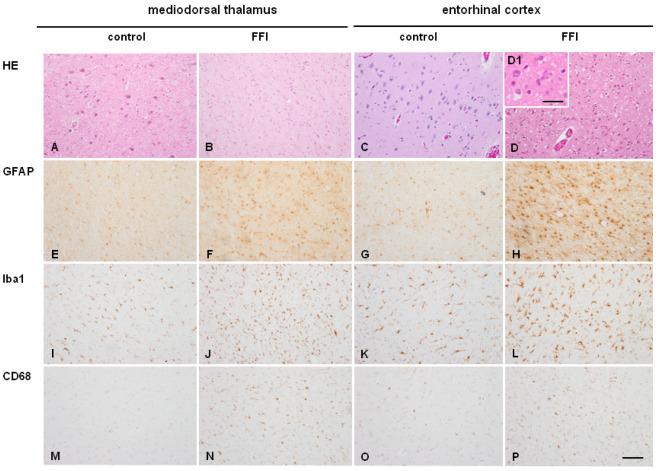
Mediodorsal thalamus (A, B, E, F, I, J, M, N) and entorhinal cortex C, D, G, H, K, L, O, P) in age‐matched control (A, C, E, G, I, K, M, O) and FFI (B, D, F, H, J, L, N, P) cases. Severe neuron loss is seen in the mediodorsal thalamus in FFI, whereas moderate neuron loss and spongiform change is observed in the entorhinal cortex in FFI. Increased numbers of GFAP‐immunoreactive astrocytes are present in mediodorsal thalamus and entorhinal cortex in FFI compared with corresponding controls. Increased numbers of Iba1‐immunoreactive cells and CD‐68‐positive cells are seen in the EC in six cases and in two of seven FFI cases (here illustrated) compared with controls. Paraffin sections, A–D. haematoxylin and eosin; E–H. GFAP immunohistochemistry; I–L. Iba1 immunohistochemistry; M–P. CD‐68 immunohistochemistry; immunohistochemical sections slightly counterstained with haematoxylin. A–P, bar in P = 55 µm. D1, H1, L1, P1, bar in P1 = 30 µm.
Immunohistochemistry using the 3F4 antibody disclosed synaptic and fine granular proteinase‐resistant PrP immunoreactivity in the entorhinal cortex, whereas PrP immunoreactivity was absent in the mediodorsal thalamus in FFI cases (Figure 2). As expected no proteinase‐resistant PrP was present in control cases (data not shown).
Figure 2.
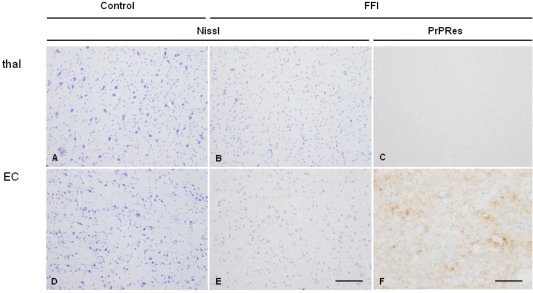
Mediodorsal nucleus of the thalamus (thal: A–C) and entorhinal cortex (EC: D–F) in control (A,B) and FFI (B,C,E,F) cases showing marked loss of neurons without spongiform change in the thalamus and moderate neuron loss and spongiform change in entorhinal cortex in FFI when compared with corresponding regions in controls. In FFI, no proteinase K‐resistant PrP (PrPRes) is observed in the mediodorsal thalamus (C) whereas punctate, synaptic‐like immunostaining is observed in the entorhinal cortex (F) processed in parallel. Paraffin sections, A, B, D, E Nissl staining, bar in E = 50 µm; C, F, PrP immunohistochemistry (3F4 antibody) following preincubation with proteinase K, bar in F = 25 µm.
Mitochondria and respiratory chain
mRNA expression
Eleven genes which codify for proteins that are part of electron transport chain complexes, one gene that encodes a translocase of the mitochondrial outer membrane (TOMM40) and two genes that codify for lysosomal ATPase (ATP6V0B and ATP6V1H1) were analyzed in the thalamus of FFI samples. Complex I was represented by NDUFA2, NDUFA7, NDUFA10, NDUFB3, NDUFB7, NDUFB10, NDUFS7, and NDUFS8; complex II by SDHB; complex III by UQCR11 and UQCRB; complex IV by COX7A2L and COX7C; and complex V by ATP5D, ATP5G2, ATP5H, ATP5L, and ATP5O. Only ATP5D was downregulated (Student's t‐test P < 0.01). Detailed data are shown in Supporting Information Table S1.
