

Abstract
Alpha‐motoneurons and muscle fibres are structurally and functionally interdependent. Both cell types particularly rely on endoplasmic reticulum (ER/SR) functions. Mutations of the ER proteins VAPB, SigR1 and HSP27 lead to hereditary motor neuron diseases (MNDs). Here, we determined the expression profile and localization of these ER proteins/chaperons by immunohistochemistry and immunoblotting in biopsy and autopsy muscle tissue of patients with amyotrophic lateral sclerosis (ALS) and other neurogenic muscular atrophies (NMAs) and compared these patterns to mouse models of neurogenic muscular atrophy. Postsynaptic neuromuscular junction staining for VAPB was intense in normal human and mouse muscle and decreased in denervated Nmd2J mouse muscle fibres. In contrast, VAPB levels together with other chaperones and autophagy markers were increased in extrasynaptic regions of denervated muscle fibres of patients with MNDs and other NMAs, especially at sites of focal myofibrillar disintegration (targets). These findings did not differ between NMAs due to ALS and other causes. G93A‐SOD1 mouse muscle fibres showed a similar pattern of protein level increases in denervated muscle fibres. In addition, they showed globular VAPB‐immunoreactive structures together with misfolded SOD1 protein accumulations, suggesting a primary myopathic change. Our findings indicate that altered expression and localization of these ER proteins and autophagy markers are part of the dynamic response of muscle fibres to denervation. The ER is particularly prominent and vulnerable in both muscle fibres and alpha‐motoneurons. Thus, ER pathology could contribute to the selective build‐up of degenerative changes in the neuromuscular axis in MNDs.
Keywords: ALS, Motor neuron disease (MND), Neurogenic muscular atrophy (NMA), Neuromuscular junction (NMJ), ER chaperones, VAPB, Sigma Receptor 1 (SigR1)
INTRODUCTION
After denervation, skeletal muscle fibres become completely atrophic within months 11, 18. Atrophy of muscle fibres that are not reinnervated is accompanied by decreased expression of structural proteins and increased expression of proteins executing muscle fibres breakdown; muscle fibre remodelling due to other causes such as disuse or cachexia also involves alterations in expression of these proteins 32, 60. NMA occurs in the context of sensorimotor neuropathies and MNDs. In fact, the pattern of atrophy differs between these groups: In the most frequent MND, ALS, fibre type grouping due to collateral re‐innervation is rather sparse 2, 62. NMA in peripheral sensorimotor neuropathy often shows a more chronic course with more prominent fibre type grouping. In addition, target fibres are relatively rare in ALS muscle 11, 57.
Both skeletal muscle fibres and motor neurons (MNs) depend on a well‐differentiated ER to ensure high levels of protein synthesis and a tightly balanced, efficient calcium metabolism. Mutations in the ER proteins Sigma receptor 1 (SigR1) and vesicle‐associated protein B (VAPB) cause familial MND with distinct ER alterations 1, 25, 46, 61. Mutations in other ER proteins affect the muscle by causing hereditary myopathies with distinctive morphological ER changes. For example, mutations in the calcium sensors stromal interaction molecule 1 (STIM1) and of the plasma membranous calcium release‐activated Ca2+ channel (ORAI1), which interacts with STIM1, cause extensive proliferations of the ER in so‐called tubular aggregate myopathy 6, 7. In addition; mutation of the ryanodine receptor (RyR), a Ca2+ release channel of the SR at triads, leads to central core myopathy characterized by focal disruption of the SR 8. These findings suggest that MNs and skeletal muscle fibres are selectively vulnerable to alterations of ER structure and function. On a more general note, defects in protein quality control are key pathomechanisms for many forms of MND and many primary myopathies that are linked to altered autophagy and other degradative processes involving the ER.
Alterations in ER proteins and functions have evolved as an important cause of ALS, implicating that the ER is a vulnerable structure in MNs. This coincides with prominent SR alterations in muscle fibres affected by this disease. Because muscle fibres and MNs are mutually interdependent within the motor unit, ER pathology and chaperonopathy evolving simultaneously and complementing each other in the two components of the unit should lead to the selective degeneration of the unit. This way, changes in the SR of affected muscle fibres in ALS could reflect primary SR degeneration in these cells as well as secondary SR changes due to denervation, and both could enhance each other in a vicious circle.
Recent experimental studies support the notion that in certain MNDs, muscle fibre alterations are not just reactive, but causative for neuronal degeneration. For example, the spinobulbar muscular atrophy (SBMA) mouse model expressing mutant androgen receptor (AR) (poly Q expansion) develops a neuromuscular pathology with early muscle, but late spinal cord disease and only minor MND pathology 69. Myopathic changes that were not merely attributed to the denervation process have also been described in muscle biopsies from human SBMA patients 59. In fact, in most animal models of SBMA only minor loss of MN cell bodies occurs 14, 37, 69. This suggests that SBMA is a lower MN degeneration of partial muscle origin. Along the same line, the predominant pathogenic event in the G93A‐SOD1 mouse model of ALS, determining the survival of the animals, is not MN death but rather the loss of muscle to neuron connectivity 27; exercise therapies targeting muscles and the neuromuscular axis as a whole in this model yield protection to MNs 17, 29, 38. Taken together, these data support the concept proposed by others already 9, 20 that in many neuromuscular disorders degeneration of one component of the motor unit, either MN or muscle fibre, leads to degeneration of the other, and that intrinsic muscle fibre defects contribute to the degeneration of the neuromuscular axis in MNDs 9, 16.
To better understand the impact of SR alterations in NMA in general and in ALS in particular, we examined the expression and localization of VAPB and of other, functionally related ER proteins (SigR1, GRP78, HSP27 and HSP70) in normal and denervated mouse and human muscle fibres. The latter were accessible to us due to the availability of muscle biopsies from affected patients, providing the opportunity to study early SR changes during the course of MND in patient tissues.
MATERIALS AND METHODS
Human samples
Human autopsy muscle samples were obtained from the Academic Medical Centre (AMC) Amsterdam, Institute of Neuropathology Brain Bank, following the guidelines of the local Ethics Committee. The intercostal muscles of these clinically confirmed sporadic and familial ALS patients as well as age‐matched controls had been removed within 6–12 h after death. All ALS patients had suffered from clinical signs and symptoms of lower and upper MND with the eventual involvement of brain stem motor nuclei. Importantly, none of these patients had cognitive impairment or dementia. Age‐matched control patients did not show any neuropathological abnormalities. Human muscle biopsies from clinically confirmed sporadic and familial ALS patients, from patients with clinically and histologically confirmed NMAs due to sensorimotor neuropathies including microangiopathic/diabetic neuropathy and neuritis as well as age‐matched control muscle biopsies were obtained from the archives of the Institute of Neuropathology, RWTH Aachen University Hospital, following the guidelines of the Ethics Committee of RWTH Aachen University Hospital. Samples of control and diseased muscles were processed in parallel. In total, muscle samples from 14 ALS and 6 control autopsy cases and muscle biopsies from 11 ALS patients, 13 NMA patients and 6 controls were examined by histology and immunohistochemistry. Muscle samples from 7 ALS patients, 7 NMA patients and 3 controls were examined by immunoblotting.
Experimental animals
All procedures were approved by the RWTH Aachen University Hospital and Würzburg University Medical School Institutional Animal Care and Use Committees. ALS mice expressing high copy numbers of human mutant G93A‐SOD1 were obtained from Jackson Laboratories [Bar Harbor ME; strain designation B6SJL‐Tg (SOD1 G93A) 1Gur/J] 30. Homozygous G93A‐SOD1 mice were maintained by mating G93A males with B6SJL/J hybrid females. Transgenic litters were genotyped using PCR for human SOD1 from tail tissue. Severely affected 18‐week‐old male mice and their corresponding control littermates were used for all experiments (n = 3 each). To ensure optimal quality of SOD1 mutant and control mouse muscles for immunohistochemical investigation, tissues were either fixed by transcardial perfusion with ice‐cold 2% paraformaldehyde (PFA) containing 2% picric acid (pH 7.4) or immediately frozen in liquid nitrogen 51.
Staining and teasing of single muscle fibres
The native gastrocnemius muscle was fixed in 4% PFA for 20 minutes. Subsequently the muscle was teased into single muscle fibres using forceps and washed for 30 minutes with PBS with 0.1 M glycine. α‐Bungarotoxin Alexa Fluor 594 (1:500, Molecular Probes) in 1× PBS was applied for 20 minutes to stain the postsynaptic parts of neuromuscular endplates. After washing for 20 minutes with PBS, the muscle fibres were permeabilized with ice‐cold methanol for 2 minutes at 20°C. The fibres were washed with PBS for 20 minutes and blocked for 1 h with 10% BSA and 0.3% Triton‐X100 in PBS. Rabbit anti‐VAPB (1:100, homemade) and guinea‐pig anti‐Synaptophysin (1:400, Synaptic Systems, Goettingen, Germany) were diluted in the blocking solution and applied overnight at 4°C. Thereafter, the muscle fibres were washed three times for 10 minutes in 1× PBS. Cy2 antiguinea‐pig (1:500, 106‐225‐003; Jackson Immunoresearch, West Grove, PA, USA) and Cy5 goat antirabbit (1:400, 111‐175‐003; Jackson Immunoresearch) as secondary antibodies were applied in the blocking solution for 1 h at room temperature. Finally, the tissue was washed three times in 1× PBS for 10 minutes and mounted with Aqua Poly/Mount (18606; Polysciences, Hirschberg an der Bergstrasse, Germany). Pictures were taken with an Olympus FluoView™ FV1000 microscope.