Protein expression of selected mitochondrial proteins
Expression levels of mitochondrial proteins in mediodorsal thalamus
NDUFB8 (complex I subunit), SDHB (complex II subunit), UQCRC2 (complex III subunit), COX2 (complex IV subunit), and ATP50 (complex V) expression levels, as revealed by western blotting, were reduced in mediodorsal thalamus in FFI, whereas UQCRB (complex III) and ATP5A (complex V subunit) expression was similar in FFI and controls. Densitometric analysis using β‐actin and VDAC for normalization showed similar results, the only exception between β‐actin and VDAC normalization was ATP5O which presented significant differences when using β‐actin between control group and FFI group but only a tendency with a P‐value 0.09 when using VDAC for normalization. Together, these results indicated that reduced protein expression of proteins of the respiratory chain was not merely the consequence of a reduced number of mitochondria, but was also related to the selective loss of certain vulnerable subunits (Figure 3). Detailed data are shown in Supporting Information Table S2.
Figure 3.
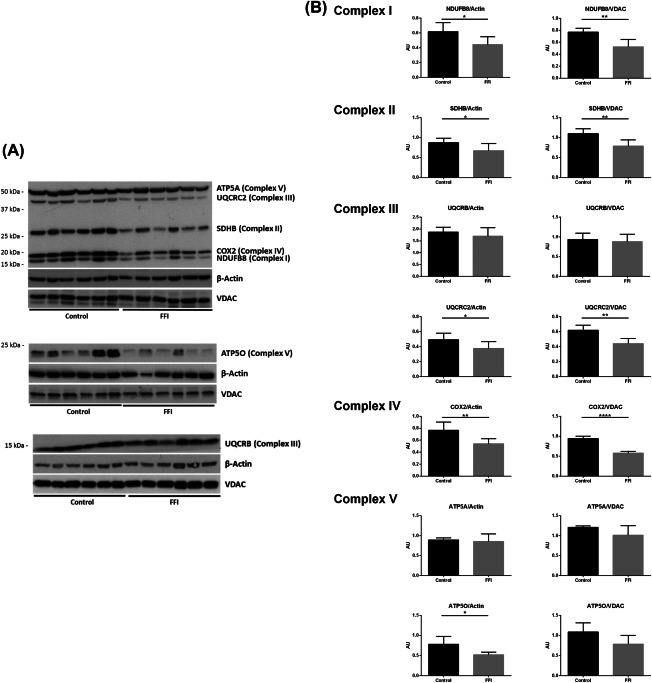
A. Western blot of subunits NADH dehydrogenase (ubiquinone) 1 beta subcomplex 8, 19 kDa (NDUFB8), succinate dehydrogenase complex, subunit B, iron sulphur (lp) (SDHB), ubiquinol‐cytochrome c reductase core protein II (UQCRC2), cytochrome c oxidase subunit II (COX2) and ATP synthase, H+ transporting, mitochondrial F1 complex, and alpha subunit 1 (ATP5A) using OXPHOS antibody; ATP synthase, H+ transporting mitochondrial F1 complex, O subunit (ATP5O) and ubiquinol‐cytochrome c reductase binding protein (UQCRB) antibodies in mediodorsal thalamus in FFI and control cases. β‐actin and VDAC were used to normalize total protein and mitochondria protein loading, respectively. B. Densitometric analysis shows significant reduction of NDUFB8, SDHB, UQCRC2, and COX2 normalized with β‐actin and VDAC in FFI compared with controls. ATP5A and UQCRB protein levels were preserved. Results were analyzed by Graphpad Prism with Student's t‐test when the distribution was normal and with Mann–Whitney test if distribution was not normal as assessed with the Kolmogorov–Smirnov normality test. Differences are considered statistically significant at *P < 0.05, **P < 0.01, and ****P < 0.0001. ATP5O shows a significant reduction when normalized with β‐actin but only a trend with VDAC.
VDAC, ATP5H, and SOD2 in mediodorsal thalamus in FFI
Consecutive sections were stained with Nissl staining and processed for immunohistochemistry to VDAC, ATP5H, and SOD2. Nissl staining served to illustrate the severe loss of neurons in the mediodorsal thalamus in FFI when compared with controls, together with the increase in the number of small nuclei corresponding, in part to astrocytes (Figures 4A,B).