Immunohistochemistry and histochemistry
We used paraffin sections of formalin‐fixed material for most of the histological analyses. The staining protocols for all antibodies used (Table S3) were optimized to achieve good results with formalin‐fixed, paraffin‐embedded tissue, because this is the most widely available for the human biopsy and autopsy cases. The same treatment was done for most of the mouse tissue to make sure that the results could be compared to the human patient tissue. As controls, we used normal healthy muscles and sections that were processed without primary antibodies.
Transverse muscle paraffin sections were placed on silane‐coated slides, deparaffinized, rehydrated and heated in citrate buffer for antigen retrieval. Processed sections were incubated with primary antibodies for 1 h at room temperature. BrightVision Poly‐HRP secondary antibodies (ready to use, IL immunologic, NL) were applied for 30 minutes, followed by DAB visualization (IL immunologic, NL). Standard histological and histochemical stains including H&E, ATPases, PAS, Oil red O, myoadenylate deaminase, modified Gomori trichrome, nonspecific esterase and NADH were performed on cryostat sections as described 18; consecutive sections that were used for immunohistochemistry (Figures 2A,G and 3A,B) were boiled for 10 minutes in citrate buffer. The histological, histochemical and DAB immunohistochemical sections were photographed using an Axioplan microscope with an Axiocam HR camera (Zeiss, Carl‐Zeiss‐Strasse, Oberkochen, Germany). For immunofluorescence secondary antibodies conjugated with Alexa fluorophore (Invitrogen, Fisher Scientific GmbH, Schwerte, Germany) were used. Staining patterns were visualized using a Zeiss LSM 700 confocal microscope. The resulting confocal images were processed using the Zeiss LSM software and Adobe Photoshop CS5.
Figure 2.
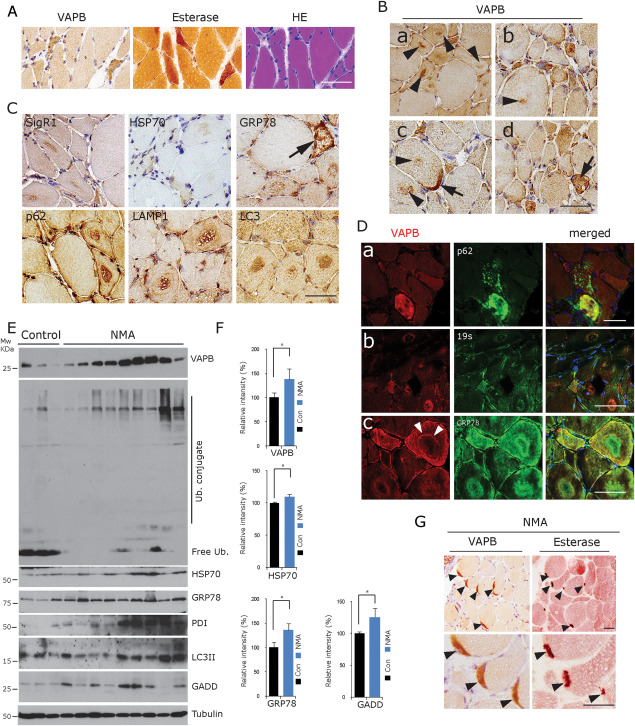
VAPB accumulation in denervated fibres in NMA. A. Accumulation of VAPB in denervated, partially atrophic and atrophic human muscle fibres. They are angular or flattened, grouped and often esterase‐positive. H&E staining, esterase histochemistry and VAPB immunohistochemistry of consecutive human muscle biopsy frozen sections from a representative case from the group of 11 NMA cases available for cryostat sectioning. Scale bar = 100 µm. B. Prominent VAPB staining of target structures in NMA cases [arrowheads in (a–c)]. The centre of the targets and ring‐like structures around targets were VAPB immunoreactive. Strong immunostaining of VAPB at an NMJ [arrow in (c)] and of a muscle fibre showing secondary myopathic alterations [arrow in (d)] (human muscle biopsy, paraffin sections). Scale bar = 50 µm. C. Staining with antibodies against other ER chaperones (SigR1, HSP70 and GRP78) and against autophagy markers (p62, Lamp1 and LC3) reveals accumulations of these proteins in the centre of target structures. Arrow: strongly GRP78‐positive lesioned muscle fibre (human muscle biopsy, paraffin sections). Scale bar = 50 µm. D. Considerable immunofluorescence colabelling of VAPB with p62 (upper panel), 19s proteasome subunit (middle panel) and GRP78 (lower panel) in lesioned fibres, targets and atrophic muscle fibres in NMA. Note the prominent VAPB‐labelling of the rim (layer 3; white arrowheads) of a target in (c), whereas layer 2 of this target is GRP78 immunoreactive (human muscle biopsy, paraffin sections). Scale bar = 50 µm. E. Western blot analysis of frozen tissue from NMA muscle biopsy specimens showing the accumulation of VAPB along with ubiquitin conjugates and with the autophagy marker LC3 and of ER stress markers in nine NMA muscles compared to three normal controls. F. Quantitative densitometric analysis of the findings depicted in (E). *P < 0.05. G. Prominent VAPB immunoreactivity of NMJs (arrowheads) labelled by esterase staining in consecutive sections from a representative case from the group of 11 NMA cases available for cryostat sectioning. Scale bar = 50 µm.
Figure 3.
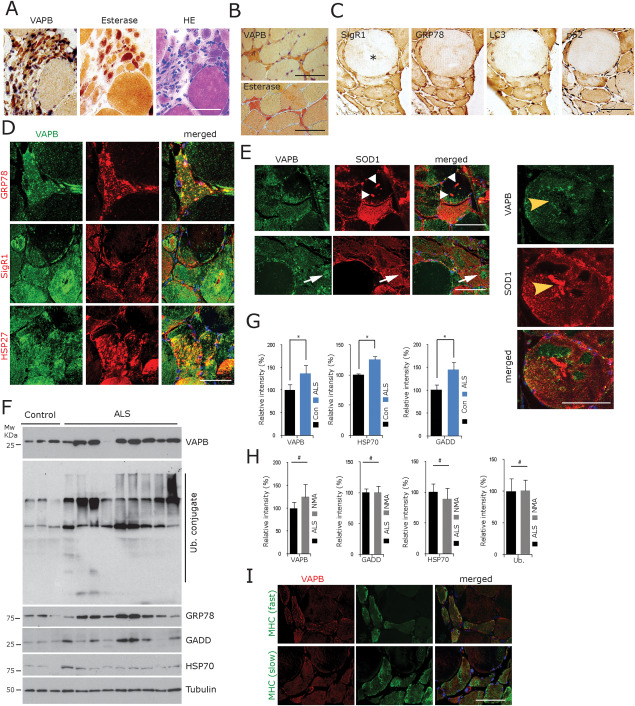
VAPB accumulation in the denervated fibres in human ALS. A. Grouped atrophic, angular or flattened, often esterase‐positive denervated muscle fibres show prominent accumulation of VAPB immunoreactivity; consecutive sections from a representative case from the group of 13 ALS cases available for cryostat sectioning. Scale bar = 100 µm. B. Another example of intense VAPB staining of atrophic, angular or flattened, esterase‐positive muscle fibres in ALS (consecutive cryostat sections). Scale bar = 50 µm. C. Considerable increase in SigR1, GRP78, LC3 and p62 immunoreactivity in the partially atrophic and atrophic fibres compared to a hypertrophic muscle fibre (asterisk) (consecutive paraffin sections). Scale bar = 50 µm. D. Partial colocalization of VAPB, GRP78 and SigR1 immunoreactivity in muscle fibres of representative ALS muscle biopsies (paraffin sections). Scale bar = 100 µm. E. There is some overlap of VAPB immunoreactivity with SOD1 staining, but several strongly SOD1‐immunoreactive structures (white and yellow arrowheads) did not label with the VAPB antiserum. Conversely, a VAPB‐immunoreactive target structure (white arrows) is not colabelled after incubation with the SOD1 antibody (consecutive paraffin sections). Scale bar = 50 µm. F. Western blot analysis of frozen tissue from nine ALS muscle biopsy specimens showing the accumulation of VAPB along with GRP78, GADD and HSP70 compared to three normal controls. G. Quantification of the findings depicted in F for VAPB, GRP78, HSP70 and GADD. *P < 0.05. H. Quantitative comparison of the levels of VAPB, GADD, HSP70 and ubiquitin conjugate normalized to tubulin levels did not reveal any significant difference between the ALS muscles vs. the muscles from patients with NMA due to other causes. I. VAPB immunoreactivity is increased in both fast and slow (labelled with specific MHC isoform antibodies) denervated muscle fibres in ALS (consecutive paraffin sections). Scale bar = 90 µm.