Figure 4.
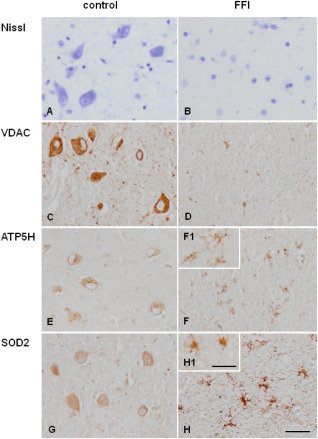
Mediodorsal nucleus of the thalamus in control (A, C, E, F) and FFI (B, D, F, H) cases. Nissl staining (A, B) shows marked decrease in the number of neurons in FFI when compared with controls. (VDAC) (C, D), (C, D) (SOD2) (E, F) immunoreactivity is found in mediodorsal thalamus in control (A, C, E) and FFI (B, D, F) cases). Decreased VDAC (C, D) and ATP synthase, H+ transporting, mitochondrial Fo complex, subunit d (ATP5H) (E, F) immunoreactivity is found in the mediodorsal thalamus in FFI compared with controls due to the dramatic decrease in the number of neurons. However, ATP5H is observed in reactive glial cells. Weak superoxide dismutase 2 (SOD2) (G, H) immunoreactivity is seen in neurons in control cases which contrasts with enhanced SOD2 immunostaining in reactive astrocytes in FFI. Paraffin sections, A, B: Nissl staining; C–H immunohistochemical sections slightly counterstained with haematoxylin. A–H, bar in H = 25 µm. F1, H1, bar in H1 = 10 µm.
In control brains, neurons presented strong VDAC immunoreactivity, punctuate ATP5H immunoreactivity and weak SOD2 immunoreactivity in the cytoplasm. Marked decrease in VDAC immunoreactivity related to the loss of neurons was observed in the mediodorsal thalamus in FFI when compared with controls (Figures 4C,D). This was accompanied by general decrease in ATP5H immunoreactivity due to neuron loss together with enhanced visualization of ATP5H immunostaining in reactive astrocytes (Figures 4E,F). Marked SOD2 immunoreactivity was found in reactive astrocytes (Figures 4G,H). Details of cell types are recognized at higher magnification (Figure 4 inserts). Quantitative data are shown in Table 4.
Table 4.
Quantitative data of cells stained with Nissl stain and immunoreactive with antibodies against voltage‐dependent anion channel (VDAC), glial fibrillary acidic protein (GFAP), CD8 (reactive microglia), nucleolin (NCL), nucleoplasmin 3 (NPM3), histone H3K9me2, ATP5H protein and superoxide dismutase 2 (SOD2) in the mediodorsal thalamus in control and FFI cases. Values are expressed as mean values ± SD. Assessed cell types are named in the left column excepting ATP5H and SOD2 immunoreactive cells which are neurons in controls and glia in FFI cases.
| Control | FFI | |
|---|---|---|
| Nissl (only neurons) | 20.5 ± 2.01 | 0.9 ± 073 |
| VDAC (only neurons) | 20.4 ± 2.06 | 1.3 ± 0.94 |
| GFAP (astrocytes) | 8.8 ± 2.09 | 35 ± 6.87 |
| CD68 (reactive microglia) | 2.3 ± 0.94 | 18.4 ± 3.40 |
| NCL (only neurons | 6.8 ± 1.81 | 0 |
| NPM3 (only neurons) | 6 ± 1.82 | 0 |
| H3K9me2 (only neurons) | 21.8 ± 2.04 | 0 |
| ATP5H | 23.7 ± 2.45 (neurons) | 23 ± 3.52 (glia) |
| SOD2 | 20.6 ± 1.83 (neurons) | 25.5 ± 2.83 (glia) |
MOLECULAR PATHWAYS INVOLVED IN PROTEIN SYNTHESIS
mRNA expression of ribosomal proteins RPL and RPS, nucleolar proteins, and 18S rRNA and 28S rRNA
Nucleolin (NCL), nucleophosmin (NPM1), and eukaryotic upstream binding factor (UBTF) gene expression levels were analyzed in FFI cases and controls. NPM1 was upregulated in the thalamus in FFI (Figure 5).
Figure 5.
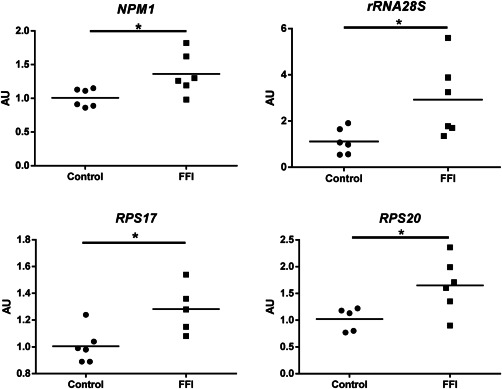
mRNA expression of ribosomal proteins (RPL and RPS) in mediodorsal thalamus in FFI and controls. RPS17, RPS20, NMP1, and 28S rRNA are upregulated in FFI cases using XPNPEP1 for normalization. Mean fold‐change values for each group are compared with Student's t‐test and differences were considered statistically significant at *P < 0.05.
28S rRNA was also upregulated in FFI cases (Figure 5).
The expression of nine RPL (RPL5, RPL7, RPL21, RPL22, RPL23A, RPL26, RPL27, RPL30, and RPL31) and seven RPS (RPS3A, RPS5, RPS6, RPS10, RPS13, RPS17, and RPS20) genes was assessed in the mediodorsal thalamus in FFI cases and controls. RPS17 and RPS20 were upregulated (Figure 5). Detailed data are shown in Supporting Information Table S3.
Immunohistochemistry of selected molecules involved in protein synthesis
In control mediodorsal thalamus, anti‐NCL antibodies weakly decorated the nucleolus and heavily the cytoplasm of neurons. NCL immunoreactivity was weak if present in glial cells under the same staining conditions. Anti‐nucleoplasmin 3 (NPM3) antibodies decorated the nucleolus of neurons. Histone H3 di‐methylated K9 (H3K9me2), which modulates NCL and NPMs function in rRNA transcription 4, 35, was strongly expressed in the nucleus of neurons (Figures 6A,C,E).
Figure 6.
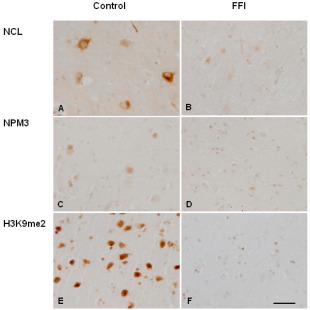
Nucleolin (NCL) (A, B), NPM3 (C, D), and H3 dimethyl K9 (H3K9me2) (E, F) immunoreactivity is observed in neurons in the mediodorsal thalamus in controls (A, C, E). Weak NCL immunoreactivity is found in the nucleolus whereas strong NCL immunoreactivity is present in the neuronal cytoplasm. NPM3 immunoreactivity is observed in the nucleolus, whereas H3K9me2 immunolabeling is localized in the nucleus. Decreased NCL, NMP3, and H3K9me2 expression is found in FFI cases (B, D, E) due to the marked decrease in the number of neurons. Paraffin sections slightly counterstained with haematoxylin; bar = 25 µm.
Immunoreactivity of NCL and (NPM3) was reduced in the mediodorsal thalamus in FFI cases when compared with controls (Figures 6B,D). H3K9me2 expression was also decreased in the thalamus in FFI (Figure 6F). NCL, NPM3, and H3K9me2 reduction in the mediodorsal thalamus was clearly due to the loss of neurons; reactive glial cells (already represented in previous figures) did not show increased immunoreactivity. Quantitative data are shown in Table 4.
Expression of nucleolin, and initiation and elongation factors, in mediodorsal thalamus
Expression levels of nucleolin were markedly decreased in the mediodorsal thalamus in FFI (Figure 7) as expected from the immunohistochemical studies. Regarding initiation and elongation factors, only eEF1A was significantly decreased in the mediodorsal thalamus of FFI cases (Figure 7). Detailed data are shown in Supporting Information Table S4.
Figure 7.