Preparation and staining of frozen sections of Nmd2J mouse gastrocnemius muscle
The gastrocnemius muscle was freshly prepared from Nmd2J mice after cervical dislocation and immediately frozen in isopentane cooled in liquid nitrogen. For immunostaining, 10 μm cryostat sections were blocked with 10% BSA and 0.3% Triton X‐100 in 1× PBS for 1 h and incubated with mouse antidystrophin (1:100, NCL‐DYS1; Leica, Leica Mikrosysteme Vertrieb GmbH, Wetzlar, Germany) or α‐bungarotoxin Alexa Fluor 594 (1:500; Molecular Probes, Fisher Scientific GmbH, Schwerte, Germany), rabbit anti‐VAPB (1:100) and chicken antineurofilament (1:500, AB5539; Millipore, Merck Chemicals GmbH, Darmstadt, Germany) diluted in blocking solution. After washing three times for 10 minutes, Cy3 goat antimouse (1:200, 115‐165‐003; Jackson Immunoresearch) or FITC swine antirabbit (1:40, F0205; Dako, Glostrup, Denmark) or Cy5 goat antichicken (1:400, ab6569‐100, Abcam, Cambridge, UK) as secondary antibodies were applied for 1 h. Finally, the sections were washed three times for 10 minutes, mounted in Aqua Poly/Mount (18606; Polysciences) and examined using an Olympus Fluo View TM FV1000 microscope. The fluorescent nucleic acid stain Hoechst 33258 and vecta‐shield mounting medium with DAPI were from Molecular Probes and Vectors Laboratories (Peterborough, UK), respectively.
Western blot analysis
Frozen biopsy and autopsy muscle tissues were homogenized in lysis buffer (0.5% Triton X‐100 in PBS, 0.5 mM PMSF and complete protease inhibitor mixture; Roche Applied Sciences, Mannheim, Germany). After incubation on ice for 30 minutes, lysates were sonicated briefly. Clear lysates were obtained after brief centrifugation for 5 minutes at 6000 rpm. Protein concentrations were determined using the BCA method (Molecular Probes). Equal amounts of protein were boiled for 5 minutes in 2× SDS‐sample buffer and subjected to 10% or 12% SDS‐PAGE prior to transfer to a polyvinylidenedifluoride (PVDF) membrane. The membranes were blocked in 4% skimmed milk in 0.05% Tween 20/Tris‐buffered saline (TBS‐T) for 30 minutes prior to incubation with primary antibodies at a dilution of 1:1000. The membranes were incubated overnight at 4°C, then washed three times in TBS‐T and incubated for 1 h in appropriate horseradish peroxidase‐conjugated secondary antibody (1:10 000; Thermo Scientific, Schwerte, Germany). Immunoreactive proteins were detected by enhanced chemiluminescence (Thermo Scientific). Densitometric quantification of the band intensity normalized to tubulin levels was performed using Adobe Photoshop CS5.
RESULTS
VAPB immunoreactivity in the SR and selective accumulation at postsynaptic sites of NMJs in normal, innervated muscle
Normal adult human skeletal muscle fibre calibers range from 40 to 70 µm 34, 42, 50, 67. In sections from 6 normal adult human muscle biopsies, staining with antibodies against VAPB, SigR1, GRP78 and HSP27 colocalized with SERCA1 and calsequestrin labelling in these muscle fibres (Figure 1A,B). As expected, and confirming the specificity of the antibodies, the labelling was consistently associated with the sarcomeric patterning. In mice, a similar pattern was also observed after colabelling with a ryanodine receptor antibody (Figure 1C). The labelling was present throughout the SR for all ER proteins tested. VAPB immunoreactivity was prominent at postsynaptic sites of neuromuscular junctions in normal adult human and adult mouse muscle as demonstrated by DAB staining (Figure 1D). This pattern was also observed in immunofluorescence staining for VAPB with synaptophysin, neurofilament protein, dystrophin and alpha‐bungarotoxin labelling (Figure 1G). Myonuclear envelopes were prominently GRP78 immunoreactive in mouse and human muscle, confirming earlier results in rabbit muscle 66; in fact, this specialized ER compartment was much more intensely GRP78 immunoreactive than the intermyofibrillar SR (Figure 1A, arrowheads and inset). In contrast, only mild, if any, preferential staining of the envelope of myonuclei was achieved with the SigR1 and VAPB antibodies (not shown).
Figure 1.
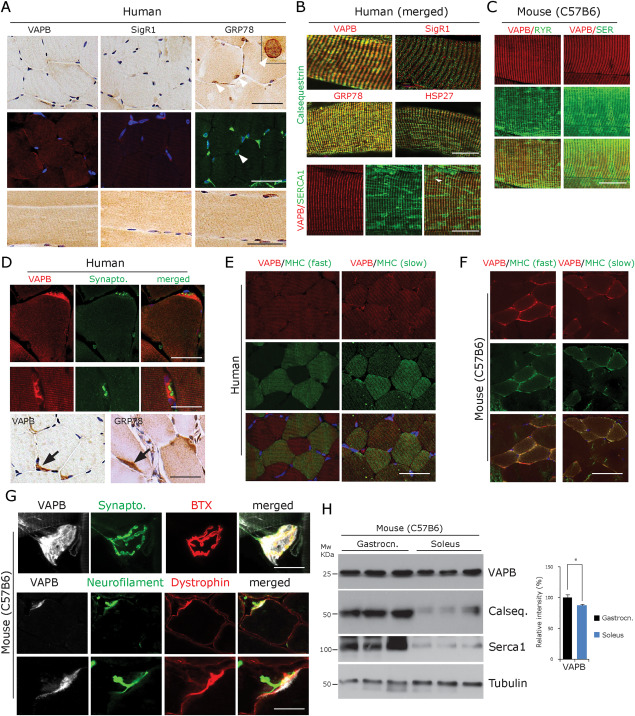
VAPB expression profile in normal innervated human and mouse muscle fibres. A. Human muscle biopsy paraffin sections of normal human control muscles (n = 6, upper and middle row: transverse section, lower row: longitudinal sections) showing a pattern of VAPB, SigR1 and GRP78 immunoreactivity associated with sarcomeric bands after diaminobezidine and immunofluorescence labelling. Note the preferential GRP78 staining of the envelope of myonuclei (arrowheads). One myonucleus is shown at higher magnification in the inset. Scale bar = 40 µm. B. Merged immunofluorescence staining of VAPB, SigR1, GRP78 and HSP27 immunoreactivities associated with sarcomeric bands. Immunofluorescence double‐labelling demonstrates colabelling with calsequestrin (upper and middle panels) and with SERCA1 (lower panel) in normal‐sized, innervated fibres (human muscle biopsy paraffin sections). Scale bar = 30 µm. C. VAPB colocalization with RyR1 (left panel) and with SERCA1 (right panel) in normal‐sized, innervated mouse gastrocnemius muscle fibres. Scale bar = 25 µm. D. Immunofluorescence and DAB staining (lower panel) showing intense VAPB labelling of the postsynaptic sarcoplasm at human NMJs costained for the presynaptic marker synaptophysin. GRP78 immunoreactivity showed a similar pattern (human muscle biopsy, paraffin sections). Scale bar = 30 µm. E. VAPB expression does not differ between slow compared to fast muscle fibres in normal human muscle; immunofluorescence using antibodies against VAPB as well as fibre type‐specific myosin heavy chain isoforms (human muscle biopsy, paraffin sections). Scale bar = 50 µm. F. VAPB is expressed at slightly decreased levels in slow compared to fast muscle fibres in normal mouse muscle; immunofluorescence using antibodies against VAPB as well as fibre type‐specific myosin heavy chain isoforms. Scale bar = 50 µm. G. VAPB immunofluorescence of NMJs of teased normal mouse muscle fibres costained for the presynaptic marker synaptophysin and for the postsynaptic marker alpha‐bungarotoxin (BTX) shows prominent VAPB immunoreactivity. Lower panel: triple labelling with antibodies against VAPB, neurofilament protein and dystrophin shows postsynaptic localization of VAPB immunoreactivity. Scale bar = 40 µm. H. Western blot analysis of VAPB protein demonstrating significantly lower expression levels in soleus vs. gastrocnemius muscles in normal mice (n = 3). There are even lower levels of the Ca2+‐binding proteins calsequestrin and SERCA1 in soleus compared to gastrocnemius muscle. The graph illustrates the results of the quantitative densitometric analysis of the VAPB levels. *P < 0.05.
There was a slightly reduced staining of slow muscle fibres compared to the fast fibres in C57Bl/6 mice (Figure 1F), in line with slightly lower levels of VAPB in the soleus muscle compared to the gastrocnemius muscle detected by immunoblotting (Figure 1H). The soleus muscle is largely composed of type I and IIA fibres, whereas the gastrocnemius muscle mostly contains type IIB and type IID fibres in mice 4, 31, 49. On the other hand, both fast and slow muscle fibres in normal, innervated human muscle fibres labelled by myosin heavy chain (MHC) isoform‐specific antibodies showed similar levels of slight VAPB immunoreactivity (Figure 1E). However, when interpreting these data we need to consider that the division of the fibre subtypes differs between adult human and mouse muscles 4, 31, 49.
VAPB accumulates in the denervated muscle fibres in NMA due to sensorimotor neuropathy
Immunohistochemistry of 11 muscle biopsies form patients with NMA due to sensorimotor neuropathy (see Table S1) revealed diffusely enhanced VAPB staining of partially atrophic (cross‐sectional diameter 20–40 µm), recently denervated, esterase‐positive muscle fibres and of completely atrophic fibres (diameters <20 µm; Figure 2A,D). Completely atrophic muscle fibres with diameters of less than 20 µm that often contained clumps of pyknotic nuclei often showed prominent SR immunoreactivity for VAPB and GRP78 as well as other proteins investigated. Arrangement of the partially atrophic and atrophic fibres in groups, flattened or angular shape and esterase positivity were additional criteria to assume that the fibres showed neurogenic atrophy. Conversely, many target fibres of various sizes, which are thought to be reinnervated 55, 56, 64, were actually immunoreactive for VAPB and other chaperones. Partially, atrophic fibres showing myopathic features such as vacuolization were also prominently VAPB immunoreactive (Figure 2B,D).