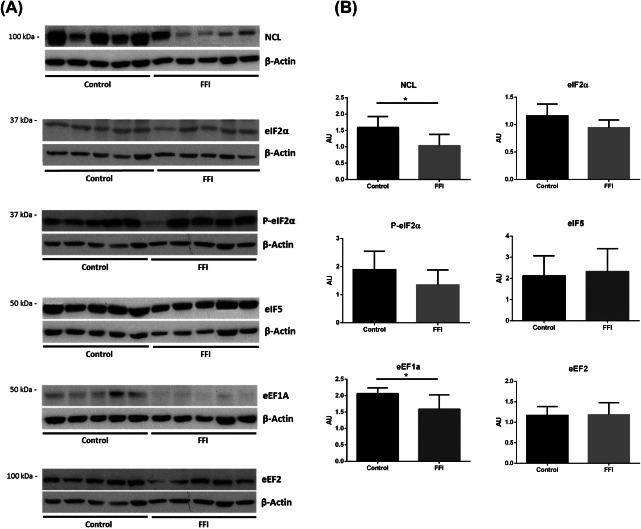
A. Western blots of nucleolin, initiation (eIF2α, P‐eIF2α, eIF5) and elongation factors (eEF1A, eEF2) in mediodorsal thalamus in FFI and control cases. β‐actin was used to normalize total protein. B. Densitometric analysis shows significant reduction of NCL and eEF1A. Results were analyzed by Graphpad Prism with Student's t‐test when the distribution was normal and with Mann–Whitney test if distribution was not normal as assessed with the Kolmogorov–Smirnov normality test. Differences are considered statistically significant at *P < 0.05.
DISCUSSION
All the cases examined in the present series are males, have the D178N mutation and are Met/Met homozygous at the codon 129 of PRNP. In spite of the variable age at onset, disease duration was between 6 and 9 months; the neuropathological findings were similar from one case to another; therefore, this series conforms a relative homogeneous population. General neuropathological findings in these series are similar to those already described in FFI 14, 21, 22, 26, 28. Based on these features and on the small number of cases, no attempt was made to divide the sample into subgroups.
The postmortem delay between death and processing was longer in the FFI group when compared with the controls. However, the RIN values were similar in both groups and protein preservation as revealed by western blotting and immunohistochemistry was similar in controls and diseased brains.
Brain hypo‐metabolism, as revealed by [18F]‐2‐fluoro‐2‐deoxy‐D‐glucose and positron emission tomography PET to study regional cerebral glucose utilization, predominates in the thalamus and cingular cortex, but the basal and lateral frontal cortex, the caudate nucleus, and the middle and inferior temporal cortex are also moderately affected 7. The neurodegenerative process starts between 13 and 21 months before the clinical symptoms and it is manifested by hypo‐metabolism in the thalamus and impaired thalamic sleep spindle formation in preclinically affected family members 6. The present findings are in line with functional studies showing down‐regulation of ATP5D mRNA expression as revealed by qRT‐PCR, and significant reduction in the expression of NDUFB8 (complex I subunit), SDHB (complex II subunit), UQCRC2 (complex III subunit), COX2 (complex IV subunit), and ATP5O (complex V subunit) protein levels, as assessed with western blotting of total homogenates of the mediodorsal thalamus. Immunohistochemistry discloses the origin of reduced mitochondrial expression as being linked to severe neuron loss, as practically no neurons were stained with antibodies against VDAC and ATP5H. These changes are accompanied by increased oxidative stress responses manifested by increased SOD2 immunostaining in reactive astrocytes.
The expression of molecules involved in the machinery of protein synthesis was also altered in the mediodorsal thalamus in FFI when compared with controls. Curiously, increased mRNA expression of NPM1, 28S rRNA, RPS17, and RPS20 is in contrast to the decreased protein expression levels of the histone‐binding chaperones nucleolin (NCL) and nucleoplasmin 3 (NPM3), which regulate rRNA transcription 2, 4, 10, 11, 16, 18, 27, and of histone H3 di‐methylated K9 (H3K9me2), which modulates NCL and NPMs function in rRNA transcription 4, 35. Changes in protein expression are clearly related to the dramatic reduction of neurons as disclosed by immunohistochemistry.
Regarding the protein expression of initiation and elongation factors of protein transcription, only eEF1A is downregulated. However, the role of this factor is crucial, as elongation occurs when eEF1A is activated following GTP binding and forms a complex with aminoacyl‐tRNA, which recognizes the specific sequence in mRNA at the ribosome. Once the interaction of the codon in mRNA with the anti‐codon in tRNA is decoded, eEF1A‐GDP is hydrolyzed, released from the ribosome, and recycled into its active form by eEF1B. eEF2 assists in the precise codon location at the ribosome 1, 8, 32, 38.