Partially atrophic and atrophic muscle fibres also displayed an increased SigR1 labelling; the centre of many targets was moderately SigR1 immunoreactive (Figure 2C). There was moderate HSP70 immunoreactivity of some atrophic fibres and of targets without a predominance of certain target zones. The autophagy markers p62, LC3 and LAMP1 preferentially accumulated in the centre of targets, LAMP1 also in the outer layer [zone 3 (26, 55); Figure 2C)]. Denervated muscle fibre labelling after incubation with antibodies against other proteins of interest showed overlapping patterns, but also differences (Figures 2D and S1). Interestingly, in colabelling studies, our VAPB antiserum and the GRP78 antibody stained different zones of some targets, whereas VAPB and 19s proteasome subunit immunoreactivities were persistently co‐localized within the same target structures (Figures 2D and S1). There is certainly a broad variability in target composition which is probably due to different stages of development and “maturation”; this may also account for the differentiation between “target” and “targetoid” structures 55, 56, 64.
NADH histochemistry depicts the distribution of mitochondria and SR, which differed from the distribution of the chaperone proteins examined by immunohistochemistry (not shown). The latter should more precisely reflect the ongoing remodelling of myofibrillar structures and organelles. Muscle fibres showing secondary myopathic features were labelled with all markers including SigR1, GRP78 and p62 (Figure 2C,D). Results of the semi‐quantitative analysis of the immunohistochemical staining patterns with all antibodies are summarized in Table S2. Significant accumulation of VAPB, ubiquitin conjugates, GRP78, LC3II, HSP70 and the additional ER stress markers PDI and GADD in the NMA biopsies was confirmed by immunoblotting (Figure 2E,F).
Neuromuscular junctions (NMJs) were found in 3/11 NMA biopsies. As in the normal biopsies (see above), these motor end plates showed markedly elevated immunoreactivity for VAPB (Figure 2G) and moderately enhanced SigR1, GRP78, HSP70, HSP27, STIM1 and p62 immunolabelling compared to the extrasynaptic regions (not shown). The junctional staining in these biopsies appeared neither increased nor decreased compared to the immunoreactivity of junctions in innervated muscle. However, as these NMJs could always be assigned to muscle fibres of normal size, they apparently corresponded to innervated junctions. These are still present in most muscles undergoing atrophy due to sensorimotor neuropathy, because the neuropathy is usually slowly progressive and does not lead to simultaneous denervation of all junctions at a given time point.
VAPB and related proteins accumulate in denervated muscle fibres in human ALS muscle
In ALS muscle biopsies the partially atrophic and atrophic fibres showed diffusely elevated VAPB SigR1, GRP78, LC3 and p62 immunoreactivities (Figure 3A–C), as seen in NMA due to sensorimotor neuropathy (see above). Completely atrophic muscle fibres harboring pyknotic nuclei often showed prominent SR immunoreactivity for VAPB and GRP78 as well as other proteins investigated (Figure 3C). However, the levels of VAPB immunoreactivity and esterase positivity showed some variability, with loss of esterase positivity and VAPB immunoreactivity especially of completely atrophic fibres that must have been denervated for longer periods of time. These fibres had probably entered a more progressed, prefinal stage of atrophy. The immunohistochemical results were confirmed by our immunoblot studies using the same antibodies which revealed a significant increase in VAPB, HSP70, GADD and GRP78 protein levels in ALS muscle compared to normal innervated control muscle (Figure 3F,G).
As described by others already, target fibres were less frequent in the ALS patient biopsies compared to the cases of NMA due to sensorimotor neuropathy 11, 57. Irrespective of these differences between denervated muscle in ALS and sensorimotor neuropathy, levels of VAPB, GADD, HSP70 and ubiquitin conjugate as detected by immunoblotting were not significantly different in ALS versus NMA muscle biopsies as judged by densitometric analysis (Figure 3H). There was no apparent difference in the staining intensities of partially atrophic and atrophic type 1 compared to partially atrophic and atrophic type 2 muscle fibres neither in the NMA nor in the ALS muscle biopsies (Figure 3I). Immunofluorescence provided a clearer picture of the subcellular distribution of our proteins of interest in muscle fibres in ALS muscle which underwent secondary myopathic changes. These were strongly immunoreactive for VAPB, SOD1, HSP27, SigR1 and GRP78 (Figures 3E and S2).
VAPB accumulates in denervated muscle fibres in familial ALS (C9orf72, FUS)
Autopsy muscles from six ALS patients harboring pathological C9orf72 hexanucleotide repeat expansions and from two ALS patients with pathogenic FUS gene mutations showed numerous flattened and angular, partially and completely atrophic denervated muscle fibres (Figure 4A,B). In the two FUS mutation cases, many partially atrophic fibres were basophilic (Figure 4B). The atrophic muscle fibres showed increased VAPB, GRP78, HSP27, LAMP1 and STIM1 immunoreactivity (Figure 4C,D) compared to the normal‐sized muscle fibres. These patterns were similar to the ones observed in the sporadic ALS (sALS) and in the non‐ALS NMA biopsy cases. The increased staining was in line with the significant increase in the levels of ubiquitin conjugate, VAPB, SigR1, GRP78, HSP27, p62 and LC3 II detected by immunoblotting (Figure 4E,F). There were no peculiar aggregates or other distinctive additional lesions in the muscle fibres of the C9ORF72 or FUS mutation patients.
Figure 4.
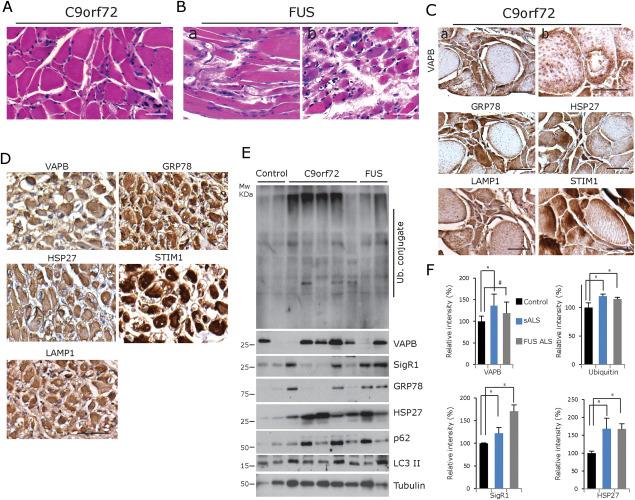
ER chaperones and autophagy markers in autopsy muscle specimens from patients with ALS and C9orf72 repeat expansion or FUS point mutation. A. Groups of angular and flattened, partially atrophic and atrophic muscle fibres of an ALS patient harboring a pathogenic expansion within the C9orf72 gene (paraffin section). Scale bar = 60 µm. B. Prominent grouped muscle fibre atrophy in a case of ALS due to the FUS mutation (a and b). The area depicted in (b) is rich in basophilic fibres. Scale bar = 50 µm. C,D. Strong labelling of the denervated muscle fibres with antibodies against VAPB, GRP78, HSP27, STIM1 and LAMP1 in both the representative C9orf72 ALS case and the representative FUS mutation case (paraffin sections). Scale bar in (C) = 50 µm and (D) = 100 µm. E. Western blot analysis confirms the increase in ubiquitin conjugate, VAPB, SigR1, GRP78, HSP27, p62 and LC3 levels in the C9orf72 and FUS mutation cases; note, however, the considerable variability in the protein levels in the C9orf72 mutation cases. F. Quantification of the results depicted in (E). *P < 0.05.
VAPB accumulates in denervated muscle fibres of G93A‐SOD1 ALS mice
In gastrocnemius muscles of G93A‐SOD1 mice 18 weeks of age (n = 6) marked grouped neurogenic muscle fibre atrophy was present (Figure 5A–D). The partially atrophic and atrophic fibres showed diffusely increased VAPB immunoreactivity compared to the nonatrophic fibres (Figure 5Aa,d). Interestingly, many partially atrophic and also several normal‐sized muscle fibres contained numerous vesicular and granular VAPB‐immunoreactive structures. These were regularly distributed over the cross‐sectional areas of the affected muscle fibres suggestive of an intermyofibrillar localization pattern associated with the SR (Figure 5Ab,c,e,f). Several of the muscle fibres had central nuclei indicative of myopathic alterations (Figure 5Ac,e), and in some of the fibres, larger, irregularly shaped clumps of VAPB‐immunoreactive material were detected (Figure 5Ab,e). We could detect diffuse as well as speckled vesicular and globular SOD1 labelling in partially atrophic and atrophic compared to the normal‐sized muscle fibres (Figure 5B). Using the B8H10 antibody which specifically recognizes misfolded SOD1 protein, we found a consistent pattern of vesicular and globular accumulation of misfolded SOD1in muscle fibres of 14‐week‐old G93ASOD1 mice (Figure 5Cb–d). This pattern of misfolded SOD1 accumulation showed partial overlap with the pattern of VAPB accumulation (Figure 5D).
Figure 5.