Present molecular studies show reduced expression of proteins linked to the mitochondrial respiratory chain involving subunits encoded by genomic DNA of the five mitochondrial complexes in line with a marked reduction in the number of neurons as revealed by morphological and immunohistochemical studies, as well as decreased metabolism as shown by PET studies in vivo in the mediodorsal nucleus of the thalamus in FFI. Altered machinery of protein synthesis is also manifested by decreased expression of nucleolar chaperones, histone modifications and decreased methylation of DNA in the mediodorsal thalamus in FFI. Alterations in the levels of proteins do not correlate with modifications in the expression of corresponding mRNA levels, which are often maintained or even upregulated. This may be explained by failed compensatory mechanisms in the remaining neurons or by increased RNA expression in reactive astrocytes. In favor of the latter, is the dramatic increase in reactive astrocytes and the increased expression of superoxide dismutase 2 which tags reactive astrocytes as crucial components of oxidative stress responses in the mediodorsal thalamus in FFI.
All these changes are observed at terminal stages of the disease, with devastating effects on selected brain regions. Little can be done at this stage but efforts must be made at preclinical stages to prevent mitochondrial and protein synthesis havoc at advanced stages of the disease.
Supporting information
Additional Supporting Information may be found in the online version of this article at the publisher's web‐site.
Table S1. Gene expression of subunits of mitochondrial complexes in the mediodorsal thalamus in FFI and control cases normalized with XPNPEP1.
Table S2. Densitometric values of expression of mitochondrial proteins as revealed by western blotting in the mediodorsal thalamus in FFI cases and controls normalized with β‐actin (A) and VDAC (B).
Table S3. Gene expression of nucleolar proteins. 18S and 28S rRNAS and ribosomal proteins in the mediodorsal thalamus in FFI and control cases normalized with XPNPEP1.
Table S4. Densitometric values of expression of initiation and elongation factors of protein transcription in the mediodorsal thalamus in FFI cases and controls normalized with β‐actin.
ACKNOWLEDGMENTS
This study was funded by the Ministerio de Ciencia e Innovación, Instituto de Salud Carlos III – Fondos FEDER, a Way to Build Europe FIS grant PI14/00757, and coordinated Intraciber 2014. We wish to thank T. Yohannan for editorial help.
Compliance with ethical standards No relevant data.
The authors declare that they have no conflicts of interest.
REFERENCES
- 1. Andersen GR, Nissen P, Nyborg J (2003) Elongation factors in protein biosynthesis. Trends Biochem Sci 28:434–441. [DOI] [PubMed] [Google Scholar]
- 2. Angelov D, Bondarenko VA, Almagro S, Menoni H, Mongelard F, Hans F et al (2006) Nucleolin is a histone chaperone with FACT like activity and assists remodeling of nucleosomes. EMBO J 25:1669–1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Capellari S, Strammiello R, Saverioni D, Krtezschmar H, Parchi P (2011) Genetic Creutzfeldt‐Jakob disease and fatal familial insomnia: insights into phenotypic variability and disease pathogenesis. Acta Neuropathol 121:21–37. [DOI] [PubMed] [Google Scholar]
- 4. Cong R, Das S, Ugrinova I, Kumar S, Mongerlard F, Wong J, Bouvet P (2012) Interaction of nucleolin with ribosomal RNA genes and its role in RNA polymerase I transcription. Nucleic Acids Res 40:9441–9454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cortelli P, Gambetti P, Montagna P, Lugaresi E (1999) Fatal familial insomnia: clinical features and molecular genetics. J Sleep Res 8:23–29. [DOI] [PubMed] [Google Scholar]
- 6. Cortelli P, Perani D, Montagna P, Gallassi R, Tinuper P, Federica P et al (2006) Pre‐symptomatic diagnosis in fatal familial insomnia: serial neurophysiological and 18FDG‐PET studies. Brain 129:668–675. [DOI] [PubMed] [Google Scholar]
- 7. Cortelli P, Perani D, Parchi P, Grassi F, Montagna P, De Martin M et al (1997) Cerebral metabolism in fatal familial insomnia: relation to duration, neuropathology, and distribution of protease‐resistant prion protein. Neurology 49:126–133. [DOI] [PubMed] [Google Scholar]
- 8. Dever TE, Green R (2012) The elongation, termination, and recycling phases of translation in eukaryotes. Cold Spring Harb Perspect Biol 4:a013706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Durrenberger PF, Fernando FS, Magliozzi R, Kashefi SN, Bonnert TP, Ferrer I et al (2012) Selection of novel reference genes for use in the human central nervous system: a BrainNet Europe study. Acta Neuropathol 124:893–903. [DOI] [PubMed] [Google Scholar]
- 10. Frehlick LJ, Eirín‐López JM, Ausio J (2007) New insights into the nucleophosmin/nucleoplasmin family of nuclear chaperones. Bioassays 29:49–59. [DOI] [PubMed] [Google Scholar]
- 11. Gadad SS, Shandilya J, Kishore AH, Kundu TK (2010) NPM3, a member of the nucleophosmin/nucleoplasmin family, enhances activator‐dependent transcription. Biochemistry 49:1355–1357. [DOI] [PubMed] [Google Scholar]
- 12. Gallassi R, Morreale A, Montagna P, Cortelli P, Avoni P, Castellani R et al (1996) Fatal familial insomnia: behavioral and cognitive features. Neurology 46:935–939. [DOI] [PubMed] [Google Scholar]
- 13. Gambetti P, Parchi P, Chen SG (2003) Hereditary Creutzfeldt‐Jakob disease and fatal familial insomnia. Clin Lab Med 23:43–64. [DOI] [PubMed] [Google Scholar]
- 14. Gambetti P, Parchi P, Petersen RB, Chen SG, Lugaresi E (1995) Fatal familial insomnia and familial Creutzfeldt‐Jakob disease: clinical, pathological and molecular features. Brain Pathol 5:43–51. [DOI] [PubMed] [Google Scholar]
- 15. Goldfarb LG, Petersen RB, Tabaton M, Brown P, LeBlanc AC, Montagna P et al (1992) Fatal familial insomnia and familial Creutzfeldt‐Jakob disease: disease phenotype determined by a DNA polymorphism. Science 258:806–808. [DOI] [PubMed] [Google Scholar]
- 16. Huang N, Negi S, Szebeni A, Olson MOJ (2005) Protein NPM3 interacts with the multifunctional nucleolar protein B23/nucleophosmin and inhibits ribosome biogenesis. J Biol Chem 280:5496–5502. [DOI] [PubMed] [Google Scholar]
- 17. Krasnianski A, Bartl M, Sanchez Juan PJ, Heinemann U, Meissner B, Varges D et al (2008) Fatal familial insomnia: clinical features and early identification. Ann Neurol 63:658–661. [DOI] [PubMed] [Google Scholar]
- 18. Lindström MS (2011) NPM1/B23: a multifunctional chaperone in ribosome biogenesis and chromatin remodeling. Biochem Res Int 195209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real‐time quantitative PCR and the 2(‐Delta Delta C(T)) method. Methods 25:402–408. [DOI] [PubMed] [Google Scholar]
- 20. Lugaresi A, Baruzzi A, Cacciari E, Cortelli P, Medori R, Montagna P et al (1987) Lack of vegetative and endocrine circadian rhythms in fatal familial thalamic degeneration. Clin Endocrinol (Oxf) 26:573–580. [DOI] [PubMed] [Google Scholar]
- 21. Lugaresi E, Medori R, Montagna P, Baruzzi A, Cortelli P, Lugaresi A et al (1986) Fatal familial insomnia and dysautonomia with selective degeneration of thalamic nuclei. N Engl J Med 315:997–1003. [DOI] [PubMed] [Google Scholar]
- 22. Manetto V, Medori R, Cortelli P, Montagna P, Tinuper P, Baruzzi A et al (1992) Fatal familial insomnia: clinical and pathologic study of five new cases. Neurology 42:312–319. [DOI] [PubMed] [Google Scholar]
- 23. Medori R, Tritschler HJ (1993) Prion protein gene analysis in three kindred with fatal familial insomnia (FFI): codon 178 mutation and codon 129 polymorphism. Am J Hum Genet 53:822–827. [PMC free article] [PubMed] [Google Scholar]