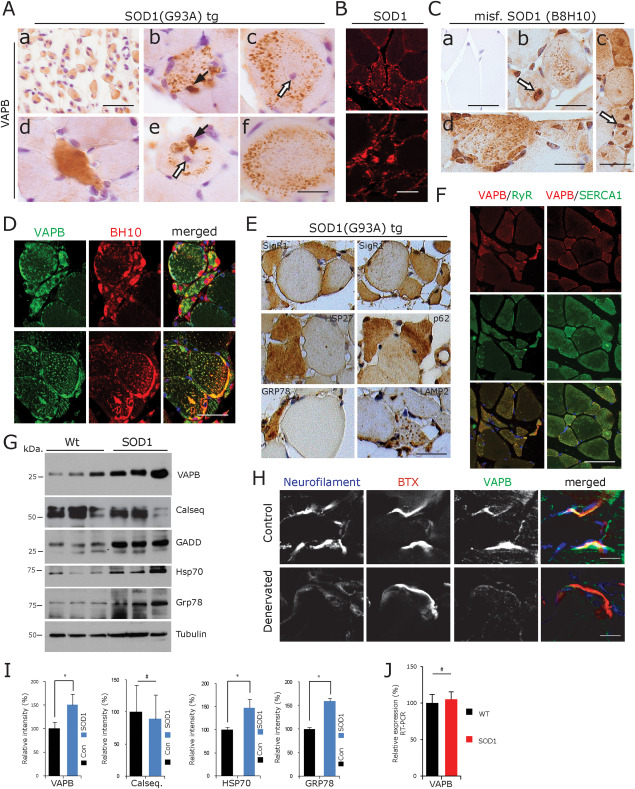
MND mouse model studies. A. VAPB staining of muscle from 18 week old G93A SOD1 mice. a. Large group of atrophic muscle fibres showing moderate diffuse VAPB immunoreactivity. d. Partially atrophic, flattened, recently denervated, strongly VAPB‐immunoreactive muscle fibre. b,c,e,f. Peculiar vesicular or granular VAPB‐immunoreactive structures in normal‐sized muscle fibres, two of which show central nuclei (white arrows) indicating secondary myopathic changes. Black arrows: large globular VAPB‐positive structure (paraffin sections). Scale bar in (a) = 30 µm and in (f) for (b–f) = 20 µm. B. Diffuse as well as speckled vesicular and globular hSOD1 labelling of muscle fibres from an 18‐week‐old G93A SOD1 mouse (SOD1 immunofluorescence). Scale bar = 40 µm. C. Vesicular and globular accumulation of misfolded hSOD1 (B8H10) in gastrocnemius muscle fibres of 14‐week‐old G93ASOD1 mice (b–d), compared to age matched control (a) (paraffin sections). Scale bar in (a,b) = 30 µm, (c) = 50 µm and (d) = 25 µm. D. Partial overlap of misfolded hSOD1 (B8H10) immunoreactivity with the pattern of VAPB immunofluorescence in gastrocnemius muscle fibres of 14‐week‐old G93ASOD1 mice (immunofluorescence, paraffin sections). Scale bar = 30 µm. E. SigR1, HSP27, p62, GRP78 and LAMP2 immunoreactivity of partially and completely atrophic, angular or flattened muscle fibres of 18‐week‐old G93ASOD1 mice. Paraffin sections, scale bar = 50 µm. F. VAPB colocalization with RyR1 (left panel) and with SERCA1 (right panel) in partially and completely atrophic, angular or flattened muscle fibres of 18‐week‐old G93ASOD1 mice (immunofluorescence, paraffin sections). Scale bar = 25 µm. G. Increased levels of VAPB, GADD, HSP70 and GRP78 protein in G93A‐SOD1 mouse gastrocnemius muscle lysates at 18 weeks of age. Note that calsequestrin levels do not differ between wt and SOD1 mouse muscle. H. Denervated motor end plates in 10 days old Nmd2J mice show absence of neurofilament staining and prominent reduction of VAPB immunofluorescence. Scale bar = 15 µm. I. Quantification of the results depicted in (G). *P < 0.05. J. No significant alteration in VAPB mRNA expression in G93ASOD1 mouse muscle compared to wt control revealed by RT‐PCR.
Staining with the SigR1, HSP27, p62, GRP78 and LAMP2 antibodies also yielded an increased labelling of the partially or completely atrophic compared to the normal‐sized fibres (Figure 5E). After incubation with the p62, LAMP1 and GRP78 antibodies (and to a lesser extent also in the SigR1 and HSP27 staining), a granulovesicular staining pattern was found (not shown) which was similar to the one depicted in Figure 5Ab,c,e,f for VAPB. Overall, this pattern was suggestive of altered autophagy in the affected muscle fibres leading to accumulation of these SR‐associated proteins. Interestingly, partially atrophic and atrophic fibres showed increased labelling of RyR1 and SERCA1 together with VAPB (Figure 5F). The increases in VAPB and GRP78 protein levels detected by immunohistochemistry were confirmed by immunoblotting of wt and G93A‐SOD1 mouse gastrocnemius muscle extracts (Figure 5G,I).
RT‐PCR analysis of gastrocnemius muscles from 18‐week‐old wt and G93A‐SOD1 mice (n = 3 each) reveled no significant increase in VAPB mRNA expression (Figure 5J). This result suggests that the increase in VAPB protein levels was probably caused by reduced degradation and was not due to elevated RNA expression.
VAPB in NMD mouse muscle
To examine the pattern of VAPB expression in clearly identifiable denervated neuromuscular junctions in a relevant MND model, we investigated Nmd2J mice, a model for the juvenile form of spinal muscular atrophy with respiratory distress type 1 (SMARD1). These mice exhibit 34% denervated NMJs in the gastrocnemius muscle already at P10. Triple staining with NF, BTX and VAPB in the gastrocnemius muscle at P10 revealed that denervated postsynapses exhibit reduced postsynaptic VAPB protein (Figure 5H).
DISCUSSION
VAPB expression has been described in both MNs and skeletal muscle fibres of various species including human, mouse and drosophila 16, 26, 51, 54. Several functions have been assigned to VAPB including protein quality control and vesicular transport 36, 39. VAPB mutations have been shown to cause familial late onset MND (ALS‐8) in patients, predominantly affecting the second motor neuron 25, 46. Studies on the consequences of mutant VAPB expression in mice have demonstrated only minor, incomplete MN degeneration associated with neuronal accumulations of abnormal VAPB protein and mild neurogenic atrophy 3, 39, 40. Recent findings suggest that gain‐of‐function mutations of the Drosophila VAPB analogue DVAP cause primary myopathic changes 54. Loss of VAPB function was studied by Kabashi et al using zebrafish and mouse models with either a decreased or a complete loss of VAPB expression 35. VAPB knockdown in zebrafish led to swimming deficits, whereas VAPB knockout mice showed mild motor deficits after 18 months of age, yet had innervated NMJs 35. VAPB aggregates were not detected in a recent study on cultured neuronal cells generated from ipS cells derived from ALS8 patient fibroblasts 44, whereas VAPB‐immunoreactive punctate structures were present in cultured fibroblasts from a P56S VAPB mutation patient described by us previously 25, 51. In the following, those experimental data are discussed in the context of our present results on normal and denervated human and mouse muscle.
We found that VAPB staining in normal mouse and human skeletal muscle fibres is associated with the sarcomeric patterning, extending previous observations in fly muscle 54. By colabelling with SERCA, calsequestrin and RyR, we demonstrated that VAPB (and also other ER chaperones such as SigR1) are localized in the SR. We also found that muscle fibres undergoing neurogenic atrophy show higher levels of VAPB and other ER chaperones including SigR1, GRP78 and HSP27 as well as the autophagy proteins LAMP, p62 and LC3. There was no significant difference in immunoreactivities between the ALS patient muscle biopsies vs. the biopsies from patients with NMA due to sensorimotor neuropathies. These findings indicate that VAPB, together with other ER chaperones (see below), is involved in SR remodelling processes and the increased proteasomal/lysosomal activity during atrophy and muscle fibre breakdown 21.
G93A‐SOD1 mice 30, a frequently used ALS model, also demonstrated increased diffuse immunohistochemical staining of VAPB in denervated muscle fibres, along with elevated VAPB levels in immunoblots. Interestingly, additional prominent focal vesicular VAPB staining of non‐atrophic fibres of the affected mouse muscles was found. This pattern is probably indicative of selective early SR changes linked to the perturbed calcium handling in SOD1 mouse muscle fibres 15; it is consistent with the observed accumulation of the ER calcium sensor STIM1 in these fibres described by us recently 26. As mentioned earlier, several studies have already reported evidence supporting a primary muscle fibre pathology in SOD1 mice that presumably contributes to neuromuscular degeneration 13, 19, 68. The prominent and intriguing vesicular changes present in nonatrophic muscle fibres of G93A‐SOD1 mice (Figure 5) could reflect such primary myopathic damage, possibly due to the high level of expression of the human SOD1 transgene under the control of the endogenous SOD1 promoter 30. Consistent with this, we found a peculiar pattern of vesicular and globular accumulation of misfolded SOD1 in muscle fibres of 14‐week‐old G93ASOD1 mice. This pattern of misfolded SOD1 accumulation showed partial overlap with the pattern of VAPB accumulation. Alternatively, the vesicular degeneration might be an early consequence of denervation in those muscle fibres that are yet to undergo atrophy. Still, the G93ASOD1 transgene is not only expressed in alpha MNs, where it leads to aggregation of misfolded SOD1 (10, 52, 70) but also expressed in muscle fibres. Recently, evidence has accumulated that aggregation of misfolded SOD1 protein is also present in alpha‐MNs of human familial and sporadic ALS cases and may play a role in the disease process 10, 53.