- 24. Medori R, Tritschler HJ, LeBlanc A, Villare F, Manetto V, Chen HY et al (1992) Fatal familial insomnia, a prion disease with a mutation at codon 178 of the prion protein gene. N Engl J Med 326:444–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Monari L, Chen SG, Brown P, Parchi P, Petersen RB, Mikol J et al (1994) Fatal familial insomnia and familial Creutzfeldt‐Jakob disease: different prion proteins determined by a DNA polymorphism. Proc Natl Acad Sci U S A 91:2839–2842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Montagna P, Gambetti P, Cortellui P, Lugaresi E (2003) Familial and sporadic fatal insomnia. Lancet Neurol 2:167–176. [DOI] [PubMed] [Google Scholar]
- 27. Okuwaki M, Matsumoto K, Tsujimoto M, Nagata K (2011) Function of nucleoplasmin/B23, a nucleolar acidic protein, as a histone chaperone. FEBS Lett 506:272–276. [DOI] [PubMed] [Google Scholar]
- 28. Parchi P, Capellari S, Gambetti P (2011) Fatal familial and sporadic insomnia. In: Neurodegeneration: The Molecular Pathology of Dementia and Movement Disorders. Dickson DW, Weller RO (eds), pp. 346–349. Wiley‐Blackwell: Oxford. [Google Scholar]
- 29. Parchi P, Castellani R, Cortelli P, Montagna P, Chen SG, Petersen RB et al (1995) Regional distribution of protease‐resistant prion protein in fatal familial insomnia. Ann Neurol 38:21–29. [DOI] [PubMed] [Google Scholar]
- 30. Parchi P, Petersen RB, Chen SG, Autilio‐Gambetti L, Capellari S, Monari L et al (1998) Molecular pathology of fatal familial insomnia. Brain Pathol 8:539–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Rodriguez‐Martinez AB, Alfonso‐Sanchez MA, Peña JA, Sanchez‐Valle R, Zerr I, Capellari S et al (2008) Molecular evidence of founder effects of fatal familial through SNP haplotypes around the D178N mutation. Neurogenetics 9:109–118. [DOI] [PubMed] [Google Scholar]
- 32. Sasikumar AN, Perez WB, Kinzy TG (2012) The many roles of the eukaryotic elongation factor 1 complex. Wiley Interdiscip Rev RNA 3:543–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Schroeder A, Mueller O, Stocker S, Salowsky R, Leiber M, Gassmann M et al (2006) The RIN: an RNA integrity number for assigning integrity values to RNA measurements. BMC Mol Biol 7:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Shi Q, Chen LN, Zhang BY, Xiao K, Zhou W, Chen C et al (2015) Proteomics analyses for the global proteins in the brain tissues of different human prion diseases. Mol Cell Proteomics 14:854–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Tamada H, Thuan NV, Reed P, Nelson D, Katoku‐Kikyo N, Wudel J et al (2006) Chromatin decondensation and nuclear reprogramming by nucleoplasmin. Mol Cell Biol 26:1259–1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Tian C, Liu D, Sun Q‐L, Chen C, Xu Y, Wang H et al (2013) Comparative analysis of gene expression profiles between cortex and thalamus in Chinese fatal familial insomnia patients. Mol Neurobiol 48:36–48. [DOI] [PubMed] [Google Scholar]
- 37. Tinuper P, Montagna P, Medori R, Cortelli P, Zucconi M, Baruzzi A, Lugaresi E (1989) The thalamus participates in the regulation of sleep‐waking cycle. A clinic‐pathological study in fatal familial thalamic degeneration. Electroencephalogr Clin Neurophysiol 73:117–123. [DOI] [PubMed] [Google Scholar]
- 38. Voorhees RM, Ramakrishnan V (2013) Structural basis of the translational elongation cycle. Annu Rev Biochem 82:203–236. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting Information may be found in the online version of this article at the publisher's web‐site.
Table S1. Gene expression of subunits of mitochondrial complexes in the mediodorsal thalamus in FFI and control cases normalized with XPNPEP1.
Table S2. Densitometric values of expression of mitochondrial proteins as revealed by western blotting in the mediodorsal thalamus in FFI cases and controls normalized with β‐actin (A) and VDAC (B).
Table S3. Gene expression of nucleolar proteins. 18S and 28S rRNAS and ribosomal proteins in the mediodorsal thalamus in FFI and control cases normalized with XPNPEP1.
Table S4. Densitometric values of expression of initiation and elongation factors of protein transcription in the mediodorsal thalamus in FFI cases and controls normalized with β‐actin.