Thus it is likely that the mutated SOD1 expressed in the muscle fibres also accumulates in the form of misfolded SOD1 aggregates over time and causes primary myopathic changes. On the other hand, denervation is very rapidly progressive in the G93A‐SOD1 ALS mice we used, even more rapid than the brisk denervation that can be encountered in human ALS muscles and may lead to a considerable secondary myopathy. Therefore, the distinct vesicular VAPB staining pattern in G93A‐SOD1 mouse muscle fibres most likely reflects the combined effects of rapid denervation and of the primary muscle fibre damage due to the expression of the mutant human SOD1 in the muscle fibres.
Like VAPB and STIM1, SigR1 is a transmembrane ER protein; mutations in SigR1 have been shown to cause ALS in human patients 1 and very recently shown to be associated with autosomal recessive distal hereditary motor neuropathy (dHMN) 28, 41. Mutations in other proteins investigated in the context of the present study have also been linked to hereditary neuromuscular diseases: p62/SQSTM1 leads to hereditary ALS/FTLD 63, Lamp‐2 mutation is the cause of Danon disease, a vacuolar myopathy 47, and HSP27 mutations cause an axonal form of Charcot–Marie–Tooth disease, CMT2F 22. In addition, mutation of GRP78 leads to defects in CNS axon growth in mice 23. These diverse mutations thus caused similar pathologies in various neuromuscular disorders, highlighting the selective vulnerability of the ER in neurons and in skeletal muscle fibres. Our results suggest that the same proteins also play a role in muscle fibre remodelling in sALS and other nonhereditary NMAs, providing evidence for yet another connection of ER pathologies in muscle fibres and MNs.
In this context, it is interesting to mention other proteins that are linked to selective damage to both the neuronal and muscle fibre component of the neuromuscular axis. For example, mutations in the ATP‐driven chaperone valosin‐containing protein (VCP) can lead to vacuolar myopathy, ALS, FTLD or combinations thereof 5. Similarly, mutation of the nuclear matrix protein Matrin‐3 may lead to either vacuolar myopathy or ALS 33, 45, 58; recent evidence suggests that Matrin‐3 binds to ER chaperones such as GRP78 48. VCP and Matrin‐3 are thus interesting target proteins to be examined in future studies on mechanisms of muscle fibre degeneration in ALS and other neurogenic atrophies.
Notably, levels of VAPB and of the other ER proteins examined were consistently elevated in two muscle fibre subpopulations in denervated muscle: (i) in the small, angular, recently denervated muscle fibres that were esterase positive in consecutive sections and (ii) in target fibres. The recently denervated fibres are a prominent feature especially of ALS muscle, whereas target fibres were more frequently found in the less rapidly progressive cases of neurogenic atrophy due to sensorimotor neuropathy. Within targets we observed a differential staining of their three zones with the various antibodies. For example, we found accumulation of VAPB immunoreactivity in all zones, whereas SigR1 staining was increased especially in the central zone. This variability probably reflects the differential involvement of the ER chaperones examined in the protein and organelle breakdown as well as SR changes within the targets. These proteins therefore appear to be involved in both acute (progressive atrophy) and chronic (target formation) stages of muscle fibre remodelling in the context of ALS and NMA.
NMJ pathology deserves special attention in the context of diseases that selectively affect the motor unit, because the end plates comprise the decisive contact points between MNs and muscle fibres. More specifically, a role for VAPB in NMJ maintenance is suggested by studies demonstrating altered sizes and numbers of boutons at motor end plates in flies expressing mutated VAPB 12. We found that VAPB immunoreactivity is accumulated considerably at motor end plates in normal human and mouse muscle, along with other ER proteins such as SigR1 and STIM1 26. Double‐labelling studies on mouse muscle confirmed that VAPB is expressed at high levels in the postsynaptic muscle fibre compartment. This selective synapse‐associated immunoreactivity decreased with denervation in Nmd2J mouse muscle (Figure 5H), possibly reflecting ER remodelling at this site. An alternative explanation can be extrapolated from results of studies in flies demonstrating that the VAPB MSP domain is cleaved and secreted by MNs and is internalized in muscle fibres via binding to Eph receptors 65. This constant flow of VAPB MSP domain across the synaptic cleft should cease after denervation, resulting in reduced postsynaptic VAPB immunoreactivity. Because the antiserum that we used for the present studies was raised against a sequence within the MSP domain, we cannot differentiate between these two intriguing alternatives at present.
Taken together, our results suggest that VAPB, SigR1 and other ER proteins causally involved in hereditary neuromuscular disease serve important ER‐associated functions in ALS/NMA muscle. In line with this, SigR1 agonists have already been reported to delay disease progression in the SOD1 ALS mice 43. Similarly, expression of the GRP78 co‐chaperone SIL1 has recently been shown to ameliorate the ALS phenotype in such mice 24. Considering the results of the present study, pharmacological manipulation of these and related proteins should inhibit progression of ER pathology simultaneously in both muscle fibres and MNs. This way, it could ameliorate MND by targeting the disease process at multiple levels.
AUTHOR CONTRIBUTIONS
A.G., C.M.J. and J.W. raised the hypotheses and designed the experiments. C.M.J., E.B., P.T., A.C., C.M.S., C.D., A.Y., A.D. and A.G. performed the experimental work. S.W., M.G., D.T. and J.W. provided ALS and NMA cases. I.K. performed immunoblots and supporting electron microscopy. D.T., S.N. and J.W. examined the ALS and NMA cases. J.A. prepared the autopsy muscle tissues. S.J. and C.B. maintained and prepared the SOD1 transgenic mice. A.G., C.M.J., M.S. and J.W. wrote the article. J.W. supervised the entire project. All authors discussed results and commented on the article.
FUNDING
This work was supported by grants of the Interdisciplinary Centre for Clinical Research (IZKF Aachen) (N1‐1 and N5‐3) and by the German Motor Neuron Disease Network (BMBF‐MND‐Net; Funds 360644) to J.W. and of the German Myopathy Society (DGM) and the Initiative Therapieforschung ALS e.V. to A.G. and J.W.
CONFLICT OF INTEREST STATEMENT
No conflict of interest.
Supporting information
Additional Supporting Information may be found in the online version of this article at the publisher's web‐site:
Figure S1. Patterns of VAPB labelling in targets as well as atrophic and partially atrophic muscle fibres. Paraffin sections in (A–H), immunofluorescence in (A–F) and (H) and DAB staining in (G). A. VAPB may selectively accumulate at the inner margin of layer 2 (a,b) or in the centre (c) of target structures (arrows). Scale bar = 30 µm. B–D. There is partial overlap of the VAPB labelling with immunohistochemical staining for GRP78, SigR1 and HSP27 in targets (arrows). Scale bar = 30 µm. E. VAPB immunoreactivity together with p62 in a target (arrow) and in lesioned muscle fibre (asterisk). Scale bar = 30 µm. F. No costaining of VAPB‐immunoreactive targets (arrows) with the cytochrome c oxidase monoclonal antibody. Scale bar = 50 µm. G. Enhanced SigR1, GRP78, p62 and LC3 immunoreactivity of partially and completely atrophic fibres and of target (arrows) fibres in ALS. Arrowheads: GRP78‐immunoreactive muscle fibre nuclei. Scale bar = 50 µm. H. Strong nuclear envelope labelling for GRP78 in partially denervated muscle fibres in ALS. DAPI counter stain for nuclei. Scale bar = 30 µm.
Figure S2. ER chaperone staining in ALS patient muscle biopsies. Partial overlap of VAPB immunoreactivity with GRP78, SigR1 and, most prominently, HSP27 staining in human ALS patient muscle fibres, some of which are sectioned longitudinally (immunofluorescence, paraffin sections). Scale bar = 20 µm.
Supporting Information
ACKNOWLEDGEMENTS
We are grateful to the patients and their relatives for their support. We thank A. Knischewski, H. Wiederholt, E. Pascual and H. Mader (Institute of Neuropathology, RWTH Aachen University Hospital) and H. Heidtmann (Institute of Physiology, RWTH Aachen University) for technical support. We thank V. Kumar (International Max Planck Research School, Tübingen, Germany) for image analysis and S. Gründer (Institute of Physiology, RWTH Aachen University Hospital) and his lab members for support with the confocal microscopy and generous help in other technical matters.
REFERENCES
- 1. Al‐Saif A, Al‐Mohanna F, Bohlega S (2011) A mutation in sigma‐1 receptor causes juvenile amyotrophic lateral sclerosis. Ann Neurol 70:913–919. [DOI] [PubMed] [Google Scholar]
- 2. Al‐Sarraj S, King A, Cleveland M, Pradat PF, Corse A, Rothstein JD et al (2014) Mitochondrial abnormalities and low grade inflammation are present in the skeletal muscle of a minority of patients with amyotrophic lateral sclerosis; an observational myopathology study. Acta Neuropathol Commun 2:165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Aliaga L, Lai C, Yu J, Chub N, Shim H, Sun L et al (2013) Amyotrophic lateral sclerosis‐related VAPB P56S mutation differentially affects the function and survival of corticospinal and spinal motor neurons. Hum Mol Genet 22:4293–4305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Augusto V, Padovani CR, Campos GER (2004) Skeletal muscle fiber types in C57BL6J mice. Braz J Morphol Sci 21:89–94. [Google Scholar]
- 5. Ayaki T, Ito H, Fukushima H, Inoue T, Kondo T, Ikemoto A et al (2014) Immunoreactivity of valosin‐containing protein in sporadic amyotrophic lateral sclerosis and in a case of its novel mutant. Acta Neuropathol Commun 2:172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bohm J, Chevessier F, Koch C, Peche GA, Mora M, Morandi L et al (2014) Clinical, histological and genetic characterisation of patients with tubular aggregate myopathy caused by mutations in STIM1. J Med Genet 51:824–833. [DOI] [PubMed] [Google Scholar]
- 7. Bohm J, Chevessier F, Maues De Paula A, Koch C, Attarian S, Feger C et al (2013) Constitutive activation of the calcium sensor STIM1 causes tubular‐aggregate myopathy. Am J Hum Genet 92:271–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Boncompagni S, Rossi AE, Micaroni M, Hamilton SL, Dirksen RT, Franzini‐Armstrong C, Protasi F (2009) Characterization and temporal development of cores in a mouse model of malignant hyperthermia. Proc Natl Acad Sci USA 106:21996–22001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Boyer JG, Ferrier A, Kothary R (2013) More than a bystander: the contributions of intrinsic skeletal muscle defects in motor neuron diseases. Front Physiol 4:356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Brotherton TE, Li Y, Cooper D, Gearing M, Julien JP, Rothstein JD et al (2012) Localization of a toxic form of superoxide dismutase 1 protein to pathologically affected tissues in familial ALS. Proc Natl Acad Sci USA 109:5505–5510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Carpenter S, Karpati G (2001) Structures and reactions: denervation. In: Pathology of Skeletal Muscle, 2nd edn., pp. 100–101. Oxford University Press: Oxford, New York. [Google Scholar]
- 12. Chai A, Withers J, Koh YH, Parry K, Bao H, Zhang B et al (2008) hVAPB, the causative gene of a heterogeneous group of motor neuron diseases in humans, is functionally interchangeable with its Drosophila homologue DVAP‐33A at the neuromuscular junction. Hum Mol Genet 17:266–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chen D, Wang Y, Chin ER (2015) Activation of the endoplasmic reticulum stress response in skeletal muscle of G93A*SOD1 amyotrophic lateral sclerosis mice. Front Cell Neurosci 9:170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chevalier‐Larsen ES, O'Brien CJ, Wang H, Jenkins SC, Holder L, Lieberman AP, Merry DE (2004) Castration restores function and neurofilament alterations of aged symptomatic males in a transgenic mouse model of spinal and bulbar muscular atrophy. J Neurosci 24:4778–4786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chin ER, Chen D, Bobyk KD, Mazala DA (2014) Perturbations in intracellular Ca2+ handling in skeletal muscle in the G93A*SOD1 mouse model of amyotrophic lateral sclerosis. Am J Physiol Cell Physiol 307:C1031–C1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. de Oliveira GP, Maximino JR, Maschietto M, Zanoteli E, Puga RD, Lima L et al (2014) Early gene expression changes in skeletal muscle from SOD1(G93A) amyotrophic lateral sclerosis animal model. Cell Mol Neurobiol 34:451–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Deforges S, Branchu J, Biondi O, Grondard C, Pariset C, Lecolle S et al (2009) Motoneuron survival is promoted by specific exercise in a mouse model of amyotrophic lateral sclerosis. J Physiol 587:3561–3572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Dubowitz V, Sewry CA, Oldfors A (2013) Muscle Biopsy. A Practical Approach, 4th edn. Saunders: Philadelphia. [Google Scholar]
- 19. Dupuis L, di Scala F, Rene F, de Tapia M, Oudart H, Pradat PF et al (2003) Up‐regulation of mitochondrial uncoupling protein 3 reveals an early muscular metabolic defect in amyotrophic lateral sclerosis. FASEB J 17:2091–2093. [DOI] [PubMed] [Google Scholar]
- 20. Dupuis L, Echaniz‐Laguna A (2010) Skeletal muscle in motor neuron diseases: therapeutic target and delivery route for potential treatments. Curr Drug Targets 11:1250–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Evans WJ (2010) Skeletal muscle loss: cachexia, sarcopenia, and inactivity. Am J Clin Nutr 91:1123S–1127S. [DOI] [PubMed] [Google Scholar]
- 22. Evgrafov OV, Mersiyanova I, Irobi J, Van Den Bosch L, Dierick I, Leung CL et al (2004) Mutant small heat‐shock protein 27 causes axonal Charcot–Marie–Tooth disease and distal hereditary motor neuropathy. Nat Genet 36:602–606. [DOI] [PubMed] [Google Scholar]
- 23. Favero CB, Henshaw RN, Grimsley‐Myers CM, Shrestha A, Beier DR, Dwyer ND (2013) Mutation of the BiP/GRP78 gene causes axon outgrowth and fasciculation defects in the thalamocortical connections of the mammalian forebrain. J Comp Neurol 521:677–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Filezac de L'Etang A, Maharjan N, Cordeiro Brana M, Ruegsegger C, Rehmann R, Goswami A et al (2015) Marinesco‐Sjogren syndrome protein SIL1 regulates motor neuron subtype‐selective ER stress in ALS. Nat Neurosci 18:227–238. [DOI] [PubMed] [Google Scholar]
- 25. Funke AD, Esser M, Kruttgen A, Weis J, Mitne‐Neto M, Lazar M et al (2010) The p.P56S mutation in the VAPB gene is not due to a single founder: the first European case. Clin Genet 77:302–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Goswami A, Jesse CM, Chandrasekar A, Bushuven E, Vollrath JT, Dreser A et al (2015) Accumulation of STIM1 is associated with the degenerative muscle fibre phenotype in ALS and other neurogenic atrophies. Neuropathol Appl Neurobiol 41:304–318. [DOI] [PubMed] [Google Scholar]
- 27. Gould TW, Buss RR, Vinsant S, Prevette D, Sun W, Knudson CM et al (2006) Complete dissociation of motor neuron death from motor dysfunction by Bax deletion in a mouse model of ALS. J Neurosci 26:8774–8786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gregianin E, Pallafacchina G, Zanin S, Crippa V, Rusmini P, Poletti A et al (2016) Loss‐of‐function mutations in the SIGMAR1 gene cause distal hereditary motor neuropathy by impairing ER‐mitochondria tethering and Ca2+ signaling. Hum Mol Genet 10.1093/ddw220 [DOI] [PubMed] [Google Scholar]
- 29. Grondard C, Biondi O, Armand AS, Lecolle S, Della Gaspera B, Pariset C et al (2005) Regular exercise prolongs survival in a type 2 spinal muscular atrophy model mouse. J Neurosci 25:7615–7622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gurney ME, Pu H, Chiu AY, Dal Canto MC, Polchow CY, Alexander DD et al (1994) Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science 264:1772–1775. [DOI] [PubMed] [Google Scholar]
- 31. Hamalainen N, Pette D (1995) Patterns of myosin isoforms in mammalian skeletal muscle fibres. Microsc Res Tech 30:381–389. [DOI] [PubMed] [Google Scholar]
- 32. Jackman RW, Kandarian SC (2004) The molecular basis of skeletal muscle atrophy. Am J Physiol Cell Physiol 287:C834–C843. [DOI] [PubMed] [Google Scholar]
- 33. Johnson JO, Pioro EP, Boehringer A, Chia R, Feit H, Renton AE et al (2014) Mutations in the Matrin 3 gene cause familial amyotrophic lateral sclerosis. Nat Neurosci 17:664–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Johnson MA, Polgar J, Weightman D, Appleton D (1973) Data on the distribution of fibre types in thirty‐six human muscles. An autopsy study. J Neurol Sci 18:111–129. [DOI] [PubMed] [Google Scholar]
- 35. Kabashi E, El Oussini H, Bercier V, Gros‐Louis F, Valdmanis PN, McDearmid J et al (2013) Investigating the contribution of VAPB/ALS8 loss of function in amyotrophic lateral sclerosis. Hum Mol Genet 22:2350–2360. [DOI] [PubMed] [Google Scholar]
- 36. Kanekura K, Nishimoto I, Aiso S, Matsuoka M (2006) Characterization of amyotrophic lateral sclerosis‐linked P56S mutation of vesicle‐associated membrane protein‐associated protein B (VAPB/ALS8). J Biol Chem 281:30223–30233. [DOI] [PubMed] [Google Scholar]
- 37. Katsuno M, Adachi H, Kume A, Li M, Nakagomi Y, Niwa H et al (2002) Testosterone reduction prevents phenotypic expression in a transgenic mouse model of spinal and bulbar muscular atrophy. Neuron 35:843–854. [DOI] [PubMed] [Google Scholar]
- 38. Kirkinezos IG, Hernandez D, Bradley WG, Moraes CT (2003) Regular exercise is beneficial to a mouse model of amyotrophic lateral sclerosis. Ann Neurol 53:804–807. [DOI] [PubMed] [Google Scholar]
- 39. Kuijpers M, van Dis V, Haasdijk ED, Harterink M, Vocking K, Post JA et al (2013) Amyotrophic lateral sclerosis (ALS)‐associated VAPB‐P56S inclusions represent an ER quality control compartment. Acta Neuropathol Commun 1:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Larroquette F, Seto L, Gaub PL, Kamal B, Wallis D, Lariviere R et al (2015) Vapb/Amyotrophic lateral sclerosis 8 knock‐in mice display slowly progressive motor behavior defects accompanying ER stress and autophagic response. Hum Mol Genet 24:6515–6529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Li X, Hu Z, Liu L, Xie Y, Zhan Y, Zi X et al (2015) A SIGMAR1 splice‐site mutation causes distal hereditary motor neuropathy. Neurology 84:2430–2437. [DOI] [PubMed] [Google Scholar]
- 42. Lie DC, Weis J (1998) GDNF expression is increased in denervated human skeletal muscle. Neurosci Lett 250:87–90. [DOI] [PubMed] [Google Scholar]
- 43. Mancuso R, Olivan S, Rando A, Casas C, Osta R, Navarro X (2012) Sigma‐1R agonist improves motor function and motoneuron survival in ALS mice. Neurotherapeutics 9:814–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Mitne‐Neto M, Ramos CR, Pimenta DC, Luz JS, Nishimura AL, Gonzales FA et al (2007) A mutation in human VAP‐B–MSP domain, present in ALS patients, affects the interaction with other cellular proteins. Protein Expr Purif 55:139–146. [DOI] [PubMed] [Google Scholar]
- 45. Muller TJ, Kraya T, Stoltenburg‐Didinger G, Hanisch F, Kornhuber M, Stoevesandt D et al (2014) Phenotype of matrin‐3‐related distal myopathy in 16 German patients. Ann Neurol 76:669–680. [DOI] [PubMed] [Google Scholar]
- 46. Nishimura AL, Mitne‐Neto M, Silva HC, Richieri‐Costa A, Middleton S, Cascio D et al (2004) A mutation in the vesicle‐trafficking protein VAPB causes late‐onset spinal muscular atrophy and amyotrophic lateral sclerosis. Am J Hum Genet 75:822–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Nishino I (2003) Autophagic vacuolar myopathies. Curr Neurol Neurosci Rep 3:64–69. [DOI] [PubMed] [Google Scholar]
- 48. Osman AM, van Loveren H (2014) Matrin 3 co‐immunoprecipitates with the heat shock proteins glucose‐regulated protein 78 (GRP78), GRP75 and glutathione S‐transferase pi isoform 2 (GSTpi2) in thymoma cells. Biochimie 101:208–214. [DOI] [PubMed] [Google Scholar]
- 49. Pette D, Staron RS (2000) Myosin isoforms, muscle fiber types, and transitions. Microsc Res Tech 50:500–509. [DOI] [PubMed] [Google Scholar]
- 50. Polgar J, Johnson MA, Weightman D, Appleton D (1973) Data on fibre size in thirty‐six human muscles. An autopsy study. J Neurol Sci 19:307–318. [DOI] [PubMed] [Google Scholar]
- 51. Prause J, Goswami A, Katona I, Roos A, Schnizler M, Bushuven E et al (2013) Altered localization, abnormal modification and loss of function of Sigma receptor‐1 in amyotrophic lateral sclerosis. Hum Mol Genet 22:1581–1600. [DOI] [PubMed] [Google Scholar]
- 52. Rakhit R, Robertson J, Vande Velde C, Horne P, Ruth DM, Griffin J et al (2007) An immunological epitope selective for pathological monomer‐misfolded SOD1 in ALS. Nat Med 13:754–759. [DOI] [PubMed] [Google Scholar]
- 53. Ruegsegger C, Maharjan N, Goswami A, Filezac de L'Etang A, Weis J, Troost D et al (2016) Aberrant association of misfolded SOD1 with Na(+)/K(+)ATPase‐alpha3 impairs its activity and contributes to motor neuron vulnerability in ALS. Acta Neuropathol 131:427–451. [DOI] [PubMed] [Google Scholar]
- 54. Sanhueza M, Zechini L, Gillespie T, Pennetta G (2014) Gain‐of‐function mutations in the ALS8 causative gene VAPB have detrimental effects on neurons and muscles. Biol Open 3:59–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Schmitt HP, Volk B (1975) The relationship between target, targetoid, and targetoid/core fibers in severe neurogenic muscular atrophy. J Neurol 210:167–181. [DOI] [PubMed] [Google Scholar]
- 56. Schotland DL (1969) An electron microscopic study of target fibers, target‐like fibers and related abnormalities in human muscle. J Neuropathol Exp Neurol 28:214–228. [PubMed] [Google Scholar]
- 57. Schroder JM (1982) Amyotrophische Lateralsklerose. In: Pathologie Der Muskulatur, pp 643–648, Springer Verlag, Berlin/Heidelberg/New York. [Google Scholar]
- 58. Senderek J, Garvey SM, Krieger M, Guergueltcheva V, Urtizberea A, Roos A et al (2009) Autosomal‐dominant distal myopathy associated with a recurrent missense mutation in the gene encoding the nuclear matrix protein, matrin 3. Am J Hum Genet 84:511–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Soraru G, D'Ascenzo C, Polo A, Palmieri A, Baggio L, Vergani L et al (2008) Spinal and bulbar muscular atrophy: skeletal muscle pathology in male patients and heterozygous females. J Neurol Sci 264:100–105. [DOI] [PubMed] [Google Scholar]
- 60. Stewart CE, Rittweger J (2006) Adaptive processes in skeletal muscle: molecular regulators and genetic influences. J Musculoskelet Neuronal Interact 6:73–86. [PubMed] [Google Scholar]
- 61. Tagashira H, Shinoda Y, Shioda N, Fukunaga K (2014) Methyl pyruvate rescues mitochondrial damage caused by SIGMAR1 mutation related to amyotrophic lateral sclerosis. Biochim Biophys Acta 1840:3320–3334. [DOI] [PubMed] [Google Scholar]
- 62. Telerman‐Toppet N, Coers C (1978) Motor innervation and fiber type pattern in amyotrophic lateral sclerosis and in Charcot‐Marie‐Tooth disease. Muscle Nerve 1:133–139. [DOI] [PubMed] [Google Scholar]
- 63. Teyssou E, Takeda T, Lebon V, Boillee S, Doukoure B, Bataillon G et al (2013) Mutations in SQSTM1 encoding p62 in amyotrophic lateral sclerosis: genetics and neuropathology. Acta Neuropathol 125:511–522. [DOI] [PubMed] [Google Scholar]
- 64. Tomonaga M, Sluga E (1969) Ultrastructure of “target fibers”. Virchows Arch A: Pathol Anat 348:89–104. [PubMed] [Google Scholar]
- 65. Tsuda H, Han SM, Yang Y, Tong C, Lin YQ, Mohan K et al (2008) The amyotrophic lateral sclerosis 8 protein VAPB is cleaved, secreted, and acts as a ligand for Eph receptors. Cell 133:963–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Villa A, Podini P, Nori A, Panzeri MC, Martini A, Meldolesi J, Volpe P (1993) The endoplasmic reticulum‐sarcoplasmic reticulum connection. II. Postnatal differentiation of the sarcoplasmic reticulum in skeletal muscle fibers. Exp Cell Res 209:140–148. [DOI] [PubMed] [Google Scholar]
- 67. Weis J, Lie DC, Ragoss U, Zuchner SL, Schroder JM, Karpati G et al (1998) Increased expression of CNTF receptor alpha in denervated human skeletal muscle. J Neuropathol Exp Neurol 57:850–857. [DOI] [PubMed] [Google Scholar]
- 68. Wong M, Martin LJ (2010) Skeletal muscle‐restricted expression of human SOD1 causes motor neuron degeneration in transgenic mice. Hum Mol Genet 19:2284–2302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Yu Z, Dadgar N, Albertelli M, Gruis K, Jordan C, Robins DM, Lieberman AP (2006) Androgen‐dependent pathology demonstrates myopathic contribution to the Kennedy disease phenotype in a mouse knock‐in model. J Clin Invest 116:2663–2672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Zetterstrom P, Graffmo KS, Andersen PM, Brannstrom T, Marklund SL (2011) Proteins that bind to misfolded mutant superoxide dismutase‐1 in spinal cords from transgenic amyotrophic lateral sclerosis (ALS) model mice. J Biol Chem 286:20130–20136. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting Information may be found in the online version of this article at the publisher's web‐site:
Figure S1. Patterns of VAPB labelling in targets as well as atrophic and partially atrophic muscle fibres. Paraffin sections in (A–H), immunofluorescence in (A–F) and (H) and DAB staining in (G). A. VAPB may selectively accumulate at the inner margin of layer 2 (a,b) or in the centre (c) of target structures (arrows). Scale bar = 30 µm. B–D. There is partial overlap of the VAPB labelling with immunohistochemical staining for GRP78, SigR1 and HSP27 in targets (arrows). Scale bar = 30 µm. E. VAPB immunoreactivity together with p62 in a target (arrow) and in lesioned muscle fibre (asterisk). Scale bar = 30 µm. F. No costaining of VAPB‐immunoreactive targets (arrows) with the cytochrome c oxidase monoclonal antibody. Scale bar = 50 µm. G. Enhanced SigR1, GRP78, p62 and LC3 immunoreactivity of partially and completely atrophic fibres and of target (arrows) fibres in ALS. Arrowheads: GRP78‐immunoreactive muscle fibre nuclei. Scale bar = 50 µm. H. Strong nuclear envelope labelling for GRP78 in partially denervated muscle fibres in ALS. DAPI counter stain for nuclei. Scale bar = 30 µm.
Figure S2. ER chaperone staining in ALS patient muscle biopsies. Partial overlap of VAPB immunoreactivity with GRP78, SigR1 and, most prominently, HSP27 staining in human ALS patient muscle fibres, some of which are sectioned longitudinally (immunofluorescence, paraffin sections). Scale bar = 20 µm.
Supporting Information